

Abstract
In the RNA world, RNA is assumed to be the dominant macromolecule performing most, if not all, core “house-keeping” functions. The ribo-cell hypothesis suggests that the genetic code and the translation machinery may both be born of the RNA world, and the introduction of DNA to ribo-cells may take over the informational role of RNA gradually, such as a mature set of genetic code and mechanism enabling stable inheritance of sequence and its variation. In this context, we modeled the genetic code in two content variables—GC and purine contents—of protein-coding sequences and measured the purine content sensitivities for each codon when the sensitivity (% usage) is plotted as a function of GC content variation. The analysis leads to a new pattern—the symmetric pattern—where the sensitivity of purine content variation shows diagonally symmetry in the codon table more significantly in the two GC content invariable quarters in addition to the two existing patterns where the table is divided into either four GC content sensitivity quarters or two amino acid diversity halves. The most insensitive codon sets are GUN (valine) and CAN (CAR for asparagine and CAY for aspartic acid) and the most biased amino acid is valine (always over-estimated) followed by alanine (always under-estimated). The unique position of valine and its codons suggests its key roles in the final recruitment of the complete codon set of the canonical table. The distinct choice may only be attributable to sequence signatures or signals of splice sites for spliceosomal introns shared by all extant eukaryotes.
Keywords: Genetic code, Purine content, Sensitivity, Codon, Amino acid
Introduction
The genetic code and its codon organization are yet to be fully understood [1–6] albeit plenteous displays, inspiring interpretations and thoughtful hypotheses in the vast literature [7–13]. There are at least two basic facets for the code to be engaged together with evolving cellular machineries with increasing complexity and efficiency [14,15]. One is informational, where the code is inscribed into mRNAs through transcription and decoded by tRNAs and aminoacyl tRNA synthetases (AARS) through translation into amino acids; the other is operational, where anticodons of tRNAs interacts directly with codons of a mRNA to make sure that the code is translated accurately. There are many other basic elements of the genetic code, which follow the “two-track” scheme. For instance, the two types of nucleotides—purines and pyrimidines—are different in shape (two-ringed vs. one-ringed) and molecular weight, but they are equivalent informationally. Another example concerns AARS that adding amino acids to the correct tRNAs for protein synthesis [16]. Since AARS do not directly recognize anticodons borne by tRNAs and thus the code, they evolve to dictate the relationship between codons and their corresponding anticodons in rather complex and dynamic ways [17]. Most strikingly, the tRNA pool and the AARS set are rather dynamic and have gone through membership change and recruiting process across taxa [18–25].
We have recently showed [3–6] that the algebraic representation of the code is structurally equivalent to a content-centric organization and that codon and amino acid usage under different classification schemes are correlated closely with GC content, implying a set of rules governing compositional dynamics across a wide variety of prokaryotic genome sequences and perhaps even eukaryotic ones. Our results also indicate that codons and amino acids are not randomly partitioned in the table [1,2,4,6], where the 6-fold degenerate codons and their amino acids play important roles not only to balance the compositional dynamics of protein-coding DNA sequences, but also the physiochemical dynamics of proteins. Therefore, the content-centric organization of the code is of great usefulness in deciphering its hitherto defined organizations and regularities as well as the dynamics of nucleotide, codon and amino acid compositions.
In this report, we describe the dynamics of codons and their encoded amino acids in relation to purine content changes by displaying their purine sensitivity in a context of GC change as a follow-up for our previous publications [3,5]. Not only is there purine sensitivity among the seemingly content-insensitive codons, but also the sensitivity has exhaustive patterns that are unique to different grouping schemes of the codon triplets. These patterns are uniformity, symmetry and ending of codon triplets (i.e., the third codon position or cp3). Surprisingly, the most insensitive 4-fold degenerate amino acid is valine (V), which is encoded by GUN (N stands for one of the four nucleotides). Based on the step-wise evolution scenario of the genetic code [1,2], the impermanent and impermissible usage of GUN suggests that in the proto-cell of the RNA world, the splicing machinery was already invented since GU serves as an irreducible dinucleotide sequence for the splice site and the genetic code might not evolve to the canonical form universal to the present-day life.
Results and discussion
Symmetric and asymmetric natures of the genetic code
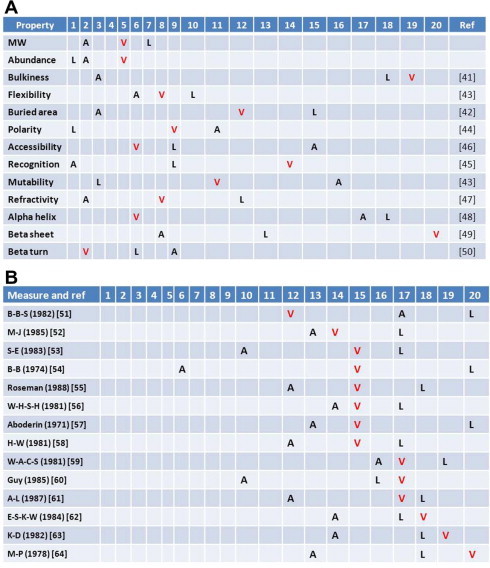
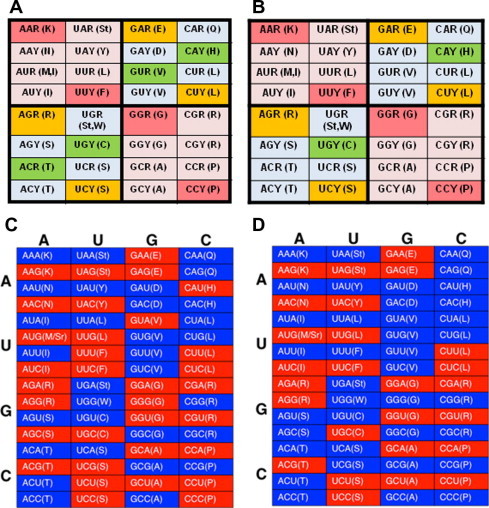
The genetic code (in this context it is the codon table—a 64 codon full-display) is organized primarily as symmetric or pairing units (Figure 1) because base pairing, albeit imperfect at times, is the primary operational force to embrace codons and their corresponding anti-codons together. These basic units should be independent from each other unless connected in the informational track, i.e., encoding the same amino acids as in the case of the three 6-fold degenerate codons for leucine (L), arginine (R) and serine (S). The table is simply divided into four quarters (Figure 1A) and each has 16 sequence-complimentary codons (Figure 1B and C). The pairing is between the heavy codons consisting of 2 purines and 1 pyrimidine (2R + 1Y) and the light codons (2Y + 1R) within the four basic units according to the operational rules (thus the operational track) other than sequence complementation that is indeed merely an informational concern (Figure 1D). In addition to this heavy-light codon pairing, the odd-numbered triplet itself always has two flanking nucleotides that are also considered as symmetry: when the two nucleotides are identical, both purines, both pyrimidines and both AU or GC. The third scheme is uniformity where all three nucleotides are considered: all identical, all purines or pyrimidines.
Figure 1.
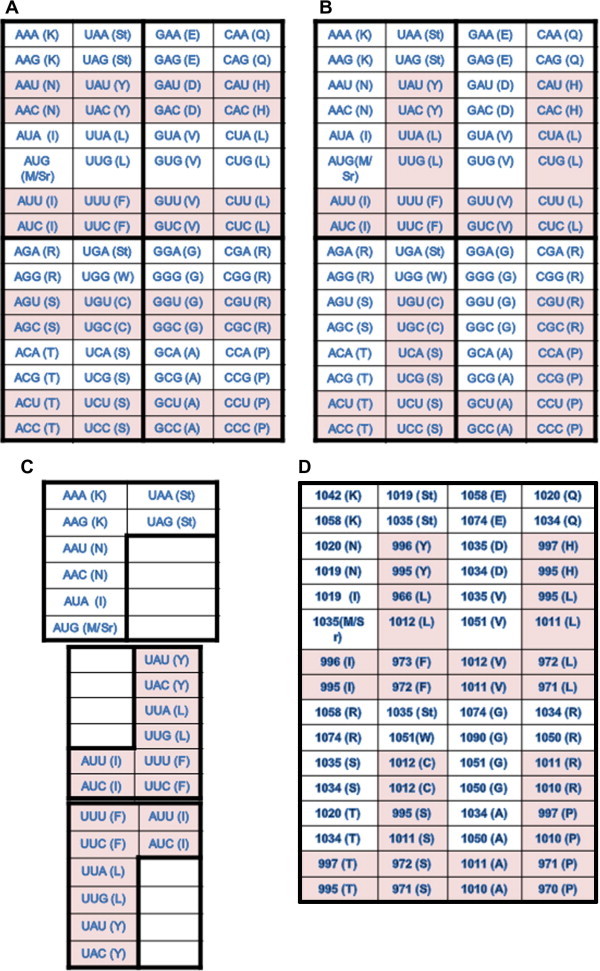
A display of 64-slot codon table A. The codons are organized in four different quarters based on GC content variation. Pyrimidine-ending codons are shaded. B. Sequence complimentary codons in the four quarters are shaded. C. The complementary codons are rearranged to show their mirror images (the top and the bottom panels). D. Codons are partitioned into high molecular weight and low molecular weight groups (low molecular weight codons are shaded), based on NMP molecular weight (AMP, 347.2; CMP, 323.2; GMP, 363.2; and UMP, 324.2).
Aside from its basic organization, there are several features where the genetic code and its codons are organized in a non-perfect symmetry, largely due to historic reasons in terms of its step-wise evolution or selection [1,2]. Although the pyrimidine-ending codons are perfectly organized in the table, i.e., all U-ending and C-ending codons are interchangeable vertically without altering their encoded amino acids, the purine-ending codons have a couple of exceptions (Figure 1A). One example is in the AU-rich box (Figure 2): there are two codon duplexes—AU (G/A) for M/I and UG (A/G) for Stop/W—encoding different amino acids or stop signals. Another is the 6-fold degenerate codon sets: L, R and S, which are proposed to provide balance between the pro-diversity and pro-robustness halves for the dominant physiochemical properties: hydrophobicity, polarity, charge, shape and size [3–6].
Figure 2.
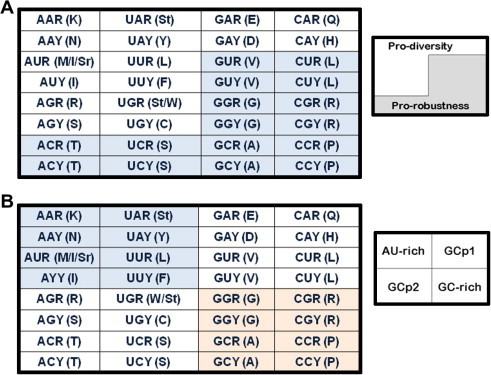
The organization of the genetic code A. The genetic code is organized in half and half, where the purine-sensitive half encodes more amino acids than the other half, whose cp3 (the third codon position) nucleotides possess diverse physiochemical properties and thus named as the pro-diversity half. The other half, however, encodes fewer amino acids than the pro-diversity half, whose cp3 nucleotides are not sensitive to compositional changes. B. The genetic code is partitioned into four quarters: AU-rich, GC-rich, GCp1 and GCp2. The GC content sensitive quarters are shaded in different colors. Sr (start) and St (stop) represent the start and stop signals, respectively.
The symmetry of codon organization is also multifold beyond the basic units organized in the four quarters (Figure 2A). The characteristics of the four-quarter organization are built on the sensitivity of GC-content [1–3]. The AU-rich and GC-rich quarters are both sensitive to GC variation but the other two are seemingly not. The second way to divide the codons is one-to-two: the pro-diversity half and the pro-robustness half (Figure 2B). The pro-robustness half is so organized that it almost suggests that it is evolved to be filled not only fast but in a perfect order. There must be a third feature or even more features in the table organization, concerning the two GC content insensitive boxes or quarters. What is it? How does purine content change manifest them in the codon organization? Is purine sensitivity ancient since the code might have started from purine-pyrimidine pairing to discrete AU and GC pairing (Figure 3A)? Most importantly, how did the code actually expand into the next and even the current form? We show one of the possible next steps in Figure 3B.
Figure 3.
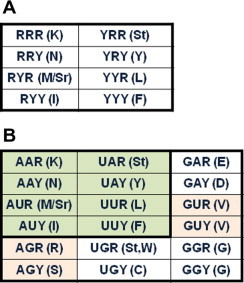
The evolutionary scenarios of the genetic code A. The assumed early code that encodes 7 amino acids with triple duplexes, one stop codon, and one start codon. Individual nucleotide may be not necessary to be distinguished other than purines (R) and pyrimidines (Y). B. As the emergence of protein synthesis machinery, the translational machinery may become more precise so that individual nucleotides are recognized. The first-phase code (shaded in green) may have expanded into the second phase where G is involved in protein-coding. Since GU and AG are involved in splice sites, AGN and GUN (shaded in pink) may not be part of the second-phase genetic code.
Predictable trends and categorizations of purine variation sensitivity of the codon triplets
Understanding the compositional dynamics of protein-coding sequences is of great significance in deciphering underlying mechanisms of sequence evolution. Towards this end, we have previously made several attempts to model sequence dynamics quantitatively. Based on an assumption that mutation and selection act at the level of nucleotide, our model takes account of diverse forces from both mutation and selection at three codon positions and factors both GC and purine contents as two essential parameters. As testified on a large collection of species across three domains of life, our model is capable of quantitatively recapturing the compositional dynamics of nucleotides, codons and amino acids with changing GC and purine contents [3–6].
Both GC and purine contents are important background parameters for composition related analyses as we have been working with a focus on prokaryotes for over a decade [26–33]. GC content is known to vary in a more dramatic way ranging from 20% to 80%, whereas purine content fluctuates narrowly around 50% with a deviation of 10% up and down [3]. Extensive studies have documented the primary contribution of GC content in codon/amino acid composition variations [34–36]. However, there is little attention paid to uncovering the contribution of purine content to composition variation as it is less impressive than GC content. Of course, purine content must have its own characteristics and indispensable influences on compositional dynamics of codons and amino acids. Most importantly, since purine content at the second codon position often controls physiochemical properties of amino acids, amino acid usage (as well as the related codon usage) may display unique codon or amino acid dynamic patterns in terms of the relationship between mutation and selection, which can be estimated based on a variety of methods [37–40]. For instance, amino acids with similar physiochemical properties may exchange at certain positions through codon-specific relationship to achieve size variations, such as among hydroxyl group-containing amino acids—Y, T and S, in addition to the largest group of hydrophobic amino acids that include P, G, A, I, V, L, F and M. Therefore, it is hypothesized that together with the changing purine content, some amino acids may be used at a constant frequency as purine variation-insensitive and others may vary accordingly as purine variation-sensitive.
To test this hypothesis, let us begin with how codons react to purine content variations when purine variation sensitivity is plotted as a function of GC variation (Figure 4). The plots are more complex than what we anticipated but very characteristic. From this large collection, we observe several obvious variables. First, the predicted trend, assuming five fixed purine contents (0.40, 0.45, 0.50, 0.55 and 0.60), provides possible sensitivity and some of the real data are deviated significantly from the predictions but limited in number. Second, the trend, where both increase and decrease are obvious as GC content varies, is mostly definable based on the GC content of the codon endings, e.g., G-ending and C-ending codons increase as genomic GC content goes up and the opposite trend belongs to A-ending or U-ending codons. Third, the trends have three categories: going with the model calculation, over-estimation and under-estimation within the purine variability boundaries. Fourth, the scattering of the data points, which may reflect the interplay of selective constraints and mutation pressures, is rather impressive as some of the codons are almost invariable but others tend to go wild. We categorize the codons, codon sets (within a slot of four) and amino acids according to predictions and their fit to the real data in two ways. The first way is to examine all plots for manual classification, and the second is to use statistics to set cut-off values for validating the primary observation. We first would like to examine the predicted trends of codons and codon sets rather than amino acids that are subjected for major consideration of selection as one of the key variables.
Figure 4.
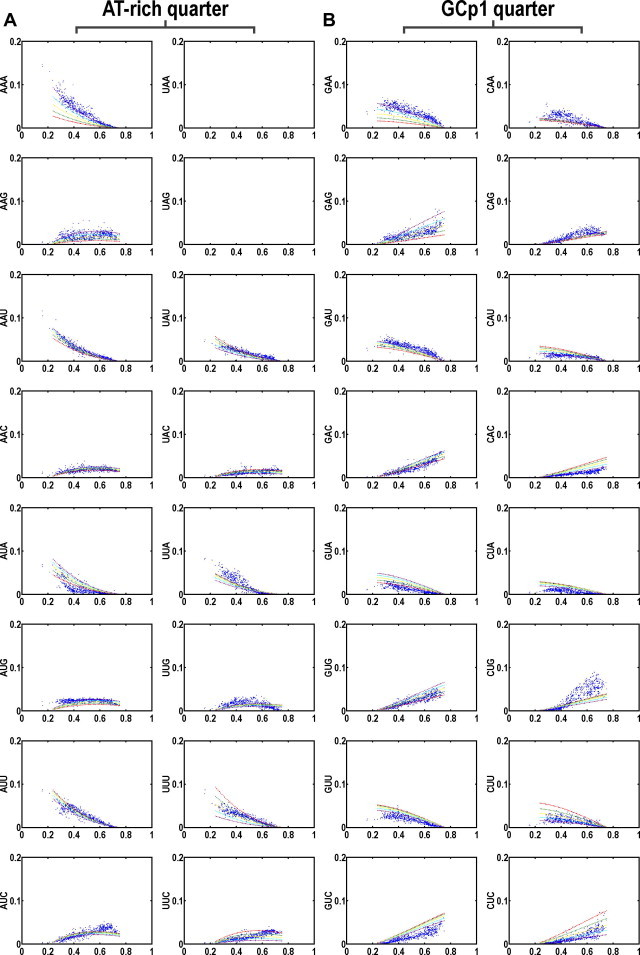
Codon usage plots based on fixed background GC and purine contents Purine contents (R) are set from 0.4 to 0.6 (color-coded; red for R = 0.4, green for R = 0.45, yellow for R = 0.5, cyan for R = 0.55, purple for R = 0.6). The genetic code is grouped into four quarters with each having 16 codons including AT-rich quarter (A), GCp1 quarter (B), GCp2 quarter (C), and GC-rich quarter (D). The scattering of the curves indicates sensitivity to purine variation in a context of GC content increase. Data from each bacterial genome is represented as a single solid circle.
Purine sensitivities of the 64 codons are essentially partitioned into four purine sensitivity categories including one purine-insensitive group and three purine-sensitive groups when uniformity is a sole concern. The purine-sensitive codons essentially fall into three obvious patterns (Figure 4). The first or pattern-I, also the most sensitive group of codons, is composed of four and the only four identical triplets (IT): AAA (K), UUU (F), GGG (G) and CCC (P). The second or the pattern-II is composed of all-purine (RT) and all-pyrimidine (YT) triplets except the four codons of pattern-I. The third or pattern-III is the all-AU (AT) and all-GC (GT) triplets except the four identical triplets of pattern-I.
When the observation goes further into the codon slot or set and within the triplet, we found other relevant patterns when within-codon symmetry is also concerned. Other than those overlapping with uniformity, including the RT, YT, AT and GT groups, we have many codons falling into symmetry groups, even within codons that are deemed insensitive as we categorized in this analysis. These symmetric codons are exceptional, i.e., they behave like the sensitive codons. For instance, within GAR and AGR, the symmetric codons GAG and AGA definitely have a stronger bias than the asymmetric codons GAA and AGG, which are perfect for uniformity. Similarly, in the YT groups, CUC and UCU are biased more than CUU and UCC due to the two perfect symmetries.
Among the codons in different patterns, the sensitivities are also not uniform. We can also examine the details within the 2-, 3- and 4-fold degenerate codons for each amino acid, where we categorize the paired purine-ending or pyrimidine-ending codons within an amino acid-to-codon slot as either R-duplexes or Y-duplexes, which are the components of the RT or YT groups. First, let us look at the trends of codon biases in the AAN box. There are three distinct patterns. One is AAA, belonging to pattern-I, also among the stronger; another is AAG, an RT group codon, whose bias is weaker than that of AAA but stronger than that of AAU that is an AT group codon. The AAC codon, which does not belong to any of the sensitivity groups, is a member of the insensitive group. In addition, since the codon biases are plotted as a function of GC content change, both the G- and C-ending codons have an up-going trajectory, whereas the AU-ending codons are either flat, near plateau-reaching, or even downward. Another observation is that the RT codons tend to have a more scattered biases overall. Similarly, everything in the UUN, CCN and GGN slots resembles what are in the AAN slot. Second, let us look at the RT or YT groups. There are only two RT-groups, AGR and GAR, both of which are expected to be weaker in the bias as compared to the four IT codons. Third, we have 12 codons in the AT and GT groups with 6 in each. The symmetric rule in the RT and YT groups is also true in the AT and GT groups, i.e., AUA, UAU, GCG and CGC are more sensitive than the asymmetric codons. Fourth, as we expected, if nucleotide symmetry is the essential element of the compositional dynamics, we can also see purine sensitivities among the insensitive codon groups within each slot that are overall “insensitive”. For instance, in the CAN slot, CAC is expected to be sensitive and it is true. Furthermore, we can also expect that CAU is also sensitive since C and U are both pyrimidines so that the pyrimidine symmetry in the two positions is also significant albeit the symmetry of the identical nucleotide is stronger than CAU, where the symmetry is YAY. Similarly, in the GUN slot, we can expect that GUG is more sensitive than GUA, where G and A satisfy purine symmetry.
There are essentially three parameters we have observed so far: triplet ending that involves only the cp3 nucleotide, triplet symmetry that concerns the two flanking nucleotides of the codon triplet, and triplet uniformity that reflects the collective effect covering the entire triplet (Figure 2). Codon ending is rather straightforward, relevant to either GC or AU contents. The ultimate uniformity is what of the IT group, followed by RT and YT groups and ended at AT and GT groups. A similar “packing order” is predictable under the symmetry framework: from identical and purine/pyrimidine to AU or GC at the two codon positions. Under the three frameworks—codon-ending, uniformity and symmetry—the defined effects are by and large additive, i.e., the effects under three frameworks as well as any of the groupings are all relevant. For instance, the effect of AGA comes from the RT group under the uniformity framework and ANA under the symmetry framework.
We now examine the rank of nucleotides in manifesting purine sensitivity. First, it appears that purines have stronger influence (of course) than pyrimidines, and this rule is true for all patterns. However, G and A behave differentially under the two-parameter frameworks: for the IT group, A has stronger effect than G under the uniformity framework, and the opposite is seen in the symmetry framework (such as ANA vs GNG). Second, the trends for U and C are similar to the case of A and G under both frameworks. It is rather clear that the two frameworks have different underlying mechanisms.
Purine variation sensitivity of codon sets and amino acids
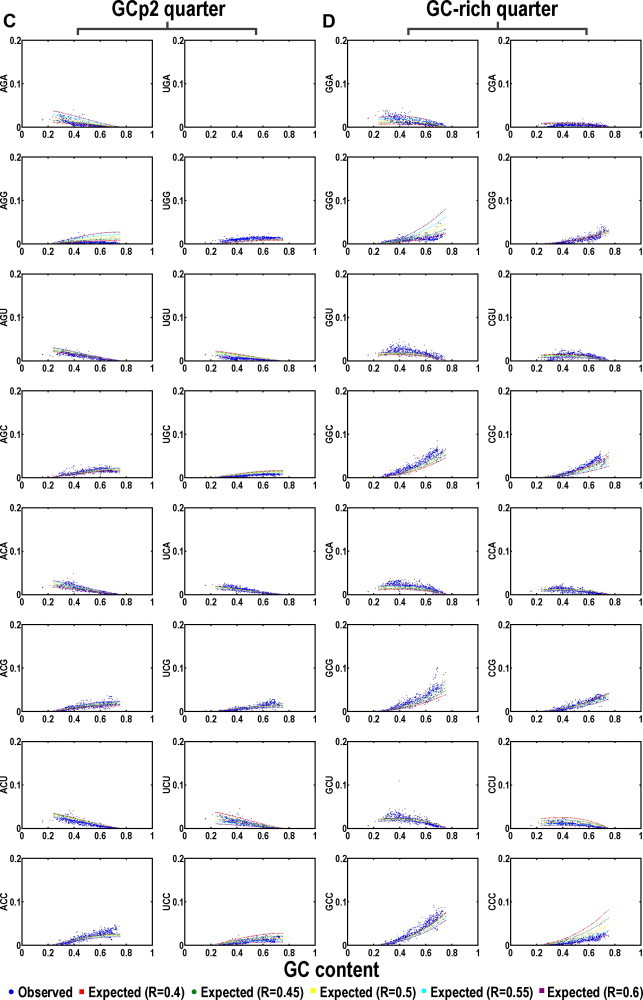
Based on the purine and pyrimidine pairing scheme among codons, we can now look into the organization of purine variation-sensitive and -insensitive codons (Figure 5). We have 16 insensitive codons that do not possess any of the purine sensitive elements (Figure 5A), and if we further ignore GC content uniformity (i.e., the two flanking nucleotides both are either AU or GC), exactly half of the codons are either sensitive or insensitive (Figure 5B). Moreover, if we eliminate purine or pyrimidine uniformities, only 24 codons are left sensitive to purine content change (Figure 5C). Once the table is simplified to show only 36 codon sets, we see a clear pattern now in the GC content variation-insensitive quarters, GCp1 and GCp2 (Figure 5D and E). This pattern fits our expectation that codons in the GCp1 and GCp2 boxes are not sensitive to GC content variation but the AU-rich and GC-rich boxes are in general. Of course, other factors may rise to become dominant.
Figure 5.

Purine sensitivity of amino acids in a context of GC content variation A. Nucleotide triplets that are not sensitive to GC content variation (shaded) due to lack of uniformity (all purines and all pyrimidines) and symmetry (RNR and YNY). B. Nucleotide triplets that have G- or CNG or C and A- or UNA or U (shaded) are assumed purine variation-insensitive when GC content varies. C. Only uniformity and true symmetric nucleotide triplets (not shaded) are considered purine variation-sensitive in a context of GC variation. D. The codon table after consolidating the third codon positions into purine and pyrimidines. The less purine variation-sensitive codons in the AU-rich and GC-rich quarters are shaded in pink. E. A schematic representation of the codon table after marking the purine variation sensitive-codons (S). Note that the insensitive codons are all in the diagonal (shaded in grey).
The amino acids involved in this scheme are: S, T, W, V, D, L and Q (Figure 6). Significantly, 4 of the 6 serine codons are all in this category but only 2 of the 6 leucine, one half of the valine, and one half of the threonine codons are. Surprisingly, if sensitivity is dominant, there are only three insensitive amino acids in the entire codon set: W, D and Q. Importantly, none of them are irreducible since there are functionally and structurally equivalent amino acids in the table if size is not set to be very stringent. If we go further, ignoring W, a single-codon-encoded amino acid, it is very suggestive as why D and Q are recruited to their current positions in the table.
Figure 6.
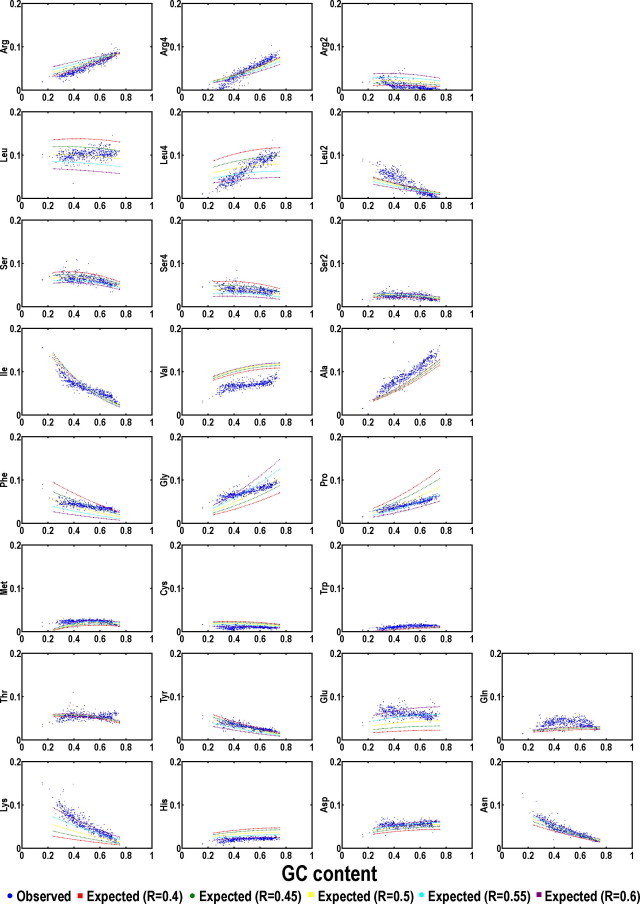
Amino acid usage plots based on fixed background GC and purine contents Purine (R) contents are set from 0.4 to 0.6 (color-coded; red for R = 0.4, green for R = 0.45, yellow for R = 0.5, cyan for R = 0.55, purple for R = 0.6). Amino acid frequencies are plotted against GC content. Data from each bacterial genome is represented as a single solid dot.
Although we are now able to predict purine sensitivity for each codon, it is still necessary to inspect each amino acid to see if they actually follow the prediction (Figure 6). The answer is surprising—not all amino acids follow the prediction. Let us look at the amino acids that are actually predictable. First, the two acidic amino acids are rule-followers, where E takes advantage of both frameworks and GAA and GAG are the RT and G symmetry groups. Along the same line, we can see that most of the 8 amino acids encoded by duplex codons appear to follow the rule quite well except C that is much weaker than the other 2-fold degenerate codons in the same class. The exception also suggests other rules to be discovered. Second, among the eight 4-fold degenerate codons, together with those from the 6-fold degenerate codons, we have 8 amino acids: A, G, L, P, R, S, T and V. Isoleucine (I) is a 3-fold degenerate amino acid but similar to a 4-fold one. In the cases of 6-fold degenerate amino acids, the trends are winner-takes-all, i.e., the 4-fold degenerate codons determine overall trajectories. In this regard, the amino acids with C-leading codons are all sensitive except the duplex encoding Q. Similarly, in the U-leading column, not only do all stop codons reside, but also the rest only contains single-codon-encoded amino acids, W and C, which are physiochemically unique. What are left include A, T, V and I, all of which fall into a category of purine-leading codons, either A- or G-leading. These 4 amino acids belong to different groups; V and T are in the GC-invariable quarter (GCp2), whereas I and A are in the AU-rich and GC-rich quarters, respectively. In this sense, T and V are the most insensitive amino acids toward compositional variations regardless if the variations are GC or AU. Therefore, V and T are the most variation-proof amino acids in the entire collection of the 20 amino acids, although one half of the codons are predicted weakly sensitive. It is rather clear that the diagonal rules are universal to all basic units of the genetic code: sensitivity to purine variations within and across the quarters (Figure 6). Even when the amino acid as a whole is considered, the rule is still obvious.
The reasons why amino acids V and T are chosen for the particular position deserves an in-depth discussion. First, both amino acids are not the kinds with unique physiochemical properties; in other words, both have functional supplements in the code, i.e., S can substitute T and several hydrophobic amino acids (such as A, I and L) can substitute V, by and large functionally. If we assume the dominant rule in the four quarters, i.e., the insensitive codons override the sensitive ones in the GCp1 and GCp2 quarters and the sensitive codons override the insensitive ones in the AU-rich and GC-rich quarters, there are two other amino acids, L and S, which have codons that both achieve uniformity and symmetry (CUY and UCY), so that they remain sensitive to purine changes, albeit in the insensitive neighborhood. Furthermore, there are three amino acids containing hydroxyl group, S, T and Y, all of which have codons in both the sensitive and insensitive categories. The trend that amino acids with similar physiochemical properties tend to spread among different quarters is very obvious. Second, V and T are the most insensitive in the two insensitive quarters due to the fact that their sensitive codons only realize symmetry but not uniformity (ACR and GUR). Third, aside from the reason why T and V are chosen to be inflexible for variability, there is another question—why ACN and GUN? One seemingly plausible explanation is that both dinucleotides are involved in RNA splicing and have been used for operational purposes. There are two essential types of spliceosomal introns; one is the U2 type and the other is the U12 type. The U2-type introns have GU-AG (5′ and 3′) splice sites whereas U12-type introns have AU-AC (5′ and 3′). If we refer them to the primitive splice site, when C is not heavily involved in any of the biological tracks (informational and operational), the irreducible splice site must have been GU-RR (GG, AG and GA when AAN has to encode amino acids). This is a rather bold assumption where we consider that the genetic code and the primitive life forms are actually originated and matured in a eukaryote-like lineage where a relatively complete compartmentalization has been achieved with a separation between the nucleus and the cytosol but DNA may have not been fully utilized as genetic material.
The next question is why valine not other amino acids becomes the “chosen one”. We have several arguments according to the principle [1,2] that the amino acids are fixed in the canonical table though intensive evolutionary selection at the dawn of the DNA Era, and it is the collective moderate physiochemical properties that triumph valine to its current position (Figure 7). Uniqueness should precede complementation in the evolution of the genetic code even though reshuffling may be unavoidable at a time before fixation. The idea of moderateness is also applicable to threonine and perhaps histidine, in the contexts of hydroxyl group and positive charge, respectively. First, valine is among the small amino acids but smaller than T and V within the peck (Figure 7A) [41–50]. Second, it is among the abundant amino acids but less abundant than L, A, G and S, ranking the fifth. Third, it is the number two ranking “bulky” amino acid but very moderate in flexibility, polarity and refractivity, among many other characteristic parameters of the protein-building amino acid set. Fourth, among the hydrophobic amino acids, valine is moderate in size, neither small – as compared to G, P and A – nor large when against F and L. Although many measurements had been devised in the past (such as some are size-sensitive and others appear not), majority of them agree with this point (Figure 7B) [51–64]. Fifth, when it comes to protein secondary structures, valine is strongly pro-beta sheet but also less repelling to alpha helix. Of course, its avoidance to beta turn is obvious due to its branched side chain.
Figure 7.
Physiochemical properties of valine The parameters of alanine (red) and leucine are specifically listed for comparative purposes and other amino acids are not indicated for simplification. All parameters are ranked in a shared order among the parameters, either from small to large or from weak to strong in the common sense for convenience; some may not be ordered in a “correct way” and truly “shared” by all but the results are not sensitive to it. All parameters are referenced with keys (abbreviations, last name initials, and last names when single authored papers are quoted) and to their corresponding literature. A. Some selected physiochemical properties of valine among all amino acids. The property measures and related references are as follows (from the first row): (1) molecular weight (MW); (2) amino acid composition (%) in the UniProtKB/Swiss-Prot data bank (release 2011_09); (3) bulkiness [41]; (4) average area buried on transfer from standard state to folded protein [42]; (5) flexibility [43]; (6) polarity [44]; (7) recognition factors [45]; (8) molar fraction (%) of 3220 accessible residues [46]; (9) relative mutability of amino acids (Ala = 100) [43]; (10) refractivity [47]; (11) conformational parameter for alpha helix (computed from 29 proteins) [48]; (12) normalized frequency for beta-sheet [49] and (13) conformational parameter for beta-turn [50]. B. Different hydrophobicity measures of valine. The hydrophobicity measures and related references are as follows (from the first row): (1) retention coefficient in TFA [51]; (2) hydrophobicity scale based on contact energy derived from 3D data [52]; (3) optimized matching hydrophobicity (OMH) [53]; (4) hydrophobicity (free energy of transfer to surface in kcal/mole) [54]; (5) hydrophobicity scale (pi-r) [55]; (6) hydrophobic constants derived from HPLC peptide retention times [56]; (7) mobilities of amino acids on chromatography paper (RF) [57]; (8) hydrophilicity [58]; (9) hydration potential (kcal/mole) at 25 °C [59]; (10) hydrophobicity scale based on free energy of transfer (kcal/mole) [60]; (11) hydrophobicity (delta G1/2 cal) [61]; (12) normalized consensus hydrophobicity scale [62]; (13) hydropathicity [63] and (14) average surrounding hydrophobicity [64].
The slight variable results from different thresholds
Our manual inspection of the purine variation sensitivity of the amino acids led to a summary in Figure 8A and B, where the diagonal rule is obvious. However, eyeballing may not be able to detect subtleties. To put things in a statistical context, we adopted the Kolmogorov–Smirnov test, a nonparametric test for estimating statistical distance between two distributions and examining whether they are drawn from the same distribution. If it is true, the K–S distance is small, indicating close similarity; otherwise, the K–S distance is large if the two distributions are different. Since we have five purine content variables (0.40, 0.45, 0.50, 0.55 and 0.60), there are accordingly five distributions for each codon. We have two versions of the K–S distance estimates between two distributions. One uses the purine contents of 0.4 and 0.6 (Figure 8C). If one codon is purine-sensitive (or purine-insensitive), its corresponding K-S distance is relatively larger (or smaller). Based on the K–S distances, we then use the K-means method to cluster codon/amino acid compositions into different purine sensitivity groups. Similar results are obtained when we choose the two distributions with the purine contents of 0.45 and 0.55 (Figure 8D).
Figure 8.
Purine-sensitive classes of codons and amino acids A. The insensitive codons and amino acids are those in the two GC-insensitive quarters and do not possess either uniformity or asymmetry in their codon triplets (see also Figure 6). Only the amino acids in the GC-insensitive diagonal line are assumed purine-insensitive other than the codons that fit to symmetric pattern of the triplets: (1) symmetry: CAY, GUR, UGY and ACR; (2) uniformity: AGR, CUY, AGR and UCY. B. If insensitivity is dominant, the only two 4-fold degenerate amino acids are V and T. C. When a numerical method is applied, the sensitive (red) and insensitive (blue) classes are defined more precisely between 0.4 and 0.6. D. Similar to what was shown in panel (C) except the window now is narrower: 0.45 and 0.55. There are several obvious rules. First, the insensitive amino acids are all in the two GC-sensitive quarters. The only insensitive 4-fold degenerate amino acid is V. Second, the differences between panels (C) and (D) are two-fold. One is that UCG and AGC in the GCp2 quarter and CAU and GUA in the GCp1 quarter are classified as sensitive; the other is that two codons—UUU and CCC—become insensitive in the AU-rich and GC-rich quarters, respectively. Third, within the AU-rich and GC-rich quarters, G/C-ending and A/U-ending determine the sensitivity in the two quarters, respectively. Therefore, uniformity (all three positions), symmetry (the two flunking positions), and codon ending (cp3) are all relevant to purine sensitivity.
From Figure 8C and D, we have several observations. First, in the two GC (or AU)-insensitive quarters where some of the codons or amino acids are actually sensitive to purine variation, both symmetry and uniformity are at work, and the narrower distributions appear to give more stringent results (Figure 8B). There are only two exceptions in the GCp2 quarter when the parameter setting is narrower (Figure 8B), ACA (threonine) and UGU (cystine), both of which should be purine variation-sensitive but not in the case when evaluated with the K-means method. Although the opposite happens in AGC (serine) and UCG (serine) (Figure 8A), they disappear under more stringent settings. In the GCp1 quarter, we observed that uniformity is dominant over symmetry (Figure 8). Second, when we look at the AU-rich and GC-rich quarters, the trend is rather clear-cut: the insensitivity is related to codon ending, i.e., when codons are in the AU-rich quarter, those ending with A or U are insensitive but those ending with G or C are sensitive. And the opposite is true for codons in the GC-rich quarters. When we examine the exceptions between the two-parameter settings, the conclusion is that symmetry dominates uniformity. Therefore, all the rules are by and large definable, depending on the weight placement on codon ending, uniformity or symmetry; all are involved. Third, all 6-fold degenerate amino acids – L, S and T – have their codons in the purine variation-sensitive category. Fourth, valine is the only 4-fold degenerate amino acid that has all its codons in the insensitive category, aside from the three 2-fold degenerate amino acids Q, H and D, as we can easily learn by visual inspection.
Deviations from the predicted values and possible interpretations
On the one hand, compositional dynamics of codons reflects how mutation pressure is eliminated to the minimum through the organization of the codons or genetic code, and the compositional dynamics of amino acids. On the other hand, compositional dynamics of codons responds to selective pressure, exhibiting how selection is at work through the relatedness of physiochemical properties of the amino acids that are mostly related in one aspect or another. The over-estimations or under-estimations of purine content sensitivities of the 20 amino acids are very characteristic of such dynamics (Figures 4 and 6).
Among the 6-fold degenerate amino acids that are all sensitive to purine variation, S is the most insensitive with very tight distributions (Figure 6), as both L and R are rather GC-sensitive, belonging to AU-rich and GC-rich in part, respectively. The most biased estimates are what for V and A; the former is one of the GCp1 amino acids and the latter is one of the GC-rich amino acids (Figure 6). The curves and plots indicate that the sensitivities of V to purine variation are mostly underestimated and those of A are mostly overestimated regardless of CG content changes. The codons of these two amino acids are also easily convertible to each other through a single transition event from T to C (GTN for V to GCN for A). The interpretation here is that there are always fewer valines and more alanines (A) than what we anticipate. Similar conversions are from V to M and I, and the trends are consistent with our explanations. Aside from hydrophobicity shared by these two amino acids, the differences are obvious. First, V-to-A or A-to-V exchanges promote size change that is very common for proteins to alter their functions or properties in some very subtle ways. Second, V is beta-sheet-prone but A is alpha-helix-prone in general, similar to L; other two 6-fold degenerate amino acids, S and R, are turn-prone and no-preference, respectively [49]. Therefore, V represents a unique physiochemical property in connecting amino acids that are capable of inter-conversion to achieve diversity in protein secondary structures. Third, there are more connections regarding to the measures of amino acid residues, such as surface area and volume [65], and what are in the GCp1 quarter are either similar in surface area or in volume, which are rather unique as we have not seen similar features in any of the other three quarters.
The biased purine sensitivities upon GC content variation are most likely contributed by both selection and mutation, which manifest as exchangeability of the amino acid residues in proteins. For instance, both C and H are under-estimated and the residues of these two amino acids in proteins may be negatively selected when mutation drives become dominant to convert them to R and Y, for instance. Another example is the case of over-estimated amino acids, such as A, Q, K and E. Other than A, which may related to the hydrophobic group as a whole, the rest of them are all critical for catalytic roles so that they may be subjected to constant positive or negative selections.
Conclusion
Once the table and codons are in the correct order, the characteristics of the genetic code become obvious. We have three essential overall organizational schemes: the one-to-four, the half-half, and the diagonal. The first partitions GC change sensitivity into four quarters, the second divides purine change sensitivity into halves, the pro-diversity half and the robustness half, and the third plots out scenarios of purine-sensitive and -insensitive codons and amino acids in their diagonals, which are closely related to triplet codon structural elements—ending, symmetry and uniformity. These characteristics and organizational features all appear operationally defined.
Genetic code has two build-in logics. One is mutation-centric and the other is selection-centric. In the mutation-centric logic, DNA or nucleotide sequence is assumed to vary freely according to Darwinian ideas. However, mechanisms of mutation occurrence may not be as straightforward as we have wished, and the results of in-depth analyses suggested a possibility of Lamarckian contributions [66–73]. In the selection-centric logic, amino acid or protein sequence varies according to nucleotide variation but sometimes is selected by external forces when the number of individuals in a population, possessing either advantageous or disadvantageous mutations, changes toward either maxima or minima. How the sequence changes at these two levels related to each other is determined by the relationship specified by the table and the functional context of the encoded proteins and their structural alterations. In addition, selection may work on non-coding sequences or sequence elements [74–78]. Therefore, such natures of the genetic code also support an operational origin.
The extension of the genetic code from its prototype to maturation is still an unsettled mystery but it might have gone through some step-wise scenarios [1,2], since the significant part of the code is believed to be operationally defined, so have tRNAs and their AARS been by and large believed as operationally flexible [18]. What is striking here is the fact that valine is placed at a unique position as the only 4-fold degenerate amino acid with a codon set of GUN. We have previously hypothesized that the reason why the GUN codon set was not used by the early code is due to its involvement in spliceosomal intron splicing as discrete sequence signal for the splice site that is still used by all eukaryotes that have intron-split genes. Therefore, we are cornered to believe that the origin of life started as a unicellular eukaryote-like organism with RNA splicing as one of the major cellular operational machineries where the genetic code was also born in a step-wise way. Nevertheless, the notion may have its scientific ground as evidence is emerging that the boundaries between RNA and DNA genomes [79] and between prokaryotes and eukaryotes may one day disappear [80]. Regardless what have been debating on the origin of life on earth, there are three minimal elements for the origin of life: macromolecules, compartmentalization and homeostasis that makes molecules, large or small, moving among the compartments in an organized fashion in the primordial Darwin’s warm little pond [81]. However, we are far from painting the right pictures, let alone understanding it.
Materials and methods
We retrieved bacterial genome sequences from National Center for Biotechnology Information (NCBI) at ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/. In order to ensure a sufficient sample size for calculating codon and amino acid compositions, we excluded bacterial chromosomes with <64 protein coding sequences. Bacteria with alternative genetic codes were also removed from this study. As a result, we obtained a total of 686 bacterial genome sequences. For each species, all protein-coding sequences were concatenated into a single contiguous sequence excluding stop codons, and observed codon and amino acid compositions were calculated from each concatenated sequence.
Based on our previous study for modeling compositional dynamics of protein-coding sequences [3–6], expected codon and amino acid compositions were quantitatively derived from background nucleotide contents (viz., GC and purine contents). The parameter settings for examining purine content sensitivity were: GC content ranging from ∼0.2 to ∼0.8 and purine contents were fixed at five different purine contents for each dataset: 0.40, 0.45, 0.50, 0.55 and 0.60.
Authors’ contributions
ZZ designed the model and collected sequence data. ZZ and JY analyzed the results and wrote the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that no competing interests exist.
Acknowledgements
This work was supported by grants from the National Basic Research Program (973 Program) (Grant No. 2011CB944100 and 2011CB944101), National Natural Science Foundation of China (Grant No. 90919024), the Special Foundation Work Program, the Ministry of Science and Technology (Grant No. 2009FY120100), the “100-Talent Program” of Chinese Academy of Sciences (Grant No. Y1SLXb1365), and the National Programs for High Technology Research and Development (863 Program; Grant No. 2012AA020409) by the Ministry of Science and Technology of China.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

References
- 1.Yu J. A content-centric organization of the genetic code. Genomics Proteomics Bioinformatics. 2007;5:1–6. doi: 10.1016/S1672-0229(07)60008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao J., Yu J. A scenario on the stepwise evolution of the genetic code. Genomics Proteomics Bioinformatics. 2007;5:143–151. doi: 10.1016/S1672-0229(08)60001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z., Yu J. Modeling genome compositional dynamics based on GC and purine contents. Biol Direct. 2010;5:63. doi: 10.1186/1745-6150-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Z., Yu J. On the organizational dynamics of the genetic code. Genomics Proteomics Bioinformatics. 2011;9:1–29. doi: 10.1016/S1672-0229(11)60004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z., Li J., Cui P., Ding F., Li A., Townsend J.P. Codon deviation coefficient: a novel measure for estimating codon usage bias and its statistical significance. BMC Bioinformatics. 2012;13:43. doi: 10.1186/1471-2105-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z., Yu J. The Pendulum Model for genome compositional dynamics: from the four nucleotides to the 20 amino acids. Genomics Proteomics Bioinformatics. 2012;10:175–180. doi: 10.1016/j.gpb.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knight R.D., Freeland S.J., Landweber L.F. Selection, history and chemistry: the three faces of the genetic code. Trends Biochem Sci. 1999;24:241–247. doi: 10.1016/s0968-0004(99)01392-4. [DOI] [PubMed] [Google Scholar]
- 8.Crick F.H. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 9.Wong J.T. Role of minimization of chemical distances between amino acids in the evolution of the genetic code. Proc Natl Acad Sci U S A. 1980;77:1083–1086. doi: 10.1073/pnas.77.2.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Giulio M. The extension reached by the minimization of the polarity distances during the evolution of the genetic code. J Mol Evol. 1989;29:288–293. doi: 10.1007/BF02103616. [DOI] [PubMed] [Google Scholar]
- 11.Ribas de Pouplana L., Turner R.J., Steer B.A., Schimmel P. Genetic code origins: tRNAs older than their synthetases? Proc Natl Acad Sci U S A. 1998;95:11295–11300. doi: 10.1073/pnas.95.19.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yarus M., Caporaso J.G., Knight R. Origins of the genetic code: the escaped triplet theory. Annu Rev Biochem. 2005;74:179–198. doi: 10.1146/annurev.biochem.74.082803.133119. [DOI] [PubMed] [Google Scholar]
- 13.Tlusty T. A colorful origin for the genetic code: information theory, statistical mechanics and the emergence of molecular codes. Phys Life Rev. 2010;7:362–376. doi: 10.1016/j.plrev.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Yu J. Challenges to the common dogma. Genomics Proteomics Bioinformatics. 2012;10:55–57. doi: 10.1016/j.gpb.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J. Life on two tracks. Genomics Proteomics Bioinformatics. 2012;10:123–126. doi: 10.1016/j.gpb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donoghue P., Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67:550–573. doi: 10.1128/MMBR.67.4.550-573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woese C.R., Olsen G., Ibba M., Söll D. Aminoacyl-tRNA synethetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ardell D.H. Computational analysis of tRNA identity. FEBS Lett. 2010;584:325–333. doi: 10.1016/j.febslet.2009.11.084. [DOI] [PubMed] [Google Scholar]
- 19.Fujishima K., Sugahara J., Tomita M., Kanai A. Sequence evidence in the archaeal genomes that tRNAs emerged through the combination of ancestral genes as 5′ and 3′ tRNA halves. PLoS One. 2008;3:e1622. doi: 10.1371/journal.pone.0001622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan P.P., Cozen A.E., Lowe T.M. Discovery of permuted and recently split transfer RNAs in Archaea. Genome Biol. 2011;12:R38. doi: 10.1186/gb-2011-12-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freyhult E., Cui Y., Nilsson O., Ardell D.H. New computational methods reveal tRNA identity element divergence between Proteobacteria and Cyanobacteria. Biochimie. 2007;89:1276–1288. doi: 10.1016/j.biochi.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Freyhult E., Moulton V., Ardell D.H. Visualizing bacterial tRNA identity determinants and antideterminants using function logos and inverse function logos. Nucleic Acids Res. 2006;34:905–916. doi: 10.1093/nar/gkj478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giegé R. Toward a more complete view of tRNA biology. Nat Struct Mol Biol. 2008;15:1007–1014. doi: 10.1038/nsmb.1498. [DOI] [PubMed] [Google Scholar]
- 24.Jakó É., Ittzés P., Szenes Á., Kun Á., Szathmáry E., Pál G. In silico detection of tRNA sequence features characteristic to aminoacyl-tRNA synthetase class membership. Nucl Acids Res. 2007;35:5593–5609. doi: 10.1093/nar/gkm598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szenes Á., Pál G. Mapping hidden potential identity elements by computing the average discriminating power of individual tRNA positions. DNA Res. 2012;19:245–258. doi: 10.1093/dnares/dss008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao Q., Tian Y., Li W., Xu Z., Xuan Z., Hu S. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X., Hu J., Yu J. Comparative analysis of eubacterial DNA polymerase III alpha subunits. Genomics Proteomics Bioinformatics. 2006;4:203–211. doi: 10.1016/S1672-0229(07)60001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X., Zhang Z., Yan J., Yu J. GC content variability of eubacteria is governed by the pol III alpha subunit. Biochem Biophys Res Commun. 2007;356:20–25. doi: 10.1016/j.bbrc.2007.02.109. [DOI] [PubMed] [Google Scholar]
- 29.Hu J., Zhao X., Zhang Z., Yu J. Compositional dynamics of guanine and cytosine content in prokaryotic genomes. Res Microbiol. 2007;158:363–370. doi: 10.1016/j.resmic.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Hu J., Zhao X., Yu J. Replication-associated purine asymmetry may contribute to strand-biased gene distribution. Genomics. 2007;90:186–194. doi: 10.1016/j.ygeno.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Qu H., Wu H., Zhang T., Zhang Z., Hu S., Yu J. Nucleotide compositional asymmetry between the leading and lagging strands of eubacterial genomes. Res Microbiol. 2010;161:838–846. doi: 10.1016/j.resmic.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 32.Wu H., Zhang Z., Hu S., Yu J. On the molecular mechanism of GC content variation among eubacterial genomes. Biol Direct. 2012;7:2. doi: 10.1186/1745-6150-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H., Qu H., Zhang Z., Hu S., Yu J. Strand-biased gene distribution and nucleotide composition. Genomics Proteomics Bioinformatics. 2012;10:186–196. doi: 10.1016/j.gpb.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hildebrand F., Meyer A., Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6:e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hershberg R., Petrov D.A. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 2010;6:e1001115. doi: 10.1371/journal.pgen.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Leuven J.T., McCutcheon J.P. An AT mutational bias in the tiny GC-rich endosymbiont genome of Hodgkinia. Genome Biol Evol. 2012;4:24–27. doi: 10.1093/gbe/evr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z., Li J., Wang J., Wong G.K., Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006;4:259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z., Yu J. Evaluation of six methods for estimating synonymous and nonsynonymous substitution rates. Genomics Proteomics Bioinformatics. 2006;4:173–181. doi: 10.1016/S1672-0229(06)60030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D., Zhang Y., Zhang Z., Zhu J., Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8:77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z., Nielsen R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol. 2008;25:568–579. doi: 10.1093/molbev/msm284. [DOI] [PubMed] [Google Scholar]
- 41.Zimmerman J.M., Eliezer N., Simha R. The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol. 1968;21:170–201. doi: 10.1016/0022-5193(68)90069-6. [DOI] [PubMed] [Google Scholar]
- 42.Rose G.D., Geselowitz A.R., Lesser G.J., Lee R.H., Zehfus M.H. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 43.Dayhoff M.O., Schwartz R.M., Orcutt B.C. A model of evolutionary change in proteins. In: Dayhoff M.O., editor. Atlas of protein sequence and structure. Silver Spring, National Biomedical Research Foundation; Washington, DC, MD: 1978. pp. 345–352. [Google Scholar]
- 44.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 45.Fraga S. Theoretical prediction of protein antigenic determinants from amino acid sequences. Can J Chem. 1982;60:2606–2610. [Google Scholar]
- 46.Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–492. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- 47.Jones D.D. Amino acid properties and side-chain orientation in proteins: a cross correlation approach. J Theor Biol. 1975;50:167–183. doi: 10.1016/0022-5193(75)90031-4. [DOI] [PubMed] [Google Scholar]
- 48.Chou P.Y., Fasman G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 49.Levitt M. Conformational preferences of amino acids in globular proteins. Biochemistry. 1978;17:4277–4285. doi: 10.1021/bi00613a026. [DOI] [PubMed] [Google Scholar]
- 50.Deléage G., Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. 1987;1:289–294. doi: 10.1093/protein/1.4.289. [DOI] [PubMed] [Google Scholar]
- 51.Browne C.A., Bennett H.P.J., Solomon S. The isolation of peptides by high-performance liquid chromatography using predicted elution positions. Anal Biochem. 1982;124:201–208. doi: 10.1016/0003-2697(82)90238-x. [DOI] [PubMed] [Google Scholar]
- 52.Miyazawa S., Jernigen R.L. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 53.Sweet R.M., Eisenberg D. Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J Mol Biol. 1983;171:479–488. doi: 10.1016/0022-2836(83)90041-4. [DOI] [PubMed] [Google Scholar]
- 54.Bull H.B., Breese K. Surface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues. Arch Biochem Biophys. 1974;161:665–670. doi: 10.1016/0003-9861(74)90352-x. [DOI] [PubMed] [Google Scholar]
- 55.Roseman M.A. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J Mol Biol. 1988;200:513–522. doi: 10.1016/0022-2836(88)90540-2. [DOI] [PubMed] [Google Scholar]
- 56.Wilson K.J., Honegger A., Stötzel R.P., Hughes G.J. The behaviour of peptides on reverse-phase supports during high-pressure liquid chromatography. Biochem J. 1981;199:31–41. doi: 10.1042/bj1990031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aboderin A.A. An empirical hydrophobicity scale for α-amino-acids and some of its applications. Int J Biochem. 1971;2:537–544. [Google Scholar]
- 58.Hopp T.P., Woods K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci U S A. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolfenden R.V., Andersson L., Cullis P.M., Southgate C.C.F. Affinities of amino acid side chains for solvent water. Biochemistry. 1981;20:849–855. doi: 10.1021/bi00507a030. [DOI] [PubMed] [Google Scholar]
- 60.Guy H.R. Amino acid side-chain partition energies and distribution of residues in soluble proteins. Biophys J. 1985;47:61–70. doi: 10.1016/S0006-3495(85)83877-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abraham D.J., Leo A.J. Extension of the fragment method to calculate amino acid zwitterion and side chain partition coefficients. Proteins. 1987;2:130–152. doi: 10.1002/prot.340020207. [DOI] [PubMed] [Google Scholar]
- 62.Eisenberg D., Schwarz E., Komarony M., Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol. 1984;179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 63.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 64.Manavalan P., Ponnuswamy P.K. Hydrophobic character of amino acid residues in globular proteins. Nature. 1978;275:673–674. doi: 10.1038/275673a0. [DOI] [PubMed] [Google Scholar]
- 65.Zamyatnin A.A. Protein volume in solution. Prog Biophys Mol Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
- 66.Chen K., Meng Q., Ma L., Liu Q., Tang P., Chiu C. A novel DNA sequence periodicity decodes nucleosome positioning. Nucleic Acids Res. 2008;36:6228–6236. doi: 10.1093/nar/gkn626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen K., Wang L., Yang M., Liu J., Xin C., Hu S. Sequence signatures of nucleosome positioning in Caenorhabditis elegans. Genomics Proteomics Bioinformatics. 2010;8:92–102. doi: 10.1016/S1672-0229(10)60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong G.K., Wang J., Tao L., Tan J., Zhang J., Passey D.A. Compositional gradients in Gramineae genes. Genome Res. 2002;12:851–856. doi: 10.1101/gr.189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cui P., Ding F., Lin Q., Zhang L., Li A., Zhang Z. Distinct contributions of replication and transcription to mutation rate variation of human genomes. Genomics Proteomics Bioinformatics. 2012;10:4–10. doi: 10.1016/S1672-0229(11)60028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J., Zhang J., Li R., Zheng H., Li J., Zhang Y. Evolutionary transients in the rice transcriptome. Genomics Proteomics Bioinformatics. 2010;8:211–228. doi: 10.1016/S1672-0229(10)60023-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cui P., Lin Q., Ding F., Hu S., Yu J. The transcript–centric mutations in human genomes. Genomics Proteomics Bioinformatics. 2012;10:11–22. doi: 10.1016/S1672-0229(11)60029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green P., Ewing B., Miller W., Thomas P.J. NISC comparative sequencing program, Green ED. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- 73.Majewski J. Dependence of mutational asymmetry on gene-expression levels in the human genome. Am J Hum Genet. 2003;73:688–692. doi: 10.1086/378134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu J., Yang Z., Kibukawa M., Paddock M., Passey D.A., Wong G.K. Minimal introns are not “junk”. Genome Res. 2002;12:1185–1189. doi: 10.1101/gr.224602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu J., He F., Wang D., Liu K., Huang D., Xiao J. A novel role for minimal introns: routing mRNAs to the cytosol. PLoS One. 2010;5:e10144. doi: 10.1371/journal.pone.0010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang D., Yu J. Both size and GC-content of minimal introns are selected in human population. PLoS One. 2011;6:e17945. doi: 10.1371/journal.pone.0017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang L., Yu J. A comparative analysis of divergently-paired genes (DPGs) among Drosophila and vertebrate genomes. BMC Evol Biol. 2009;9:55. doi: 10.1186/1471-2148-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cui P., Liu W., Zhao Y., Lin Q., Ding F., Xin C. The association between H3K4me3 and antisense transcription. Genomics Proteomics Bioinformatics. 2012;10:74–81. doi: 10.1016/j.gpb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diemer G.S., Stedman K.M. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol Direct. 2012;7:13. doi: 10.1186/1745-6150-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamaguchi M., Mori Y., Kozuka Y., Okada H., Uematsu K., Tame A. Prokaryote or eukaryote? A unique microorganism from the deep sea. J Electron Microsci (Tokyo) 2012;61:423–431. doi: 10.1093/jmicro/dfs062. [DOI] [PubMed] [Google Scholar]
- 81.Darwin C. vol. 3. John Murray; London: 1887. (The life and letters of Charles Darwin, including an autobiographical chapter). [Google Scholar]