


Abstract
The Cdk12/CycK complex promotes expression of a subset of RNA polymerase II genes, including those of the DNA damage response. CDK12 is among only nine genes with recurrent somatic mutations in high-grade serous ovarian carcinoma. However, the influence of these mutations on the Cdk12/CycK complex and their link to cancerogenesis remain ill-defined. Here, we show that most mutations prevent formation of the Cdk12/CycK complex, rendering the kinase inactive. By examining the mutations within the Cdk12/CycK structure, we find that they likely provoke structural rearrangements detrimental to Cdk12 activation. Our mRNA expression analysis of the patient samples containing the CDK12 mutations reveals coordinated downregulation of genes critical to the homologous recombination DNA repair pathway. Moreover, we establish that the Cdk12/CycK complex occupies these genes and promotes phosphorylation of RNA polymerase II at Ser2. Accordingly, we demonstrate that the mutant Cdk12 proteins fail to stimulate the faithful DNA double strand break repair via homologous recombination. Together, we provide the molecular basis of how mutated CDK12 ceases to function in ovarian carcinoma. We propose that CDK12 is a tumor suppressor of which the loss-of-function mutations may elicit defects in multiple DNA repair pathways, leading to genomic instability underlying the genesis of the cancer.
INTRODUCTION
Gene transcription by RNA polymerase II (RNAPII) is a sophisticated process involving numerous factors that enable regulated progression of the polymerase through sequential stages of the transcription cycle (1). Therein, the C-terminal domain (CTD) of the Rbp1 subunit of RNAPII, consisting of multiple heptapeptide repeats with consensus sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser, undergoes a dynamic cycle of post-translational modifications and cis/trans isomerizations, providing a platform for mRNA biogenesis and export factors (2). Among various modifications, Ser2 phosphorylation (Ser2-P) of the CTD is most strongly linked to productive transcription and pre-mRNA processing, the steps following promoter-proximal pausing of RNAPII (3,4). In addition to the well-established P-TEFb kinase, which consists of the catalytic Cdk9 and regulatory cyclin (Cyc) T subunits, recent evidence from fruit fly and human cells indicates that the Cdk12/CycK complex catalyzes the Ser2-P mark as well (5–9). Likewise, Ctk1 and Lsk1, the yeast orthologs of Cdk12, are major Ser2-P kinases in Saccharomyces cerevisiae and Schizosaccharomyces pombe, respectively (10,11). Illustrating the significance of this novel transcriptional kinase, genetic inactivation of CycK in mice is embryonic lethal (6), possibly due to the role of Cdk12/CycK in maintaining embryonic stem cell self-renewal (12). In contrast to the broad requirement for P-TEFb in transcription (13,14), depletion of Cdk12/CycK in human cells leaves expression of most genes unaltered (6). However, a prominent group among the downregulated genes is the one directing DNA-damage response (DDR), which includes many core players that ensure genomic stability, such as BRCA1, ATR, FANCI and FANCD2. Accordingly, knockdown of Cdk12/CycK induces DNA damage, as well as sensitivity to various DNA damaging agents (6). This important function of Cdk12/CycK seems to be evolutionarily conserved as mutations of CTK genes in S. cerevisiae prevent upregulation of several DNA repair genes, rendering yeast cells incapacitated in the face of genotoxic insult (15,16).
Mutations in factors controlling transcription underlie many diseases including cancer (17). While contribution of misregulated transcription elongation to tumorigenesis through P-TEFb has been documented (13,18,19), the relationship between mutations in Cdk12/CycK, altered gene expression and relevance to cancer has not been defined. Importantly, CDK12 is perturbed in several cancers, including breast, gastric and ovarian cancer. In breast cancer, CDK12 was found to be co-amplified with ERBB2, the tyrosine kinase receptor gene, which is deregulated in about 20% of breast tumors (20,21). Furthermore, out-of-frame rearrangements of CDK12 were identified in micropapillary breast carcinoma, 13% of ERBB2 positive cancers (22) and gastric cancers (23). Finally, recent work by The Cancer Genome Atlas (TCGA) on the high-grade serous ovarian carcinoma (HGS-OvCa) has provided the most compelling evidence for a possible role for mutated CDK12 in cancer (24). Employing whole-exome DNA sequencing, the TCGA study reported a catalog of somatic gene mutations for 316 HGS-OvCa tumor samples. Whereas TP53 dominated the mutation spectrum, CDK12 was identified as one of only eight further genes with statistically recurrent somatic mutations. Detailed re-analyses of the CDK12 mutations from the TCGA work and the Catalog of somatic mutations in cancer (COSMIC) database defined 7 out of 12 mutations as homozygous, highlighting CDK12 as a novel candidate tumor suppressor in ovarian carcinoma (25). Importantly, approximately half of all HGS-OvCa cases display defects in homologous recombination (HR) (24), the pathway that repairs DNA double-strand breaks (DSBs) most faithfully (26). Since depletion of Cdk12/CycK downregulates many components of DDR that function in HR (27), it is possible that CDK12 mutations in HGS-OvCa could be detrimental to the efficacy of HR.
Despite the available genetic evidence, the significance of CDK12 mutations for the assembly and function of Cdk12/CycK remains to be defined. Furthermore, it is unclear how mutations in this novel transcriptional kinase might influence cancerogenesis. To address these questions, we focused on CDK12 mutations identified in HGS-OvCa. Collectively, our results show that the CDK12 mutations impair the transcriptional role of Cdk12/CycK in DNA damage repair by HR, elucidating an important link between the non-functional Cdk12 proteins and cancer.
MATERIALS AND METHODS
Cell culture
HEK 293, HEK 293 Flp-In T-REx (Life Technologies), Caov-3 (ATCC) and HeLa DR-GFP cell lines were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin. HCT116 cell line was grown in DMEM supplemented with 5% FBS. HEK 293 Flp-In T-REx cell lines expressing 3X-FLAG peptide or Cdk12-F proteins were generated according to the manufacturer's instructions (Life Technologies). All cell lines were maintained at 37°C with 5% CO2. Media and supplements were from Sigma-Aldrich.
Plasmid DNAs and siRNAs
The plasmids used in this study are listed in Supplementary Table S4. Rev-Cdk12 and Cdk12-F proteins encoded by the pRev-Cdk12 and pcDNA3.1-Cdk12-F expression plasmids, respectively, were described previously (6). Cdk12 #1 siRNA (sc-44343), CycK siRNA (sc-37600) and Cdk13 siRNA (sc-72835) were from Santa Cruz Biotechnology. Cdk12 #2 siRNA has the sense sequence 5′-rCrArGrArUrGrArCrCrCrUrUrGrArArGrCrUrUdTdT-3′. Whereas Cdk12 #1 siRNA was used in Figure 5A and B, and Supplementary Figure S7, Cdk12 #2 siRNA was used in Figure 5C and Supplementary Figure S8. Control siRNAs (sc-37007 and SIC001) were from Santa Cruz Biotechnology and Sigma-Aldrich, respectively. The plasmids encoding the wild-type and mutant Cdk12 proteins that were used to generate the HEK 293 Flp-In T-REx cell lines were mutated using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) to become resistant to Cdk12 #2 siRNA using the following primer pair: 5′-CAGCAGAACAGACGACACTCGAGGCTTCAAGCACACCAG-3′; 5′-CTGGTGTGCTTGAAGCCTCGAGTGTCGTCTGTTCTGCTG-3′.
Figure 5.
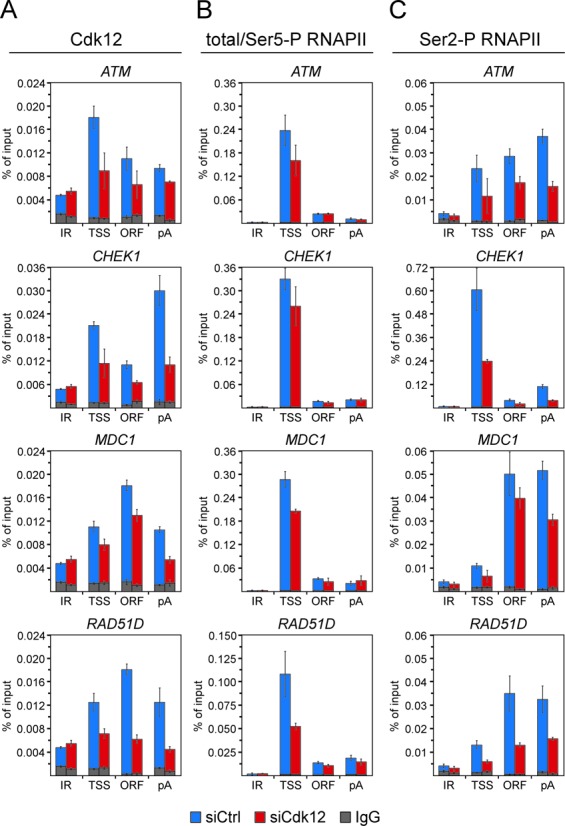
Cdk12/CycK is present at the novel HR genes to promote phosphorylation of the CTD of RNAPII at Ser2. (A, B and C) Control (blue bars) or Cdk12 knockdown HeLa DR-GFP cells (red bars) were subjected to ChIP-qPCR analysis to determine the levels of Cdk12 (panel A), total/Ser5-P RNAPII (panel B) and Ser2-P RNAPII (panel C) occupancy at RAD51D intergenic region (IR) and three gene-specific regions as indicated below the graphs. The identity of genes analyzed is indicated on top of each graph. Levels of IgG signals at IR and gene regions are presented as gray bars. Where these levels were significantly lower than those obtained with specific antibody, the error bars were omitted for simplicity reasons. Results are presented as percent of input DNA and plotted as the mean ± SD.
Immunoprecipitation assay and in vitro kinase assay
HEK 293 Flp-In T-REx cell lines expressing 3X-FLAG and Cdk12-F proteins upon induction with 1 μg/μl of tetracycline for 24 h and transiently transfected HEK 293 cells (Supplementary Figure S2 and Figure 4) were lysed in buffer C containing 20 mM Tris–HCl (pH.7.9), 150 mM NaCl, 10 mM KCl, 1.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% NP-40 and protease/phosphatase inhibitors (Pierce). Upon centrifugation at 20 000 g for 30 min at 4°C, protein complexes containing Cdk12-F proteins were immuno-purified from whole cell extracts (WCEs) using 10 μl of packed FLAG-M2 agarose (Sigma-Aldrich) for 1 h at 4°C. The immuno-purified complexes were washed 3× with 1 ml of buffer C, eluted by boiling in Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, resolved by SDS-PAGE and examined by Western blotting using CycK (NBP1-06519, Novus Biologicals) and FLAG (F3165, Sigma-Aldrich) antibodies. For in vitro kinase assays, immuno-purifications of Cdk12-F-containing complexes were performed as above with the following modifications. Cell pellets were lysed in buffer B containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40 and protease/phosphatase inhibitors (Pierce). The complexes were washed two times with buffer B, followed by five washes with the kinase buffer A containing 50 mM Tris–HCl, 100 mM NaCl, 10 mM MgCl2 and 0.1% NP-40. The beads with the purified complexes were resuspended in 30 μl of kinase buffer A supplemented with 100 mM adenosine triphosphate, 1 mM dithiothreitol (DTT) and 300 ng of recombinant Glutatione S-transferase (GST)-CTD substrate (9) and incubated for 10 min at room temperature, followed by 1 h at 30°C in Bioer mixing block MB-102 at 300 rpm. Reactions were stopped by boiling in SDS-PAGE sample buffer, resolved by SDS-PAGE and analyzed by Western blotting using CycK (NBP1-06519, Novus Biologicals), Ser2-P RNAPII (ab5095, Abcam), FLAG (F3165, Sigma-Aldrich) and GST (sc-138, Santa Cruz Biotechnology) antibodies.
Figure 4.
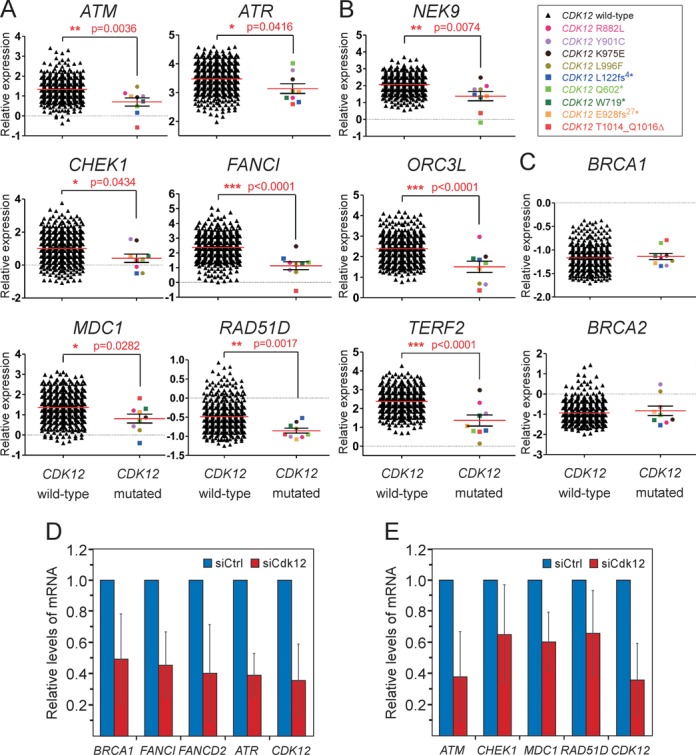
The crucial DDR genes are downregulated in HGS-OvCa patient samples with mutations in CDK12. (A, B and C) Graphs show comparisons of relative expression levels between the HGS-OvCa samples with the wild-type or mutated CDK12. The identity of genes is indicated on top of each graph. The data was generated using the following microarray probes: ATM (212672_at), ATR (209903_s_at), CHEK1 (205394_at), FANCI (213007_at), MDC1 (203062_s_at), RAD51D (209965_s_at), NEK9 (212299_at), ORCL3 (210028_s_at), TERF2 (203611_at), BRCA1 (211851_x_at), BRCA2 (208368_s_at). Whereas samples with the wild-type CDK12 are plotted as black triangles, those containing individual missense or nonsense/indel CDK12 mutations are depicted as colored circles or squares, respectively, as indicated by the legend in the top right corner. Results are presented as mean (red line) with standard error of the mean (SEM) (black whiskers). P-values are given next to red asterisks (*) and the number of asterisks indicates the degree of significance as follows: * = P ≤ 0.05; ** = P ≤ 0.01; *** = P ≤ 0.001. Panels A and C show genes related to the HR pathway, while other affected DDR genes are shown in panel B. (D and E) Depletion of Cdk12 decreases the mRNA levels of HR genes in Caov-3 cells. Relative mRNA levels of genes indicated below the bars were determined by RT-qPCR using total RNA samples isolated from Caov-3 cells treated with the control (blue bars) or Cdk12 #1 siRNA (red bars). The error bars represent the mean ± SD.
RNA-tethering gene reporter assay
HEK 293 cells were plated in 12-well format at 100 000 cells per well and transfected with 0.3 μg of pSLIIB-CAT and 1.5 μg of Rev-Cdk12, Rev or Cdk12-F expression plasmids as indicated by the FuGENE6 reagent (Roche) After 48 h, cells were lysed in 125 μl of lysis buffer (250 mM Tris–HCl, 0.5% Triton-X, pH 7.5) for 30 min on ice. Finally, 100 μl of the heat-inactivated cell lysates was supplemented with 50 μl of CAT assay buffer (0.5 μl of 3H-acetyl CoA (250 μCi, Amersham), 30 μl of 1.66 mg/ml chloramphenicol, 5 μl of 250 mM Tris (pH 7.5), 14.5 μl ddH2O) and CAT activity was measured by a scintillation counter. For normalization, total protein content in whole-cell lysate was determined using Bio-Rad DC kit. Experiments were done twice in biological triplicates. Statistical analysis was performed using one-way ANOVA. Values are plotted as mean ± SD. P-value between groups is statistically significant (P < 0.05). Levels of Rev-Cdk12 chimeras were detected by Western blotting using Cdk12 (sc-32643, Santa Cruz Biotechnology) antibody.
RNAi, RT-qPCR and Western blotting
Thirty to fifty percent confluent Caov-3 or HCT116 cells were transfected with 10 pmol of control (sc-37007), Cdk12 #1, Cdk12 #2, CycK or Cdk13 siRNA using Lipofectamine RNAiMax reagent (Life Technologies). Total RNA was isolated using RNAzol (Molecular Research Center) 72 h post-transfection. Reverse transcription was performed with M-MLV reverse transcriptase system using random hexamers (Life Technologies). For qPCR, 1 μl of cDNA was mixed with 2× Light Cycler 480 Sybr Green Master (Roche) and 0.3 μM forward and reverse primer (for sequences of the primers, see Supplementary Table S5) in a final volume of 20 μl. Amplifications were run on the Light Cycler 480II (Roche) using the following cycle conditions: 94°C for 2 min (1 cycle); 95°C for 30 s, 60°C for 30 s, 72°C for 30 s (45 cycles). HPRT expression levels were used for normalization and gene expression differences were calculated using the threshold cycle method. Values are plotted as mean ± SD. Levels of the analyzed proteins in control, Cdk12, CycK and Cdk13 siRNA-treated HCT116 cells were determined by Western blotting using ATM (sc-23921), Chek1 (sc-8408), Rad51D (sc-366363) and GAPDH (sc-32233) antibodies from Santa Cruz Biotechnology.
ChIP-qPCR assay
The assay was performed as described previously (28) with the following modifications. Hela DR-GFP cells were plated in 15 cm plates, transfected with 400 pmol of Cdk12 #2 or control siRNA using Lipofectamine RNAiMAX reagent (Life Technologies) at 70% confluency, plated in two 15 cm plates 24 h later and grown for additional 24 h before proceeding with the assay. Crude nuclear extract obtained from the confluent 15 cm plate was sonicated in 800 μl of RIPA buffer (150 mM NaCl, 1% NP40, 0.5% DOC, 0.1% SDS, 50 mM Tris.Cl pH 8.0, 5 mM EDTA pH 8.0) 10× for 11 s at output power of 11 Watt. Chromatin was clarified by centrifugation at 13 000 g for 15 min, 400 μl of supernatant added to 12 μl of antibody-coupled protein G Dynabeads (Life Technologies) and the mixture supplemented with additional 600 μl of RIPA buffer rotated overnight at 4°C. Before adding the chromatin, the beads were pre-blocked with bovine serum albumin and salmon sperm DNA overnight at a final concentration of 0.2 μg/μl, pre-incubated in 500 μl RIPA buffer for 4 h with the antibody and collected by magnet to remove the unbound antibody. We used 3 μg of Cdk12 (ab57311), 2 μg of total/Ser5-P RNAPII (ab5408) and 1 μg of Ser2-P RNAPII (ab5095) antibodies from Abcam. Normal mouse (sc-2025) and rabbit (sc-2027) IgG control antibodies were from Santa Cruz Biotechnology. Generally, 1/60th of the precipitated ChIP sample was used for each qPCR reaction. For input DNA, 2.5% of the cleared chromatin was used and 1/100th of the DNA dissolved in 100 μl of water was used for each qPCR reaction. For sequences of the primers, see Supplementary Table S6. Samples were analyzed by the Stratagene Mx3005P real-time polymerase chain reaction system and FastStart Universal SYBR Green QPCR Master (Rox) (Roche). Results were obtained from three independent experiments. Values are plotted as mean of percentage of input DNA ± SD.
DR-GFP assay
Hela DR-GFP cells were plated in 6 cm plates at 800 000 and transfected with 100 pmol of control (SIC001) or Cdk12 #2 siRNA using Lipofectamine RNAiMAX reagent (Life Technologies). Twenty-four hours after siRNA transfection, the cells were co-transfected with 2 μg of I-SceI and 3 μg of Cdk12-F expression vectors or a parental pcDNA5/FRT/TO/3XFLAG vector as indicated by using Lipofectamine 3000 reagent (Life Technologies). After 48 h, the cells were trypsinized, collected by centrifugation at 300 x g for 5 min, resuspended in 500 μl of DMEM and analyzed for GFP fluorescence using BD Accuri flow cytometer. Experiments were performed in biological triplicates and statistical analysis was done using one-way ANOVA. The error bars represent the mean ± SD. P-value between groups is statistically significant (P < 0.05). Levels of Cdk12-F proteins were detected by Western blotting using Cdk12 (sc-32643, Santa Cruz Biotechnology) antibody.
Statistical analysis of TCGA study
The expression levels of DDR genes in TCGA were downloaded from the Oncomine database (TCGA_Ovarian dataset). The information about CDK12 mutation status was obtained from the COSMIC database. Data from these repositories was retrieved on February 26, 2014. Samples for which CDK12 mutation status was not available in COSMIC were discarded from the TCGA_Ovarian dataset similarly to the five samples ‘variants of unknown origin’ and ‘previously reported’ (1 sample) resulting in 462 HGS-OvCa samples. Out of these samples, there are 453 CDK12 wild-type samples, four samples with missense mutation in CDK12 and five samples with nonsense/indel mutation in CDK12 (see also Supplementary Table S1). Data were analyzed with the Statistica software (Version 12.0) and visualized using GraphPad Prism 5. Results are expressed as mean with standard error of the mean. The Shapiro-Wilk test was used to assess the normal distribution of the variables (P > 0.05). Data without a normal distribution were analyzed with non-parametric test (Mann–Whitney test), and data with a normal distribution were analyzed with parametric test (unpaired t-test). Unpaired t-test with Welsch's correction was applied for variables with significantly different variances (F-test, P < 0.05). The statistical analysis was conducted at 95% confidence level. Symbols used to express statistical significance: non-significant (ns) = P > 0.05; * = P ≤ 0.05; ** = P ≤ 0.01; *** = P ≤ 0.001.
Structural analyses
Structural analysis of the CDK12 mutations was performed on the human Cdk12/CycK structure determined at 2.2 Å resolution (4NST, (9)). The molecular diagrams were drawn with PyMOL (http://www.pymol.org/).
RESULTS
Defective interaction between CycK and Cdk12 is the predominant consequence of CDK12 mutations in HGS-OvCa
In this study, we focused our attention on nine validated CDK12 mutations, which were identified in the TCGA work (24), including five nonsense and insertion/deletion mutations, as well as four missense mutations (Supplementary Table S1 and Figure 1A). Among the nonsense mutations, the L122fs4* and Q602* are the most drastic, since they yield the mutant Cdk12 proteins without most predicted regions. Next, the nonsense W719* and the insertion E928fs27* mutation terminate Cdk12 at the start and in the middle of its kinase domain (KD), respectively. Finally, the four missense mutations, including R882L, Y901C, K975E and L996F and an internal deletion T1014_Q1016Δ mutation are all positioned within the KD of Cdk12. Of note, by inspecting the COSMIC database, we found that among the nine HGS-OvCa patient cases with the mutated CDK12, each sample contains one CDK12 mutation (data not shown), providing the rationale for examining the impact of single CDK12 mutations.
Figure 1.
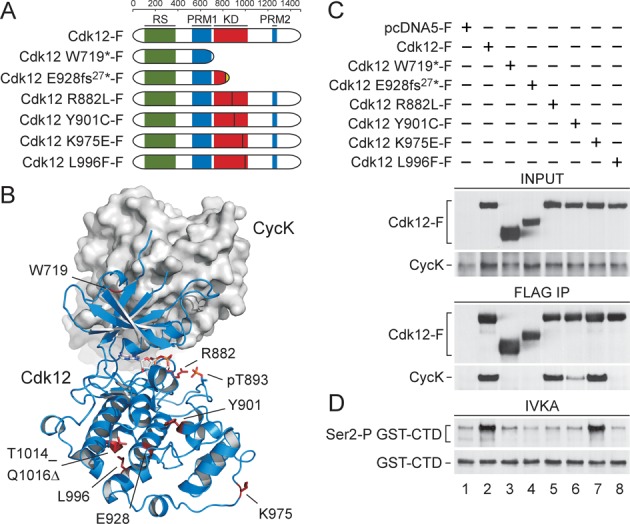
CDK12 mutations in HGS-OvCa abrogate the activity of Cdk12 predominantly by impairing the interaction between Cdk12 and CycK. (A) Schematic depiction of the wild-type and mutant Cdk12 proteins containing individual CDK12 mutations analyzed in this study. Highly structured kinase domain (KD; red), arginine/serine rich region (RS; green) and two regions with proline-rich motifs (PRM1 and PRM2; blue) are depicted. The ruler on top indicates the length of Cdk12 protein in amino acids. Vertical lines denote sites of individual missense and insertion mutations. Finally, ‘fs’ stands for a frame-shift mutation and the associated number indicates the number of altered amino acids at the C-terminus of mutant Cdk12 protein (dotted). (B) Overall structure of human Cdk12/CycK and positions of the mutations. Cdk12 is shown as cartoon representation in blue and CycK as surface representation in grey. CDK12 missense, insertion and internal deletion mutations are located in the C-terminal lobe of the KD. The mutated amino acid residues are highlighted in red. pT893 is highlighted in orange. (C) Effects of the CDK12 mutations on the interaction between Cdk12 and CycK. The indicated wild-type and mutant FLAG epitope-tagged Cdk12 proteins (Cdk12-F) were immuno-purified from whole cell extracts (WCEs) of the individual HEK 293 Flp-In T-Rex cell lines using FLAG-M2 agarose (FLAG IP) and examined for their interaction with endogenous CycK. Levels of Cdk12-F and CycK proteins in WCEs (INPUT, 5% of WCEs; top) and IPs (FLAG IP; bottom) were detected by Western blotting using FLAG and CycK antibodies. (D) CDK12 mutations abrogate the kinase activity of Cdk12. The indicated wild-type and mutant Cdk12-F proteins were immuno-purified (IP) as in panel C and the complexes were examined for their kinase activity by in vitro kinase assay (IVKA) toward the recombinant GST-CTD. Levels of Ser2-P GST-CTD isoforms and input GST-CTD (30%) were detected by Western blotting using Ser2-P-specific RNAPII and GST antibodies.
We analyzed W719* and E928fs27* as the two most representative CDK12 nonsense and insertion mutations, as well as all four CDK12 missense mutations (Figure 1A). Whereas the deletions of the Cdk12 KD brought about by the former mutations are expected to inactivate Cdk12, it is the evolutionary conservation of the amino acids altered by the latter mutations that likely reflects their functional importance. In fact, except for the K975 residue which is conserved from fruit flies to humans, the R882, Y901 and L996 residues are all conserved from budding yeast to humans (Supplementary Figure S1). To get a structural perspective on these mutations, we took advantage of the crystal structure of the human Cdk12/CycK complex encompassing the KD of Cdk12 and the cyclin box domain of CycK (9). An important characteristic of this structure is that CycK interacts only with the N-terminal lobe of the KD. However, upon positioning the CDK12 mutations on the structure, we found that the insertion and missense mutations are all located within the C-terminal lobe of the KD (Figure 1B). This observation suggested that possible effects of the mutations on the Cdk12/CycK complex could be exerted via allosteric mechanisms.
Activation of any Cdk is a two-step process wherein the binding between the Cdk KD and the cyclin reorients the catalytic cleft of the kinase, setting the stage for its full activation by T loop phosphorylation (29,30). Thus, we next asked whether the CDK12 mutations identified in HGS-OvCa affect the interaction between Cdk12 and CycK (Figure 1C). Here, we employed the Flp-In T-REx system to generate stable HEK 293 cell lines each expressing the wild-type or mutant 3XFLAG epitope-tagged Cdk12 (Cdk12-F) proteins upon tetracycline induction. This system allows accurate comparison between the CDK12 mutations because the integration of individual Cdk12 expression vectors to the defined locus ensures the same genetic background of different cell lines. Upon immuno-purifying the wild-type and mutant Cdk12-F proteins from the WCEs, we examined the levels of endogenous CycK in these isolations by Western blotting (Figure 1C, bottom panels). As expected, the wild-type Cdk12 interacted with CycK, and the mutant Cdk12 W719* and E928fs27* proteins failed to do so (Figure 1C, lanes 1–4). In contrast, while the mutant Cdk12 R882L and K975E proteins displayed unperturbed CycK binding, this interaction was greatly reduced or lost in the case of the Y901C or L996F mutations, respectively (Figure 1C, lanes 5–8). We also asked whether the binding between Cdk12 and CycK is impacted by the panel of CDK12 mutations found in lung, skin, ovarian, and large intestine cancers (23,31,32) (Supplementary Figure S2). Of note, a relative majority of all currently identified CDK12 mutations in human cancers clusters within the KD, while the rest of them are located within other Cdk12 regions, such as those with many arginine/serine (RS) residues and proline-rich motifs (PRM) (Supplementary Table S2). Thus, we selected S325N and S352F as the Cdk12 mutations representing the RS region, and the G909R and E1024* mutations found within and right after the Cdk12 KD, respectively. While the mutations within the RS region did not show any defects in CycK binding, the mutant Cdk12 G909R protein lost the ability to bind CycK, and the E1024* mutation decreased this interaction considerably (Supplementary Figure S2B, top panels, lanes 3–6). Collectively, these findings demonstrate that individual CDK12 mutations in HGS-OvCa and other cancers that delete or alter the KD can have detrimental effects on the formation of the Cdk12/CycK complex. However, some mutations did not interfere with the binding between Cdk12 and CycK, the presumed obligatory step in the activation of Cdk12, raising a possibility that activity of those mutant Cdk12 proteins is not compromised.
CDK12 mutations in HGS-OvCa abrogate the kinase activity of Cdk12
To investigate the effects of the mutations on the catalytic activity of Cdk12, we next performed in vitro kinase assays. We immuno-purified the wild-type and mutant Cdk12-F-containing complexes from WCEs of the HEK 293 Flp-In cell lines as above and used their comparable amounts in the kinase assays employing the recombinant GST-CTD protein as a substrate (Figure 1D and Supplementary Figure S2C). Since Cdk12/CycK catalyzes the formation of Ser2-P in human cells (6–8), we followed kinase activity of the Cdk12-F-containing complexes by Western blotting using the antibody specific for this RNAPII CTD mark. Compared with our control immuno-purification, which was prepared using WCEs of HEK 293 Flp-In cell line expressing 3XFLAG peptide, the wild-type Cdk12-F protein phosphorylated the GST-CTD chimera effectively (Figure 1D, lanes 1–2). However, except for the mutant Cdk12 K975E protein, the entire set of the proteins containing the CDK12 mutations failed to phosphorylate the GST-CTD above background levels (Figure 1D, lanes 3–8). Using the same experimental approach, we also examined catalytic activity of the panel of Cdk12 proteins carrying the mutations found in various cancers (Supplementary Figure S2B). While we observed that the S325N and S352F mutations had no effect, the mutant Cdk12 G909R and E1024* proteins displayed impaired kinase activity (Supplementary Figure S2C, bottom panels, lanes 3–6). Thus, except for one, all the CDK12 mutations that delete or alter the KD of Cdk12 ablate the activity of the kinase. Whereas deficient CycK binding correlated completely with the loss of Cdk12 activity, the R882L mutation inactivated the kinase without impairing CycK binding, suggesting that a subsequent defect prevents activation of this mutant Cdk12 protein.
Insertion and missense CDK12 mutations in HGS-OvCa likely elicit structural defects that perturb formation and activity of the Cdk12/CycK complex
These findings prompted us to analyse why the CDK12 mutations provoke such profound effects on the formation and activity of the Cdk12/CycK complex (Figure 2). Given that the mutant Cdk12 W719* protein lacks the entire KD, its failure to bind CycK and phosphorylate the CTD is not surprising. Even though we did not examine them experimentally, the same applies to the rest of the CDK12 nonsense mutations that generate more drastically truncated Cdk12 L122fs*4 and Q602* proteins. However, we were particularly eager to understand those CDK12 mutations in HGS-OvCa that alter the KD within its C-terminal lobe but still compromise the ability of Cdk12 to bind CycK or phosphorylate the CTD of RNAPII (Figure 2A).
Figure 2.
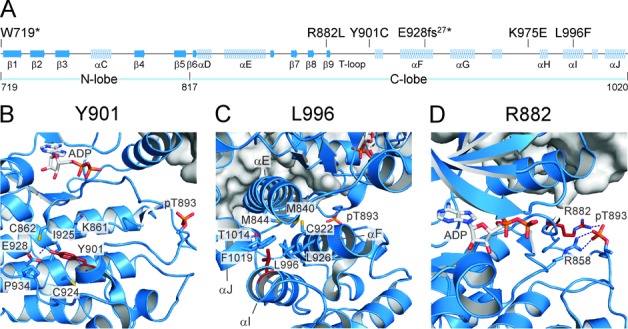
Structural analysis of the CDK12 mutations. (A) Schematic representation of secondary structure elements within the KD of Cdk12. Canonical α-helices and β-strands as well as boundaries of N- and C-terminal lobes are labeled. Positions of the mutated Cdk12 amino acids analyzed in this study are indicated on top of the schematic. (B) The hydroxyl group of the Y901 aromatic ring mediates a tight hydrogen bond (2.7 Å) to the carboxyl group of E928. Interestingly, the tyrosine is closely surrounded by two cysteines, C862 and C924, which could make the Y901C mutation unfavorable. Hydrophobic contacts of Y901 are formed to I925 and K861, with P934 stabilizing the conformation of E928. (C) L996 resides on helix αI. It is surrounded by T1014 and F1019 on helix αJ, by C922, G923 and L926 of helix αF, and M840 and M844 of helix αE. The replacement of L996 by the spacious aromatic ring of a phenylalanine might preclude the tight assembly of the four helical bundle. (D) R882 is part of the canonical arginine network in CDKs that stabilize the T-loop upon phosphorylation of the critical threonine. Together with R858 of the HRD motif, R882 mediates salt bridges to the phosphate group of pT893. Its mutation to leucine might prohibit the conformational arrangement of the T loop required for full activation of the kinase.
For these analyses, we again employed the structural information of Cdk12/CycK (9). First, the E928fs27* mutation, which causes the appearance of 27 irregular residues before reaching the stop codon, truncates Cdk12 in the middle of the C-terminal lobe, most likely resulting in misfolding of the remaining part of the KD including the N-terminal lobe, leading to the observed defects in CycK binding and kinase activity. Next, the Y901C mutation substitutes the large aromatic residue located in the core of the C-terminal lobe and beneath the catalytic center of the kinase with a much smaller cysteine residue, creating a ‘void’ in the local environment and prohibiting multiple bonds of Y901 with the residues nearby (Figure 2B). In turn, the rearrangements within the C-terminal lobe are likely to follow, affecting the conformation of the N-terminal lobe and thus leading to the decreased assembly and activity of this mutant Cdk12/CycK complex. Furthermore, the L996F mutation is located on helix αI in the core of the C-terminal lobe and surrounded by helices αE, αF and αJ, forming a tight four helical bundle (Figure 2C). It is likely that the spacious aromatic ring of this Cdk12 mutation demolishes the helical assembly. In turn, this could provoke misfolding of the C- as well as N-terminal lobes, rendering the N-terminal lobe refractory to CycK binding, thus inactivating the kinase. In contrast, the R882L mutation did not affect the binding of CycK to Cdk12 but did result in the inactive kinase, indicating that this mutation precludes the activation of Cdk12 following CycK binding. In fact, R882 is one of the three canonical arginine residues in Cdks that are coordinated upon activation by the phosphorylated threonine 893 (pT893) in the T loop via salt bridges (9,29). Subsequently, the arginine residues contact other Cdk12 and CycK groups, extending the organizing influence of the phosphate group, which completes reorganization of the catalytic cleft and substrate recognition site initiated by the cyclin binding. Given that the R882L mutation prevents the contact to pT893, the subsequent series of events may not ensue, leading to inactive Cdk12 (Figure 2D). Finally, K975 is located in the exposed region of the C-terminal lobe, suggesting no structural role for this residue. That the K975E mutation did not show any effects on CycK binding and Cdk12 activity but did compromise the function of Cdk12/CycK in cells (see Figures 3, 4 and 6 below) may suggest the failure of our biochemical assays to reveal subtle defects of this mutation. Alternatively, K975 could play a stimulatory role in Cdk12-dependent gene expression via a mechanism that has yet to be determined.
Figure 3.
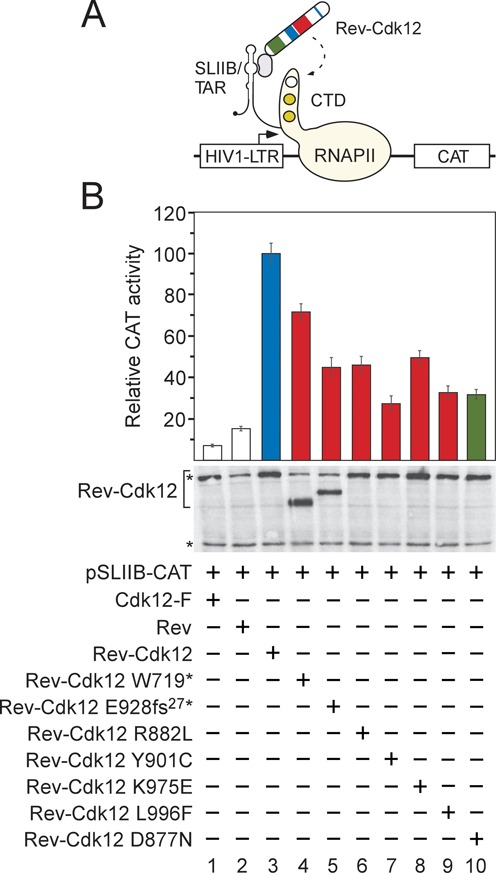
CDK12 mutations in HGS-OvCa decrease transcriptional activation by Cdk12. (A) Schematic depiction of the heterologous RNA tethering assay. Plasmid reporter pSLIIB-CAT contains modified HIV1-LTR promoter in which the apical region of transactivation response RNA element (TAR) was substituted with 29-nucleotide stem-loop IIB (SLIIB) subdomain of the HIV-1 Rev response element (RRE), the Rev binding RNA sequence. The interaction between Rev (pink oval) and SLIIB within the TAR/SLIIB stem-loop structure at the 5′ end of nascent RNA tethers the Rev-Cdk12 chimeric protein to RNAPII engaged in transcription, resulting in elevated transcription of CAT reporter gene. CTD of the biggest RNAPII subunit Rbp1 is represented as a tail of RNAPII (yellow), wherein white circle depicts Ser2 residue to be phosphorylated by Rev-Cdk12 (dashed arrow) and gold circles depict Ser5 and Ser7 residues in an already phosphorylated form. Arrow within HIV1-LTR indicates transcription start site. (B) CDK12 HGS-OvCa mutations compromise stimulation of transcription by Rev-Cdk12. HEK 293 cells were co-transfected with pSLIIB-CAT reporter gene and plasmids encoding the proteins indicated below the graph. Transcriptional activities of Cdk12-F (white bar 1), Rev (white bar 2), the mutant Rev-Cdk12 chimeras (red bars) and catalytically dead Rev-Cdk12 D887N chimera (green bar) are represented as CAT activities relative to the activity of wild-type Rev-Cdk12 chimera (blue bar), which was set to 100%. Results are presented as the mean ± SD. Levels of the Rev-Cdk12 chimeras and endogenous Cdk12 protein are shown below the graph and were detected by Western blotting using Cdk12 antibody. The top asterisk (*) indicates migration of the endogenous Cdk12 protein and the bottom one indicates the position of an unspecific band recognized by the Cdk12 antibody, which serves as a loading control.
Figure 6.
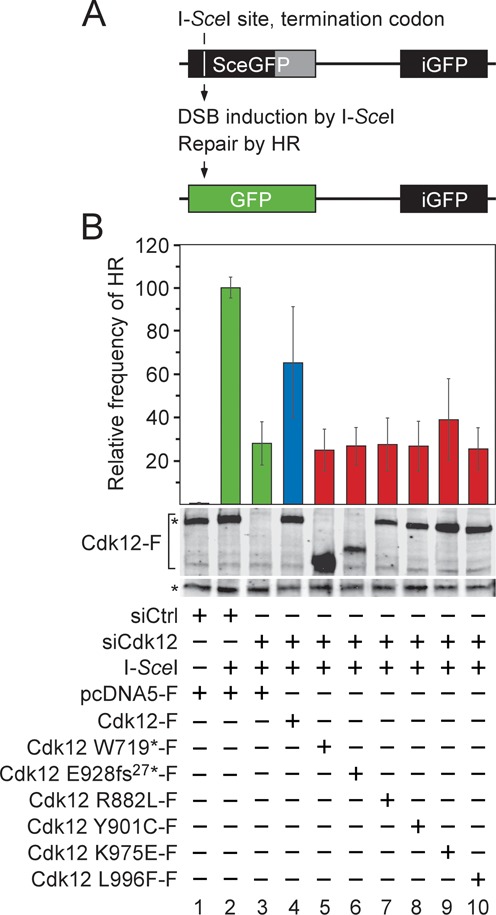
CDK12 mutations in HGS-OvCa abrogate the ability of Cdk12/CycK to stimulate the repair of DNA double-strand breaks by HR. (A) Schematic representation of the DR-GFP recombination substrate. The defective EGFP genes of the cassette, separated by 3.7 kb, are shown (top). The first one (SceGFP) contains an I-SceI endonuclease site and an in-frame termination codon (white vertical line), while the second one (iGFP) is an internal EGFP fragment, yielding GFP− cells. The homologous EGFP sequences are depicted as black rectangles and the non-repetitive EGFP sequence is depicted as gray rectangle. Induction of DSB by I-SceI triggers the repair of SceGFP by HR using the iGFP sequence as a donor DNA, resulting in wild-type EGFP (green rectangle) and GFP+ cells (bottom). (B) The mutant Cdk12 proteins fail to promote the repair of the DSB by HR. HeLa DR-GFP cells were treated with the control or Cdk12 #2 siRNA and transfected with the I-SceI expression plasmid together with plasmids encoding the wild-type (blue bar) or mutant (red bars) Cdk12-F proteins as indicated below the graph. The HR frequency for each experimental condition is represented as the frequency relative to the one reached by the I-SceI expression in the control siRNA-treated cells, which was set to 100% (green bar). Results are presented as the mean ± SD. Levels of the endogenous Cdk12 and Cdk12-F proteins are shown below the graph and were detected by Western blotting using Cdk12 antibody. The top asterisk (*) indicates migration of the endogenous Cdk12 protein and the bottom one indicates the position of an unspecific band recognized by the Cdk12 antibody, which serves as a loading control.
We also attempted to shed light on how the G909R and E1024* mutations could lead to the defects in CycK binding and the activity of Cdk12. We found that the mutated residues are located within the C-terminal lobe and right after the end of the KD, respectively (Supplementary Figure S3). We cannot offer a probable scenario for the effects of the G909R mutation. The E1024* mutation, however, truncates the C-terminal extension of the KD that otherwise promotes the kinase activity of Cdk12 by stabilizing the conformation of the KD (9). Thus, the E1024* mutation may impair the latter, leading to the decreased CycK binding and diminished Cdk12 activity. Overall, we conclude that the CDK12 mutations within the C-terminal lobe of KD or right after it likely elicit conformational changes in the KD. In turn, the CycK interacting surface in the N-terminal lobe is altered, preventing the interaction between Cdk12 and CycK, and the subsequent activation of the kinase.
CDK12 mutations in HGS-OvCa decrease transcriptional activation by Cdk12
The Cdk12/CycK complex is a transcriptional kinase, which can stimulate expression of target genes (33,34). Since the CDK12 mutations led to such pronounced defects in the biochemical experiments, we next asked whether they also compromise transcriptional activation by Cdk12 (Figure 3). Here, we employed a classical RNA tethering gene reporter assay, composed of the chimeric Rev-Cdk12 protein and the SLIIB-CAT reporter gene, which has been used to follow transcriptional activation by Cdk12 (6) (Figure 3A). Therein, the interaction between Rev and the SLIIB RNA element of nascent transcripts tethers the Rev fusion protein to RNAPII engaged in transcription, resulting in the activation of the SLIIB-CAT reporter gene (35).
We first determined whether transcriptional activation by Rev-Cdk12 requires the kinase activity of Cdk12. Hence, we constructed the mutant Cdk12 D877N protein, in which the aspartic residue within the activation segment ‘DFG’ motif was substituted with asparagine, the mutation which can generate a kinase-dead form of Cdk (36). Indeed, while the mutation did not affect the interaction between Cdk12 and CycK, it did abolish the activity of Cdk12 in vitro (Supplementary Figure S4A, lanes 1–3). Importantly, it also led to a significant decrease in the ability of the chimeric Rev-Cdk12 D877N protein to activate transcription (Figure 3B, bars 3 and 10), setting the stage for examining the effects of the CDK12 mutations.
We hypothesized that the CDK12 mutations in HGS-OvCa, which abrogated the formation of the Cdk12/CycK complex and/or activity of the kinase, would hamper the ability of the Rev-Cdk12 chimera to activate the reporter gene expression effectively. Indeed, the Rev-Cdk12 chimeric proteins containing all these CDK12 mutations showed compromised activation of the reporter gene compared to the activity of the wild-type Rev-Cdk12 chimera (Figure 3B, bars 3–7 and 9), correlating with our biochemical findings. The mutant Rev-Cdk12 chimera harboring the K975E mutation, which did not affect CycK binding and activity of Cdk12, also failed to activate the reporter gene expression strongly (Figure 3B, bar 8). This result suggests that the K975E mutation may indeed interfere with an important event, which is in addition to the kinase activity required for the transcriptional activation by Cdk12/CycK. Of note, among the mutant proteins, the Rev-Cdk12 719* chimera displayed the highest activity (Figure 3B, bar 4). This result could reflect that functional regions that remained in this truncated protein, such as the RS region (see below), become more active at stimulating gene expression compared to the same regions that reside in the context of the full-length Cdk12 protein.
Using this reporter assay, we also examined transcriptional properties of the mutant Cdk12 proteins carrying the CDK12 mutations found in other cancers (Supplementary Figure S4B). Again, all mutant proteins displayed the decreased activation of the reporter gene (Supplementary Figure S4B, bars 3–6). Although the activity of the mutant Rev-Cdk12 G909R and E1024* proteins was compromised the most, the S325N and S352F mutations also decreased the activity considerably, arguing for a contributing role for the RS region in the stimulation of gene expression by Cdk12/CycK. Collectively, these findings demonstrate that CDK12 mutations found in HGS-OvCa and other cancers compromise the ability of Cdk12 to promote gene expression, raising the possibility of decreased transcription of Cdk12-dependent target genes in cancer cells.
HGS-OvCa patient samples with CDK12 mutations exhibit downregulation of genes of the HR repair pathway
In a previous study that used a non-ovarian HeLa cell line and RNAi approach, the Cdk12/CycK complex has been found to promote the expression of ∼30 DDR genes (6). Among these were key effectors in DNA repair mechanisms, including BRCA1, ATR, FANCD2 and FANCI proteins, implicating the Cdk12/CycK complex in the maintenance of genomic stability. However, whether the CDK12 mutations affect mRNA levels of these and additional DDR genes in HGS-OvCa remains an open question. To address this issue, we took advantage of the mRNA expression data sets of the HGS-OvCa patient samples reported by the TCGA study (24). We compared gene expression levels between the samples that harbor either the wild-type CDK12 or CDK12 with the nine validated mutations (Figure 4). Given that defects within the HR pathway occur in approximately half of all the HGS-OvCa cases (24), and that several identified Cdk12-dependent DDR genes are required for HR (37), we hypothesized that the CDK12 mutations in HGS-OvCa may trigger downregulation of genes of this most faithful DNA repair pathway. Our comparison of mRNA levels of genes encoding the known components of HR (for the list of thirty eight examined genes, see Supplementary Table S3) identified six genes, including ATM, ATR, CHEK1, FANCI, MDC1 and RAD51D, which were significantly downregulated (P < 0.05) in tumor samples with mutated CDK12 in comparison to the samples carrying the wild-type CDK12 (Figure 4A, Supplementary Figure S5A, and Supplementary Figure S6). Among these, only ATR and FANCI were defined previously as Cdk12-dependent (6). To our surprise, we found that expression of BRCA1, a key effector of HR and a known Cdk12-dependent gene, as well as of BRCA2, another important HR player, was not affected in the HGS-OvCa samples with the CDK12 mutations (Figure 4C). Of note, due to the absence of the array probes for FANCD2 and MMS22L, two known Cdk12-dependent genes implicated in the HR pathway, we were unable to determine whether their expression was compromised by the CDK12 mutations.
In addition to the HR genes, depletion of Cdk12/CycK led to decreased expression of many other DDR genes (6). To determine whether this group of genes is also affected by the CDK12 mutations, we analyzed their mRNA levels as above (for the list of twenty three examined genes, see Supplementary Table S3). We found three DDR genes unrelated to HR, including NEK9, ORC3L and TERF2, to be significantly downregulated (P < 0.05) in the patient samples with mutated CDK12 (Figure 4B, Supplementary Figure S5B, and Supplementary Figure S6).
To confirm that expression of the previously characterized and the new HR genes identified above requires Cdk12/CycK in human cells of ovarian origin, we lowered CDK12 mRNA levels by RNAi in the ovarian carcinoma Caov-3 cell line. Indeed, compared to cells treated with a non-targeting control short interfering RNA (siRNA), cells treated with an siRNA specific to CDK12 displayed decreased expression of BRCA1, FANCI, FANCD2 and ATR, the HR genes previously characterized as Cdk12-dependent (6) (Figure 4D). We observed these effects in the colon cancer HCT116 cell line as well (Supplementary Figure S7A). Furthermore, mRNA levels of ATM, CHEK1, MDC1 and RAD51D, the downregulated HR genes in the HGS-OvCa cases with mutated CDK12 that have not been previously defined as Cdk12-dependent, also decreased upon the knockdown of CDK12 in both Caov-3 and HCT116 cell lines (Figure 4E and Supplementary Figure S7B). Of note, the depletion of Cdk12 and CycK but not Cdk13, which forms the separate Cdk13/CycK complex, led to modest but reproducible reduction of ATM, Chek1 and Rad51D protein levels (Supplementary Figure S7C). To rule out possible off-target effects of the CDK12 siRNA, we performed RNAi in same cell lines by targeting a different region in CDK12. Again, we observed similar defects on the HR gene expression (Supplementary Figure S8 and data not shown). Finally, to provide additional evidence that the downregulation of all HR genes analyzed here is specific to the Cdk12/CycK complex, we treated Caov-3 cells with siRNA targeting CCNK and CDK13. Whereas the knockdown of CCNK, the gene encoding CycK, led to decreased expression of the HR genes, that of CDK13 did not (Supplementary Figure S8). Together, these analyses provide important evidence that in HGS-OvCa patient samples, the CDK12 mutations lead to decreased expression of a subset of DDR genes, particularly those encoding key components of the HR pathway. Moreover, considering our biochemical and gene reporter findings, we conclude that the observed downregulation of DDR genes in patient samples is a consequence of the disabled kinase activity of the mutant Cdk12 proteins.
The Cdk12/CycK complex occupies HR genes and promotes Ser2 phosphorylation of RNAPII
Given our above findings on the stimulatory role of Cdk12/CycK in the expression of HR genes, it is important to determine whether these DDR genes are direct targets of Cdk12/CycK and whether this complex promotes the levels of Ser2-P of RNAPII at them. To address these possibilities, we employed the quantitative chromatin immunoprecipitation (ChIP-qPCR) assay to measure the occupancy of Cdk12, total/Ser5-P RNAPII and Ser2-P RNAPII at select genes at three different regions: the vicinity of transcription start site (TSS), the interior of the open reading frame (ORF) and the vicinity of the polyadenylation site (pA) (Figure 5). We analyzed all four of the newly identified HR genes, including ATM, CHEK1, MDC1 and RAD51D, as well as ATR and FANCD2 representing the previously established Cdk12-dependent genes. To assess whether the signals at these positions were specific, we used RAD51D and FANCI intergenic regions which showed minimal differences between the occupancy of Cdk12, both forms of RNAPII and IgG antibody control (Supplementary Figure S9A). Because of the subsequent functional studies involving the HR pathway, we performed the ChIP-qPCR assay using HeLa DR-GFP cell line, a HeLa cell line derivative containing one copy of the HR substrate, the direct repeat green fluorescence protein (DR-GFP) cassette (38).
Following confirmation of successful depletion of Cdk12 (Supplementary Figure S9B), we proceeded with the assay on control and Cdk12 knockdown cells. As shown in Figure 5A and Supplementary Figure S9C, Cdk12 is present at all three regions of the newly identified as well as the established Cdk12-dependent HR genes. At the novel set of genes, Cdk12 enrichment over the intergenic region ranged from 1.9-fold (at ATM pA region) to 6.1-fold (at CHEK1 pA region) and averaged 3.1-fold (SD = 1.2). Although these levels were not very robust, they were specific given that the Cdk12 enrichment over IgG was substantially higher at genes compared to that at the intergenic region (Figure 5A, compare blue and gray bars). Whereas the former were from 7.6-fold (at MDC1 TSS region) to 55.7-fold (at RAD51D ORF region) and averaged 15.7-fold (SD = 13.2), the latter equaled 3.2-fold. Confirming specificity of the Cdk12 signals, depletion of Cdk12 decreased the occupancy of Cdk12 at the novel genes by an average of 44.8% (SD = 14.0), while the levels of Cdk12 at the intergenic regions were similar (Figure 5A, compare blue and red bars, and Supplementary Figure S9A). To find out whether the Cdk12/CycK complex facilitates Ser2 phosphorylation at these genes, we next monitored the occupancy of total/Ser5-P and Ser2-P forms of RNAPII at them in control and Cdk12-depleted cells (Figure 5B and C and Supplementary Figure S9D and E). The knockdown of Cdk12 reduced the occupancy of total/Ser5-P RNAPII by an average of 16.4% (SD = 21.3) and primarily at TSSs of the genes, where the average decrease was 34.1% (SD = 12.3) (Figure 5B, compare blue and red bars). This effect may reflect in part the reported ability of Cdk12/CycK to phosphorylate the CTD of RNAPII at Ser5 in vitro (9). Importantly, however, decrease in the levels of the Ser2-P form of RNAPII was much more pronounced, averaging 48.9% (SD = 13.5) (Figure 5C, compare blue and red bars). In addition, this reduction was evident at all three gene regions, thus very likely spanning the entire length of the genes. We conclude that the HR genes are direct targets of the Cdk12/CycK complex, which is present at them to facilitate the CTD Ser2 phosphorylation of transcriptionally engaged RNAPII.
CDK12 mutations in HGS-OvCa disable the stimulatory role of the Cdk12/CycK complex in the repair of DSBs by HR
In light of our demonstration that the absence of functional Cdk12/CycK leads to coordinated downregulation of critical HR genes, we finally asked whether the CDK12 mutations disable the repair of DSBs in DNA by HR (Figure 6). To this end, we employed the DR-GFP assay, a classical chromosomal DSB repair assay that has been widely used to measure HR in living cells (Figure 6A) (38). The genomic DR-GFP cassette contains two direct repeats of differentially mutated enhanced green fluorescent protein (EGFP) genes both of which fail to produce a functional EGFP protein. Whereas the upstream repeat has been modified to contain the recognition site for the rare-cutting I-SceI endonuclease and an in-frame termination codon, the repeat that follows is an EGFP fragment truncated at the 3′ end. Expression of I-SceI leads to a DSB in the upstream repeat, triggering the repair by HR using the wild-type sequence of the downstream repeat as a DNA donor, resulting in EGFP positive cells.
To determine whether the repair of DSB by HR relies on Cdk12/CycK, we used RNAi to deplete endogenous Cdk12 in HeLa DR-GFP cells. Next, we transiently expressed the I-SceI endonuclease in these cells and in those treated with the non-targeting control siRNA. In agreement with recent reports (39,40), depletion of Cdk12 decreased the frequency of the HR-mediated repair by ∼3.5-fold (Figure 6B, bars 1–3). To find out whether the CDK12 mutations in HGS-OvCa result in deficient HR-mediated DSB repair, we next co-expressed the I-SceI endonuclease together with the Cdk12-siRNA resistant wild-type and mutant Cdk12-F proteins in HeLa DR-GFP cells depleted of the endogenous Cdk12 protein. Importantly, the wild-type Cdk12-F protein rescued the EGFP signal by ∼2.3-fold over the diminished levels provoked by the Cdk12 depletion, reaching 65% of the HR frequency in cells with endogenous Cdk12/CycK (Figure 6B, bars 2–4). In contrast, despite their comparable expression to the wild-type protein, all Cdk12-F proteins carrying the CDK12 mutations but one failed at increasing the frequency of HR-mediated repair of the DSB (Figure 6B, bars 5–10). The only exception was Cdk12 carrying the K975E mutation, which showed an ∼1.4-fold increase in the HR frequency over the background levels (Figure 6B, bar 9). Thus, these functional findings correlate completely with the damaging impacts of the CDK12 mutations on the activity of the Cdk12/CycK complex that we observed in our gene expression studies. We conclude that the CDK12 mutations found in HGS-OvCa disable the faithful repair of DSBs by HR, likely contributing to the pervasive instability of the genome in this cancer.
DISCUSSION
To clarify the relationship between the genetic alterations in Cdk12 and cancerogenesis, we focused here on the mutations identified in HGS-OvCa. We provide biochemical, structural, gene expression and functional evidence that the recurrent somatic mutations in CDK12 are not inert. Rather, they are loss-of-function mutations that disable the ability of the Cdk12/CycK complex to promote the expression and function of genes critical to the HR DNA repair pathway. Thus, we uncovered an important mechanism connecting the defects in Cdk12/CycK to cancerogenesis, supporting the notion that CDK12 is a potential tumor suppressor that ceases to function in this most deadly form of ovarian cancer.
Our findings on the mechanism by which CDK12 mutations incapacitate Cdk12/CycK provide new insights into the deregulation of Cdks in cancer. It is established that hyperactivation of the cell cycle Cdks enable cancerogenesis (41). Mechanistically, the increased kinase activity stems from overexpression or amplification of the Cdks or their corresponding cyclins. In addition, the same can be accomplished by diminished levels of Cdk inhibitors (CKI) in cancers or by missense mutations within the KD of Cdks that disrupt CKI binding. Similarly, transcriptional Cdks, including the Cdk8/CycC and Cdk9/CycT complexes, promote tumorigenesis by elevated kinase activity (17). Whereas CDK8 is overexpressed or amplified in cancers (42), P-TEFb can enable cancerogenesis in concert with the amplified c-Myc oncogene (13), by stimulating the improper target gene expression within mixed lineage leukemia (MLL) fusion protein complexes (18) or via the direct upregulation of transcription factors promoting the epithelial-mesenchymal transition (19). In contrast, we demonstrate that the CDK12 mutations in HGS-OvCa and other cancers disable the kinase activity of Cdk12. Our results on the Cdk12 inactivation in ovarian carcinoma are in perfect agreement with the recent report by Karnitz laboratory (40). Importantly, we further disclose that the loss of the interaction between Cdk12 and CycK is the predominant mechanism underlying this defect. Of note, among transcriptional Cdks, Cdk12 and its paralogue Cdk13 are mutated most heavily in human cancers (data not shown). We propose that at least with regard to Cdk12, the KD mutations are selected for during the genesis of human cancers because of their damaging impacts on the Cdk12 activation process. Further investigations of the non-KD mutations in Cdk12 and of those in other transcriptional Cdks are needed to understand their mechanisms and contributions to cancer.
Our results from the gene reporter assay and the mRNA expression analysis of HGS-OvCa patient samples indicate that all CDK12 mutations hamper the ability of Cdk12 to promote target gene expression. Importantly, by using ChIP-qPCR assay, we find that all examined HR genes are direct targets of Cdk12/CycK, at which this transcription elongation-associated kinase facilitates optimal levels of RNAPII Ser2 phosphorylation. Together, these findings suggest that rather than through an indirect mechanism, the CDK12 mutations in HGS-OvCa directly downregulate DDR gene expression by precluding the Cdk12/CycK complex to exert its stimulatory action on transcriptionally engaged RNAPII at the genes. However, further work is needed to reveal which steps in the DDR gene expression fail to operate due to the inactive Cdk12 proteins. Since the knockdown of Cdk12/CycK lowers the levels of nascent transcripts of several DDR genes (6), regulates alternative splicing (43), and interferes with effective 3′-end formation of model pre-mRNAs (8,44), the mutations could hamper the synthesis and maturation of DDR gene transcripts. Because all CDK12 HGS-OvCa mutations except one inactivate Cdk12 activity in vitro, these possible defects could stem from the poorly phosphorylated Ser2 residues of the transcribing RNAPII, preventing efficient recruitment of elongation and RNA processing factors (3,4). Alternatively, the mutations might antagonize the cooperation between Cdk12/CycK and some of its recently reported interacting factors (44,45), resulting in the reduction of gene expression.
A previous report has found that half of the HGS-OvCa tumor cases display genetic and epigenetic defects in the components of the HR repair pathway (24). Our findings on the causal relationship between the CDK12 mutations and the failure of the HR-mediated DNA repair shed a new light on the source of the HR defects in this cancer. Efficient HR is mediated by several groups of factors including components of the ATM/ATR signaling pathway, BRCA1/2 proteins, Fanconi anemia (FA) proteins and members of the Rad51 protein family (46–49). Our analysis of the HGS-OvCa patient samples harboring CDK12 mutations demonstrates significant decrease in mRNA levels of several factors from these groups. In particular, the ATM/ATR signaling pathway, which responds to DNA damage and transduces signals to downstream effectors (50), seems to be predominantly compromised. We find deficient mRNA levels of ATM, ATR, Chek1 and Mdc1 proteins, out of which only ATR was previously characterized as a Cdk12-dependent gene (6). Next, we also reveal decreased expression of mRNA for FANCI, an FA protein that functions in the repair of DNA inter strand crosslinks (ICLs) together with FANCD2 (51), the factor vital to efficient HR. Finally, our analysis shows the decrease in mRNA levels of Rad51D, a paralogue of Rad51 which plays a role in the assembly and maintenance of the Rad51 nucleoprotein filament, the critical intermediate for strand invasion during HR (26). Despite the fact that BRCA1, a known Cdk12-dependent gene (6,39,40), is not affected by the CDK12 mutations in HGS-OvCa, we speculate that expression of this crucial HR protein may be diminished in earlier stages of this cancer containing CDK12 mutations. Importantly, we noted that HGS-OvCa patient samples contain somatic and/or germline mutations in all of the Cdk12-dependent HR genes revealed here. Moreover, by inspecting the COSMIC database, we found that mutations in these HR genes are mutually exclusive with the CDK12 mutations (data not shown). This also holds true for the majority of BRCA1 and BRCA2 mutations, which are absent in 78% of HGS-OvCa cases that contain CDK12 mutations (39). Together, these revelations suggest that mutating CDK12 equips the developing cancer cells with an alternative source of defects in the HR repair pathway, which is achieved by the collective downregulation of critical HR genes. Finally, three recent studies showed that depletion of Cdk12 sensitizes ovarian cancer cell lines to PARP1/2 inhibitors (22,39,40). Because this sensitivity is a hallmark of the defective HR pathway, our work is in agreement with these findings and also supports the possibility that tumors with CDK12 mutations might be particularly susceptible to the therapy with these inhibitors.
Together, our study provides a framework for understanding how mutations in Cdk12/CycK could promote cancerogenesis. By impairing HR, the most error-free cellular repair mechanism of DSBs, CDK12 mutations may elicit the usage of error-prone DNA repair pathways, including non-homologous end joining and single-strand annealing (52). As a result, spontaneous and DNA damage-induced gene and chromosomal aberrations are likely to accumulate, giving rise to the unstable genome, the enabling characteristic of any cancer (53). Because HR plays a central role in repairing ICLs that stall replication forks (54), an increase in errors during DNA replication, which is an important source of genomic instability, can also stem from the HR deficiency provoked by the CDK12 mutations. In support of this possibility, depletion of Cdk12/CycK sensitizes cells to the ICL-inducing drug mitomycin C (6), and CycK is one of the major proteins underlying the resistance to camptothecin, the drug that creates an obstruction in the DNA, thereby blocking its replication (55). Finally, unlike ATM, which is activated by DSBs, ATR responds to a broad spectrum of DNA aberrations. In addition to DSBs, it is activated primarily by replication protein A-coated single stranded DNA, an intermediate in many DNA repair pathways, such as nucleotide excision repair, mismatch repair and base excision repair (50). Thus, we speculate that the loss-of-function CDK12 mutations may trigger malfunctions in a variety of DNA repair pathways, contributing to the remarkable genomic disarray in HGS-OvCa.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank all the members of Barboric and Blazek laboratories for discussions and critical comments on the manuscript; Junya Kobayashi and Kenshi Komatsu for generously providing the HeLa DR-GFP cell line and I-SceI expression plasmid; Heini Hakala and Liisa Kauppi for technical assistance and critical reading of the manuscript and Petra Ovesná for help with statistical analysis of Oncomine data.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
The authors wish it to be known that, in their opinion, these authors should be regarded as Joint Third Authors.
FUNDING
Academy of Finland [1263825 to M.B., 1273842 to T.L.]; Marsha Rivkin Center for Ovarian Cancer Research [to M.B. and D.B.]; Sigrid Juselius Foundation [4702687 to M.B.]; University of Helsinki Three-year Research Grant [490123 to M.B.]; Project ‘CEITEC-Central-European Institute of Technology’ [CZ.1.05/1.1.00/02.0068 to D.B.]; GACR [14-09979S to D.B., GAP301/11/0747 to V.B.]; Deutsche Forschungsgemeinschaft [GE-976/9 to M.G.]; European Social Fund and the state budget of Czech Republic ‘Postdoc I’ Grant [CZ.1.07/2.3.00/30.0009 to V.P. and V.B.]; DFG Excellence Cluster ImmunoSensation [EXC 1023 to M.G.]. Funding for open access charge: Academy of Finland [1263825 to M.B.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Fuda N.J., Ardehali M.B., Lis J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461:186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eick D., Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev. 2013;113:8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Q., Li T., Price D.H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenasi T., Barboric M. P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol. 2010;7:145–150. doi: 10.4161/rna.7.2.11057. [DOI] [PubMed] [Google Scholar]
- 5.Bartkowiak B., Liu P., Phatnani H.P., Fuda N.J., Cooper J.J., Price D.H., Adelman K., Lis J.T., Greenleaf A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng S.W., Kuzyk M.A., Moradian A., Ichu T.A., Chang V.C., Tien J.F., Vollett S.E., Griffith M., Marra M.A., Morin G.B. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 2012;32:4691–4704. doi: 10.1128/MCB.06267-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davidson L., Muniz L., West S. 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 2014;28:342–356. doi: 10.1101/gad.231274.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosken C.A., Farnung L., Hintermair C., Merzel Schachter M., Vogel-Bachmayr K., Blazek D., Anand K., Fisher R.P., Eick D., Geyer M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014;5:3505. doi: 10.1038/ncomms4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho E.J., Kobor M.S., Kim M., Greenblatt J., Buratowski S. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 2001;15:3319–3329. doi: 10.1101/gad.935901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coudreuse D., van Bakel H., Dewez M., Soutourina J., Parnell T., Vandenhaute J., Cairns B., Werner M., Hermand D. A gene-specific requirement of RNA polymerase II CTD phosphorylation for sexual differentiation in S. pombe. Curr. Biol. 2010;20:1053–1064. doi: 10.1016/j.cub.2010.04.054. [DOI] [PubMed] [Google Scholar]
- 12.Dai Q., Lei T., Zhao C., Zhong J., Tang Y.Z., Chen B., Yang J., Li C., Wang S., Song X., et al. Cyclin K-containing kinase complexes maintain self-renewal in murine embryonic stem cells. J. Biol. Chem. 2012;287:25344–25352. doi: 10.1074/jbc.M111.321760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahl P.B., Lin C.Y., Seila A.C., Flynn R.A., McCuine S., Burge C.B., Sharp P.A., Young R.A. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao S.H., Price D.H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 15.Ostapenko D., Solomon M.J. Budding yeast CTDK-I is required for DNA damage-induced transcription. Eukaryot. Cell. 2003;2:274–283. doi: 10.1128/EC.2.2.274-283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winsor T.S., Bartkowiak B., Bennett C.B., Greenleaf A.L. A DNA damage response system associated with the phosphoCTD of elongating RNA polymerase II. PLoS One. 2013;8:e60909. doi: 10.1371/journal.pone.0060909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee T.I., Young R.A. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith E., Lin C., Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji X., Lu H., Zhou Q., Luo K. LARP7 suppresses P-TEFb activity to inhibit breast cancer progression and metastasis. Elife. 2014;3:e02907. doi: 10.7554/eLife.02907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benusiglio P.R., Pharoah P.D., Smith P.L., Lesueur F., Conroy D., Luben R.N., Dew G., Jordan C., Dunning A., Easton D.F., et al. HapMap-based study of the 17q21 ERBB2 amplicon in susceptibility to breast cancer. Br. J. Cancer. 2006;95:1689–1695. doi: 10.1038/sj.bjc.6603473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sircoulomb F., Bekhouche I., Finetti P., Adelaide J., Hamida A., Bonansea J., Raynaud S., Innocenti C., Charafe-Jauffret E., Tarpin C., et al. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer. 2010;10:539. doi: 10.1186/1471-2407-10-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natrajan R., Wilkerson P.M., Marchio C., Piscuoglio S., Ng C.K., Wai P., Lambros M.B., Samartzis E.P., Dedes K.J., Frankum J., et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J. Pathol. 2014;232:553–565. doi: 10.1002/path.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zang Z.J., Ong C.K., Cutcutache I., Yu W., Zhang S.L., Huang D., Ler L.D., Dykema K., Gan A., Tao J., et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res. 2011;71:29–39. doi: 10.1158/0008-5472.CAN-10-1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jasin M., Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blazek D. The cyclin K/Cdk12 complex: an emerging new player in the maintenance of genome stability. Cell Cycle. 2012;11:1049–1050. doi: 10.4161/cc.11.6.19678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenasi T., Peterlin B.M., Barboric M. Cap-binding protein complex links pre-mRNA capping to transcription elongation and alternative splicing through positive transcription elongation factor b (P-TEFb) J. Biol. Chem. 2011;286:22758–22768. doi: 10.1074/jbc.M111.235077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavletich N.P. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J. Mol. Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 30.Larochelle S., Amat R., Glover-Cutter K., Sanso M., Zhang C., Allen J.J., Shokat K.M., Bentley D.L., Fisher R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012;19:1108–1115. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M.D., Cibulskis K., Sougnez C., Greulich H., Muzny D.M., Morgan M.B., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kan Z., Jaiswal B.S., Stinson J., Janakiraman V., Bhatt D., Stern H.M., Yue P., Haverty P.M., Bourgon R., Zheng J., et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 33.Drogat J., Hermand D. Gene-specific requirement of RNA polymerase II CTD phosphorylation. Mol. Microbiol. 2012;84:995–1004. doi: 10.1111/j.1365-2958.2012.08071.x. [DOI] [PubMed] [Google Scholar]
- 34.Kohoutek J., Blazek D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012;7:12. doi: 10.1186/1747-1028-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiley L.S., Madore S.J., Malim M.H., Cullen B.R. The VP16 transcription activation domain is functional when targeted to a promoter-proximal RNA sequence. Genes Dev. 1992;6:2077–2087. doi: 10.1101/gad.6.11.2077. [DOI] [PubMed] [Google Scholar]
- 36.Garriga J., Mayol X., Grana X. The CDC2-related kinase PITALRE is the catalytic subunit of active multimeric protein complexes. Biochem. J. 1996;319:293–298. doi: 10.1042/bj3190293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.San Filippo J., Sung P., Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 38.Pierce A.J., Johnson R.D., Thompson L.H., Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajrami I., Frankum J.R., Konde A., Miller R.E., Rehman F.L., Brough R., Campbell J., Sims D., Rafiq R., Hooper S., et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi P.M., Sutor S.L., Huntoon C.J., Karnitz L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014;289:9247–9253. doi: 10.1074/jbc.M114.551143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malumbres M., Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 42.Firestein R., Bass A.J., Kim S.Y., Dunn I.F., Silver S.J., Guney I., Freed E., Ligon A.H., Vena N., Ogino S., et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen H.H., Wang Y.C., Fann M.J. Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol. Cell. Biol. 2006;26:2736–2745. doi: 10.1128/MCB.26.7.2736-2745.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eifler T.T., Shao W., Bartholomeeusen K., Fujinaga K., Jager S., Johnson J.R., Luo Z., Krogan N.J., Peterlin B.M. Cyclin-dependent kinase 12 increases 3′ end processing of growth factor-induced c-FOS transcripts. Mol. Cell. Biol. 2015;35:468–478. doi: 10.1128/MCB.01157-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartkowiak B., Greenleaf A.L. Expression, purification, and identification of associated proteins of the full length hCDK12/CyclinK complex. J. Biol. Chem. 2014;290:1786–1795. doi: 10.1074/jbc.M114.612226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrison C., Sonoda E., Takao N., Shinohara A., Yamamoto K., Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moynahan M.E., Chiu J.W., Koller B.H., Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 48.Nakanishi K., Yang Y.G., Pierce A.J., Taniguchi T., Digweed M., D'Andrea A.D., Wang Z.Q., Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCabe N., Turner N.C., Lord C.J., Kluzek K., Bialkowska A., Swift S., Giavara S., O'Connor M.J., Tutt A.N., Zdzienicka M.Z., et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 50.Marechal A., Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013;5:a012716. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., 3rd, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D'Andrea A.D., et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moynahan M.E., Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 54.Sirbu B.M., Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013;5:a012724. doi: 10.1101/cshperspect.a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Connell B.C., Adamson B., Lydeard J.R., Sowa M.E., Ciccia A., Bredemeyer A.L., Schlabach M., Gygi S.P., Elledge S.J., Harper J.W. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol. Cell. 2010;40:645–657. doi: 10.1016/j.molcel.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.