

Key Points
MERIT40-deficient mice harbor an expanded HSC pool with increased quiescence, enhanced self-renewal, and reconstitution potential.
MERIT40 negatively controls HSC homeostasis through regulating the Tpo/Mpl pathway.
Abstract
Hematopoietic stem cell (HSC) self-renewal and multilineage reconstitution are controlled by positive and negative signaling cues with perturbations leading to disease. Lnk is an essential signaling adaptor protein that dampens signaling by the cytokine thrombopoietin (Tpo) to limit HSC expansion. Here, we show that MERIT40 (Mediator of RAP80 Interactions and Targeting 40 kDa [M40]), a core subunit of an Lnk-associated Lys63 deubiquitinating (DUB) complex, attenuates HSC expansion. M40 deficiency increases the size of phenotypic and functional HSC pools. M40−/− HSCs are more resistant to cytoablative stress, and exhibit superior repopulating ability and self-renewal upon serial transplantation. M40−/− HSCs display increased quiescence and decelerated cell cycle kinetics accompanied by downregulation of gene sets associated with cell division. Mechanistically, M40 deficiency triggers hypersensitivity to Tpo stimulation and the stem cell phenotypes are abrogated on a background null for the Tpo receptor Mpl. These results establish M40-containing DUB complexes as novel HSC regulators of HSC expansion, implicate Lys63 ubiquitination in HSC signaling, and point to DUB-specific inhibitors as reagents to expand stem cell populations.
Introduction
Hematopoietic stem cells (HSCs) comprise a rare population of cells residing in the bone marrow (BM). They have the unique capability to maintain a balance between quiescence, self-renewal, and proliferation/differentiation into multiple blood lineages. This dynamic equilibrium is essential for preserving stem cell pools throughout the life of the organism, while constantly supplying blood cells at the steady state and under stress conditions such as infection or bleeding. Cell-intrinsic regulation of signal transduction, cell cycle progression, and gene expression, as well as extrinsic factors from the microenvironment, have been implicated in regulating HSC self-renewal vs differentiation decisions. Importantly, quiescence is required to preserve HSC stemness and their long-term reconstitution ability. However, intrinsic mechanisms that regulate HSC homeostasis and cell cycle state to promote stemness remain incompletely understood.
Cytokines signaling through their cognate receptors play important roles in hematopoiesis. One such signaling axis is thrombopoietin (Tpo) and its receptor, Mpl. Tpo is the primary cytokine that regulates megakaryocyte development and platelet production.1-3 Tpo activates Mpl in HSCs to maintain HSC quiescence and self-renewal,4,5 and Mpl−/− or Tpo−/− mice exhibit reduced HSC numbers and self-renewal capability.6-9 Furthermore, Mpl loss-of-function mutations are responsible for congenital amegakaryocytic thrombocytopenia and progressive BM failure.10 These findings established a critical role for Tpo/Mpl signaling in HSC development and functions in mice and humans.
Tpo binding to Mpl activates Janus kinase 2 (JAK2), triggering a cascade of signaling events, involving signal transducer and activator of transcription 5 (Stat5), phosphatidylinositol 3-kinase/Akt, and p44/42 mitogen-activated protein kinase.1,11 JAK2-deficient hematopoietic cells fail to respond to Tpo and an array of hematopoietic cytokines, revealing JAK2’s essential role in cytokine receptor signaling.12 We and others have previously shown that the adaptor protein Lnk (also called SH2B3) negatively regulates the Tpo/Mpl/JAK2 pathway.13-15 Lnk−/− mice harbor a markedly expanded HSC pool, with superior reconstitution ability due to an increase in HSC self-renewal.13,16 The effects of Lnk in HSCs are negated upon deletion of Mpl,13 further cementing the role of the Tpo/Mpl/JAK2 signaling axis in regulating HSC cell cycle and self-renewal.
To delineate mechanisms for Lnk function, we previously used a proteomic strategy to identify Lnk-interacting proteins.17 This approach revealed a novel interaction between Lnk and a deubiquitinating enzyme (DUB) complex, Brcc36 isopeptidase complex (BRISC).17 The BRISC DUB complex specifically hydrolyzes lysine 63–ubiquitin (K63-Ub) conjugates, a nondegradative form of Ub that has been implicated in hematopoiesis and cytokine receptor signaling.18-20 There are 8 different possible linkages for Ub chains. K48-Ub is the canonical form that targets proteins for degradation through the proteasome.21 In contrast, K63-Ub does not target proteins to the proteasome, but rather, mediates various biological processes, including DNA repair,22,23 protein trafficking,24 autophagy,25 and signal transduction.26 An in vivo role of K63-Ub in early stages of hematopoiesis has been previously suggested based on the observation that the loss of Ubc13 (the Ub-conjugating enzyme specific for K63-Ub chains) in mice leads to hematopoietic failure owing to loss of HSCs and progenitor cells (HSPCs).27 However, how K63-Ub affects hematopoiesis or HSC function has not been well established.
BRISC was first biochemically purified as a major K63-DUB activity in the cytoplasm.28 The BRISC complex has recently been implicated in inflammatory cytokine signaling20; however, a role for BRISC in hematopoiesis has not been reported. BRISC is composed of the enzyme BRCC36 and 3 core complex constituents (KIAA0157, MERIT40 [Mediator of RAP80 Interactions and Targeting 40 kDa (M40)], and BRCC45).18,19 M40 (also called BABAM1) is the scaffold protein critical for the complex stability and DUB activity.22,29,30 The Lnk-BRISC interaction suggests a potential role of BRISC in hematopoiesis. In this study, we investigated the potential role of M40 in regulating HSC cell cycle and self-renewal, and the underlying mechanism. Our studies revealed a novel contribution of M40 to stem cell homeostasis, thus suggesting an unappreciated role for nondegradative deubiquitination in regulating stem cell function.
Methods
Animals
C57/BL6 (CD45.2), SJL (CD45.1), and F1 (CD45.1/CD45.2) breeding pairs were from The Jackson Laboratory and maintained at the in-house facility. M40-gene trap (M40−/−) mice on the C57/B6 background were generated by the Texas A&M Institute for Genomic Medicine. Mpl−/− mice2 were provided by Dr Frederic de Sauvage (Genentech, South San Francisco, CA). This work was conducted under an approved protocol from the institutional animal care and use committee of Children’s Hospital of Philadelphia.
Flow cytometric analysis of HSPC subsets
The staining procedure was performed as previously described.13,17,31 Briefly, for the HSPC subset, BM cells were first stained with a biotinylated-lineage cocktail that contains anti-CD4, CD8, CD5, Ter119, Gr-1, Mac-1, B220, CD19, and interleukin-7 receptor (IL-7R) antibodies (eBioscience), followed by streptavidin–phycoerythrin (PE)–Texas Red secondary antibodies (Caltag Laboratories, Invitrogen) and PE-Cy5.5–conjugated anti-Sca-1, allophycocyanin (APC)–Alexa 750–Kit, APC-CD34, and PE-Flk2 (eBioscience) antibodies. For signaling lymphocyte activation molecule (SLAM) markers, PE-Cy7-CD150 and fluorescein isothiocyanate (FITC)–CD48 antibodies (Biolegend) were used. Cells were subsequently collected on a Fortessa flow cytometer (BD Biosciences), and data were analyzed using FlowJo software.
Limiting dilution BM transplantation
Serially diluted unfractionated total BM donor cells (CD45.2) from 2 to 3 age-matched wild-type (WT) or M40−/− mice were mixed with 3 × 105 competitor BM cells (CD45.1) and injected retro-orbitally into lethally irradiated (a split dose of 10 Gy of orthovoltage x-ray; Precision X-ray Inc) F1 recipient mice. Sixteen weeks after injection, the percentage of donor-derived cells in peripheral blood was determined by flow cytometry as previously described.13 Mice with donor-derived cells in the blood higher than 1% were counted as “positive reconstitution.” Data from 3 to 5 independent experiments were combined and competitive repopulation units were calculated using L-Calc software (Stemcell Technologies).
BMT of purified HSCs
Fluorescence-activated cell sorting of HSCs for BM transplantation (BMT) was performed as described in “Flow cytometric analysis of HSPC subsets”.32 Briefly, Lin− (lineage-negative) BM cells were first enriched with Dynabeads (Invitrogen), followed by antibody staining as described above. Fifty CD34−CD150+CD48− Lineage−Sca-1+Kit+ (LSK) HSCs were double-sorted using FACSAria (BD Biosciences) high-speed sorters, and directly deposited into 96-well plates, and subsequently transplanted with 4 × 105 Sca-1–negative competitors (CD45.1) and injected into lethally irradiated F1 recipients. The percent of chimerism in peripheral blood was assessed by flow cytometry 16 weeks after BMT. Primary transplanted mice were sacrificed at 4 months, and 3000 donor LSK cells were purified and transplanted into lethally irradiated secondary recipients along with competitor cells. Cells from each primary recipient were injected into 1 to 4 secondary recipients.
5-FU assay
WT and M40−/− mice were injected intraperitoneally with 150 mg/kg 5-Fluorouracil (5-FU) once a week for the indicated time and moribund animals were killed according to the protocol. Identification of moribund mice for euthanasia was conducted with the assistance of the veterinarian technicians in a blinded fashion. Briefly, anorexia, lethargy, pallor, and weight loss were used as signs of stress. For the hematopoietic recovery assay, mice received a single injection of 5-FU. Complete blood counts were measured at indicated time points. HSC frequency was determined by flow cytometry 7 days after a single injection of 5-FU. Long-term HSCs (LT-HSCs) were defined as the CD34−Flk2−CD150+ LSK population.
Colony assays
To quantify colony-forming cells (CFCs), 8 × 104 BM cells from WT and M40−/− mice 1 day or 10 days after 5-FU treatments were plated in duplicate in semisolid methylcellulose (M3434; StemCell Technologies) according to the manufacturer’s protocol.
Microarray and reverse transcription–quantitative polymerase chain reaction
Purified CD150+CD48−LSK HSCs from WT and M40−/− mice were sorted directly into TRIzol LS (Invitrogen). RNA was isolated using the microRNeasy kit (QIAGEN), and the microarray analysis was performed at the PENN Molecular Profiling/Genomics Facility using the GeneChip Mouse Gene 2.0ST array (Affymetrix). Resulting expression data were normalized using robust multichip analysis directly from the CEL files. Significant differential expression between the 2 groups was analyzed, and genes with significance analysis of microarray data P values < .05 were selected. Microarray data were also tested for gene set enrichment analysis (GSEA) using MSigDb c2.cp v3.0.33
BrdU and Pyronin/Hoechst staining
WT and M40−/− mice were injected with BrdU (5′-Bromodeoxyuridine) for 2 hours or fed with BrdU (0.5 mg/mL) in drinking water for 7 days before analysis. Total BM cells were isolated and Lin−ckit+ cells were sorted using Aria. Cells were then stained with ckit-APC-Cy7, Sca1-PE, CD48-FITC, CD150-PE-Cy7, and BrdU-APC antibodies following the manufacturer’s instructions (BD Biosciences). Cells were resuspended in buffer containing DAPI (4,6 diamidino-2-phenylindole) for flow cytometry. Sorted SLAM LSK cells were incubated with 5 μg/mL Hoechst 33342 (Molecular Probes) in Hank balanced salt solution containing 20 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid), 5 mM glucose, and 10% fetal bovine serum at 37°C for 45 minutes, followed by an additional 45 minutes of incubation with 1 μg/mL Pyronin Y (Sigma-Aldrich). Cells were subsequently analyzed on a Fortessa flow cytometer (BD Biosciences).
Western blot analysis
LSK cells from WT and M40−/− mice were starved for 2 hours in RPMI medium (Invitrogen) supplemented with 0.5% bovine serum albumin and stimulated with 1 or 10 ng/mL mouse recombinant Tpo (PeproTech) for 10 minutes. Cell lysates were subjected to standard western blot (WB) analysis with antibodies to pStat5, pAkt, and Akt (Cell Signaling Technology), Stat5, and JAK2 (Santa Cruz Biotechnology). Cell lysates from WT and M40−/− BM cells were subjected to WB with anti-β-actin (Santa Cruz Biotechnology), Brcc3634 (Bethyl Laboratory), KIAA 0157 (Aviva), and M4022 antibodies.
Results
Loss of M40 expands HSC pool
We previously found that the BRCC36-containing DUB complex BRISC associates with Lnk,17 which is an important negative regulator of Tpo/Mpl-mediated signaling in HSCs.13 To determine whether this interaction is involved in cytokine signaling in hematopoiesis, we generated mice deficient in M40, the scaffold protein that is critical for the integrity of this DUB complex.22 M40−/− mice showed complete loss of M40 expression at both messenger RNA and protein levels (supplemental Figure 1, available on the Blood Web site). As predicted, protein levels of BRCC36 were markedly reduced in the BM from M40−/− mice (supplemental Figure 1), in agreement with our published studies showing that M40 plays a scaffolding and stability function for the M40-BRCC36 containing DUB complexes.22
M40−/− mice were born at the predicted Mendelian ratio with an appearance indistinguishable from their WT littermates. Complete blood counts in 8- to 12-week-old animals revealed significantly increased platelet numbers in M40-deficient mice (supplemental Figure 2). Although M40−/− mice showed normal myeloid, T-lineage, and B-lineage distributions in the BM and spleen (supplemental Figure 2), they exhibited an elevation in the primitive hematopoietic compartment. Flow cytometric analysis demonstrated that M40−/− mice had increased phenotypic LT-HSCs as determined by the established and most stringent HSC markers, CD150+CD34−Flk2−LSK (LSK) (Figure 1A), but not lineage-committed progenitors (supplemental Figure 3). Because the total BM cells remain unchanged (supplemental Figure 2), our results suggest that M40−/− mice have increased numbers of phenotypic HSCs.
Figure 1.

M40 deficiency leads to an expansion of phenotypic and functional HSCs. (A) Frequency of LT-HSCs in WT and M40−/− mice as determined by flow cytometry. LT-HSC is defined as CD34−Flk2−CD150+LSK, and representative plots and gating strategy are shown. Each symbol represents an individual mouse. P value was determined by the 2-tailed Student t test. (B) Functional HSC frequency in WT and M40−/− mice as determined by limiting dilution BMT. Percentage of donor-derived cells in peripheral blood 16 weeks after BMT were analyzed by flow cytometry. Data from 3 to 5 independent experiments were combined, and reconstitution frequency and statistical analysis were calculated using L-Calc software. The results are presented as number of positively engrafted mice vs total number of mice analyzed for the indicated doses. Positive engraftment was defined as >1% donor-derived cells in the peripheral blood. (C-D) Donor chimerism in total leukocytes (C) and different lineages (D) in peripheral blood of the recipient mice transplanted with 3 × 104 and 1 × 105 donor cells along with competitors are shown. Each symbol represents an individual mouse; horizontal lines indicate the mean value in each group. P values were calculated using the 2-tailed Student t test. *P < .05; **P < .01; ***P < .005. 1SE, 1 standard deviation; CRU, competitive repopulating unit.
To functionally quantify HSC frequency and assess whether M40 affects HSC reconstitution potential, we used competitive limiting dilution BMT. The results revealed that M40−/− mice indeed harbored increased numbers of functional HSCs (Figure 1B). Importantly, M40−/− BM cells gave rise to greater donor chimerism in all blood lineages in the recipient mice (Figure 1C-D), suggesting that M40−/− BMs have superior long-term repopulating abilities. Of note, M40 deficiency did not affect HSPC homing efficiency (supplemental Figure 4). Hence, our data strongly support that M40 deficiency leads to an expansion of phenotypic and functional HSC pool.
Enhanced repopulating and self-renewal ability of M40−/− HSCs
We transplanted purified HSCs to investigate whether the increase in repopulation observed in M40−/− transplanted animals is due to intrinsic HSC properties rather than that of the committed progenitors. HSCs (CD34−CD150+CD48−LSK) from WT and M40−/− mice were double-sorted and 50 HSCs were directly transplanted into lethally irradiated animals along with competitors. Our results demonstrate that M40−/− HSCs exhibited superior reconstitution ability to that of WT HSCs in the primary transplants (Figure 2A).
Figure 2.

Purified M40−/− HSCs exhibit enhanced repopulation and self-renewal ability. (A) Fifty CD34−CD150+CD48−LSK cells from WT and M40−/− mice were mixed with 0.4 × 106 Sca1-depleted competitor BM cells and transplanted. Donor percent reconstitutions in the blood 16 weeks post-BMT are shown. (B) At the end of primary transplant, 3000 donor-derived LSK cells from each primary recipient were sorted and individually transplanted into 1 to 4 secondary recipients. Donor chimerisms in peripheral blood of secondary recipient mice are shown. Each symbol represents an individual mouse; horizontal lines are the mean values in each group. P values were calculated using the 2-tailed Student t test.
Donor-derived HSCs are subjected to significant stress during BMT.35 These normally quiescent cells must self-renew, proliferate, and differentiate to support the increased number of lineage-restricted progenitors required to reconstitute myeloid and lymphoid populations after ablation of the recipient BM. HSC concentrations and their repopulating capacity drop significantly after each round of transplant.36-38 Thus, we performed secondary BMT to analyze stem cell self-renewal. We purified donor-derived LSKs from the primary transplants and injected them into lethally irradiated recipients. M40−/− HSCs again displayed superior reconstitution in the secondary transplants (Figure 2B). Our data strongly suggest that M40 deficiency augments HSC intrinsic repopulating activity by increasing HSC self-renewal.
Enhanced regenerative potential of M40−/− HSCs following cytotoxic stress
M40 deficiency increases platelet counts and HSC homeostasis at the steady state. We next subjected M40−/− mice to cytoablative stress induced by 5-FU. M40−/− mice were more resistant to 5-FU treatment with increased platelet recovery (supplemental Figure 5), and increased survival upon repetitive 5-FU administration (Figure 3A). One day after 5-FU treatment, M40−/− mice showed increased clonogenic survival in comparison with WT mice (Figure 3B), suggesting that M40−/− HSPCs are more resistant to cytoablative stress. 5-FU depletes cycling hematopoietic cells and forces primitive hematopoietic cells including HSCs to regenerate. Analysis of the stem cell compartment after 5-FU injection demonstrated that M40−/− HSCs regenerated faster than WT HSCs upon cytotoxic stress at day 7 (Figure 3C) with increased cell proliferation as determined by BrdU incorporation assays (Figure 3D). After 2 injections of 5-FU, WT mice showed marked hypoplastic BM with progenitor cell numbers being substantially lower than those of M40−/− mice (Figure 3E), which explains the prolonged survival due to loss of M40. Collectively, these data suggest that M40 deficiency protects HSPCs from cytotoxic stress and promotes HSC regeneration.
Figure 3.
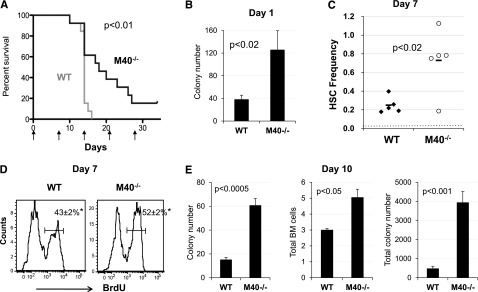
M40−/− mice are more resistant to cytotoxic stress and M40−/− HSPCs show superior regenerative ability upon stress. (A) Kaplan-Meier survival curve of WT and M40−/− mice after weekly administration of 5-FU (150 mg/kg). Data from 2 independent experiments are pooled and shown here; n = 13 per genotype. P < .01, log-rank nonparametric test comparing WT and M40−/− groups. (B) CFC numbers per 8 × 104 BM cells 1 day after 5-FU injection were quantified in semisolid methylcellulose media with cytokines (M3434; Stemcell Technologies Inc). (C) Frequency of phenotypic LT-HSCs (CD34−CD150+CD48−LSK) at day 7 following single injection of 5-FU. P value was calculated by the Student t test. Dotted red line represents the frequency of steady-state LT-HSCs. (D) Representative histograms depicting BrdU incorporation in SLAM LSK cells 7 days after 1 single 5-FU injection are shown. BrdU was injected 2 hours prior to BM harvest. BrdU+ fractions were quantified (mean ± SE). *P < .05 as determined by the Student t test. (E) Colony numbers per 8 × 104 BM cells (left), total BM cells (middle), and total colony numbers in the BM (right) 10 days after 5-FU injection (ie, 3 days after the second 5-FU injection) are shown. P values were calculated by the 2-tailed Student t test. SE, standard error.
Decelerated cell cycle and increased quiescence in M40−/− HSCs
The proper control of HSC quiescence and cell proliferation/differentiation is critical for HSC maintenance and self-renewal ability. We thus examined HSC cell cycle kinetics using BrdU incorporation assays. In both long-term (7 days) and short-term (2 hours) BrdU labeling, M40−/− SLAM LSKs had a marked decrease in the BrdU+ population compared with WT HSCs (Figure 4A). Lin−Kit+Sca-1− progenitor cells as well as CD48+LSK multipotential progenitors were also analyzed at the same time, and the cell cycle kinetics were similar between WT and M40−/− progenitors (data not shown). Thus, our data suggest that M40 specifically controls HSC cell cycle progression. The decreased BrdU+ populations observed in M40−/− HSCs imply an increase in quiescence. To directly measure the quiescent HSC population, we stained HSCs (SLAM LSKs) with an RNA-specific dye, Pyronin Y (Py), in conjunction with a DNA-specific dye, Hoechst 33342 (Ho), to differentiate G0 from G1 populations. The G0 fraction is defined by PylowHolow cells representing low RNA amounts and 2N DNA content.39 Consistent with our BrdU experiment, M40−/− HSCs exhibited a larger G0 population than WT HSCs (Figure 4B). Together, our data suggest that loss of M40 slows the cell cycle and acquires HSC quiescence.
Figure 4.

M40−/− HSCs show decelerated cell cycle progression, increased quiescence, and downregulation of gene expression associated with cell division. (A) BrdU incorporation analysis in SLAM LSK cells. WT and M40−/− mice were either fed with water containing BrdU for 7 days or intraperitoneally injected 2 hours prior to sacrifice. Representative flow cytometric plots of SLAM HSCs and histograms depicting BrdU labeling are shown. BrdU+ fractions were quantified (mean ± SD); n = 6. (B) Sorted SLAM LSK HSCs were stained with Py and Ho, and representative flow cytometric plots are shown. The quiescence G0 populations defined as Py−Ho− are indicated (mean ± SE); n = 4. P values were calculated by the Student t test. *P < .01; **P < .05. (C) Enrichment plot of GSEA analysis using WT vs M40−/− HSC expression data against a cell cycle signature from the MSigDB c2.cp database. (D) RT-qPCR analysis of cell cycle regulatory genes in WT and M40−/− HSCs. Relative expressions to GAPDH (mean ± SE) are shown. P values were calculated by the Student t test. *P < .001; **P < .02. FDR, false discovery rate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Ho, Hoechst; NES, normalized enrichment score; Nom, nominal; Py, Pyronin Y; SD, standard deviation; SE, standard error.
To investigate the molecular mechanisms by which M40 regulates the HSC cell cycle, we performed genome-wide transcriptome analysis using sorted SLAM LSKs from WT and M40−/− mice. GSEA33 revealed, and reverse transcription–quantitative polymerase chain reaction (RT-qPCR) confirmed, that M40−/− HSCs showed downregulation in gene sets associated with cell cycle, mitosis, DNA replication pathways (Figure 4C-D, supplemental Table 1). Furthermore, the top 5 most significantly downregulated Gene Ontology (GO) terms in the biological process in M40−/− HSCs were associated with cell division (supplemental Table 2). Thus, the gene expression signature is consistent with a role for M40 in controlling the HSC cell cycle.
Hyperactivation of Tpo signaling in M40−/− HSPCs
Lnk deficiency stimulates the major Mpl-controlled signaling pathways involving Stats and Akt in megakaryocytes40 and HSCs.13 Given the physical association of Lnk and M40, we examined whether the HSPC response to Tpo is affected by the loss of M40. Purified LSKs from WT and M40−/− animals were stimulated with varying concentrations of Tpo for 10 minutes following serum starvation. Western blot analysis with antibodies specific for activated phospho-JAK2, Stat5, and Akt demonstrated that M40−/− HSPCs were more sensitive to Tpo than WT HSPCs in activating Tpo downstream signaling pathways (Figure 5). These results indicate that M40 negatively regulates Tpo-mediated signaling in HSCs consistent with a Lnk-associated function.
Figure 5.

M40−/− HSCs show enhanced signaling in response to Tpo. Purified LSK cells from WT and M40−/− mice were starved and stimulated for 10 minutes with 1 or 10 ng/mL Tpo. Lysates from LSK cells were resolved on Tris-Glycine gels and blots were probed with antibodies specific for phosphorylated and total JAK2, Stat5, and Akt proteins. The ratios of phosphoprotein to total protein levels are indicated on top of individual bands.
M40 functions in HSCs are mediated through the Tpo/Mpl pathway
Mice deficient for M40 harbor elevated numbers of LT-HSCs and circulating platelets, both of which depend on Mpl signaling triggered by Tpo. Therefore, our finding of increased sensitivity to Tpo in M40−/− LSK cells is consistent with a role for M40 in regulating Tpo/Mpl signaling. To genetically test this hypothesis, we generated mice deficient in both Mpl and M40 (Mpl−/−M40−/−). We found that Mpl and M40 double nullizygous mice resembled Mpl single null mice showing a marked decrease in platelet counts and HSC numbers (Figure 6), attesting that M40 functions through Tpo/Mpl signaling in HSCs.
Figure 6.

The effect of M40 on HSCs is mediated through Tpo/Mpl. (A) Peripheral blood platelet counts in Mpl−/−, M40−/−, and Mpl−/−M40−/− mice. (B) Phenotypic LT-HSC frequencies in Mpl−/−, M40−/−, and Mpl−/−M40−/− mice as determined by CD34−Flk2−CD150+LSK surface markers. Each symbol represents an individual mouse. The mean in each group is indicated by horizontal lines. P value is calculated by the Student t test.
Discussion
In this study, we investigated the role of the adaptor protein M40 in hematopoiesis and cytokine signaling in HSPCs. M40 deficiency increases the numbers of phenotypic and functional LT-HSCs. Purified M40−/− HSCs exhibit a superior reconstituting potential to WT HSCs, indicating that M40 plays a cell-intrinsic role in HSC expansion. Loss of M40 provides a survival advantage in response to cytotoxic challenge, demonstrating M40 is critical for HSC regeneration under both steady-state and stress conditions. Similar to Lnk deficiency,41 M40−/− mice harbor an expanded HSC pool with increased quiescence, superior multilineage reconstitution, and serial transplantability. This phenotype is rare among mouse models with elevated HSC activity. Therefore, understanding M40 regulatory functions in HSCs will provide significant insights into stem cell biology.
It is of interest that M40−/− mice harbor a more quiescent and yet expanded pool of HSCs. One plausible explanation is that quiescent M40−/− HSCs undergo increased self-renewal divisions than WT HSCs. The BrdU incorporation assay does not differentiate self-renewal from non-self-renewal divisions. Quantification of the functional stem cell pool by both phenotypic and transplant assays suggests that M40−/− HSCs have increased self-renewal divisions to achieve increased HSC numbers under homeostatic conditions. Because M40 deficiency leads to a larger stem cell pool, a smaller proportion of HSCs contributes to non-self-renewing divisions or proliferation/differentiation to maintain hematopoiesis. This interpretation is compatible with the decelerated cell cycle observed in M40−/− HSCs. Genome-wide gene expression profiling in M40−/− HSCs showed downregulation of genes important for cell cycle progression, DNA replication, and mitosis, which is consistent with its role in controlling HSC cell cycle. M40−/− HSCs are hypersensitive to Tpo in activating downstream signaling. Importantly, the effect of M40 on HSCs was abrogated on an Mpl null background, indicating a role for M40 in Tpo/Mpl-mediated HSC homeostasis. It has been shown that Tpo provides an osteoblastic niche signal that keeps HSCs in quiescence.4,5 The Tpo/Mpl axis is reminiscent of Ang-1/Tie2 function in HSCs, in that Ang-1 enhances HSC quiescence and fosters the interaction with the BM niche to protect the HSC compartment from myelosuppressive stress.42 However, the pathways by which these 2 signaling systems impact on HSCs are likely distinct.
HSCs are largely dormant under steady-state conditions. Quiescent or slow-cycling HSCs are poised and ready to proliferate upon stress. Dormant HSCs display superior reconstitution potential in transplant experiments.43-49 HSC engraftment capacity is a function of the cell cycle such that quiescent HSCs engraft better than their counterparts in S/M/G2.50 Moreover, HSC divisional history correlates with HSC activity, and temporal quiescence is a better predictor of function than cell-surface phenotype.51 Hence we infer that the increased numbers of quiescent HSCs found in M40−/− mice account for their enhanced response to challenges such as 5-FU treatment or transplantation into lethally irradiated hosts. The protection of mice from 5-FU–induced lethality afforded by M40 deficiency may not be solely attributed to HSC quiescence. In fact, we showed that M40−/− HSPCs exhibited better colonogenic survival. Furthermore, M40−/− HSCs are quiescent but yet poised to undergo rapid proliferation upon 5-FU stress. Additional insights on the relationship between HSC cell cycle status and self-renewal can be gleaned from other mouse models. Mice deficient for Mpl,4,5 Foxo3a,52 PTEN,53 HIF,54 Fbw7,55 Necdin,56 or p5757 all exhibit defective maintenance of quiescence and increased cycling, leading to impaired self-renewal and a loss of competitive repopulating capacity. To our knowledge, Lnk−/− and M40−/− mice are the only mouse models showing increased quiescence but enhanced self-renewal and superior reconstitution ability. We found that both Lnk and M40 act through the Tpo/Mpl pathway, attesting to the importance of Tpo/mpl in protecting HSC quiescence and promoting self-renewal.
Cytokine signaling is regulated by posttranslational modifications, such as phosphorylation and ubiquitination, to enable rapid transduction of extracellular cues. A role for protein ubiquitination in stem cell homeostasis has begun to emerge.58 Characterization of mouse models with deletion of Ub ligases showed hematopoietic perturbations often caused by failure to target selected proteins for proteosomal degradation54,59-61 and causing dysregulation of the stem cell compartment. For example, lack of Cbl led to elevated HSPC numbers with increased repopulating activities and augmented proliferation in response to Tpo.62 Similarly, increased numbers of HSPCs with high proliferative potential were reported in mice deficient in Itch, an E3 ligase for Notch.63 Furthermore, mice deficient in Fbw7, an E3 ligase for Myc, have functionally impaired HSPC, an increased fraction of cycling HSCs, and decreased expression of genes associated with HSC self-renewal phenotype.55,64 Ubiquitination is a reversible process, as DUBs can rapidly deconjugate ubiquitinated substrates.65 Deletion of Mysm, a DUB for histone H2A, results in loss of HSC quiescence and stem cell exhaustion.66 These examples illustrate the complex effects of protein ubiquitination on stem cell maintenance and functions.
M40 is essential for the integrity of complexes with K63-DUB activity, suggesting a previously unrecognized role for nondegradative ubiquitination in regulating stem cell activity. Consistent with a role for K63-ubiquitination in protein activity rather than proteosomal degradation, we detected increased phosphorylation of JAK2, STAT5, and Akt but not total protein levels. Increased Tpo-mediated signaling in M40−/− HSPCs suggests that component(s) of Tpo/Mpl/JAK2 signaling pathway are likely subjected to regulation by M40-associated DUB activities. Our genetic experiments showed that M40 negatively regulates Mpl signaling, and that M40’s effects on HSCs are dependent upon the Mpl pathway. Moreover, our previous biochemical studies showed physical interaction between Lnk, JAK2, and the M40 complex.13,17 Together, these results are consistent with a role of M40 in the Tpo/Mpl/JAK2/Lnk signaling pathway. We recently reported that one of the M40-associated complexes, BRISC, localizes to and deubiquitinates actively engaged interferon receptor, thus limiting its K63-Ub–mediated internalization.20 However, we failed to detect any changes in cell surface expression or endocytosis of the Mpl receptor in M40 null HSPCs (data not shown). Future investigation is warranted to pinpoint the exact targets of the M40 complex in HSCs.
Allogeneic HSPC transplantation is standard of care for a variety of hematopoietic malignancies and congenital blood diseases.67 However, a majority of patients remain ineligible for conventional allogeneic HSPC transplantation due to the lack of appropriately matched donors. The use of umbilical cord blood as a source of allogeneic HSPC has expanded the donor pool. However, it is associated with delayed engraftment and failure to engraft. Despite extensive research efforts, the process of stem cell expansion is not fully understood. Cytokine therapy may not only enhance the production of mature hematopoietic cells, but also improve the engraftment and expansion of HSCs. DUBs are specialized proteases that have emerged as potential “druggable” targets. Thus, our studies might yield novel pharmacologic strategies that could be used for controlled stem cell expansion for transplantation without malignant transformation. In conclusion, this study has identified M40 as a novel HSC regulator, suggesting an unappreciated role for nondegradative deubiquitination in regulating stem cell function.
Acknowledgments
The authors thank the Human Hematopoietic Stem Cell Center of Excellence P30DK090969 for its support of microarray analysis, and are grateful to Dr Gerd Blobel for critical review of the manuscript.
W.T. was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL095675 and R01HL110806, as well as awards from the Myeloproliferative Neoplasm Research Foundation and Gabrielle’s Angel Foundation for Cancer Research. R.A.G. is supported by National Institutes of Health, National Cancer Institute grant R01CA138835. K.R. is supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant T32HL007150.
W.T. is a Leukemia & Lymphoma Society Scholar. K.R. and J.J. were American Heart Association postdoctoral fellows.
Footnotes
The microarray data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE65245).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.R. and W.T. designed and performed the experiments, analyzed the data, and wrote the manuscript; J.J. and R.D. performed experiments; and B.A. and R.A.G. generated M40−/− mice and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wei Tong, Children’s Hospital of Philadelphia, Abramson Building, Suite 316A, 3615 Civic Center Blvd, Philadelphia, PA 19104-4318; e-mail: tongw@email.chop.edu.
References
- 1.Kaushansky K. Thrombopoietin: a tool for understanding thrombopoiesis. J Thromb Haemost. 2003;1(7):1587–1592. doi: 10.1046/j.1538-7836.2003.00273.x. [DOI] [PubMed] [Google Scholar]
- 2.Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265(5177):1445–1447. doi: 10.1126/science.8073287. [DOI] [PubMed] [Google Scholar]
- 3.de Sauvage FJ, Carver-Moore K, Luoh SM, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183(2):651–656. doi: 10.1084/jem.183.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1(6):671–684. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685–697. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 6.Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci USA. 1998;95(3):1195–1200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solar GP, Kerr WG, Zeigler FC, et al. Role of c-mpl in early hematopoiesis. Blood. 1998;92(1):4–10. [PubMed] [Google Scholar]
- 8.Fox N, Priestley G, Papayannopoulou T, Kaushansky K. Thrombopoietin expands hematopoietic stem cells after transplantation. J Clin Invest. 2002;110(3):389–394. doi: 10.1172/JCI15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carver-Moore K, Broxmeyer HE, Luoh SM, et al. Low levels of erythroid and myeloid progenitors in thrombopoietin-and c-mpl-deficient mice. Blood. 1996;88(3):803–808. [PubMed] [Google Scholar]
- 10.Ballmaier M, Germeshausen M, Schulze H, et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97(1):139–146. doi: 10.1182/blood.v97.1.139. [DOI] [PubMed] [Google Scholar]
- 11.Watowich SS, Wu H, Socolovsky M, Klingmuller U, Constantinescu SN, Lodish HF. Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu Rev Cell Dev Biol. 1996;12:91–128. doi: 10.1146/annurev.cellbio.12.1.91. [DOI] [PubMed] [Google Scholar]
- 12.Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 13.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest. 2008;118(8):2832–2844. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buza-Vidas N, Antonchuk J, Qian H, et al. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 2006;20(15):2018–2023. doi: 10.1101/gad.385606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seita J, Ema H, Ooehara J, et al. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc Natl Acad Sci USA. 2007;104(7):2349–2354. doi: 10.1073/pnas.0606238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ema H, Sudo K, Seita J, et al. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev Cell. 2005;8(6):907–914. doi: 10.1016/j.devcel.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Jiang J, Balcerek J, Rozenova K, et al. 14-3-3 regulates the LNK/JAK2 pathway in mouse hematopoietic stem and progenitor cells. J Clin Invest. 2012;122(6):2079–2091. doi: 10.1172/JCI59719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419(6905):403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 19.Verma R, Aravind L, Oania R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298(5593):611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 20.Zheng H, Gupta V, Patterson-Fortin J, et al. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Reports. 2013;5(1):180–193. doi: 10.1016/j.celrep.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137(1):133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao G, Patterson-Fortin J, Messick TE, et al. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23(6):740–754. doi: 10.1101/gad.1739609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sobhian B, Shao G, Lilli DR, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316(5828):1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauwers E, Jacob C, André B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol. 2009;185(3):493–502. doi: 10.1083/jcb.200810114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12(9):836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 26.Deng L, Wang C, Spencer E, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103(2):351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Yamamoto M, Akira S, Sun SC. Regulation of hematopoiesis by the K63-specific ubiquitin-conjugating enzyme Ubc13. Proc Natl Acad Sci USA. 2009;106(49):20836–20841. doi: 10.1073/pnas.0906547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28(6):621–631. doi: 10.1038/emboj.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23(6):719–728. doi: 10.1101/gad.1770609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23(6):729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ema H, Morita Y, Yamazaki S, et al. Adult mouse hematopoietic stem cells: purification and single-cell assays. Nat Protoc. 2006;1(6):2979–2987. doi: 10.1038/nprot.2006.447. [DOI] [PubMed] [Google Scholar]
- 32.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci USA. 2009;106(9):3166–3171. doi: 10.1073/pnas.0807485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauch P, Rosenblatt M, Hellman S. Permanent loss in stem cell self renewal capacity following stress to the marrow. Blood. 1988;72(4):1193–1196. [PubMed] [Google Scholar]
- 36.Harrison DE, Astle CM, Delaittre JA. Loss of proliferative capacity in immunohemopoietic stem cells caused by serial transplantation rather than aging. J Exp Med. 1978;147(5):1526–1531. doi: 10.1084/jem.147.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison DE, Astle CM. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J Exp Med. 1982;156(6):1767–1779. doi: 10.1084/jem.156.6.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrison DE, Stone M, Astle CM. Effects of transplantation on the primitive immunohematopoietic stem cell. J Exp Med. 1990;172(2):431–437. doi: 10.1084/jem.172.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shapiro HM. Flow cytometric estimation of DNA and RNA content in intact cells stained with Hoechst 33342 and pyronin Y. Cytometry. 1981;2(3):143–150. doi: 10.1002/cyto.990020302. [DOI] [PubMed] [Google Scholar]
- 40.Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200(5):569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bersenev A, Rozenova K, Balcerek J, Jiang J, Wu C, Tong W. Lnk deficiency partially mitigates hematopoietic stem cell aging. Aging Cell. 2012;11(6):949–959. doi: 10.1111/j.1474-9726.2012.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 44.Foudi A, Hochedlinger K, Van Buren D, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27(1):84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 46.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lerner C, Harrison DE. 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol. 1990;18(2):114–118. [PubMed] [Google Scholar]
- 48.Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116(10):2808–2816. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nygren JM, Bryder D, Jacobsen SE. Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J Immunol. 2006;177(1):201–208. doi: 10.4049/jimmunol.177.1.201. [DOI] [PubMed] [Google Scholar]
- 50.Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Reports. 2014;2(4):473–490. doi: 10.1016/j.stemcr.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 53.Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 54.Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 55.Thompson BJ, Jankovic V, Gao J, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205(6):1395–1408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asai T, Liu Y, Di Giandomenico S, et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood. 2012;120(8):1601–1612. doi: 10.1182/blood-2011-11-393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsumoto A, Takeishi S, Kanie T, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9(3):262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 58.Moran-Crusio K, Reavie LB, Aifantis I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012;33(7):357–363. doi: 10.1016/j.it.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reavie L, Della Gatta G, Crusio K, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol. 2010;11(3):207–215. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez S, Wang L, Mumaw C, et al. The SKP2 E3 ligase regulates basal homeostasis and stress-induced regeneration of HSCs. Blood. 2011;117(24):6509–6519. doi: 10.1182/blood-2010-11-321521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Onoyama I, Nakayama KI. Fbxw7 in cell cycle exit and stem cell maintenance: insight from gene-targeted mice. Cell Cycle. 2008;7(21):3307–3313. doi: 10.4161/cc.7.21.6931. [DOI] [PubMed] [Google Scholar]
- 62.Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22(8):992–997. doi: 10.1101/gad.1651408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rathinam C, Matesic LE, Flavell RA. The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat Immunol. 2011;12(5):399–407. doi: 10.1038/ni.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsuoka S, Oike Y, Onoyama I, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22(8):986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 66.Wang T, Nandakumar V, Jiang XX, et al. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood. 2013;122(16):2812–2822. doi: 10.1182/blood-2013-03-489641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weissman IL. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science. 2000;287(5457):1442–1446. doi: 10.1126/science.287.5457.1442. [DOI] [PubMed] [Google Scholar]