

Gβγ regulation of the perinuclear Golgi PI4P pathway and a separate pathway at the PM is required for ET-1–stimulated hypertrophy, and the efficacy of Gβγ inhibition in preventing heart failure may be due, in part, to its blocking both of these pathways.
Abstract
We recently identified a novel GPCR-dependent pathway for regulation of cardiac hypertrophy that depends on Golgi phosphatidylinositol 4-phosphate (PI4P) hydrolysis by a specific isoform of phospholipase C (PLC), PLCε, at the nuclear envelope. How stimuli are transmitted from cell surface GPCRs to activation of perinuclear PLCε is not clear. Here we tested the role of G protein βγ subunits. Gβγ inhibition blocked ET-1–stimulated Golgi PI4P depletion in neonatal and adult ventricular myocytes. Blocking Gβγ at the Golgi inhibited ET-1–dependent PI4P depletion and nuclear PKD activation. Translocation of Gβγ to the Golgi stimulated perinuclear Golgi PI4P depletion and nuclear PKD activation. Finally, blocking Gβγ at the Golgi or PM blocked ET-1–dependent cardiomyocyte hypertrophy. These data indicate that Gβγ regulation of the perinuclear Golgi PI4P pathway and a separate pathway at the PM is required for ET-1–stimulated hypertrophy, and the efficacy of Gβγ inhibition in preventing heart failure maybe due in part to its blocking both these pathways.
INTRODUCTION
G protein–coupled receptors (GPCRs) regulate many aspects of cardiac function, including cardiac contractility and hypertrophic growth. One of the key GPCR-regulated pathways in cardiomyocytes is the phosphoinositide-specific phospholipase C (PI-PLC) signaling pathway, which hydrolyzes plasma membrane (PM) phosphatidylinositol 4,5-bisphosphate (PIP2) to produce inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3-dependent calcium release in the nucleus and DAG-dependent activation of protein kinase D (PKD) are two of the critical signals involved in regulation of cardiac hypertrophic growth (Vega et al., 2004; Wu et al., 2006). Gq has been shown to drive cardiac hypertrophy (Knowlton et al., 1993; D'Angelo et al., 1997) and is known to activate PLCβ (Smrcka et al., 1991; Taylor et al., 1991). Indeed, PLCβ1 has been directly implicated in α1-adrenergic receptor–dependent hypertrophic growth of neonatal rat ventricular myocytes (NRVMs; Filtz et al., 2009).
Our laboratory has identified unique roles for a distinct isoform of PI-PLC, PLCε, in regulation of various aspects of cardiac function (Smrcka et al., 2012). PLCε provides a link between β-adrenergic receptor activation and type 2 ryanodine receptor (Ryr2)–dependent calcium release in the regulation of cardiac contractility (Wang et al., 2005; Oestreich et al., 2007; Oestreich et al., 2009). PLCε is also required for development of the pressure overload model of cardiac hypertrophy in mice and is downstream of multiple receptor-dependent stimuli in isolated ventricular myocytes (Zhang et al., 2011, 2013). Like all PI-PLCs, PLCε hydrolyzes PIP2, but it has distinct regulatory features, in that it is directly activated by small GTPases, including Ras, Rap, and Rho (Smrcka et al., 2012). PLCε can also be activated by G protein βγ subunits, although direct interactions have not been demonstrated (Wing et al., 2001). These regulatory features potentially place it downstream of any signals that can activate these G proteins.
We recently identified a novel mechanism for PLCε-dependent regulation of cardiac hypertrophy (Zhang et al., 2013). PLCε scaffolded at the nuclear envelope in cardiac myocytes by direct binding to muscle-specific A kinase anchoring protein (mAKAP) hydrolyzes perinuclear Golgi phosphatidylinositol 4-phosphate (PI4P) to generate local DAG and regulate nuclear PKD. PKD is a key kinase that phosphorylates histone deacetylase (HDAC) to stimulate hypertrophic gene expression, and deletion of PKD inhibits development of pressure overload and angiotensin II–induced cardiac hypertrophy (Vega et al., 2004; Fielitz et al., 2008). In cardiac myocytes, the Golgi apparatus intimately surrounds the nuclear envelope, and local signals generated at the Golgi in close proximity to the nucleus can regulate nuclear PKD activation. We recently proposed that Golgi-localized perinuclear DAG generated by mAKAP-scaffolded PLCε is necessary to maintain activation of PKD at the nucleus, since DAG is not diffusible from membranes (Zhang et al., 2013). In these studies, treatment of cardiac myocytes with endothelin-1 (ET-1) stimulated the PLCε pathway to regulate nuclear PKD. The question that arises is how the signal is transmitted from PM ET-1A receptors to PLCε at the perinuclear Golgi apparatus. ET-1AR can couple to multiple different G proteins, including G12/13 and Gq, which in turn lead to activation of intracellular signaling proteins such as Ras or Rho that could directly activate PLCε.
A pathway involving PKD activation at the Golgi has been shown to be involved in vesicular transport and secretion in cultured cell lines and involves G protein βγ subunits (Jamora et al., 1999; Diaz Anel and Malhotra, 2005; Campelo and Malhotra, 2012). Some of these studies suggest that Gβγ directly binds and activates PKD, but there is evidence that Gβγ-dependent regulation of PLC activity is required to produce DAG at the Golgi for PKC and PKD activation (Diaz Anel, 2007; Irannejad and Wedegaertner, 2010). The substrate for PLC in this reaction was presumed to be PIP2 but was not defined. Exactly how Gβγ is regulated at the Golgi is unclear, but it has been shown that Gβγ can translocate from the PM to the Golgi to regulate Golgi fission (Chisari et al., 2007; Saini et al., 2007, 2010).
The similarities between the Golgi trafficking and cardiac hypertrophy pathways and the fact that PLCε can be regulated by Gβγ suggests the idea that Gβγ mediates the signaling downstream of ET-1 to regulate PLCε-dependent PI4P hydrolysis, PKD activation, and cardiomyocyte hypertrophy. Blocking Gβγ signaling with either protein- or small molecule–based inhibitors has profound salutary effects on heart failure in vivo and in cardiomyocytes in culture (Koch et al., 1995; Rockman et al., 1998; Casey et al., 2010; Rengo et al., 2011). Here we provide evidence that Gβγ is required for ET-1–dependent perinuclear PI4P hydrolysis, nuclear PKD activation, and NRVM hypertrophy. This indicates that one of the potential mechanisms by which Gβγ blockade inhibits development of heart failure is through inhibition of this novel pathway.
RESULTS
ET-1–dependent perinuclear PI4P hydrolysis relies on Gβγ signaling
As we previously reported, in NRVMs transfected with the PI4P-specific probe green fluorescent protein–four-phosphate adapter protein–pleckstrin homology domain (GFP-FAPP-PH), a perinuclear fluorescent ring is observed by confocal fluorescence microscopy that strongly colocalizes with the Golgi apparatus and is identified with an antibody recognizing a Golgi-specific 58K protein (Figure 1A; Zhang et al., 2013). The localization of PI4P also appears to overlap with mAKAP associated with the nuclear envelope (Figure 1B). Treatment with brefeldin A disrupts the localization of the perinuclear PI4P probe (Figure 1B) and the Golgi-specific 58K protein (Figure 1C) but does not affect the distribution of mAKAP, supporting the idea that PI4P and mAKAP are in distinct but closely associated compartments—Golgi and nuclear envelope—that are not readily distinguished by conventional light microscopy.
FIGURE 1:
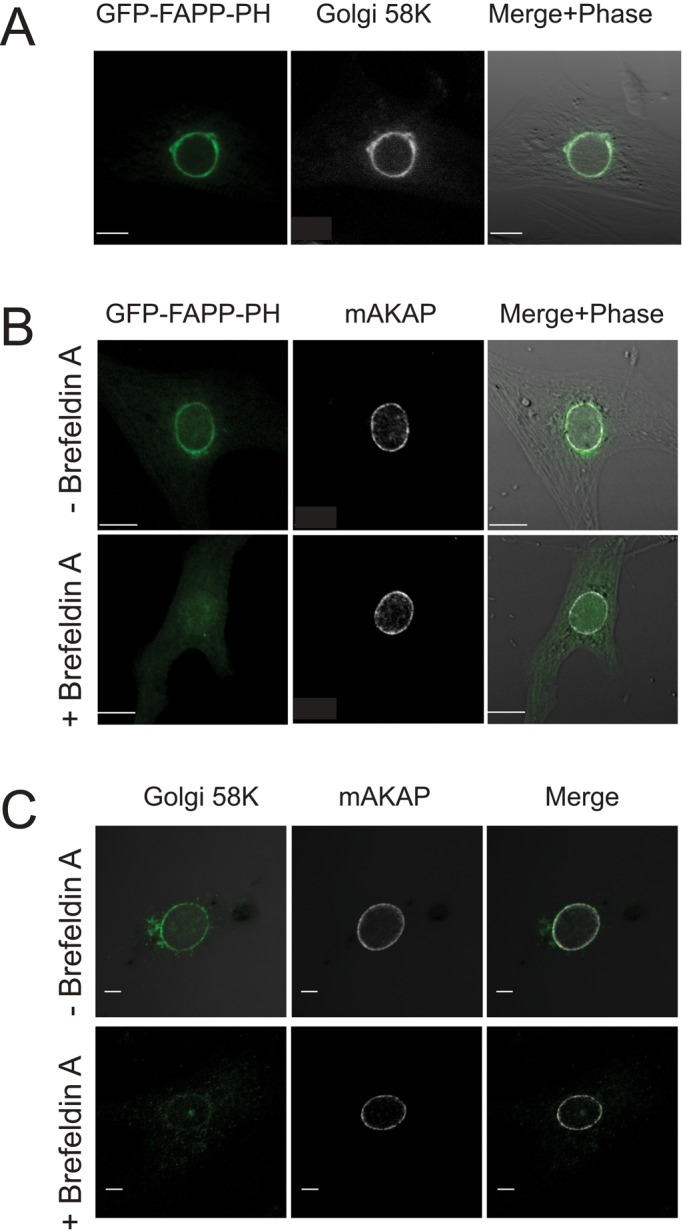
PI4P-associated GFP-FAPP-PH fluorescence colocalizes with a Golgi marker and is closely associated with nuclear envelope–localized mAKAP. (A) NRVMs were transduced with adenovirus expressing GFP-FAPP-PH. Cells were fixed and stained for Golgi-specific 58K protein (Alexa Fluor 546 secondary antibody) and imaged for GFP and red fluorescence. (B) Same as A, except that cells were stained for mAKAP instead of Golgi and treated with and without 10 μM brefeldin A for 20 min as indicated. (C) Cells were treated for the indicated times with brefeldin A, fixed, and costained for mAKAP and Golgi-specific 58K protein. Scale bars, 10 μm.
To determine whether Gβγ is involved in perinuclear Golgi PI4P hydrolysis, we monitored perinuclear fluorescence intensity associated with GFP-FAPP-PH in NRVMs in the presence and absence of ET-1. Stimulation with ET-1 leads to a time-dependent decrease in perinuclear PI4P–associated GFP-FAPP-PH fluorescence, as previously reported (Figure 2A). Pretreatment of NRVMs with 10 μM gallein, a Gβγ blocker (Bonacci et al., 2006), completely inhibited ET-1–dependent decrease in PI4P fluorescence. To control for potential nonspecific effects of gallein, we treated NRVMs with the Epac activator cpTOME to stimulate PLCε. cpTOME binding to Epac leads to activation of Rap, which in turn directly activates PLCε, resulting in perinuclear PI4P depletion (Bos, 2003; Zhang et al., 2013). Because this pathway is independent of Gβγ signaling, gallein would be predicted to have no effect on cpTOME-dependent depletion of perinuclear PI4P. Treatment of NRVMs with cpTOME leads to PI4P depletion, as previously reported, and pretreatment with 10 μM gallein does not alter this response (Figure 2B), indicating relative specificity of gallein for ET-1–dependent PI4P depletion.
FIGURE 2:

Gβγ inhibitors block ET-1–stimulated perinuclear PI4P depletion in NRVMs. (A) NRVMs transfected with GFP-FAPP-PH were pretreated with 10 μM gallein or PBS vehicle control, followed by stimulation with 100 nM ET-1. Perinuclear FAPP fluorescence intensity was measured over time by confocal microscopy. (B) NRVMs were treated as in A, except that 10 μM cpTOME was added to stimulate PI4P hydrolysis. (C) NRVMs were cotransfected with GFP-FAPP-PH and GRK2ct or vehicle control, followed by ET-1 stimulation as in A. (D) NRVMs were transfected as in A were pretreated for 18 h with 100 ng/ml pertussis toxin. All traces represent pooled data from at least three cells from three separate myocyte preparations ± SEM. The last five values from each time course were averaged and tested for significance using Student's t test. ***p < 0.005; ns, not significant.
To provide further evidence for Gβγ signaling controlling perinuclear PI4P hydrolysis, we transfected NRVMs with the C-terminus of GRK2 (GRK2ct), a well-established Gβγ blocker. In cells transfected with GRK2ct, ET-1–dependent PI4P depletion was completely abolished (Figure 2C). Taken together, these data indicate that Gβγ signaling is required for ET-1–dependent stimulation of perinuclear PI4P hydrolysis. Gβγ signaling is often associated with signaling via Gi proteins, but ET-1A receptors couple primarily to Gq. To determine whether Gi proteins were involved in the ET-1–mediated response, we pretreated cells with pertussis toxin (PTX) before ET-1. PTX had no effect on ET-1–stimulated PI4P hydrolysis, indicating that Gi is not involved in this response.
Subcellular requirement for Gβγ-dependent PI4P hydrolysis
ET-1A receptors are present in the sarcolemmal membrane, whereas ET-1B receptors are present on intracellular membranes in ventricular myocytes (Bkaily et al., 2011). Acute stimulation of NRVMs with the ET-1 peptide, however, should stimulate only receptors that are at least initially present on the cell surface. Previous data with the ET1A receptor–selective blocker BQ-123 indicate that ET-1–dependent PI4P depletion is dependent on the ET-1A receptor (Zhang et al., 2013). Thus the question arises of whether Gβγ at the PM or at the Golgi is required for ET-1–dependent PI4P depletion. The Wedegaertner laboratory developed Golgi- and PM-targeted GRK2ct by fusing GRK2ct with the Golgi-targeting sequence KDELrD193N or the 66–amino acid PM-targeting motif of Rit GTPase (Irannejad and Wedegaertner, 2010). These constructs have been extensively characterized to localize to the appropriate compartments in HeLa cells. When transfected into NRVMs, Golgi GRK2 was clearly targeted to the perinuclear Golgi apparatus and colocalized exclusively with a Golgi marker (Figure 3A). PM-targeted GRK2ct was more diffuse, but because of the flat nature of these cells, a clear PM-associated fluorescent ring around the periphery of the cell is difficult to observe (Figure 3B); however, PM-targeted GRK2ct had similar distribution as PM-targeted myristoylated and palmitoylated–yellow fluorescent protein (lyn-YFP), which is used extensively as a PM marker in other cell types (Teruel et al., 1999; Figure 3B). It is also clear that the Golgi-targeted GRK2ct shows no PM association and the PM-targeted GRK2ct shows no Golgi localization, speaking to the specificity of these inhibitors for these compartments. When NRVMs were transfected with PM-targeted GRK2ct, there was no effect on ET-1–stimulated Golgi PI4P hydrolysis (Figure 3, C and E). This is in stark contrast to the data in Figure 2C showing that cytosolic GRK2ct expression blocks ET-1–dependent PI4P depletion, indicating that restriction of GRK2ct to the PM prevents access to the pool of Gβγ that is relevant to stimulation of PI4P hydrolysis. On the other hand, transfection of NRVMs with Golgi-GRK2ct significantly inhibited ET-1–dependent PI4P hydrolysis, indicating that Gβγ signaling is required at the Golgi to stimulate perinuclear PI4P hydrolysis (Figure 3, D and E). Western blots of HEK293 cells or NRVMs transfected with PM or Golgi GRK2ct show similar levels of expression (Supplemental Figure S1).
FIGURE 3:

Gβγ signaling at the Golgi apparatus stimulates perinuclear PI4P depletion in NRVMs and nuclear PKD activation. (A) NRVMs were transfected with Golgi-GRK2ct. GRK2ct localization was analyzed by immunocytochemistry with a GRK2 antibody with an Alexa Fluor 546–conjugated secondary antibody and imaged in the red channel. The Golgi-specific antibody (anti-Golgi 58K protein) was used to identify the Golgi apparatus, with Alexa Fluor 488 secondary antibody imaged in the green channel. (B) Cells cotransfected with PM-GRK2ct, and myristoylated YFP to mark the PM, were analyzed for YFP fluorescence and GRK2ct localization as in A. (C) Cells were cotransfected with GFP-FAPP-PH and either LacZ or PM-GRK2ct constructs. Perinuclear GFP-FAPP-PH fluorescence was monitored with time after ET-1 addition. (D) Same as C, except that Golgi-targeted GRK2ct was used. (E) Quantitation of data in C and D. Curves in C and D were fitted with single-exponential decay curves using GraphPad Prism 6 and analyzed for statistical significance. *p < 0.05 and ****p < 0.001 relative to ET-1 control. (F) NRVMs were cotransfected with nDKAR and LacZ, PM-GRK2ct, or Golgi-GRK2ct, and the nDKAR YFP/CFP ratio in the nucleus was monitored over time after addition of ET-1. (G) Changes in YFP/CFP ratio, normalized to time 0, ± SEM for each trace in F were pooled from 35 to 40 min, averaged, and analyzed by a one-way analysis of variance (ANOVA). (H) Curves from F were fitted and analyzed as in E. All traces (C, D, F, G) are pooled data from at least three cells from three separate myocyte preparations ± SEM. Scale bars, 10 μm.
We proposed that the role of perinuclear PI4P hydrolysis is to generate local DAG to maintain activation of a nuclear pool of PKD. PKD is then involved in phosphorylating HDAC and activation of hypertrophic gene expression. Because Golgi-GRK2ct blocked ET-1–dependent PI4P hydrolysis, we predicted that Golgi-GRK2ct would inhibit activation of the nuclear pool of PKD. NRVMs were cotransfected with a fluorescence resonance energy transfer (FRET)–based reporter of PKD activity that is specifically targeted to the nucleus, nDKAR (Kunkel et al., 2007), with either Golgi- or PM-targeted GRK2ct. ET-1 treatment of NRVMs led to a decrease in nDKAR FRET (indicative of phosphorylation by nuclear PKD), which was completely inhibited by cotransfection with Golgi-targeted GRK2ct (Figure 3, F–H). Targeting GRK2ct to the PM partially inhibited nuclear PKD activation (Figure 3, F–H). These data indicate that Gβγ at the perinuclear Golgi in cardiac myocytes is required for nuclear PKD activation and that Gβγ activation at the PM is also partially required for nuclear PKD activation.
Rapamycin-induced γ translocation to the Golgi induces PI4P hydrolysis and PKD activation
As an alternate approach to examining the role of Gβγ in different compartments, we used a rapamycin-inducible translocation system to specifically direct Gβγ to either the PM or the Golgi (Irannejad and Wedegaertner, 2010). In this system, FRB is fused to the C-terminus of the Gγ subunit and FKB is fused to either amino acids 1–11 of Lyn for PM targeting or KDELr-D193N for Golgi targeting. To examine the effects of Gβγ targeting on perinuclear PI4P hydrolysis, we cotransfected NRVMs with Gβ1γ2-FRB, GFP-FAPP-PH, and either PM or Golgi FKBP. Rapamycin-inducible translocation of Gβ1γ2-FRB to either PM (with Lyn-FKBP–cyan fluorescent protein [CFP]) or Golgi (KDELr-D193N-FKBP-CFP) has been demonstrated in HeLa cells (Irannejad and Wedegaertner, 2010). Figure 4A, top, shows that GFP-Gβ becomes enriched in the perinuclear Golgi with rapamycin treatment. Figure 4A, bottom, shows localization of Golgi-targeted FKBP before and after 20 min of rapamycin treatment. PM-dependent translocation was more difficult to observe, perhaps because of the diffuse PM localization observed in NRVMs (unpublished data; see Figure 2B). For measurement of PI4P depletion, 24 h after cotransfection of FRB and FKBP constructs, rapamycin was added, and perinuclear PI4P-associated GFP-FAPP-PH fluorescence was monitored. Targeting Gβ1γ-FRB to the Golgi produced a strong stimulation of PI4P hydrolysis, whereas PM targeting of Gβγ had no effect (Figure 4B). In other cell types, forced translocation of Gβγ to the Golgi caused Golgi breakdown and vesiculation that could affect apparent PI4P-associated GFP-FAPP-PH fluorescence (Jamora et al., 1999; Irannejad and Wedegaertner, 2010). As shown in Figure 4A, rapamycin-induced translocation to the Golgi in NRVMs does not affect overall Golgi structure, since the Golgi-targeted KDELr-D193N-FKBP-CFP localization and associated morphology are not altered by rapamycin treatment under conditions in which PI4P fluorescence is dramatically decreased.
FIGURE 4:

Targeting Gβγ to the Golgi apparatus stimulates PI4P hydrolysis and nuclear PKD activation. (A) Cells were transfected with mCherry-Gβ1, Gγ2FRB, and CFP-Golgi-FKBP. mCherry fluorescence (top two) and CFP fluorescence (bottom two) before and 10 min after rapamycin addition. (B) NRVMs were transfected with GFP-FAPP-PH, Gβ1, Gγ2FRB, and either CFP-Golgi-FKBP or PM-FKBP. At time 0, 24 h after transfection, 10 μM rapamycin was added, and perinuclear PI4P fluorescence was monitored with time. (C) Cells were transfected with Gβ1, Gγ2FRB, and nDKAR and either PM-FKBP or Golgi FKBP. At time 0, 24 h after transfection, 10 μM rapamycin was added, and nDKAR YFP/CFP fluorescence in the nucleus was monitored. All traces represent pooled data from at least three cells from three separate myocyte preparations ± SEM. Scale bars, 10 μm.
Targeting of Gβ1γ-FRB to the Golgi, but not the PM, also stimulated nuclear PKD activation (Figure 4C). Rapamycin alone had no effect on PI4P hydrolysis or nuclear PKD activation (unpublished data). Although rapamycin dependent recruitment of Gβγ to the PM is not sufficient to activate nuclear PKD, it is possible that rapamycin-dependent recruitment of Gβγ to the PM may not be capable of generating Gβγ signaling at the PM. Together these data indicate that targeting Gβγ to the Golgi is sufficient to stimulate perinuclear PI4P hydrolysis and PKD activation.
Golgi-targeted GRK2ct blocks ET-1–stimulated cardiomyocyte hypertrophy
Overexpression of GRK2ct in cardiac myocytes has been well documented to block development of cardiomyocyte hypertrophy in vitro and in vivo (Koch et al., 1995; Rockman et al., 1998; Rengo et al., 2011). We showed that mAKAP-scaffolded PLCε hydrolyzes perinuclear PI4P at the perinuclear Golgi apparatus in cardiac myocytes, and this is required for activation of nuclear PKD and cardiomyocyte growth. The data presented thus far indicate that Gβγ can mediate activation of this hypertrophic pathway at the Golgi. This suggests the possibility that one of the mechanisms for the antihypertrophic actions of GRK2ct is to block the Golgi perinuclear PLCε/PI4P/PKD pathway. To test this, we transfected NRVMs with plasmids expressing YFP alone or YFP plus GRK2ct, Golgi GRK2ct, or PM-GRK2ct. Twenty-four hours after infection, cells were treated with ET-1 for 24 h to induce hypertrophy (Figure 5). Because transfection efficiency of NRVMs was low, fluorescent cells were identified and hypertrophy was assessed by staining for increases in intracellular atrial natriuretic factor (ANF) levels with an anti-ANF antibody. ET-1 stimulated a robust increase in the level of ANF staining (Figure 5). In Figure 5A, the GRK2ct-transfected cell (green) shows no ET-1–induced ANF staining (red), whereas a neighboring untransfected cell shows robust ET-1–dependent ANF induction. Figure 5B shows representative images of cells transfected with each of the constructs, and Figure 5C shows quantitation of these data. Surprisingly, expression of either Golgi-GRK2ct or PM-GRK2ct prevented ET-1–dependent ANF induction. As another measure of hypertrophy, the increase in cell size associated with ET-1–stimulated hypertrophy was assessed after adenoviral transduction of NRVMs with YFP, PM-GRK2ct, or Golgi-GRK2ct. Both Golgi- and PM-targeted GRK2ct blocked the ET-1–stimulated increase in cell size (Figure 6).
FIGURE 5:

Blocking Gβγ signaling in the Golgi or PM prevents ET-1–stimulated hypertrophic gene expression in NRVMs. (A) NRVMs were cotransfected with YFP and GRK2ct, followed by treatment with 100 nM ET-1 or vehicle. After 24 h, cells were fixed and stained for ANF (red). With ET-1 treatment, the intensity of red staining increases in untransfected cells but not in cells expressing YFP and GRK2ct. Scale bar, 40 μm. (B) Cells were transfected with YFP alone or YFP plus GRK2ct, Golgi GRK2ct, or PM-GRK2ct. Cells were treated as in A. Scale bar, 20 μm. (C) The intensity of ANF staining was quantitated with and without ET-1 only in transfected cells identified by YFP fluorescence. Quantitation is based on combined data (mean ±SEM) from four separate experiments. ****p < 0.0001, statistically different from YFP control. None of the other samples is statistically different from YFP control (one-way ANOVA).
FIGURE 6:
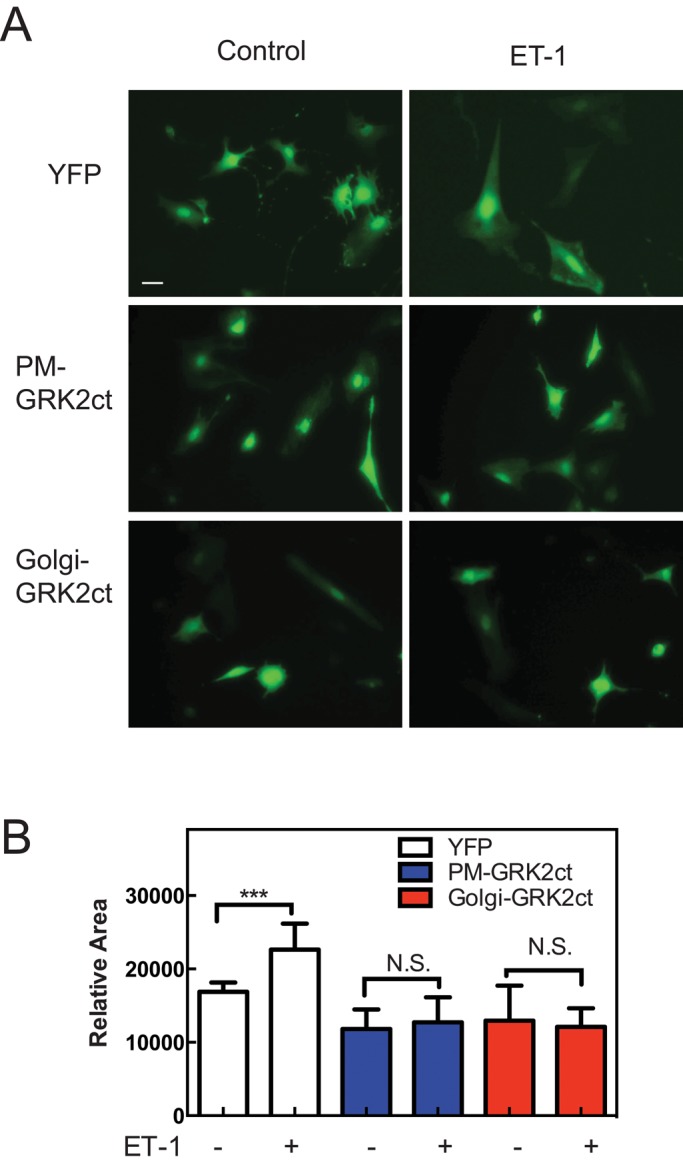
Blocking Gβγ signaling in the Golgi or PM prevents ET-1–stimulated cardiomyocyte hypertrophic cell growth. (A) NRVMs were infected with adenoviruses expressing YFP or YFP and the indicated targeted GRK2ct constructs. Cells were treated with 100 nM ET-1 or vehicle. After 48 h, cells were visualized in the YFP channel. Scale bar, 40 μm. (B) Cells treated as in A quantitated for cell area using ImageJ (National Institutes of Health, Bethesda, MD). Quantitation is based on combined data (mean ±SEM) from three separate experiments; 50 cells each experiment. Data were analyzed by one-way ANOVA; ***p < 0.005.
Neonatal cardiac myocytes are a standard model for examining signaling pathways associated with cardiac hypertrophy, but a more physiological model is the adult ventricular myocyte (AVM). AVMs were isolated from C57Bl6 mice and infected with adenoviruses expressing a PIP2 detector, tubby-GFP, or a PI4P detector, FAPP-PH-GFP, as was done previously for NRVMs. Cells transfected with Tubby GFP showed localized PIP2-associated fluorescence at the sarcolemma (Figure 7A, top), whereas FAPP-PH-GFP detected PI4P at the perinuclear Golgi (Figure 7A, bottom). Some FAPP-PH-GFP fluorescence was also observed in a pattern corresponding to Z-lines or T-tubules (Figure 7A, bottom). Thus, as in NRVMs, PIP2 is enriched at the PM in AVMs and is not observable at the perinuclear Golgi, indicating that the available PLC substrate at the Golgi is PI4P. We tested whether ET-1 treatment would lead to depletion of perinuclear PI4P as seen with NRVMs. ET-1 stimulated a time-dependent decrease in perinuclear PI4P-associated fluorescence that was inhibited by treatment with gallein (Figure 7B).
FIGURE 7:

Blocking Gβγ signaling in the Golgi or PM prevents ET-1–stimulated ANF expression in AVMs. (A) AVMs were infected with adenoviruses expressing either Tubby-GFP (top) or GFP-FAPP-PH (bottom). (B) AVMs were infected with GFP-FAPP-PH and stimulated with 100 nM ET-1, and perinuclear GFP-FAPP-PH was monitored over time as indicated. Each trace represents pooled data from at least five cells from five separate myocyte preparations ± SEM. (C) AVMs were infected with viruses (50 MOI) expressing YFP, Golgi-GRK2ct, or PM-GRK2ct (only cells expressing YFP or Golgi-GRK2ct are shown). Viruses expressing Golgi-GRK2ct and PM-GRK2ct also expressed YFP from a separate cassette. Cells were stimulated for 24 h with 100 nM ET-1 and fixed and stained for ANF expression. (D) ANF expression was quantitated as mean ± SEM for all treatments as described in Materials and Methods. ****p < 0.0001, statistically different from YFP control. None of the other samples is statistically different from YFP control (one-way ANOVA). Scale bars, 10 μm.
To determine whether Golgi-GRK2ct can block ET-1–stimulated hypertrophy in AVMs, we infected cells with adenoviruses expressing either PM- or Golgi-targeted GRK2ct, followed by treatment with ET-1 for 24 h. Hypertrophy was assessed by ANF staining. As in the NRVMs, stimulation with ET-1 caused a robust increase in ANF expression that was inhibited by expression of either PM- or Golgi-targeted GRK2ct (Figure 7, C and D).
Because either Golgi or PM GRK2ct can block hypertrophy, Gβγ-mediated pathways at the PM or the Golgi are both required for cardiomyocyte hypertrophy. Golgi-targeted GRK2ct would not be expected to influence PM GPCR desensitization mechanisms such as GRK2 recruitment and instead involve inhibition of the hypertrophic Golgi PLCε/PI4P signaling pathway. PM-targeted GRK2ct has the potential block the Golgi PLCε/PI4P pathway by preventing Gβγ translocation from the PM to the Golgi, but the data in Figure 2C indicate that PM-GRK2ct has no effect on Golgi PI4P hydrolysis. This indicates that two independent PM and Golgi Gβγ-mediated pathways are required for hypertrophy development in cardiac myocytes.
DISCUSSION
Expression of GRK2ct in cardiomyocytes to block Gβγ signaling inhibits development of hypertrophy in vivo and in vitro (Koch et al., 1995; Rockman et al., 1998; Casey et al., 2010). Our laboratory has developed small molecule–based inhibitors of Gβγ that block interactions between Gβγ and various downstream protein targets, including GRK2 (Bonacci et al., 2006). Gallein administration prevents development of heart failure in mice in several different models of hypertrophy (Casey et al., 2010). A model for the action of gallein and GRK2ct involves inhibition of GRK2 recruitment by Gβγ downstream of the βAR. On the other hand, GRK2ct and gallein/M119 are general Gβγ inhibitors that block multiple signaling pathways. This raises the possibility that the efficacy of these reagents in prevention of heart failure may rest, at least in part, in their ability to inhibit other signaling pathways downstream of Gβγ. Indeed, pathways involving Gβγ-dependent regulation of ERK phosphorylation (Lorenz et al., 2009) and phosphoinositide 3-kinase γ (PI3Kγ) recruitment (Naga Prasad et al., 2000; Nienaber et al., 2003; Prasad et al., 2003) have potential involvement in regulation of cardiac hypertrophy.
Here we identified another Gβγ pathway that is required for cardiac hypertrophy, which we propose involves Gβγ-dependent activation of PLCε at the perinuclear Golgi, downstream of the Gq- and G13-coupled ET-1A receptor. This proposal is based on the following observations: 1) Two different blockers of hypertrophy in vivo and in vitro—gallein and GRK2ct—block ET–1-dependent activation of Golgi PI4P hydrolysis in cardiac myocytes. 2) ET-1–dependent PI4P depletion at the Golgi and subsequent PKD activation requires PLCε (Zhang et al., 2013) and is blocked by a Golgi-targeted Gβγ blocker (Golgi-GRK2ct). 3) Gβγ is a known upstream regulator of PLCε (Wing et al., 2001). 4) PLCε is required for development of cardiac hypertrophy in vivo and in vitro (Zhang et al., 2011, 2013). Although it is formally possible that blockade of Gβγ signaling at the Golgi inhibits hypertrophy development through a parallel pathway that does not involve regulation of PLC, the simplest model that can account for the data involves activation of PLCε by Gβγ at the Golgi.
We also show that expression of either PM- or Golgi-targeted GRK2ct inhibits development of cardiac hypertrophy but that PM targeting of GRK2ct does not affect perinuclear PI4P hydrolysis. This indicates that there are independent roles for Gβγ at the PM and the Golgi and that requirement for Gβγ at the PM is not to regulate the Golgi PI4P hydrolysis pathway. We showed that the PLC/PI4P hydrolysis pathway at the Golgi is required for ET-1–dependent hypertrophy but not that it is sufficient to drive hypertrophy. We propose that a second parallel signal is dependent on Gβγ activity at the PM and is also required for stimulation of hypertrophy. This could involve GRK2, PLC, or an undefined effector at the PM.
A major open question is how ET-1, through activation of ET-1AR at the PM, leads to Gβγ signaling at the perinuclear Golgi apparatus. One possibility is diffusion-based translocation of Gβγ from the PM to the Golgi. Translocation of fluorescently labeled G protein βγ subunits from the PM to the Golgi was demonstrated (Saini et al., 2007). This was proposed to rely on dissociation of Gα from Gβγ, where modification of both Gα and Gγ with lipids is sufficient to maintain PM localization of a Gαβγ heterotrimer. The model suggests that when these subunits dissociate, the single lipid modification on the γ subunit is not sufficient to retain the Gβγ complex at the PM, and a new equilibrium is established in which Gβγ can equilibrate to internal membranes such as the Golgi (O'Neill et al., 2012). Alternatively, transport of Gβγ in endosomes with or without internalization of cell surface ET-1A receptors could result in accumulation of free Gβγ in the Golgi. Finally, it is possible that activation of ET1A signaling at the PM could generate a signal at the PM that could diffuse to the Golgi to activate heterotrimers resident at the Golgi. A possible candidate for this might be an activator of G protein signaling (AGS) protein that can stimulate Gβγ release from Gα subunits in a receptor-independent manner (Blumer et al., 2007). It was recently that AGS3 can translocate from the PM to the Golgi in response to receptor activation (Oner et al., 2013). Defining which, if any, of these mechanisms mediates this signal requires further investigation.
These observations have clinical significance with respect to design of inhibitors to inhibit development of heart failure. Recent efforts have focused on development of inhibitors of GRK2 that improve cardiac performance and may prevent development of heart failure (Thal et al., 2011, 2012). The data presented here suggest that although directly targeting GRK2 may be effective, a strategy that targets Gβγ may have greater efficacy due to its ability to block multiple hypertrophic signaling pathways. The list of Gβγ-dependent pathways that regulate cardiac hypertrophy continues to grow and include PI3Kγ (Prasad et al., 2003; Perrino et al., 2006) and ERK activation (Lorenz et al., 2009; Ruppert et al., 2013), to which we now add Gβγ-regulated PLC activity at the Golgi. We also observed that Gβγ inhibition at the PM inhibits hypertrophy in NRVMs but have not characterized the underlying signaling mechanism. As new therapeutic approaches to heart failure are considered, a global Gβγ inhibition strategy might have high efficacy due to multiple target inhibition, but there may be advantages to targeting Gβγ signaling at specific cellular locations such as the Golgi apparatus that could avoid potential side effects of global Gβγ inhibition or even Gβγ signaling at the PM. This remains to be explored in more-physiological animal models of heart failure.
MATERIALS AND METHODS
Materials
ET-1 was purchased from Sigma-Aldrich (St. Louis, MO). Gallein was from TCI America (Portland, OR), dissolved in phosphate-buffered saline (PBS), and titrated to pH 7.8 with NaOH to create a 15 mg/ml stock that was stored in aliquots at −20°C. Rapamycin and cpTOME were from EMD Millipore (Billerica, MA).
Adenoviral constructs and plasmids
The EGFP-FAPP-PH domain (N-terminal EGFP fusion) for detection of PI4P was as previously described (Balla et al., 2005, 2009). GFP-Tubby (Santagata et al., 2001; Balla et al., 2009) and GFP-PLCδ-PH (Balla and Varnai, 2002) for detection of PIP2 were also previously described. nDKAR (Kunkel et al., 2007) was provided by Alexandra Newton (University of California, San Diego, CA). nDKAR was inserted into an adenovirus vector by standard methods. Gγ2-C68S-FRB, PM-FKBP (LF2C, Lyn1-11-FKBP(X2)-CFP), and Golgi-FKBP (KDELr-D193N-FKBP(x2)-CFP), Golgi-GRK2ct (KEDLrD193N-GRK2(495-689)), and PM-GRK2ct (GRK2(495-689)-Rit) were constructed as previously described (Irannejad and Wedegaertner, 2010). Adenoviruses were constructed using a vector expressing YFP from one promoter and PM-GRK2ct, Golgi-GRK2ct, or GFP-Tubby from a second cytomegalovirus promoter.
Antisera
Anti-GRK2 was from Santa Cruz Biotechnology (Dallas, TX; 1:100); rabbit polyclonal affinity-purified anti-ANF was from EMD Millipore. Golgi protein, 58K protein (1:250), monoclonal antibody (58K-9) was from Thermo Fisher Scientific (Waltham, MA). mAKAP antibody was kindly provided by Michael Kapiloff (University of Miami School of Medicine, Miami, FL).
nDKAR FRET
NRVMs were transduced with an nDKAR-expressing adenovirus. After 48 h, FRET was analyzed by confocal microscopy on an Olympus FV1000MP microscope. Transduced cells were identified by exciting CFP at 440 nm and emission monitored at 480 nm. FRET was determined by measuring the ratio of YFP emission at 535 nm to CFP emission at 480 nm after CFP excitation at 440 nm. Because the YFP and CFP are fused in this construct, it is not necessary to correct for expression or spectral bleedthrough of the various constructs. Data were normalized to the initial YFP/CFP ratio in each set at an arbitrary value of 1.
Isolation of cardiac myocytes
NRVMs were isolated from 2- to 3-d-old Sprague Dawley rats and cultured essentially as described (Zhang et al., 2011). Adult myocytes were isolated from 4- to 6-mo-old wild-type mice as previously described (Oestreich et al., 2009).
Transfection and transduction of NRVMs and AVMs
NRVMs cultured for 2 d were transfected with 1–2 μg of the indicated plasmid constructs or transduced with 50 multiplicity of infection (MOI) of the indicated viruses. Adenoviral transduction efficiency was >90%, and plasmid-based transfection efficiency was ∼5%. Cells were examined 24 h later by microscopy or as indicated for measurement of nDKAR FRET or GFP-FAPP-PH fluorescence.
Confocal microscopy
Myocytes transfected with plasmids or transduced adenoviruses expressing enhanced GFP (EGFP)–, YFP-, or mCherry-tagged proteins were analyzed in an Olympus FV1000MP microscope in confocal mode with a LUMPLFL 40×/0.8 numerical aperture W (Olympus, Tokyo, Japan) lens. EGFP was excited at 488 nm and emission monitored at 510 nm, YFP was excited at 515 nm and emission monitored at 527 nm, mCherry and monomeric red fluorescent protein were excited at 559 nm and emission monitored at 618 nm, CFP was excited at 440 and emission monitored at 476 nm, and 4′,6-diamidino-2-phenylindole was excited at 405 nm and emission monitored at 461 nm. During imaging, cells were in culture medium containing serum.
Western blotting
Membranes were blocked in 5% milk in Tris-buffered saline/0.1%Tween 20 (TBST) for 2 h at room temperature. After three washes (5 min each) with TBST, the membranes were incubated with anti-GRK2 antibody (1:3000) in 3% bovine serum albumin (BSA) in TBST overnight at 4°C. After incubation, membranes were washed four times with TBST (10 min each) and incubated with anti-rabbit secondary antibody (1:10,000, IR-800; LI-COR Biosciences, Lincoln, NE) in 5% milk in TBST for 1 h at room temperature. Blocking was for 1 h at room temperature, and primary antibody was incubated for 2 h at room temperature and secondary antibody for 1 h at room temperature. The membranes were washed three times with TBST (10 min each) and scanned on a LI-COR Odyssey infrared imaging system.
Immunocytochemistry for ANF induction
NRVMs were transfected with 1 μg of empty vector, GRK2ct, PM-GRK2ct, or Golgi-GRK2ct and 1 μg of a YFP-expressing plasmid using Lipofectamine 2000 to allow identification of transfected cells. Sixteen hours after transfection 100 nM ET-1 was added for 24 h to induce hypertrophy. Cells were briefly washed with PBS and fixed for 25 min in a solution of 2% paraformaldehyde (Sigma-Aldrich) with 0.025% glutaraldehyde (J.T. Baker, Center Valley, PA) in PBS. After permeabilization in 0.2% Triton X-100 for 15 min at room temperature, cells were blocked in a solution of 10% BSA in PBS for 1 h at room temperature. Primary antibody (rabbit anti-ANF [EMD Millipore]) was incubated at a dilution of 1:2500 in PBS overnight at 4°C. After three washes in PBS, cells were incubated with the secondary antibody (goat anti-rabbit Alexa Fluor 546 [Life Technologies, Grand Island, NY]) at a 1:2500 dilution in PBS for 3 h at room temperature. Fluorescence images were captured by confocal microscopy and analyzed for ANF expression.
For AVMs, freshly isolated adult myocytes from mice were infected with adenoviruses expressing YFP, PM-GRK2ct, or Golgi-GRK2ct for 2 h, after which the virus was removed by washing and the contraction inhibitor blebbistatin was added to the culture medium. At 16 h later, cells were treated with either vehicle or 100 nM ET-1 for 24 h. Cells were fixed and stained for ANF as described.
To quantitate ANF induction, fluorescence images were captured by confocal microscopy, and transfected YFP fluorescent cells were identified and analyzed for ANF fluorescence intensity using Olympus FluoView software, version 2.0a. Perinuclear area was traced, and peak ANF intensity was measured for each cell minus background subtraction from area in the cytosol. Data were averaged based on individual cells (>40 cells total) pooled from three separate experiments for NRVMs and AVMs.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants R01GM53536 and R01GM81772 (A.V.S.) and R01 GM56444 (P.B.W.).
Abbreviations used:
- ANF
atrial natriuretic factor
- AVM
adult ventricular myocyte
- cpTOME
8-4-(chlorophenylthio)-2′-O-methyladenosine-3′,5′-monophosphate
- DAG
diacylglycerol
- Epac
exchange factor activated by cAMP
- ET-1
endothelin-1
- FAPP-PH
four-phosphate adaptor protein–pleckstrin homology domain
- GPCR
G protein–coupled receptor
- GRK2ct
C terminus of G protein–coupled receptor kinase 2
- mAKAP
muscle-specific A kinase anchoring protein
- nDKAR
nuclear targeted D kinase reporter
- NRVM
neonatal rat ventricular myocyte
- PI4P
phosphatidylinositol 4-phosphate
- PIP2
phosphatidylinositol 4,5-bisphosphate, inositol 1,4,5 trisphosphate
- PI-PLC
phosphatidylinositol-specific phospholipase C
- PKC
protein kinase C
- PKD
protein kinase D
- PM
plasma membrane
- PTX
pertussis toxin
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-10-1476) on January 21, 2015.
REFERENCES
- Balla T, Szentpetery Z, Kim YJ. Phosphoinositide signaling: new tools and insights. Physiology. 2009;24:231–244. doi: 10.1152/physiol.00014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Tuymetova G, Tsiomenko A, Várnai P, Balla T. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-iii α: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell. 2005;16:1282–1295. doi: 10.1091/mbc.E04-07-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T, Varnai P. Visualizing cellular phosphoinositide pools with GFP-fused protein-modules. Sci STKE. 2002;2002:pl3. doi: 10.1126/stke.2002.125.pl3. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Avedanian L, Al-Khoury J, Provost C, Nader M, D'Orléans-Juste P, Jacques D. Nuclear membrane receptors for ET-1 in cardiovascular function. Am J Physiol Regul Integr Comp Physiol. 2011;300:R251–R263. doi: 10.1152/ajpregu.00736.2009. [DOI] [PubMed] [Google Scholar]
- Blumer JB, Smrcka AV, Lanier SM. Mechanistic pathways and biological roles for receptor-independent activators of G-protein signaling. Pharmacol Ther. 2007;113:488–506. doi: 10.1016/j.pharmthera.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of G βγ-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- Campelo F, Malhotra V. Membrane fission: the biogenesis of transport carriers. Annu Rev Biochem. 2012;81:407–427. doi: 10.1146/annurev-biochem-051710-094912. [DOI] [PubMed] [Google Scholar]
- Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CM, et al. Small molecule disruption of G βγ signaling inhibits the progression of heart failure. Circ Res. 2010;107:532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari M, Saini DK, Kalyanaraman V, Gautam N. Shuttling of G protein subunits between the plasma membrane and intracellular membranes. J Biol Chem. 2007;282:24092–24098. doi: 10.1074/jbc.M704246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Anel AM. Phospholipase C β3 is a key component in the G βγ/PKC η/PKD-mediated regulation of trans-Golgi network to plasma membrane transport. Biochem J. 2007;406:157–165. doi: 10.1042/BJ20070359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Anel AM, Malhotra V. PKC η is required for β1 γ2/β3 γ2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J Cell Biol. 2005:169, 83–91. doi: 10.1083/jcb.200412089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielitz J, Kim M-S, Shelton JM, Qi X, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci USA. 2008;105:3059–3063. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filtz TM, Grubb DR, McLeod-Dryden TJ, Luo J, Woodcock EA. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 2009;23:3564–3570. doi: 10.1096/fj.09-133983. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Wedegaertner PB. Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein βγ subunits. J Biol Chem. 2010;285:32393–32404. doi: 10.1074/jbc.M110.154963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V. G βγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Knowlton KU, Michel MC, Itani M, Shubeita HE, Ishihara K, Brown JH, Chien KR. The α 1A-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy. J Biol Chem. 1993;268:15374–15380. [PubMed] [Google Scholar]
- Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the b-adrenergic receptor kinase or a β ARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- Kunkel MT, Toker A, Tsien RY, Newton AC. Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. J Biol Chem. 2007;282:6733–6742. doi: 10.1074/jbc.M608086200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA. G βγ-dependent phosphoinositide 3-kinase activation in hearts with in vivo pressure overload hypertrophy. J Biol Chem. 2000;275:4693–4698. doi: 10.1074/jbc.275.7.4693. [DOI] [PubMed] [Google Scholar]
- Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA. Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112:1067–1079. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cε regulate Ca2+ release in the heart by activation of protein kinase Cε and calcium-calmodulin kinase II. J Biol Chem. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase Cε plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- O'Neill PR, Karunarathne WKA, Kalyanaraman V, Silvius JR, Gautam N. G-protein signaling leverages subunit-dependent membrane affinity to differentially control βγ translocation to intracellular membranes. Proc Natl Acad Sci USA. 2012;109:E3568–E3577. doi: 10.1073/pnas.1205345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oner SS, Vural A, Lanier SM. Translocation of activator of G-protein signaling 3 to the Golgi apparatus in response to receptor activation and its effect on the trans-Golgi network. J Biol Chem. 2013;288:24091–24103. doi: 10.1074/jbc.M112.444505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Rockman HA, Chiariello M. Targeted inhibition of phosphoinositide 3-kinase activity as a novel strategy to normalize β-adrenergic receptor function in heart failure. Vasc Pharmacol. 2006;45:77–85. doi: 10.1016/j.vph.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Prasad SVN., Perrino C, Rockman HA. Role of phosphoinositide 3-kinase in cardiac function and heart failure. Trends Cardiovasc Med. 2003;13:206–212. doi: 10.1016/s1050-1738(03)00080-x. [DOI] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, Weidemann F, Lohse MJ, Lorenz K. Interference with ERKThr188 phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci USA. 2013;110:7440–7445. doi: 10.1073/pnas.1221999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini DK, Kalyanaraman V, Chisari M, Gautam N. A family of G protein βγ subunits translocate reversibly from the plasma membrane to endomembranes on receptor activation. J Biol Chem. 2007;282:24099–24108. doi: 10.1074/jbc.M701191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini DK, Karunarathne WK, Angaswamy N, Saini D, Cho JH, Kalyanaraman V, Gautam N. Regulation of Golgi structure and secretion by receptor-induced G protein βγ complex translocation. Proc Natl Acad Sci USA. 2010;107:11417–11422. doi: 10.1073/pnas.1003042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Boggon TJ, Baird CL, Gomez CA, Zhao J, Shan WS, Myszka DG, Shapiro L. G-protein signaling through tubby proteins. Science. 2001;292:2041–2050. doi: 10.1126/science.1061233. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal. 2012;29:1333–1343. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Hepler JR, Brown KO, Sternweis PC. Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science. 1991;251:804–807. doi: 10.1126/science.1846707. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, Chae HZ, Rhee SG, Exton JH. Activation of the β1 isozyme of phospholipase C by α subunits of the G q class of G proteins. Nature. 1991;350:516–518. doi: 10.1038/350516a0. [DOI] [PubMed] [Google Scholar]
- Teruel MN, Blanpied TA, Shen K, Augustine GJ, Meyer T. A versatile microporation technique for the transfection of cultured CNS neurons. J Neurosci Methods. 1999;93:37–48. doi: 10.1016/s0165-0270(99)00112-0. [DOI] [PubMed] [Google Scholar]
- Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, Chuprun JK, Song J, Gao E, Cheung JY, et al. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol. 2012;7:1830–1839. doi: 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Yeow RY, Schoenau C, Huber J, Tesmer JJ. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol Pharmacol. 2011;80:294–303. doi: 10.1124/mol.111.071522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase Cε modulates β-adrenergic receptor–dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res. 2005;97:1305–1313. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]
- Wing MR, Houston D, Kelley GG, Der CJ, Siderovski DP, Harden TK. Activation of phospholipase C-ε by heterotrimeric G protein βγ-subunits. J Biol Chem. 2001;276:48257–48261. doi: 10.1074/jbc.C100574200. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Phospholipase Cε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286:23012–23021. doi: 10.1074/jbc.M111.231993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Malik S, Pang J, Wang H, Park K, Yule D, Blaxall B, Smrcka A. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153:216–227. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.