

Abstract
Unlike other Hsp70 molecular chaperones, those of the eukaryotic cytosol have four residues, EEVD, at their C-termini. EEVD(Hsp70) binds adaptor proteins of the Hsp90 chaperone system and mitochondrial membrane preprotein receptors, thereby facilitating processing of Hsp70-bound clients through protein folding and translocation pathways. Among J-protein co-chaperones functioning in these pathways Sis1 is unique, as it also binds the EEVD(Hsp70) motif. However, little is known about the role of the Sis1:EEVD(Hsp70) interaction. We found that deletion of EEVD(Hsp70) abolished the ability of Sis1, but not the ubiquitous J-protein Ydj1, to partner with Hsp70 in in vitro protein refolding. Sis1 co-chaperone activity with Hsp70ΔEEVD was restored upon substitution of a glutamic acid of the J-domain. Structural analysis revealed that this key glutamic acid, which is not present in Ydj1, forms a salt bridge with an arginine of the immediately adjacent glycine-rich region. Thus, restoration of Sis1 in vitro activity suggests that intramolecular interaction(s) between the J-domain and glycine-rich region controls co-chaperone activity, which is optimal only when Sis1 interacts with the EEVD(Hsp70) motif. Yet, we found that disruption of the Sis1:EEVD(Hsp70) interaction enhances the ability of Sis1 to substitute for Ydj1 in vivo. Our results are consistent with the idea that interaction of Sis1 with EEVD(Hsp70) minimizes transfer of Sis1-bound clients to Hsp70s that are primed for client transfer to folding and translocation pathways by their preassociation with EEVD-binding adaptor proteins. These interactions may be one means by which cells triage Ydj1- and Sis1-bound clients to productive and quality control pathways, respectively.
Keywords: molecular chaperone, protein folding, Hsp40, J domain, EEVD motif, yeast
Introduction
By interacting with a wide variety of client proteins, molecular chaperones function in many cellular processes, ranging from the folding of nascent polypeptides to the remodeling of multimeric protein complexes [1, 2]. Ubiquitous Hsp70 systems are central players in cellular chaperone networks, performing a particularly important role in protein folding and translocation, as well as in prioritizing proteins for folding pathways and the quality control systems of protein aggregation and proteolysis [3]. All Hsp70s, regardless of the cellular compartment in which they reside, have a C-terminal peptide binding domain, which interacts with short segments of client proteins, and an N-terminal adenine nucleotide-binding domain, which regulates client interaction [4]. Obligatory co-chaperones, called J-proteins, interact at the domain interface, stimulating ATP hydrolysis, thereby stabilizing Hsp70’s interaction with client [5]. This role is carried out by the highly conserved helical J-domain found in all J-proteins. Some J-proteins also bind client proteins, serving to target them for Hsp70 binding.
Unlike other Hsp70s, eukaryotic cytosolic Hsp70s (called Ssa in Saccharomyces cerevisiae) also have a conserved EEVD tetrapeptide at their extreme C-terminus [6]. This EEVD motif, called EEVD(Hsp70) throughout, interacts with targeting/adaptor factors that facilitate trafficking of associated client proteins along Hsp90 folding pathways and assists targeting clients to mitochondria [7–11]. Sis1 co-chaperone, one of the two most abundant soluble cytosolic J proteins, also binds EEVD(Hsp70); in contrast, the other, Ydj1 does not [12, 13]. Although Ydj1 has a Zn binding domain, but Sis1 does not, the two proteins have many similarities [5]. Both have an N-terminal J-domain, followed by a glycine-rich region, two structurally similar domains with ß-barrel topology (CTD1 and CTD2), which contain the client-binding cleft of both proteins and the EEVD(Hsp70) interaction site of Sis1, and a C-terminal dimerization domain. Sis1 and Ydj1, play partially overlapping roles in determining the fate of polypeptide chains, consistent with their distinct, yet overlapping, specificity for client binding [14–16]. Ydj1 plays well-established roles in driving client proteins into Hsp90 folding pathways and across cellular membranes into organelles. On the other hand, recent data indicate that Sis1 is important in protein quality control, targeting clients to centers of aggregation and proteolysis [17–19].
As the functional relevance of the Sis1:EEVD(Hsp70) interaction is unknown, we analyzed the consequences of disrupting it. On one hand, we found the Sis1:EEVD(Hsp70) interaction to be critical for Sis1 in Hsp70-mediated protein refolding in vitro. However, this requirement is relieved by alteration of a single residue in the J-domain, which was shown to interact with the glycine-rich region. These results suggest that Sis1 partnership with Hsp70 is optimized through intramolecular and intermolecular interactions. On the other hand, we found that disruption of the Sis1:EEVD(Hsp70) interaction enhanced the ability of Sis1 to substitute for Ydj1 in vivo. Our results suggest that interaction of Sis1 with EEVD(Hsp70) may diminish the chances of Sis1-bound clients being transferred to folding and translocation pathways, providing fine-tuning of client targeting between folding and quality control pathways.
Results
EEVD(Hsp70) motif is required for refolding activity of Sis1, but not Ydj1 and Xdj1
As a first step in our analysis of the importance of the interaction of Sis1 with EEVD(Hsp70), we compared the ability of the full-length Hsp70 Ssa1 and Ssa1 lacking the C-terminal four residues (called Hsp70 and Hsp70ΔEEVD, respectively, throughout) to function with Sis1 and Ydj1 as J-protein partners in facilitating protein refolding. Sis1 and Ydj1 were similarly efficient in refolding of urea-denatured luciferase when partnering with full-length Hsp70. However, while Ydj1 facilitated refolding nearly as efficiently when partnering with Hsp70ΔEEVD as with full-length Hsp70, Sis1 did not promote refolding with Hsp70ΔEEVD (Fig. 1a). Within 45 min 100% of luciferase was reactivated with Hsp70, but only 19% with Hsp70ΔEEVD. To test whether Ydj1 was unique in being able to function efficiently with Hsp70ΔEEVD we also tested Xdj1, a paralog of Ydj1 arising from a gene duplication during the fungal lineage [20]. Xdj1 was also able to promote luciferase refolding with Hsp70ΔEEVD.
Fig. 1. EEVD Hsp70) important for Sis1, but not Ydj1/Xdj1, driven protein refolding.
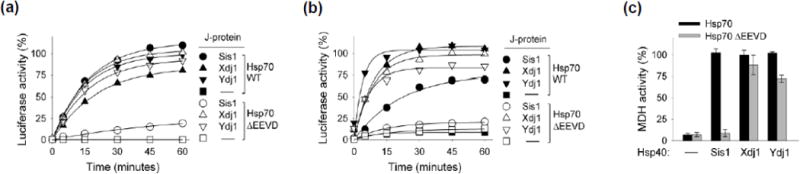
Luciferase or malate dehydrogenase (MDH) was denatured and diluted into various chaperone mixtures. Aliquots were removed at the indicated times and enzymatic activity determined. Activity of non-denatured enzyme was taken as 100%. (a) Urea-denatured luciferase or (c) heat-denatured MDH was diluted into refolding buffer containing Sis1, Xdj 1 or Ydj1 as the J-protein co-chaperone, in addition to Hsp104, Sse1 and either Ssa1 (Hsp70) or Ssa1 having EEVD deleted (Hsp70ΔEEVD). MDH activity at 90 min is shown; see Supplementary Fig. 1a for additional time points. (b) Luciferase treated with guanidinium hydrochloride was diluted into folding buffer containing only Sis1, Xdj 1 or Ydj1 and Hsp70 or Hsp70ΔEEVD. In (a) the two controls, Hsp70 without J-protein (black square) and Hsp70 ΔEEVD without J-protein (white square), overlap. Therefore, black squares are not visible.
The luciferase refolding reactions discussed above included Hsp104, a chaperone specialized in resolubilization of aggregates, as well as the nucleotide exchange factor Sse1. To ascertain whether these two components played a major role in the difference between Sis1 and Ydj1/Xdj1-driven refolding by Hsp70 and Hsp70ΔEEVD, we carried out refolding assays in which luciferase was partially denatured by guanidinium hydrochloride. This is a milder treatment than the urea denaturation method, and, as expected [21], refolding occurred when only Hsp70 and J-protein were included in the reaction (Fig 1b). As with the assay containing Hsp104 and Sse1, Sis1 did not partner with Hsp70ΔEEVD. We conclude that, independent of other chaperone proteins, the EEVD motif of Hsp70 plays an important role in partnering with Sis1, but not with Ydj1 or Xdj1, in protein refolding.
To determine whether this difference between Sis1 and Ydj1/Xdj1 in ability to partner with Hsp70 and Hsp70ΔEEVD extended to other clients, we tested refolding of malate dehydrogenase (MDH) after denaturation by heat treatment. All three J-proteins partnered with full-length Hsp70, reaching nearly 100 % refolding within 90 min (Fig. 1c; Supplementary Fig. 1a). However, when Sis1 was paired with Hsp70ΔEEVD, MDH activity did not rise above background levels. On the other hand, in reactions combining Xdj1 and Ydj1 with Hsp70ΔEEVD, MDH activity reached 88 and 72%, respectively within that time. Our results support the idea that interaction between Sis1 and EEVD(Hsp70) is important for Sis1’s ability to function efficiently in protein refolding in vitro.
Substitution of J-domain of Ydj1 or Xdj1 overcomes Sis1’s dependence on (EEVD)Hsp70
To better understand the basis of the requirement of EEVD(Hsp70) in the function of Sis1, but not Ydj1/Xdj1, we constructed chimeras. We swapped Ydj1’s and Xdj1’s J-domains for that of Sis1, thereby generating JYdj1Sis1 and JXdj1Sis1. Surprisingly, both JYdj1Sis1 and JXdj1Sis1 partnered with Hsp70ΔEEVD substantially better than Sis1 in refolding of luciferase and MDH (Fig. 2a,b; Supplementary Fig. 1b). For example, in reactions containing JYdj1Sis1 and JXdj1Sis1, 100 and 85 % of luciferase activity, respectively, was recovered, compared to only 19% for wild type Sis1 within 60 min.
Fig. 2. Substitution of J domain of Ydj1/Xdj1 for that of Sis1 overcomes Sis1 dependence on EEVD interaction.
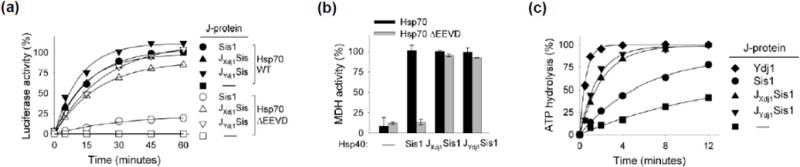
(a) Luciferase or (b) MDH was denatured by treatment with urea or heat, respectively, and diluted into chaperone-containing refolding buffer containing the indicated J-protein co-chaperone and either Hsp70 or Hsp70ΔEEVD, as indicated, in addition to Hsp104 and Sse1. MDH activity at 90 min is shown; see Supplementary Fig. 1b for additional time points. In (a) the two controls, Hsp70 without J-protein (black square) and Hsp70 ΔEEVD without J-protein (white square), overlap. Therefore, black squares are not visible. (c) Isolated Hsp70-[32P α-ATP] complex was incubated with the indicated J-protein (0.25 μM) and the release of 32P determined with time.
We next compared the ATPase stimulatory ability of Sis1, Ydj1 and the chimeras using a preformed 32P-ATP-Hsp70 complex (Fig. 2c). Ydj1 stimulated the ATPase activity of Hsp70 more effectively than Sis1, resulting in 95% hydrolysis within two minutes. 78% of the ATP was hydrolyzed by 12 min in reactions containing Sis1, at which point 41% hydrolysis had occurred in the control reaction having no J-protein. Both the JYdj1Sis1 and JXdj1Sis1 chimera stimulated ATP hydrolysis more effectively than wild type Sis1, with 74 and 63 % hydrolysis achieved within two minutes. Thus swapping of Ydj1’s or Xdj1’s J-domain for that of Sis1 both overcame Sis1’s dependence on the EEVD motif of Hsp70 for refolding and increased Sis1’s ability to stimulate Hsp70’s ATPase activity, suggesting a functional difference between these J-domains.
Alteration of Sis1 J-domain overcomes dependence on EEVD(Hsp70) for in vitro refolding
To find clues as to what residues might be responsible for these functional differences between J-domains, we compared their sequences. We found three regions (labeled A, B and C in Fig. 3a) having substantial sequence differences. Two of these, segments A and B, mainly encompass loops between helices; the majority of residues of the third, segment C, is mainly part of helix 3. Constructs were made such that A, B and C segments of Ydj1 (residues 14–20, 38–44 and 52–61, respectively) were substituted individually for those of Sis1. The activities of the resulting variants were then tested. The two constructs having exchanged segments of loops, JYdj1-ASis1 and JYdj1-BSis1 were, like Sis1, unable to partner with Hsp70ΔEEVD (Fig. 3b Supplementary Fig. 1c). On the other hand, JYdj1-CSis1 containing the 52–61 segment of Ydj1 functioned in reactivation of luciferase and MDH with Hsp70ΔEEVD, as well as it did with wild type Hsp70. Thus, we hypothesized that helix 3 is responsible for the functional difference between the Sis1 and Ydj1/Xdj1 J-domains.
Fig. 3. Segment of helix 3 of J-domain is important for overcoming EEVD dependence.
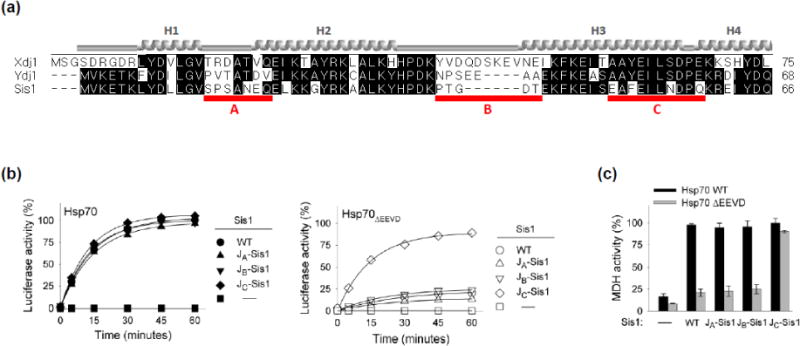
(a) Alignment of J-domain of Xdj1, Ydj1 and Sis1, performed using DNASTAR (Madison, WI). Red lines (A, B, C) indicate regions of particular divergence and were changed in Sis1. Helices (1–4), based on homology with J-domain of Escherichia coli DnaJ (PDB ID: 1XBL) indicated. (b) Luciferase or (c) MDH was denatured by treatment with urea or heat, respectively, and diluted into chaperone-containing refolding buffer containing wt Sis1 or variants having indicated alterations in Sis1 J-domain making it more similar to Ydj1, with either full-length Hsp70 or Hsp70ΔEEVD: JYdj1-ASis1 (JA-Sis1), JYdj1-BSis1 (JB-Sis1) and JYdj1-CSis1 (JC-Sis1). MDH activity at 90 min is shown; see Supplementary Fig. 1c for additional time points.
The sequence of Ydj1 and Xdj1 are identical over the 10 residue C segment. However, four differ in Sis1 (E50, F52, N56 and Q59). Thus, we substituted the Ydj1/Xdj1 residues individually into Sis1’s J-domain and tested the variants for refolding activity when partnering with Hsp70ΔEEVD. While all four variants were active with full-length Hsp70, three (Sis1F52Y, Sis1N56S and Sis1Q59E) behaved similarly to wild type Sis1, that is, they were inactive with Hsp70ΔEEVD. However, nearly 90% of the denatured luciferase was refolded within 60 min in reactions containing Sis1E50A and Hsp70ΔEEVD (Fig. 4a). Similar results were obtained in MDH refolding assays. The activity of Sis1E50A with Hsp70ΔEEVD was indistinguishable from that with full-length Hsp70, while the activities of the other three variants were substantially less (Fig 4b, Supplementary Fig. 1d). Thus, alteration of a single residue, E50, in Sis1’s J-domain6 compensates for the deficiency in Sis1’s protein refolding activity caused by the absence of the Hsp70 EEVD motif.
Fig. 4. Sis1 residue E50 of Sis1 J domain is key for overcoming dependence on EEVD binding.
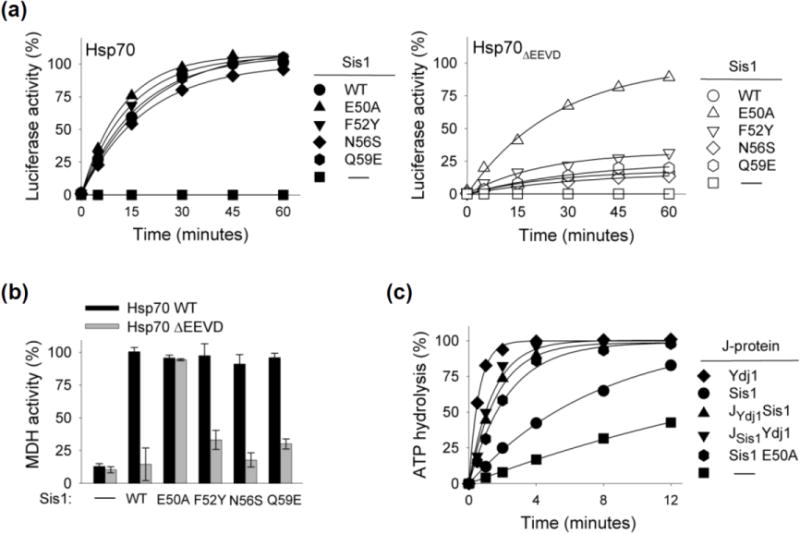
(a) Luciferase (a) or MDH (b) was denatured by treatment with urea or heat, respectively, and diluted into chaperone-containing refolding buffer containing wt Sis1 or variants having indicated alterations in Sis1 J-domain making it more similar to Ydj1, with either full-length Hsp70 or Hsp70ΔEEVD. MDH activity at 90 min is shown; see Supplementary Fig. 1d for additional time points. (c) Isolated Hsp70-[32P α-ATP] complex was incubated with the indicated J-protein (0.25 μM) and the release of 32P determined with time.
To better understand the relationship between alteration of E50 alteration and activity of the Sis1 J-domain we tested the ability of two additional constructs to stimulate the ATPase activity of Hsp70: full-length Sis1 with the E50A substitution in its J-domain (Sis1E50A) and a chimera substituting the wild type Sis1 J-domain for that of Ydj1 (JSis1Ydj1). The E50A alteration enhanced Sis1’s ability to stimulate Ssa1’s ATPase activity (Fig. 4c). This enhancement was similar to that resulting from substitution of the entire J-domain of Ydj1 (i.e. JSis1Ydj1), suggesting that the J-domain of Sis1 is not inherently less efficient than the J-domain of Ydj1.
Interaction of Sis1 E50 with R73 of glycine-rich region
To understand how E50 might affect Sis1 function we carried out structural analysis of the J-domain of S. cerevisiae Sis1 using X-ray crystallography. To increase the odds of crystallization, we tested several N-terminal fragments of Sis1, varying in length from 74 to 125 residues. We obtained several protein crystal forms for the 74- and 89-residue fragments. The best quality X-ray diffraction was collected for the 89-residue N-terminal domain construct. We obtained atomic resolution diffraction data (1.25 Å) with 60% reflection completeness in the highest resolution shell due to the crystal anisotropicity and 90% completeness at 1.37 Å resolution (Supplementary Table 1,2). The final model consists of Sis1 residues 1–81, which includes the key α-helix containing E50, as well as a portion of the glycine-rich region. It lacks the 8 C-terminal residues, which were not defined in electron density maps, as well as the three N-terminal residues remaining after cleavage of the purified protein fusion with TEV protease.
The overall structure of the 81-residue N-terminal Sis1 fragment is comprised of five α-helices (Fig. 5a). The first four helices form a typical J-domain structure with the conserved HPD motif residing between helices H2 and H3 [22]. The same structural futures have been observed in X-ray structures of other J domains, the closest being the Sis1 homolog from Caenorhabditis elegans, Dnj 12J (PDB ID: 2OCH), as well as in NMR structures of other J-domains (e.g. PDB ID: 1BQZ and 2DN9). The fifth helix, residues 68–74, is part of the glycine-rich region. The unique feature of the structure is the presence of a salt bridge between the side chains of E50 of α-helix 3 of the J-domain and R73 of α-helix 5 of the glycine-rich region (Fig. 5a). The residues are very well defined in the structure with side-chains B-factors in the 14–19 Å2 range and outstanding electron density maps. The E50/R73 contacts are the only hydrogen bonds between the J-domain and glycine-rich region in the structure and undoubtedly stabilize interaction of the J-domain with glycine-rich region.
Fig. 5. Sis1 intramolecular interaction between E50 of J domain and R73 of glycine-rich region.
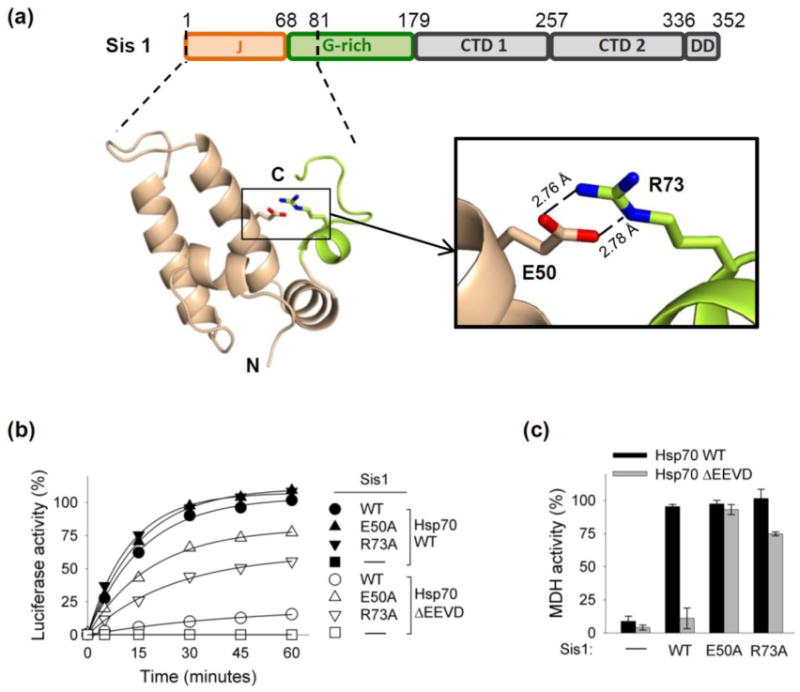
(a) Top: Line diagram of Sis1 domains, with residues at endpoints of domains indicated: J-domain (J); glycine-rich (G-rich), C-terminal domains (CTD) and dimerization domain (DD). Bottom-left: Crystal structure of S. cerevisiae Sis1 N-terminal 1–81 residue fragment (PDB ID: 4RWU), as ribbon representation generated using Pymol (DeLano Scientific LL). The side chains of E50 and R73 residues are shown as sticks. Bottom-right: Blow-up of the double salt-bridge region formed by E50 (oxygen in red) and R73 (nitrogen in blue). Hydrogen bonds are shown as dashed lines with lengths as indicated. (PDB#: 4RWU) (b, c). Luciferase (b) or MDH (c) was denatured by treatment with urea or heat, respectively, and diluted into chaperone-containing refolding buffer containing either wt Sis1, no Sis1 (−) or Sis1variants having E50A or R73A substitutions, in addition to Hsp104, Sse1 and Ssa1. In (b) the two controls, Hsp70 without J-protein (black square) and Hsp70 ΔEEVD without J-protein (white square), overlap. Therefore, black squares are not visible. MDH activity at 90 min is shown; see Supplementary Fig. 1e for additional time points.
We reasoned that if the salt-bridge observed between E50 and R73 revealed by our structural analysis is functionally relevant, alteration of R73 would be expected to affect Sis1 function similarly to that caused by alteration of E50. Therefore, we purified Sis1R73A and tested its activity in in vitro refolding assays. We found that alteration of R73 to A partially restored the ability of Sis1 to function in refolding of luciferase and MDH with Hsp70ΔEEVD (Fig. 5b,c; Supplementary Fig 1e) consistent with functional importance of the E50:R73 salt bridge.
Sis1 variants defective in EEVD(Hsp70) interaction have reduced in vitro folding activity
The experiments described above underline the importance of EEVD(Hsp70) for functional interaction with Sis1, but do not establish that it is the physical interaction between the two proteins per se that is important. To address this question, we purified Sis1 variants having alterations of CTD1 residues demonstrated to directly interact with the EEVD of the Hsp70 Ssa1 [12]. We then assessed both binding to Hsp70 and activity in in vitro refolding assays. Three residues were changed: Sis1 residue K199, which interacts with Ssa1 residue D642, as well as Sis1 residues K202 and K214, both of which interact with E640 of Ssa1 Hsp70 (Fig. 6a). To compare the interaction of wild type and variant Sis1 proteins with Hsp70, we used an ELISA assay in which the amount of Sis1 bound to Hsp70 immobilized in wells was measured using Sis1-specific antibodies. At a concentration of wild type Sis1 of 200 nM, binding to Hsp70 approached saturation, with 50% maximal binding achieved at approximately 30 nM. The variant having all three alterations, Sis1K199N/K202N/K214N, was severely affected; negligible binding above background levels was detected at the highest concentration tested, 200 nM. Binding of each single alteration variant was detected, but all were significantly defective. The ability of variants to refold luciferase and MDH followed a similar pattern. The activity of Sis1K199N/K202N/K214N was negligible in both the luciferase and MDH refolding assays (Fig 6b; Supplementary Fig. 1f).
Fig. 6. Analysis of Sis1 variants defective in EEVD interaction.
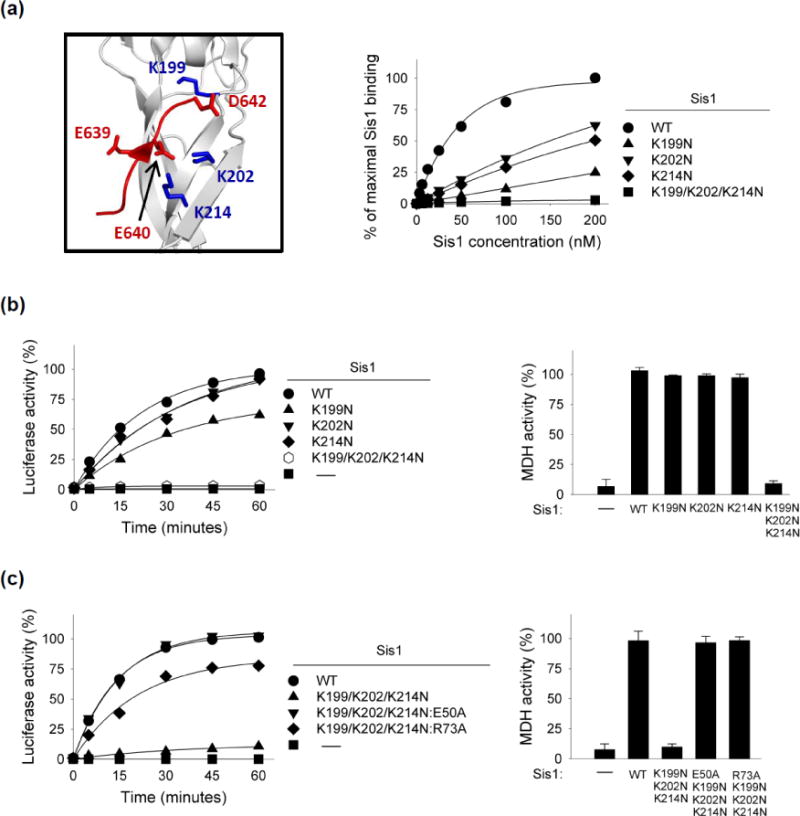
(a) Left: Three-dimensional structure of Sis1 CTD1 (gray) in complex with extreme C-terminus of Ssa1 Hsp70 (red) (PDB: 2B26). Key lysines on Sis1 for Ssa1 interaction in blue. Right: interaction of indicated Sis1 variants with Hsp70 using ELISA assay. Ssa1 Hsp70 was bound in wells and incubated with increasing concentrations of Sis1 variants. After washing, bound Sis1 was detected using anti-Sis1 antibody. Maximum binding observed for wt Sis1 was set at 100%. (b, c) Luciferase or MDH was denatured by treatment with urea or heat, respectively, and diluted into chaperone-containing refolding buffer that included Hsp104 and Sse1. MDH activity at 90 min is shown; see Supplementary Fig. 1f, g for additional time points.
As described above (Fig. 4), the E50A substitution overcame Sis1’s defect in refolding by Hsp70ΔEEVD. As a test of whether the lack of refolding function of Sis1K199N/K202N/K214N was also overcome by the E50A alteration we combined the four alterations, generating Sis1K199N/K202N/K214N:E50A. Sis1K199N/K202N/K214N:E50A was as effective as wild type Sis1 in partnering with Hsp70 in refolding of both luciferase and MDH (Fig. 6c; Supplementary Fig. 1g), supporting the idea that the functional defect of Sis1K199N/K202N/K214N is due to its lack of interaction with Hsp70 via its EEVD motif. We also tested the effect of altering R73, which interacts with E50 (Fig. 6c; Supplementary Fig. 1g). Sis1K199N/K202N/K214N:R37A was more active in both refolding assays than Sis1K199N/K202N/K214N, but did not restore activity as well as E50A, similar to what was seen for the ability of the two alterations to overcome the folding defect of Hsp70ΔEEVD (Fig. 5b, c).
Disruption of Sis1:EEVD(Hsp70) interaction enhances Sis1’s ability to substitute for Ydj1
Having in hand a Sis1 variant, Sis1K199N/K202N/K214N, which does not bind EEVD(Hsp70) provided us the opportunity to assess in vivo effects of the lack of interaction between EEVD(Hsp70) and Sis1. We tested the effect of the sis1K199N/K202N/K214N mutation in two genetic backgrounds: 1) in an otherwise wild type background, and 2) in the presence of a partial loss of function YDJ1 allele, ydj11–134. The first test was to determine if loss of EEVD(Hsp70) interaction substantially affected Sis1’s ability to carry out its housekeeping functions. We found, no obvious growth defect of sis1K199N/K202N/K214N at a variety of temperatures and culture conditions (Fig. 7a) even though Sis1 is essential [23].
Fig 7. Sis1 variant defective in EEVD interaction suppresses growth defect caused by reduced Ydj1 function.

(a–c) 10-fold serial dilutions of the indicated strains were plated on minimal medium and incubated at indicated temperatures. (a) Δsis1 expressing Sis1 (WT) or Sis1K199N/K202N/K214N (K199/202/204N): 2 days (23°, 30°, 37° C). (b) ydj11–134 Δsis1 expressing either Sis1 (WT) or Sis1K199N/K202N/K214N (K199/K202/K204N): 2 days (30°, 34° C) or 3 days (23°C). (c) as in (b) except cells contained an additional plasmid having a wild type copy of SIS1 to test dominance of Sis1K199N/K202N/K214N. (d) Total protein isolated from ydj1-134 cells expressing either Sis1 (WT) or Sis1K199N,K202N,K214N (K199,202,204N) was resolved by electrophoresis, electroblotted to nitrocellulose, and probed with antibody to Sis1, and, as a loading control, Ssa (control).
The second test was set up to determine if interaction of Sis1 with EEVD(Hsp70) might impede its ability to substitute for Ydj1 in vivo. We reasoned that an Hsp70 whose EEVD is interacting with a receptor/adaptor might be less likely to partner with Sis1 than one whose EEVD is free to interact with Sis1. Because Δydj1 cells grow so poorly, making genetic manipulation difficult, we used cells expressing Ydj11–134. The presence of Ydj1’s J-domain and glycine-rich region partially overcome the very severe growth defect of Δydj1. Cells expressing Ydj11–134 along with wt Sis1 are temperature sensitive for growth [14], failing to form colonies at 34°C (Fig 7). However, ydj11–134 cells expressing Sis1K199N/K202N/K214N supported robust growth at 34°C (Fig. 7b), even though the levels of Sis1 protein were very similar (Fig. 7d).
The observed suppression is consistent with the idea that elimination of the Sis1:EEVD(Hsp70) interaction allows Sis1 to more effectively partner with an Hsp70 that has a EEVD binding receptor bound (Fig. 8), rather than selectively interact with Hsp70s having their EEVD free. If, indeed, the elimination of the EEVD(Hsp70) binding site on Sis1 is the cause of the suppression then the suppression would be expected to occur whether or not wild type Sis1 was present, that is the mutation results in a “gain of function”. To test this idea, we expressed wt Sis1 and Sis1K199N/K202N/K214N simultaneously in the test strain expressing Ydj11–134. We found that suppression of the temperature-sensitive growth defect of ydj11–134 cells was similar in the presence or absence of wild type Sis1 (Fig. 7b,c). Thus, the absence of EEVD(Hsp70) binding by Sis1 results in partial suppression of the growth defects associated with partial loss of function of Ydj1, whether or not wild type Sis1 is present.
Fig 8. Model: EEVD(Hsp70) interaction with Sis1 and EEVD receptor of Hsp90 pathway.
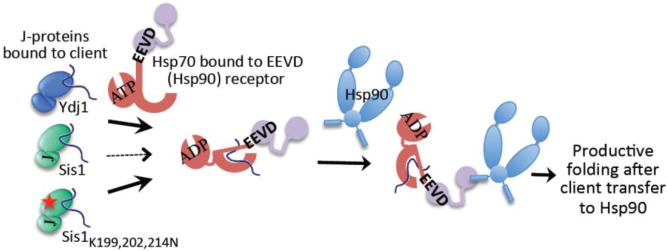
EEVD(Hsp70) binds both Sis1 and EEVD receptors, such as that of the Hsp90 system, but does not bind Ydj1. Thus, Ydj1 can bind, and efficiently transfer its client, to an Hsp70 already bound to an EEVD receptor (heavy solid arrow). However, since binding of EEVD(Hsp70) to Sis1 and to EEVD receptors is mutually exclusive, Sis1-bound clients are more poorly transferred to Hsp70 prebound to EEVD receptors (thin dotted arrow) and thus into the Hsp90 pathway. The EEVD(Hsp70)-binding defective Sis1 variant (Sis1 K199,202,214N) is able to interact with Hsp70 prebound to receptors without competition (heavy solid arrow), and thus able to compensate better than wild-type Sis1 for disruption of Ydj1 function (see Fig 7). Not shown: EEVD receptors also exist on the mitochondrial outer membrane, acting in an analogous manner to Hsp90 EEVD binding adaptor proteins.
Discussion
Several results reported here point to the conclusion that the interaction of the Sis1 co-chaperone with the EEVD motif at Hsp70’s C-terminus is important for the cooperation of these two chaperones in in vitro protein refolding. First, deletion of EEVD(Hsp70) dramatically affects Hsp70’s ability to partner with Sis1, but not with two other J-proteins, Ydj1 and Xdj1, which do not interact with EEVD(Hsp70). Second, alteration of the EEVD binding site of Sis1 also dramatically reduced the refolding activity of Sis1, even with full-length Hsp70. Third, substitution of a single residue, E50, of the Sis1 J-domain substantially overcame the folding deficiency caused by the absence of the EEVD of Hsp70, as well as a defective EEVD-interaction site on Sis1. This result is particularly informative because it addresses potential concerns that modification of the EEVD binding site of Sis1 affects other important activities (e.g. client binding). The fact that a single residue can be substituted and suppress activities of both the Hsp70 and Sis1 variants argues strongly for an important role of the Sis1:EEVD(Hsp70) interaction.
The fact that substitution of either interacting residues, that is E50 or R73, has similar effects strongly points to the importance of this salt bridge between the J-domain and the glycine-rich region. That either exchanging the Sis1 J-domain for that of Ydj1 or altering E50 increases the ATPase stimulatory ability of the full-length protein suggests that the salt-bridge reduces J-domain activity. Perhaps this E50:R73 interaction decreases the flexibility of the J-domain, constraining its activity. What might be the consequence of the Sis1 interaction with the EEVD motif of Hsp70? One possibility is a role in positioning Hsp70’s ATPase domain and the J-domain in a productive conformation. It is also possible that a lower stimulatory capacity is sufficient for Sis1 function, because the Sis1:EEVD(Hsp70) interaction itself serves to increase the local concentration of the J-domain. Further structural studies will be needed to answer such questions.
At first glance it seems surprising that we found no obvious growth defect caused by a defective Sis1:EEVD(Hsp70) interaction in otherwise wild type cells, as SIS1 is an essential gene [23]. However, it has been known for some time that yeast cells grow quite vigorously, at least under standard laboratory conditions, even when Sis1 is expressed at a substantially lower level than normal [24]. In addition, a fragment containing only the J-domain and glycine-rich region is sufficient to carry out the essential functions of Sis1, as it supports viability [25, 26]. We favor the idea that protein folding per se is not an essential function of Sis1 under normal conditions when full-length Ydj1, which is normally more abundant than Sis1 [27], is present. Indeed, we reported previously that CTD1/CTD2 of either Sis1 or Ydj1, but not both, was required for cell viability [14]. However, as the glycine-rich region of Sis1, but not Ydj1, is specifically required for cell viability, we think that this region carries out critical roles, perhaps in regulating J-domain function. A part of that regulation is likely due to the interactions between the J-domain and the glycine-rich region as illustrated by our finding, described here, of the E50:R73 salt bridge.
Our finding that disruption of the EEVD(Hsp70):Sis1 interaction enhances Sis1’s ability to compensate for diminished Ydj1 activity is consistent with the idea that a role of this interaction is to divert Sis1-bound client proteins away from entering the “productive” pathways of Hsp90 driven protein folding and import into mitochondria (Fig. 8). This interpretation is consistent with recent results [17–19] indicating that Sis1 may be more involved in triaging its clients to quality control centers, that is aggregation and proteolytic pathways, than in protein folding in vivo. Targeting proteins for centers of aggregation and proteolysis, such as Btn2 and Cur1 [28, 29], which interact with Sis1, but not Ydj1, have been identified. Such mechanisms that act to target Sis1 and its clients to such centers, coupled with the diversion away from productive pathways via the Sis1:EEVD(Hsp70) interaction discussed here, are complementary in promoting protein homeostasis. Interestingly, as the human Sis1 ortholog also interacts with EEVD(Hsp70) [30] such mechanisms may well be conserved.
Materials and Methods
Protein purification, plasmids and yeast strains
Ssa1 was purified from yeast using Ni chromatography, as described in Pfund et al [31]. SIS1, YDJ1 and XDJ1 were cloned into pMAL-His-TEV [32], and expressed in Rosetta 2 (DE3) pLys E. coli cells. Cells were grown at 37°C until A600 0.6 then expression induced at 18°C by addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) overnight. Cell lysates in phosphate buffer (20 mM phosphate, pH 7.4, 0.5 M NaCl) were prepared using a French press. Proteins were purified using Ni-NTA His-Bind Resin (Novagen Madison, WI) and a step elution with phosphate buffer containing 300 mM imidazol. Purified proteins were incubated with His-tagged Tev protease at 30°C for 30 min to remove the MBP (Maltose binding protein)-His tag. After the tags and Tev protease were removed using Ni chromatography, the purified J proteins were dialyzed against two changes of dialysis buffer (20 mM Tris-HCl, pH 8, 150 mM NaCl, 3 mM dithiothreitol (DTT), 10% glycerol). Ssa1ΔEEVD and variant J protein plasmids were constructed using the QuikChange Site-Directed mutagenesis kit from Stratagene (La Jolla, CA). Hsp104 (pMal His Tev Hsp104) was purified in a manner similar to that described for Sis1 and Sse1 (Smt3 (SUMO)-His 6 fusion protein) was purified in a manner similar to that described for Apj1 in Sahi et al [20].
Previously described yeast strains of the W303 genetic background were used: JJ1146 (ydj1::HIS3 sis1::LEU2) [14] and WY26 (sis1::LEU2) [25]. To partially rescue the severe Δydj1 growth defect JJ1146 was transformed with pRS317-ydj1-N134 [14], which includes the codons for the N-terminal 134 residues and promoter of YDJ1, and thus encodes the entire J-domain and glycine-rich region. Strains WY26 and JJ1146 [pRS317-ydj1-N134] were transformed with YCp50 carrying wild type or mutant SIS1. Resulting TRP+ colonies were grown on synthetic defined media containing 5-fluoroorotic acid (5-FOA) (Toronto Research Chemicals, Inc.) to counterselect for YCp50-SIS1 [14]. To test for dominance (Fig. 7c) cells were plated prior to counterselection with (5-FOA), and thus were expressing wild type Sis1 from YCp50 and either wild type or variant Sis1 from pRS314.
Luciferase refolding assay
Unless otherwise stated luciferase refolding assays were carried out as follows: Firefly luciferase (2.5 μM, Sigma-Aldrich, St. Louis, MO) was denatured for 30 min at 30°C in buffer A (25 mM HEPES, pH 7.4, 50 mM KCl, 5 mM MgCl2) containing DTT (5 mM) and urea (6M). The denatured luciferase was diluted, to give a final concentration of 30 nM, in buffer A containing 2 mM ATP, J protein (Sis1, 1.6 μM; Xdj1, 1.6 μM; Ydj1, 3.2 μM), Ssa1 (0.8 μM when with Sis1 or Xdj1;1.6 μM when with Ydj1), Sse1 (0.2 μM with Sis1, 0.1 μM with Xdj1 or Ydj1), and Hsp104 (1 μM) and incubated at 30°C for 1 h. The amount of J protein (Xdj1, Ydj1, and Sis1), Ssa1 and Sse1 were set at the concentrations indicated above based on optimization tests over a range of concentrations: J proteins from 0.8 μM to 6.4 μM, Ssa1 from 0.4 μM to 3.2 μM; Sse1 from 0.05 μM to 0.4 μM. To measure refolding 1 μl of the refolding mixture was mixed with 24 μl of buffer A supplemented with DTT to 1 mM and bovine serum albumin (BSA) at 0.1 mg/ml. 50 μl of luciferase assay system (Promega, Madison, WI) was added. Measurements were taken in a BioTek synergy2 plate-reader.
Refolding was also carried out after denaturation of luciferase by incubation in buffer A containing 6M guanidinium-HCl, in which case folding was achieved in the absence of Hsp104 or Sse1. The refolding mixture contained J protein (Sis1, Xdj 1 or Ydj1) and Ssa1 at 3.2 μM and 0.8 μM, respectively.
MDH refolding assay
Porcine heart MDH (5mg/ml, Roche, Nutley, NJ)) was denatured at 48°C for 30 min in refolding buffer (25 mM HEPES, pH 7.4, 50 mM KCl, 5 mM MgCl2, 2 mM ATP). After denaturation, MDH was diluted, to a final concentration of 500 nM, in refolding buffer containing chaperones (J proteins (Sis1, 3.2 μM; Xdj1, 1.6 μM; Ydj1, 1.6 μM), Ssa1 (1.6 μM for Sis1; 0.8 μM for Xdj1 and Ydj1), Sse1 (0.2 μM for Sis1; 0.1 μM for Xdj1 and Ydj1), Hsp104 (1 μM)), ADP (0.1 mM), creatine kinase (87.5 U/ml), creatine phosphate (25 mM), and incubated at 30°C. As for luciferase refolding assays, the amount of J proteins (Xdj1, Ydj1, and Sis1), Ssa1 and Sse1 were set at the concentrations indicated above based on optimization tests over a range of concentrations: J proteins from 1.6 μM to 6.4 μM; Ssa1 from 0.8 μM to 3.2 μM; Sse1 from 0.05 μM to 0.4 μM. MDH activity (absorbance at 340 nm) was determined in a BioTek synergy2 plate-reader using β-NADH (0.28 mM) and oxaloacetate (0.5 mM)) in MDH activity buffer (150 mM potassium phosphate, pH 7.5, 10 mM DTT, BSA (1mg/ml)).
Protein-Protein interaction ELISA assays
Ssa1 Hsp70 (70 μl of 0.4 μM) in 100 mM NaHCO3, pH 8.6 was bound to each well of a high binding plate (Costar 3369, Corning, NY). Wells were washed with phosphate-buffer saline (PBS), blocked with 0.5% BSA in PBS, and subsequently washed with PBS containing 0.2% tween 20 (PBST). Increasing amounts of Sis1 wild type and variants in PBST containing 0.25% BSA were added and then incubated at room temperature (RT) for 2 h. After incubation, the wells were washed with PBST and 1:1000 dilution of Sis1 polyclonal antisera was applied and incubation continued for 1.5 h at RT. After extensive washing with PBST and subsequent addition of a 1:4000 dilution of donkey anti-rabbit IgG, horseradish peroxidase linked whole antibody (GE Healthcare, Piscataway, NJ) was added, incubation was continued for 1h. The wells were then washed again using PBST and the reaction was developed by peroxidase substrate, ABTS (2, 2′-azino-bis (3-ethylbenzthiazoline-6-sulphonic acid)). Absorbance measurements were taken at 415 nm.
Single-turnover ATPase assays
Single-turnover ATPase assays of Ssa1 were performed as described in Sahi et al [20]. 32P-α-ATP-Ssa1 complexes were incubated with a 2-fold molar excess of J proteins at 24°C. Aliquots were applied to a TLC plate for detection of ATP and ADP. The amount of ATP hydrolyzed to ADP over time was determined using Imagequant TL (GE).
Protein crystallization
Several N-terminal fragments of Sis1 protein from S. cerevisiae, varying in length over the range of 67–127 residues, were designed to test protein expression and crystallization. The ORFs were amplified with KOD DNA polymerase using conditions and reagents provided by Novagen, Madison, WI. The DNA fragments were cloned into the pMCSG7 vector [33] using a modified LIC protocol [34]. This process generated expression clones producing a fusion protein with an N-terminal His6 affinity tag and a TEV protease recognition site. The proteins were expressed and purified using standard procedures on an AKTAxpress automated purification system (GE/Amersham) [35]. The concentration of purified protein was determined utilizing a ND-1000 spectro-photometer system (NanoDrop Technologies). The tag was removed as described above, except incubation with TEV protease was carried out for 49 hours at 4°C. The purified proteins were dialyzed in crystallization buffer (20 mM HEPES pH 8.0, 250 mM NaCl, 2 mM DTT) for 24 hours and concentrated using a Centricon Plus-20 concentrator with a MW cut off of 5,000 Da (Millipore Corp.).
Crystallization conditions were determined with a sparse crystallization matrix at 4°C and 16°C using the sitting-drop vapor-diffusion technique. The best crystals were obtained for the construct expressing 89-residue protein from 2.4 M sodium malonate pH 7.0 after 7 days of incubation at 16°C. Crystals selected for data collection were soaked in the crystallization buffer supplemented with 28% sucrose and flash-cooled in liquid nitrogen.
Data collection, structure determination and refinement
Single-wavelength X-ray diffraction data were collected at 100 K temperature at the 19-ID beamline of the Structural Biology Center [36] at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect. The intensities were integrated and scaled with the HKL3000 suite [37].
The structure was determined by molecular replacement using MOLREP program [38] from CCP4 suite using the J-domain of human DnaJ homolog subfamily B member 8 as a search model (PDB ID: 2DMX) [22]. This NMR structure was modified by manual truncation and was used as the starting model for model building and refinement. Several rounds of manual adjustment of structure models using COOT [39] and refinement with Refmac program [40] from CCP4 suite [22] were performed. The stereochemistry of the structure was validated with PHENIX suite [41] incorporating MOLPROBITY tools [42]. Atomic coordinates and structure factors were deposited into the Protein Data Bank as 4RWU.
Supplementary Material
Highlights.
J—protein co—chaperone Sis1, but not Ydj1, interacts with Hsp70 C—terminal EEVD Sis1:EEVD interaction important for efficient partnership in protein folding Sis1 J—domain/gly—rich region salt bridge disruption overcomes Sis1:EEVD requirement Disruption of Sis1:EEVD interaction aids Sis1 in substituting for Ydj 1 in vivo Sis1:EEVD interaction may divert Sis1—bound clients from productive folding pathways
Acknowledgments
We thank Jaroslaw Marszalek for insightful discussions, Minyi Gu for providing Sis1 clones and and all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting X-ray diffraction data collection, which is operated by the University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. This work was supported by National Institutes of Health Grants GM31107 (EAC) and GM094585 (AJ). The work of M.B. was supported by Polish National Science Center Grant 2013/09/N/NZ2/01979.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nature reviews Molecular cell biology. 2013;14:630–42. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annual review of biochemistry. 2013;82:323–55. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 3.Sontag EM, Vonk WI, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Current opinion in cell biology. 2014;26:139–46. doi: 10.1016/j.ceb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer MP. Hsp70 chaperone dynamics and molecular mechanism. Trends in biochemical sciences. 2013;38:507–14. doi: 10.1016/j.tibs.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nature reviews Molecular cell biology. 2010;11:579–92. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freeman BC, Myers MP, Schumacher R, Morimoto RI. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. The EMBO journal. 1995;14:2281–92. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- 8.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Sha B. Crystal structure of yeast mitochondrial outer membrane translocon member Tom70p. Nature structural & molecular biology. 2006;13:589–93. doi: 10.1038/nsmb1106. [DOI] [PubMed] [Google Scholar]
- 10.Odunuga OO, Hornby JA, Bies C, Zimmermann R, Pugh DJ, Blatch GL. Tetratricopeptide repeat motif-mediated Hsc70-mSTI1 interaction. Molecular characterization of the critical contacts for successful binding and specificity. The Journal of biological chemistry. 2003;278:6896–904. doi: 10.1074/jbc.M206867200. [DOI] [PubMed] [Google Scholar]
- 11.Alvira S, Cuellar J, Rohl A, Yamamoto S, Itoh H, Alfonso C, et al. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nature communications. 2014;5:5484. doi: 10.1038/ncomms6484. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Wu Y, Qian X, Sha B. Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. The Biochemical journal. 2006;398:353–60. doi: 10.1042/BJ20060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aron R, Lopez N, Walter W, Craig EA, Johnson J. In vivo bipartite interaction between the Hsp40 Sis1 and Hsp70 in Saccharomyces cerevisiae. Genetics. 2005;169:1873–82. doi: 10.1534/genetics.104.037242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson JL, Craig EA. An essential role for the substrate-binding region of Hsp40s in Saccharomyces cerevisiae. The Journal of cell biology. 2001;152:851–6. doi: 10.1083/jcb.152.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan CY, Lee S, Ren HY, Cyr DM. Exchangeable chaperone modules contribute to specification of type I and type II Hsp40 cellular function. Molecular biology of the cell. 2004;15:761–73. doi: 10.1091/mbc.E03-03-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caplan AJ, Douglas MG. Characterization of YDJ1: a yeast homologue of the bacterial dnaJ protein. The Journal of cell biology. 1991;114:609–21. doi: 10.1083/jcb.114.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–45. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Molecular biology of the cell. 2013;24:2076–87. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Summers DW, Wolfe KJ, Ren HY, Cyr DM. The Type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PloS one. 2013;8:e52099. doi: 10.1371/journal.pone.0052099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahi C, Kominek J, Ziegelhoffer T, Yu HY, Baranowski M, Marszalek J, et al. Sequential duplications of an ancient member of the DnaJ-family expanded the functional chaperone network in the eukaryotic cytosol. Molecular biology and evolution. 2013;30:985–98. doi: 10.1093/molbev/mst008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Z, Cyr DM. Protein folding activity of Hsp70 is modified differentially by the hsp40 co-chaperones Sis1 and Ydj1. The Journal of biological chemistry. 1998;273:27824–30. doi: 10.1074/jbc.273.43.27824. [DOI] [PubMed] [Google Scholar]
- 22.Szyperski T, Pellecchia M, Wall D, Georgopoulos C, Wuthrich K. NMR structure determination of the Escherichia coli DnaJ molecular chaperone: secondary structure and backbone fold of the N-terminal region (residues 2–108) containing the highly conserved J domain. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:11343–7. doi: 10.1073/pnas.91.24.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luke MM, Sutton A, Arndt KT. Characterization of SIS1, a Saccharomyces cerevisiae homologue of bacterial dnaJ proteins. The Journal of cell biology. 1991;114:623–38. doi: 10.1083/jcb.114.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aron R, Higurashi T, Sahi C, Craig EA. J-protein co-chaperone Sis1 required for generation of [RNQ+] seeds necessary for prion propagation. The EMBO journal. 2007;26:3794–803. doi: 10.1038/sj.emboj.7601811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan W, Craig EA. The glycine-phenylalanine-rich region determines the specificity of the yeast Hsp40 Sis1. Molecular and cellular biology. 1999;19:7751–8. doi: 10.1128/mcb.19.11.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sondheimer N, Lopez N, Craig EA, Lindquist S. The role of Sis1 in the maintenance of the [RNQ+] prion. The EMBO journal. 2001;20:2435–42. doi: 10.1093/emboj/20.10.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, et al. PaxDb, a database of protein abundance averages across all three domains of life. Molecular & cellular proteomics: MCP. 2012;11:492–500. doi: 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Molecular biology of the cell. 2012;23:3041–56. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alberti S. Molecular mechanisms of spatial protein quality control. Prion. 2012;6:437–42. doi: 10.4161/pri.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki H, Noguchi S, Arakawa H, Tokida T, Hashimoto M, Satow Y. Peptide-binding sites as revealed by the crystal structures of the human Hsp40 Hdj1 C-terminal domain in complex with the octapeptide from human Hsp70. Biochemistry. 2010;49:8577–84. doi: 10.1021/bi100876n. [DOI] [PubMed] [Google Scholar]
- 31.Pfund C, Huang P, Lopez-Hoyo N, Craig EA. Divergent functional properties of the ribosome-associated molecular chaperone Ssb compared with other Hsp70s. Molecular biology of the cell. 2001;12:3773–82. doi: 10.1091/mbc.12.12.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer AE, Hoover LA, Craig EA. The cytosolic J-protein, Jjj1, and Rei1 function in the removal of the pre-60 S subunit factor Arx1. The Journal of biological chemistry. 2010;285:961–8. doi: 10.1074/jbc.M109.038349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein expression and purification. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 34.Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR. High throughput methods for gene cloning and expression. Protein expression and purification. 2002;25:1–7. doi: 10.1006/prep.2001.1602. [DOI] [PubMed] [Google Scholar]
- 35.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, et al. Automation of protein purification for structural genomics. Journal of structural and functional genomics. 2004;5:111–8. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, et al. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. Journal of synchrotron radiation. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta crystallographica Section D, Biological crystallography. 2006;62:859–66. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 38.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta crystallographica Section D, Biological crystallography. 2010;66:22–5. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta crystallographica Section D, Biological crystallography. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 42.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic acids research. 2004;32:W615–9. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.