

Abstract
Heat Shock Factor 1 (HSF1) is critical for defending cells from both acute and chronic stresses. In aging cells the DNA binding activity of HSF1 deteriorates correlating with the onset of pathological events including neurodegeneration and heart disease. We find that DNA binding by HSF1 is controlled by lysine deacetylases with HDAC7, HDAC9, and SIRT1 distinctly increasing the magnitude and length of a Heat Shock Response (HSR). In contrast, HDAC1 inhibits HSF1 in a deacetylase-independent manner. In aging cells the levels of HDAC1 are elevated and the HSR is impaired, yet reduction of HDAC1 in aged cells restores the HSR. Our results provide a mechanistic basis for the age-associated regulation of the HSR. Besides HSF1, the deacetylases differentially modulate the activities of unrelated DNA binding proteins. Taken together, our data further support the model that lysine deacetylases are selective regulators of DNA binding proteins.
Keywords: Heat shock response, Heat Shock Factor 1, Lysine deacetylase, Aging
Introduction
Homeostasis relies on a dynamic cellular environment to aptly react to fluctuating physiological demands [1]. Inducible pathways culminating in gene expression programs exemplify the need for prompt action. For example, physiological stressors such as elevated temperatures or steroid hormones trigger signaling systems that cascade into transcriptional responses. In general, these multistep paths are driven forward by cooperative interactions between select protein partners including the activated transcription factors, histone acetyltransferases, chromatin remodelers, and preinitiation factors [2]. As stress-signaling pathways require quick transduction of information, the systems are reliant on efficient transitions between the cooperatively assembled structures as well as guidance to the next step [3]. Recent studies highlight the utility of molecular chaperones in mediating the rapid disassembly of transcription complexes as well as the prominent use of post-translational modifiers for guiding transcription pathways [4] and [5]. Here we investigated how lysine deacetylases control stress-signaling pathways by directly modulating the DNA binding activities of transcription factors. As a molecular model we focus on the mammalian Heat Shock Response (HSR).
The HSR is an evolutionarily conserved stress-signaling process adapting cells to pathological challenges and is a paradigm for investigating systems directly reactive to physiological cues [6]. Significantly, HSR dysfunction has dire consequences. When organisms age the capacity of the HSR declines impairing protein homeostasis and correlating with the onset of numerous ailments including neurodegeneration, type II diabetes, and heart disease [7]. Heat Shock Factor 1 (HSF1) is the principal mediator of the eukaryotic HSR as it coordinates a cell’s counteractions to both acute and chronic stressors [6]. Notably, HSF1 also is a key modulator of lifespan and a potent driver of oncogenesis [8] and [9]. Central to these events is HSF1 DNA occupancy, which is requisite to govern the gene programs driving the different cell fates. Therefore, it is imperative to understand how the DNA binding activity of HSF1 is controlled.
In transitioning from a quiescent monomer to an activated trimer the post-translational modification façade of HSF1 is revamped [10]. For instance, correlations between serine/threonine phosphorylation levels and acquisition of HSF1 trimerization, nuclear localization, and transcriptional potency have been found [6]. Besides phosphorylation, HSF1 is acetylated, sumoylated, and ubiquitinated [11]. While the significances of these modifications are not completely understood, lysine acetylation within the DNA binding domain can inhibit DNA interactions whereas acetylation of the central regulatory domain deters degradation of HSF1 [12], [13], and [14]. Hence, modification of select HSF1 lysines is a potent means to control the HSR and support cell vitality.
Lysine acetylation within the binding cleft of HSF1 is sufficient to block DNA interactions. Prior studies have shown that the GCN5 acetyltransferase selectively targets HSF1 lysine 80 (K80), which stabilizes HSF1 to DNA, whereas K80 is one of many residues modified by the P300 acetyltransferase [13] and [14]. In either case, acetylation of K80 impedes HSF1 DNA binding activity and limits the HSR. Minimally, the Sirtuin 1 (SIRT1) deacetylase relieves the inhibitory mark thereby enabling stress-induced HSF1 DNA binding and extending a HSR [12]. The role of SIRT1 is not universal since blockage of SIRT1 does not affect neuroprotection yet inhibiting type I/II histone deacetylases (HDACs) hinders neuronal heat-defenses [15]. While the responsible HDAC has not been identified, the work illustrates that the reprieve of the constraining HSF1 modifications will involve cell and disease specific lysine deacetylases (KDACs).
Intertwined within the acetylation process controlling transcription factor DNA binding activities is the p23 molecular chaperone [13]. Before the lysines coordinating DNA interactions can be acetylated p23 must dissociate the DNA-bound complex, as DNA binding conceals the target lysine [13]. The involvement of p23 in transcription factor acetylation events was founded in a synthetic genetic array linking the chaperone to both acetylases and deacetylases along with the discovery that the loss of p23 triggers increased expression of GCN5 and HDAC1 [13] and [16]. In our early model we suggested that the increase in HDAC1 balanced the elevation in GCN5—extra HDAC1 might remove the superfluous GCN5 acetylation modifications thereby reestablishing the DNA binding activities of target proteins [13]. Here, we tested this basic hypothesis and discovered that regulation through lysine deacetylase action is complicated as KDACs both positively and negative modulate DNA binding activities using deacetylase-dependent and -independent mechanisms.
Results
Select lysine deacetylases remove the inhibitory GCN5 mark on HSF1
To test the impact of HDAC1 on GCN5-mediated inhibition of the HSR we exploited the heat-induced DNA binding activity of HSF1 in the human embryonic kidney cell line 293T. A priori, we had anticipated that most, if not all, lysine deacetylases (KDACs) would suffice in removing the inhibitory K80 acetyl group. Given our previous genetic data linking p23/GCN5/HDAC1, we believed HDAC1 would certainly counter the GCN5 mark on HSF1 [13]. While overexpression of GCN5 by transient transfection effectively diminished the binding of HSF1 to the proximal heat shock element (HSE) of the HSP70 promoter following exposure to heat-stress, coexpression of HDAC1 did not rescue binding activity (Fig. 1a). Yet, joint expression of SIRT1 was adequate to recover stress-induced HSF1 DNA binding (Fig. 1a). Hence, the HDAC1 and SIRT1 deacetylase display discriminate regulation of HSF1 in conjunction with GCN5.
Fig. 1.

Lysine deacetylases selectively relieve GCN5-inhibition of HSF1 DNA binding activity. (a) Human 293T embryonic kidney cells were transiently transfected with expression constructs for human GCN5, SIRT1, or HDAC1, as marked. Cells were incubated at 37°C (-) or 42°C (+) for 20 min and the HSF1 DNA binding activity was visualized by EMSA using a radiolabeled HSE oligonucleotide. (b) 293T cells were transiently transfected with expression vectors for GCN5 and the indicated lysine deacetylases. Cells were untreated (-) or exposed to 42°C for 20 min (+) and the HSF1 DNA binding was monitored by EMSA. The DNA binding assays were performed a minimum of 6 replicates and representative data is presented.
To further explore the selectivity of KDACs with a non-histone protein target we screened a panel of 12 deacetylases with representatives from the three main KDAC groups. Based on protein structure and catalytic activity there are 18 known mammalian KDACs divided into four clusters: class I (HDAC1-3, and HDAC8); class II (HDAC4-7, HDAC9-10); class III (SIRT1-7); and class IV (HDAC11) [17] and [18]. Only 3 KDACs (HDAC7, HDAC9, and SIRT1) countered the GCN5 inhibitory effect on heat-induced HSF1 DNA binding (Fig. 1b).
Lysine deacetylases discriminately regulate HSF1 activities
To determine if the specificity of the deacetylases was particular to the GCN5 mark, we checked the intrinsic heat-triggered HSF1 pathway. Acetylation of HSF1 increases under heat shock conditions thereby dampening the response [12] and [13]. As heat shock promoters are maintained in nucleosome depleted states in vivo, we continued to exploit EMSAs as a means to monitor HSF1 DNA interactions. Similar to the relief of GCN5 inhibition, overexpression of HDAC7, HDAC9, and SIRT1 by transient transfection stimulated heat-triggered HSF1 DNA binding (Fig. 2a). Hence, these 3 KDACs can counter acetylation marks that are repressive to HSF1 DNA binding activity independent of the modifying source.
Fig. 2.

HSF1 functions are positively and negatively regulated by lysine deacetylases. Human 293T cells were transfected with expression vectors for HSF1 and the indicated lysine deacetylases. Cells were incubated at 37°C (-) or 42°C (+) for 2 h and the HSF1 DNA binding activity was assessed by EMSA (a), the RNA levels of endogenous HSP70 were measured by RT-qPCR (b), and the HSF1 steady-state protein levels and acetylation status were determined by immunoblot analysis following immunoprecipitation of HSF1 (c). The EMSA and immunoblot data are representative of experiments done in triplicate. The RT-PCR data are the averages of 6 independent replicas and the error bars are S.E.M..
Unexpectedly, HDAC1 overexpression in 293T cells was sufficient to impair HSF1 DNA interactions (Fig. 2a). The impact of the KDACs is independent of cell context, as heat-activated HSF1 was differentially modulated by the deacetylases in both rabbit reticulocyte lysate and mouse embryonic fibroblasts (Supplementary Fig. 1). Since only the individual KDACs were expressed with HSF1 the effects likely do not involve large deacetylase-complexes (e.g., NuRD or SIN3). Rather, the individual KDACs appear sufficient to control HSF1 DNA binding activity.
Importantly, the stress-induced activation of the endogenous HSF1-target gene HSP70 paralleled the changes in the DNA binding effects imposed by HDAC1, HDAC7, HDAC9, and SIRT1. In 293T cells overexpression of HDAC1 decreased HSP70 RNA production whereas HDAC7, HDAC9, and SIRT1 elevated transcription of HSP70 (Fig. 2b). Hence, KDAC modulation of HSF1 DNA binding imposes direct effects on gene expression.
Conversely, HSF1 acetylation levels did not correspond to the functional impact of the KDACs. Using an antibody recognizing acetyl-lysine it was apparent that HDAC9 and SIRT1 deacetylated HSF1 (Fig. 2c). However, either HDAC1 or HDAC7 overexpression elevated HSF1 pan-acetylation (Fig. 2c) despite the contrasting functional effects of HDAC1 and HDAC7 on HSF1 activities (Fig. 2a and 2b). No matter the extent of acetylated lysine, HSF1 steady-state protein levels remained relatively constant (Fig. 2c). As the antibody we employed recognizes just acetylated lysine and HSF1 has lysines throughout its primary sequence, we mapped the affected area(s). Overexpression of either HDAC1 or HDAC7 increased lysine acetylation in the carboxyl-terminus of HSF1 but only HDAC1 prompted acetylation of the DNA binding domain (DBD) (Supplementary Fig. 2).
KDACs use distinct mechanisms to differentially control HSF1 DNA binding
To understand the differential regulation we mapped the HSF1 domains associating with the different deacetylases. Full-length HSF1 or truncation mutants representing the amino-terminal DBD, central regulatory domain (RD), or carboxyl-terminal activation domain (AD) of HSF1 were produced in rabbit reticulocyte lysate. The HSF1 derivatives were incubated with HDAC1, HDAC7, HDAC9, or SIRT1 and then immunoprecipitated. All three positively acting KDACs (HDAC7, HDAC9, and SIRT1) associated with the AD (Fig. 3a). Only SIRT1 bound to the RD, which might explain the more robust SIRT1-influence (Fig. 2). In contrast, HDAC1 interacted with the DBD of HSF1 (Fig. 3a).
Fig. 3.

The HSF1 domains associating with the KDACs varied. (a) His-tagged HSF1 derivatives comprising the full-length protein (FL), the amino-terminal DNA binding domain (DBD), central regulatory domain (RD), or carboxyl-terminal activation domain (AD) were in vitro transcribed/translated in the presence of 35S trans-label, and combined with 35S trans-labeled HDAC1, HDAC9, SIRT1, or HDAC7, as marked. The HSF1 proteins were immunoprecipitated with α-His antibody, the precipitants were resolved by denaturing gel electrophoresis, and the radiolabeled proteins were visualized using a PhosphorImager. In the upper left panel displaying the FL HSF1 pull-downs the positions of the various full-length KDACs are marked with asterisks. During the IP step SIRT1 was often proteolytically cleaved, as can be seen in the RD and AD panels (the combined molecular size of the proteolytic fragments is equivalent to full length SIRT1). (b) The DBD or DBD-AD fusion fragments were in vitro translated, combined with aliquots of in vitro prepared HDAC1, HDAC9, SIRT1, or mock treated reticulocyte lysate along with radio-labeled HSE oligonucleotide, incubated at 37°C (-HS) or 43°C (+HS) for 30 min, resolved by native gel electrophoresis, and the protein-DNA complexes were visualized using a PhosphorImager.
To confirm if these modes of association functionally impact HSF1 we tested the influence of HDAC1, HDAC7, HDAC9, or SIRT1 on the DNA binding activities of the DBD or a fusion of the DBD and AD (DBD-AD). HDAC1 inhibited both the DBD and DBD-AD derivatives yet HDAC7, HDAC9, and SIRT1 only stimulated the activity of the DBD-AD fusion (Fig. 3b). It is possible, however, that the DNA binding activity of the DBD fragment is already maximal and therefore further stimulation isn’t feasible. Nevertheless, we suspect HDAC1 binds to the DBD and sterically blocks DNA binding whereas HDAC7, HDAC9, and SIRT1 use the AD (SIRT1 likely incorporates the RD too) as a platform for deacetylating K80 without interfering with the DBD.
The KDAC domains associating with and regulating HSF1 varied. For the positively acting KDACs the regions interacting with HSF1 contained the deacetylase catalytic domains (Supplementary Fig. 3). SIRT1 relied on its catalytic core whereas HDAC9 utilized the core plus flanking areas (Supplementary Fig. 3). In contrast, the non-catalytic amino-terminus of HDAC1 bound HSF1 and impeded DNA binding (Fig. 3a and Supplementary Fig. 3). Thus, supporting our contention that HDAC1 inhibits HSF1 through steric interference independent of its deacetylase function.
HSF1 Inhibition by HDAC1 is dependent on the p23 molecular chaperone
The ability of HDAC1 to foster the free state of HSF1 might explain its genetic relationship with the p23 chaperone [16]. Previously we found that GCN5 maintains HSF1 DNA-separation after p23 releases HSF1 from DNA [13]. To test whether HDAC1 shares a similar p23-reliance we tested the effect of HDAC1 in the presence and absence of p23. Addition of HDAC1 to reticulocyte lysate immunodepleted of p23 led to enhanced HSF1 DNA binding rather than inhibition (Fig. 4a lane 6). We suspect that HDAC1 can deacetylate K80 in the absence of p23. Under these conditions HSF1 favors binding to DNA rather than interacting with HDAC1. Restoration of p23 to normal reticulocyte lysate levels triggered release of HSF1 from DNA thereby allowing HDAC1 to block HSF1 DNA binding (Fig. 4a lane 7). Importantly, addition of the deacetylase defective HDAC1 mutant H141A was effectively inhibited HSF1 DNA binding in the presence of p23 but did not promote DNA interactions in the absence of p23, as H141A is unable to deacetylate any free HSF1 (Fig. 4b). Hence, the genetic links between p23, HDAC1, and GCN5 likely highlight a regulatory system for controlling the disassembly of protein-DNA complexes.
Fig. 4.
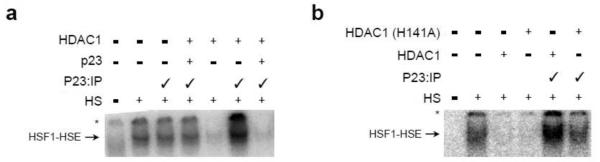
In the absence of p23 HDAC1 promotes the DNA binding activity of HSF1. (a) Aliquots of in vitro translated HSF1 were mock treated or immunodepleted of p23 (P23:IP ✓), incubated at 43°C for 30 min with radiolabeled HSE, in the presence or absence of HDAC1 with or without supplementation of p23 to normal reticulolysate levels. (b) Aliquots of in vitro translated HSF1 were mock treated or immunodepleted of p23 (P23:IP ✓), incubated at 43°C for 30 min with radiolabeled HSE, in the presence or absence of wild type HDAC1 or the deacetylase point mutant H141A [43]. The reactions were resolved by native gel electrophoresis and the protein-DNA complexes were visualized using a PhosphorImager.
KDACs tune the timing of the Heat Shock Response
In addition to the mechanisms of KDAC action, we wanted to establish the physiological relevance of the KDACs with the HSR. The fundamental purpose of HSF1 is to transduce environmental signals into select gene programs by binding HSEs at or near regulated genes [6]. Mammalian cells cultured at 37°C will undergo a transient response when exposed to 42°C in which HSF1 binds DNA maximally after 1-2 h with little to no association following 4 h [19]. Significantly, the KDACs differentially altered the HSF1 pathway. In 293T cells HDAC1 shortened the HSR while HDAC7, HDAC9, and SIRT1 all extended the response—SIRT1 increased DNA binding throughout the time course whereas HDAC7 raised binding at early times and HDAC9 altered later binding events (Fig. 5). In contrast, HDAC6 had no apparent influence on the HSR (Fig. 5). Despite the provoked variances in the HSR, the KDACs localized to the HSP70 promoter in parallel with HSF1 (Supplementary Fig. 4). The distinct regulatory effects suggest the KDACs might be a mechanism for tuning the stress-signaling pathway in a cell or disease specific manner.
Fig. 5.

KDACs differentially modulate the HSR. The influence of SIRT1, HDAC1, HDAC7, HDAC9, or HDAC6 overexpression on the HSF1 DNA binding attenuation period was determined. 293T cells transfected with the indicated KDACs were shifted from 37°C to 42°C to initiated the experiment, samples were removed at the indicated times, and HSF1 DNA binding activity was determined by EMSA. The data are representative of experiments done in triplicate.
HDAC1 impairs the HSR in aged cells
To explore this possibility we examined the HSR in young and old cells along with the expression of select KDACs. Pertinently, physical and psychological stressors fail to induce HSF1 DNA binding activity in aged cells despite comparable steady-state protein levels, heat-induced phosphorylation, trimerization capacity, and nuclear localization of HSF1 [20], [21], [22], and [23]. Additionally, extracts from high-passage cell lines or older human (ages 76-88 years) donor cells contain a heat-labile, transacting inhibitory factor of HSF1’s DNA binding activity [24] and [25].
As an aging model we used serially passaged mouse embryonic fibroblasts (MEFs) checking cells with population doublings (PDs) of either 12 or 26. While the PD-12 MEFs (i.e., young cells) had a robust HSE binding activity following exposure to 42°C the older PD-26 cells did not (Fig. 6a). The protein levels of HSF1 and HDAC9 remained constant, yet SIRT1 declined and HDAC1 increased in the PD-26 MEFs (Fig. 6b). Similar age-dependent changes to SIRT1 and HDAC1 have been noted previously [26], [27], and [28]. Given the expression changes to SIRT1 and HDAC1, we manipulated the levels of these two KDACs in an attempt to restore the HSR in aged cells.
Fig. 6.

HDAC1 inhibits the HSR in aged cells. Mouse embryonic fibroblasts (MEFs) were passaged for 12 or 26 population doublings (PDs). The attenuation responses of the PD-12 or PD-26 cells to a 42° heat-stress were visualized by EMSA (a) and the steady-state protein levels of HDAC1, SIRT1, HDAC9, and HSF1 were measured by immunoblot analysis (b). The impact of knocking down SIRT1 in PD-12 or knocking down HDAC1 in PD-26 cells on the heat-induced DNA binding activity of HSF1 was assessed by EMSA using straight extracts or equal mixtures of the PD-12 and PD-26 fractions, as marked. The relative levels of HSF1, HDAC1 and SIRT1 were monitored by immunoblot analysis as shown. The EMSA and immunoblot data are representative of experiments done in triplicate.
Knock down of HDAC1 in the PD-26 MEFs reestablished the heat-triggered DNA binding activity of HSF1 (Fig. 6c). Notably, mixing cell extracts from the old and new MEFs impeded HSF1-HSE interactions unless HDAC1 had been knocked down in the older cells (Fig. 6c). Although the decline in SIRT1 likely contributes to the lowered HSR of aged cells, HDAC1 appears to be the age-dependent, trans-acting HSF1 inhibitory protein.
KDACs display selective regulation of DNA binding proteins
Besides HSF1, we tested the influence of the KDACs on the DNA binding activities of the Glucocorticoid Receptor (GR) and Cell Division Cycle 6 (CDC6) proteins to weigh the target range of the deacetylases. Like HSF1, GR is a central mediator of a stress-signaling pathway culminating in a transcriptional response. GR reacts to steroid-hormone fluctuations prompted by programed patterns (e.g., diurnal cycle) and physiological stressors (e.g., starvation) [3]. Comparable to HSF1, overexpressed HDAC1 inhibited while SIRT1 overexpression enhanced the signal-induced DNA binding and transcriptional activities of GR (Fig. 7a and 7b). The observed increase in the transcription response mediated by GR following HDAC6 overexpression (Fig. 7b) likely resulted from the established HDAC6-modulation of HSP90 chaperone activity, which impacts HSP90-clients such as GR [29].
Fig. 7.

The lysine deacetylases selectively regulate DNA binding proteins. The impact of the indicated KDACs on the hormone (100 nM Dexamethasone)-induced DNA binding activity of the Glucocorticoid Receptor (GR) (a), transcriptional activity of GR (b), or DNA binding activity of the Cell Division Cycle 6 (CDC6) protein (c) was determined. The DNA binding activities were assessed by EMSA using oligonucleotides select for either GR or CDC6 and the RNA levels of endogenous GR target gene APA1a were measured by RT-qPCR and normalized to GAPDH. The RT-PCR data are the averages of 6 independent replicas and the error bars are S.E.M..
On the other hand, CDC6 supports the loading of DNA replication complexes in late M and early G1 phases of the cell cycle [30]. Unlike the transcription factors, CDC6 was not significantly affected by HDAC1. Rather, SIRT1 overexpression inhibited CDC6 DNA interactions while HDAC4, HDAC6, and SIRT6 overexpression stimulated (Fig. 7c). As both SIRT1 and SIRT6 impact organismal longevity and genome stability [31], our data provide additional insights into the Sirtuin-mechanisms influencing cells. Taken together, our results show that lysine deacetylases selectively target proteins to deliver distinct regulatory effects.
Discussion
The presented study demonstrates that lysine deacetylases support a network for effectively controlling the DNA binding activities of diverse proteins. The physiological relevance of the deacetylase system was highlighted through our focused work on the HSF1-controlled Heat Shock Response. The HSR coordinates protein homeostasis (i.e., proteostasis) which is the central process mediating a cell’s reaction to biological challenges by maintaining the optimal function of the proteome [7]. As cells age the proteostasis machinery falters correlating with the pathogenesis of numerous geriatric disorders including neurodegeneration, type II diabetes, and heart disease. Although an age-dependent impairment of the HSR has long been linked to the loss of HSF1 DNA binding activity [21], [22], [24], and [25], the mechanistic basis for the breakdown was not previously understood. In showing the connection between KDACs, HSF1, and cellular aging our studies support a model in which the impact of lysine deacetylases extend beyond histone modifications under both normal and disease conditions.
Post-translational modifications, including acetylation, are an effective means to respond to internal and external cues. Lysine acetylation often occurs at conserved sites within structured domains to connect signaling pathways to a cell’s metabolic status [32]. Commonly, acetylation is synonymous with epigenetics yet high-throughput studies have shown the generality of the target proteins [33]. The acetylation process classically involves 3 classes of factors—writers, readers and erasers [18]. Yet, the mark may also be used for direct functional effects, as shown here with DNA binding activities. Under these circumstances the regulatory events are balanced by the intersection between lysine acetylases and deacetylases.
The acetylation process was first shown to impinge on a transcription factor by the discovery that p53 is acetylated in its carboxyl-terminus thereby leading to stimulation of the central DNA binding domain [34]. In contrast, targeting lysines within a DNA binding domain inhibits DNA interactions of various proteins including HSF1, GR, CDC6, and YY1 [12], [13], [14], and [35]. Commonly, SIRT1 has been shown to be the eraser of transcription factor acetyl-groups, as its substrates include p53, HSF1, p73, and KU70 [9], [12], [36], [37], and [38]. However, with the exception of HSF1, SIRT1 does not typically remove DBD acetyl-marks.
Our data confirm the impact of SIRT1 with HSF1 and extend the number and types of deacetylases to HDAC7 and HDAC9 that are able to remove inhibitory acetyl-groups within a DBD. While a prior study had shown that HDAC1/2 associate with the DBD of the YY1 transcription factor, the HDACs did not remove the DBD acetylation [35]. Perhaps comparably, we observed an association between HDAC1 and the HSF1 DBD (Fig. 3). Unexpectedly, HDAC1 sterically interfered with HSF1 DNA binding in a deacetylase-independent manner. Given the capacity of HDAC1 to also impede GR DNA interactions, we suspect that HDAC1 blocks the DNA binding activities of proteins working within signaling pathways that culminate in a gene program. This simple mechanism would provide a two-fold strategy to quickly silence a response, as it prevents the activator from engaging induced promoters and it places a deacetylase near the activated promoters to quell the response through histone deacetylation.
In demonstrating the capacity of the various lysine deacetylases to differentially control the DNA binding properties of heterologous proteins our study significantly broadens the model of KDAC-action along the genome. Although the involvement of the acetylation process in the control of transcription factors has been established, the work has focused on SIRT1 and HDAC1/2 [39]. Our studies expand the range of deacetylases involved in DNA binding regulation thereby implicating all KDACs in the process. Interestingly, KDAC inhibitors are an expanding class of anticancer agents that trigger cell growth arrest and apoptosis in a variety of human cancers by means not solely attributable to histone acetylation levels [40]. Our findings that KDACs contribute to the activity of numerous DNA binding factors might fully explain the efficacy of deacetylase therapeutics. Perhaps notably, recent studies showed the capacity of activated HSF1 to drive cancerous growth [9], [41], and [42]. Given the apparent changes to deacetylase expression patterns in cancer cells [18] and [39], it is plausible that the KDACs are triggering the heat-independent DNA binding activity of HSF1 during cellular transformation. Overall, our work provides novel insights into the molecular machinery regulating protein DNA binding activities.
Materials and Methods
Plasmids
Several individuals kindly provided expression vectors including Dr. R.I. Morimoto (Northwestern University) with human HSF1 (pcDNA3c-myc-HSF1) and SIRT1 (pcDNA3flag-SIRT1), Dr. B.S. Pace (Georgia Health Sciences University) and Dr. T.K. Chatterjee (University of Cincinnati) with human HDAC9 (pTarget-HDAC9 and pCMVflag-HDAC9, respectively), and Dr. S. Prasanth (University of Illinois) with human CDC6 (CMVflag-CDC6). The vectors for human HDAC1 (pcDNA3FlaghHDAC1), HDAC3 (pcDNA3FlaghHDAC3), HDAC4 (pcDNA3FlaghHDAC4), HDAC6 (pcDNA3FlaghHDAC6), HDAC7 (pcDNA3FlaghHDAC7), HDAC8 (pcDNA3FlaghHDAC8), HDAC9 (pTargethHDAC9), SIRT1 (pcDNA3hSIRT1), SIRT2 (pcDNA3FlaghSIRT2), SIRT3 (pcDNA3FlaghSIRT3), SIRT4 (pcDNA3FlaghSIRT4), and SIRT6 (pcDNA3FlaghSIRT6) were obtained from Addgene. The plasmids for expressing human p23 (pET23a-p23), GCN5 (pcDNA3FlaghGCN5), and GR (pcDNA3hGR) were described previously [13]. The truncation derivatives of HDAC1, HDAC9, SIRT1, and HSF1 were generated by PCR and cloned into pET23a.
Antibodies
The antibodies used in this study were α-p23 (JJ3), α-HSF1 (Enzo ADI-SPA-901), α-HDAC1 (SCBT sc-81598), α-HDAC9 (Abcam ab59718), α-SIRT1 (Abcam ab110304), α-GCN5 (SCBT sc-9078), α-Acetylated Lysine (Cell Signaling 9441), α-Myc (SCBT sc-40), α-Flag M1 (Sigma F3040), and α-His (SCBT sc-8036).
Mammalian cells, DNA transfections, and Lentiviral infections
Human 293T embryonic kidney, A549 lung carcinoma, HeLa cervical adenocarcinoma, or mouse embryonic fibroblast (MEF) cells, growing on 6-well plates in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) were transfected with 1.5 μg of pcDNA3hGR or pcDNA3hHSF1 and 1.5 μg of pcDNA3hGCN5, and/or pcDNA3FlaghHDAC1, 3, 4, 6-8, pcDNA3FlaghSIRT1-4, 6 by lipid-mediated transfection (Lipofectamine 2000) according to manufactures instructions (Invitrogen); DNA amounts were standardized to 3 μg using pcDNA3. The FBS used in the GR assays was charcoal-stripped to remove existing hormones. After 24 h, fresh media was applied and the cells were either heat-stressed then collected at the indicated times or supplemented with 100 nM dexamethasone (USB) then harvested after 24 h. For extract preparation, phosphate-buffered saline washed cells were lysed (20 min, 4 °C) in Buffer C or RIPA Buffer. For HDAC1 and SIRT1 knockdown, the Lentiviruses were prepared in 293T cells by cotransfecting HDAC1 or SIRT1-selective shRNAs (Sigma Aldrich MISSION shRNA SHCLND NM_004964 and NM_012238, respectively), VSV-G packaging plasmid, and dR8.2 envelope plasmid using TransIT-LT1 transfection reagent (Mirus) according to manufactures instructions. MEF cells growing on 6-well plates in DMEM with 10% FBS and 8 μg/mL polybrene were infected with medium containing 100 μL of Lentiviral particles.
Quantitative real time RT-PCR analysis
To assess the expression of endogenous HSP70 and APA1A genes, cells were harvested and RNA was collected with Trizol (Ambion) RNA isolation reagent according to manufacturer’s instructions. For the analysis of HSP70 gene, cells were heat-shocked at 42 °C for 2 h prior to cell collection and for APA1A gene, cells were treated with 100 nM of dexamethasone for 24 h. The cDNA was generated using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). Real time qPCR was performed using the FastStart Universal SYBR Green Master (Rox) and oligonucleotides select for Hsp70 RNA (AGAGCCGAGCCGACAGAG and CACCTTGCCGTGTTGGAA). Relative quantities of the tested loci were normalized against the levels of GAPDH transcripts using select primers (CCACTCCTCCACCTTTGAC and ACCCTGTTGCTGTAGCCA).
Chromatin Immunoprecipitation
ChIP reactions were performed as previously described [13]. Briefly, samples were prepared from 3×107 HeLa cells. Immunoprecipitation was performed using 5 μL of HSF1, HDAC1, HDAC9, or SIRT1 antibody at 4°C overnight. qRT-PCR was performed with FastStart Universal SYBR Green Master (Rox) (Roche) using primers for the HSP70 promoter near proximal HSE (GGCGAAACCCCTGGAATATTCCCGA and AGCCTTGGGACAACGGGAG) and to the GAPDH promoter (TACTAGCGGTTTTACGGGCG and TCGAACAGGAGGAGCAGAGAGCGA). All reactions were done in triplicate with samples derived from experimental repeats and error bars represent the standard error of the mean.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA analysis was performed using Buffer C extracts (10 μg) from 293T, MEFs or in vitro transcribed/translated rabbit reticulocyte lysate (Promega) fractions (5 μL). The proteins were incubated with poly dI-dC (Sigma) and 32P-end labeled oligonucleotides: HSE (5′CTAGAAGCTTCTAGAAGCTTCTAG), GRE (5′AGTAGCTAGAAACCCTGTACCGTCGA), CDC6 DNA (5′CTGTGGCCGGCAGGTGAAC) along with the complimentary primer. The protein-DNA complexes were resolved by native polyacrylamide gel (4%) electrophoresis and the dried gels were visualized using a PhosphorImager (Molecular Dynamics).
Immunoprecipitation and immunodepletion assays
Rabbit reticulocyte lysate in vitro transcribed/translated Myc-HSF1 was immunoprecipitated with α-Myc agarose beads (Santa Cruz Biotechnology). After the addition of immobilized antibodies the precipitation step was carried out for 2 h at 4°C, the eluted proteins were separated by SDS-PAGE prior to visualizing the 35S-labeled proteins using a PhosphorImager. To immunodeplete rabbit p23 from the reticulocyte lysate three rounds of immunoprecipitations were performed using Protein A/G plus agarose coupled with α-p23 antibody; control lysates were incubated with uncoupled Protein A/G plus agarose.
Supplementary Material
Research Highlights.
Lysine deacetylases differentially regulate the DNA binding function of Heat Shock Factor 1
HDAC7, HDAC9, and SIRT1 promote HSF1 DNA binding activity
HDAC1 sterically interferes with HSF1 DNA interactions
HDAC1 is an age-dependent inhibitor of the Heat Shock Response
Lysine deacetylases distinctly modulate the DNA binding activities of heterologous proteins
Acknowledgements
We are grateful to Drs. R.I. Morimoto (Northwestern University), B.S. Pace (Georgia Health Sciences University), T.K. Chatterjee (University of Cincinnati), and S. Prasanth (University of Illinois) for sharing valuable reagents. The presented work was supported by the Public Service grant CA155333.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Misteli T. The concept of self-organization in cellular architecture. J. Cell Biol. 2001;155:181–185. doi: 10.1083/jcb.200108110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hager GL, McNally JG, Misteli T. Transcription dynamics. Mol. Cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman BC, Yamamoto KR. Continuous recycling: A mechanism for modulatory signal transduction. Trends Biochem. Sci. 2001;26:285–290. doi: 10.1016/s0968-0004(01)01834-5. [DOI] [PubMed] [Google Scholar]
- 4.Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010;17:666–672. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 5.Echtenkamp FJ, Freeman BC. Molecular Chaperone-Mediated Nuclear Protein Dynamics. Curr. Prot. Pept. Sci. 2014;15:216–224. doi: 10.2174/1389203715666140331112230. [DOI] [PubMed] [Google Scholar]
- 6.Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balch WE, Morimoto RI, Dillin AJ, Kelly W. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 8.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calderwood SK, et al. Signal Transduction Pathways Leading to Heat Shock Transcription. Sign. Transduct. Insights. 2010;2:13–24. doi: 10.4137/STI.S3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vihervaara A, Sistonen L. HSF1 at a glance. J. Cell Sci. 2014;127:261–266. doi: 10.1242/jcs.132605. [DOI] [PubMed] [Google Scholar]
- 12.Westerheide SD, Anckar J, Stevens SM, Jr., Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zelin E, Zhang Y, Toogun OA, Zhong S, Freeman BC. The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol. Cell. 2012;48:459–470. doi: 10.1016/j.molcel.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raychaudhuri S, et al. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell. 2014;156:975–985. doi: 10.1016/j.cell.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 15.Verma P, Pfister JA, Mallick S, D’Mello SR. HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J. Neurosci. 2014;34:1599–1612. doi: 10.1523/JNEUROSCI.3039-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Echtenkamp FJ, et al. Global functional map of the p23 molecular chaperone reveals an extensive cellular network. Mol. Cell. 2011;43:229–241. doi: 10.1016/j.molcel.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 19.Mosser DD, Theodorakis NG, Morimoto RI. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol. Cell Biol. 1988;8:4736–4744. doi: 10.1128/mcb.8.11.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hueng-Sik C, Lin Z, Boshan L, Liu AYC. Age-dependent decrease in the heat-inducible DNA sequence-specific binding activity in human diploid fibroblasts. J. Biol Chem. 1990;265:18005–18011. [PubMed] [Google Scholar]
- 21.Fawcett TW, Sylvester SL, Sarge KD, Morimoto RI, Holbrook NJ. Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J. Biol. Chem. 1994;269:32272–32278. [PubMed] [Google Scholar]
- 22.Locke M, Tanguay RM. Diminished heat shock response in the aged myocardium. Cell Stress Chaperones. 1996;1:251–60. doi: 10.1379/1466-1268(1996)001<0251:dhsrit>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heydari AR, et al. Age-related alterations in the activation of heat shock transcription factor 1 in rat hepatocytes. Exp. Cell Res. 256:83–93. doi: 10.1006/excr.2000.4808. [DOI] [PubMed] [Google Scholar]
- 24.Choi HS, Lin Z, Li BS, Liu AY. Age-dependent decrease in the heat-inducible DNA sequence-specific binding activity in human diploid fibroblasts. J. Biol. Chem. 2000;265:18005–18011. (1990) [PubMed] [Google Scholar]
- 25.Jurivich DA, Qiu L, Welk JF. Attenuated stress responses in young and old human lymphocytes. Mech. Ageing Dev. 1997;94:233–249. doi: 10.1016/s0047-6374(96)01856-8. [DOI] [PubMed] [Google Scholar]
- 26.Bandyopadhyay D, et al. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002;62:6231–6239. [PubMed] [Google Scholar]
- 27.Sasaki T, Maier B, Bartke A, Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006;5:413–422. doi: 10.1111/j.1474-9726.2006.00235.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang GL, et al. HDAC1 cooperates with C/EBPalpha in the inhibition of liver proliferation in old mice. J. Biol. Chem. 2008;283:26169–26178. doi: 10.1074/jbc.M803544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovacs JJ, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 30.O’Donnell M, Langston L, Stillman B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013;5:a010108. doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 32.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 33.Kim GW, Yang XJ. Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem. Sci. 2011;36:211–220. doi: 10.1016/j.tibs.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 35.Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo J, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 37.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 38.Cohen HY, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 39.Peng L, Seto E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011;206:39–56. doi: 10.1007/978-3-642-21631-2_3. [DOI] [PubMed] [Google Scholar]
- 40.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendillo ML, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherz-Shouval R, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;58:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu Y, et al. Dynamic interaction of HDAC1 with a glucocorticoid receptor-regulated gene is modulated by the activity state of the promoter. J. Biol. Chem. 2011;286:7641–7647. doi: 10.1074/jbc.M110.185488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.