


Abstract
Aminoacyl-tRNA synthetases (aaRSs) play a crucial role in protein translation by linking tRNAs with cognate amino acids. Among all the tRNAs, only tRNAHis bears a guanine base at position -1 (G-1), and it serves as a major recognition element for histidyl-tRNA synthetase (HisRS). Despite strong interests in the histidylation mechanism, the tRNA recognition and aminoacylation details are not fully understood. We herein present the 2.55 Å crystal structure of HisRS complexed with tRNAHis, which reveals that G-1 recognition is principally nonspecific interactions on this base and is made possible by an enlarged binding pocket consisting of conserved glycines. The anticodon triplet makes additional specific contacts with the enzyme but the rest of the loop is flexible. Based on the crystallographic and biochemical studies, we inferred that the uniqueness of histidylation system originates from the enlarged binding pocket (for the extra base G-1) on HisRS absent in other aaRSs, and this structural complementarity between the 5′ extremity of tRNA and enzyme is probably a result of coevolution of both.
INTRODUCTION
Aminoacyl-tRNA synthetases (aaRSs) play a central role in protein synthesis by establishing the linkage between cognate amino acid and the tRNA anticodon triplet (1). This reaction proceeds as a two-step process where in the first step, the specific amino acid is condensed with adenosine triphosphate (ATP) to produce an aminoacyl-adenylate intermediate (aa-AMP); and in the second, the amino acid is covalently linked to the cognate tRNA acceptor stem from aa-AMP (2). aaRSs can be divided into two major classes based on their sequence homology and oligomeric structures (3,4). Class I aaRSs contain a Rossmann fold and two highly conserved sequence motifs, and aminoacylate the 2′ OH group of the 3′ terminal ribose ring of tRNAs. Class II aaRSs are characterized by three conserved signature motifs at the active site and the primary site of aminoacylation is the 3′ OH group of A76 (5). Both classes can be further divided into finer subclasses (1,5). Subclass IIa enzymes form homodimers and with the exception of seryl-tRNA synthetase (SerRS), possess the homologous C-terminal extension that acts as the anticodon binding domain (1). Subclass IIb enzymes harbor a homologous N-terminal anticodon binding domain (6–8). Subclass IIc enzymes are tetramers and their amino acids and tRNA binding elements are less conserved than other class II aaRSs (9).
Histidyl-tRNA synthetase (HisRS) belongs to class IIa category (1,4,10). It forms the typical active site of IIa enzymes and the common anticodon binding domain. On the other hand, the histidylation system displays distinct features that are only unique to this synthetase. Histidine-specific tRNAs are well known for the extra guanosine at the 5′ terminus of the acceptor stem, designated as G-1 (11), either genetically encoded or added post-transcriptionally (12–15). Successful histidylation depends on the recognition of the essential G-1:N73 base pair by HisRS. The base across from G-1 is the discriminator base, which is well conserved as A in eukaryotes and C in prokaryotes (11,16). Mutations, deletions or substitutions of this pair causes a significant drop on Vmax/Km (17,18) and this observation is in agreement with in vivo studies (19). Additionally, the 5′ phosphate group of G-1 has also been shown to influence the enzymatic activity significantly (17,20,21). Substitution of the 5′ monophosphate of G-1 by a triphosphate group reduces histidylation efficiency remarkably (17) while deletion of the phosphate group causes a 510-fold decrease (20). Although both the accepter stem and the anticodon of tRNAHis participate in and contribute to the recognition by HisRS, the anticodon is a much weaker identity determinant or element, a specific motif in tRNAs that triggers aminoacylation, especially in Escherichia coli (18,22). For this reason, HisRS is capable of aminoacylating a wide variety of RNA substrates (23–25). In addition to the full-length tRNAHis, fragments of tRNAHis (minihelical or microhelical RNAs) also make good substrates for both the Escherichia coli and yeast enzymes. The -1:73 pair in these shorter RNAs still serves as the major determinant for recognition while the sequence context brings additional specificity (24,26,27). However, tRNAHis in Caulobacter crescentus lacks the critical G-1 residue. The major identity elements are the anticodon, where mutations lead to a dramatic loss of histidylation activity. Conversely the addition of G-1 does not improve aminoacylation efficiency (28).
Crystal structures of several prokaryotic as well as eukaryotic HisRSs have been reported previously. The first structure of HisRS solved is from Escherichia coli (EcHisRS), which is in complex with histidyl-adenylate (HAM, PDB code 1HTT) (29). Each monomer of EcHisRS contains three conserved domains: an N-terminal catalytic domain consisting of seven antiparallel β-strands, a mixed α+β C-terminal domain, and an insertion domain. Soon, crystal structures of Thermus thermophilus HisRS (TtHisRS) in complex with histidine (PDB code 1ADJ) or HAM (PDB code 1ADY), and in the apo form (PDB code 1H4V) also became available (30,31). Different from the EcHisRS structure, the insertion domain in TtHisRS becomes fully ordered and forms four α-helices. It is proposed to clamp onto the acceptor stem once tRNA binds. Besides bacteria, crystal structures of eukaryotic HisRSs have also been determined from trypanosomes and humans (PDB codes 3HRI, 3LC0, 3HRK, 4G84 and 4G85) (32,33). Eukaryotic HisRSs feature an extra N-terminal whep-TRS domain and nuclear magnetic resonance studies have shown that this domain forms an antiparallel two-helix bundle (PDB code 1X59) (33). Structural comparison with prokaryotic HisRSs indicates that eukaryotic HisRSs adopt a different fold for the insertion domain as well as a different HAM-binding mode. Along with biochemical and in vivo studies, past crystallographic research reveals the activation mechanism of histidine by the enzyme, but the aminoacylation details are still missing due to the lack of the key structure of the HisRS-tRNA complex. Here we report the crystal structure of the complex between TtHisRS and tRNAHis in the presence of histidine and a nonhydrolyzable inhibitor adenylyl-imidodiphosphate (AMPPNP). The cocrystal structure provides the first model of HisRS-tRNA complexes and addresses the recognition problem of the identity elements on the tRNAHis substrate at a molecular level.
MATERIALS AND METHODS
Cloning, expression and purification of TtHisRS
The gene encoding TtHisRS (hisS, Accession NO. AAS80708) was amplified by PCR from the genomic DNA of Thermus thermophilus HB27 strain (ATCC Number BAA-163D-5). After double digestion by the NdeI and NotI restriction enzymes, the PCR product was ligated into a modified pET-28a (+) vector (Novagen) in which the thrombin site in the pET-28a (+) vector (recognition peptide LVPRGS) was replaced by a PreScission protease cleavage site (recognition peptide LEVLFQGP). The resulting construct contains a His6-tag at the N-terminus, and was transformed into Escherichia coli BL21(DE3) for overexpression. The cells were grown in LB medium to A600 = 0.8 at 37°C, and subsequently induced for overnight with 0.1 mM IPTG. The cells were harvested by centrifugation at 4000 rpm for 20 min and resuspended in the lysis buffer containing 40 mM Tris-HCl (pH 8.0), 250 mM NaCl, 10 mM imidazole, 1 mM PMSF and 1 mM β-mercaptoethanol. After sonication, the cell lysate was cleared by centrifugation for 40 min at 14 000 rpm. The supernatant was mixed with Ni-NTA resin (Qiagen) at 4°C for 1 h and loaded onto an empty column. The bound protein was washed with ∼10 column volumes of lysis buffer and was eluted in the elution buffer containing 40 mM Tris-HCl (pH 8.0), 250 mM NaCl, 250 mM imidazole and 1 mM PMSF. The protein of interest was then treated with the PreScission protease overnight in the presence of 1 mM DTT to cleave off the his6-tag and subsequently applied onto a Histrap column (GE Healthcare) to remove uncut TtHisRS. The unbound portion was pooled, and further purified by gel-filtration chromatography with a Superdex 200 column (GE healthcare) in a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl and 1 mM DTT. The purity of the protein was assessed by SDS-PAGE and the pure protein was flash-frozen in liquid nitrogen before being stored at −80°C.
All point mutations for TtHisRS activity assays were created through QuikChange (Stratagene, CA, USA). The primers used for cloning and mutagenesis were listed in Supplementary Table S1. The TtHisRS mutants used for aminoacylation assays were purified by a single step of Ni-NTA affinity chromatography. The human SerRS (hSerRS) and human GlyRS-SF (hGlyRS) proteins (an active form without the whep-TRS domain) were purified through a three-step purification process consisting of affinity (Ni-NTA, Qiagen), anion-exchange (Histrap, GE Healthcare) and gel-filtration chromatography (Superdex 200, GE Healthcare). The protein was finally eluted in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl and 1 mM DTT. Most proteins achieved 90% purity as assessed by SDS-PAGE and their structural integrity was checked with the Superdex 200 column. Most mutants are well folded except for the deletion mutant with the Glu325-Gly328 internal truncation. 5% Glycerol was added to mutants intended for activity assays before storage.
In vitro Transcription of the tRNA Substrates
The preparation of the tRNA transcripts was described in a previous protocol (34). Thermus thermophilus tDNAHis(GUG) (Supplementary Table S2) plus the T7 promoter was ligated into the pUC18 vector using complementary primers with the restriction sites HindIII and XbaI. The transcription template was obtained by PCR amplification of the ligated DNA fragments followed by phenol extraction. The in vitro transcription reaction of tRNAHis (with the 5′ terminal GTP) for crystallization was performed at 37°C for 3 h with 0.3 μM T7 RNA polymerase in 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine and 2 mM each NTP. tRNAHis transcript for aminoacylation assays, which starts with the 5′-GMP, was produced in the reaction mix with identical recipe supplemented with 20 mM GMP. The transcript was purified by a 12%, denaturing urea-polyacrylamide gel (UREA-PAGE). After ethanol precipitation, the RNA pellet was redissolved in TE buffer (20 mM Tris-HCl (pH 8.0) and 1 mM EDTA). tRNA was annealed by heating to 65°C and allowed to cool to room temperature after addition of 10 mM MgCl2. The annealed RNA was aliquoted and stored at −80°C for further use. Wild type (WT) tRNAGly/Ser (Supplementary Table S2) and their G-1-added mutants were prepared in the same fashion and also start with 5′ GMP. The G-1 base was inserted into the pUC18/tDNASer(TGA) or pUC18/tDNAGly(CCC) plasmids through QuikChange, and G73 in tRNASer was further changed to C73 to pair with G-1.
Crystallization, data collection and structure determination
The cocrystals of HisRS-tRNAHis-histidine-AMPPNP complex were grown using the hanging drop vapor diffusion method at room temperature. To form the complex, protein was mixed with tRNAHis at a molar ratio 1:1.1 in a buffer containing 20 mM HEPES (pH 7.0), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 2 mM histidine and 2 mM AMPPNP. The final protein concentration present in the complex was 2.0 mg/ml. The sample was filtered with a 0.22 μm centrifugal device (Millipore) prior to crystallization. The drops were equilibrated against a reservoir solution containing 5% PEG 4000 and 0.1 M NaOAc (pH4.5). The cocrystals of the quaternary complex appeared after two or three days. Fully-grown crystals were flash frozen in liquid nitrogen after being soaked in a cryoprotectant containing all the reservoir solution components supplemented with 20% glycerol (v/v).
A 2.55 Å diffraction data set was collected using beamline 17U (BL17U) at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, P.R.China) and was processed with the program HKL2000 (35). Cell content analysis suggested that the asymmetric unit contains one HisRS monomer in complex with one tRNA molecule. Molecular replacement (MR) was first performed with Phenix using the apo TtHisRS structure (PDB code 1H4V) and a partial structure of Candida albicans tRNAHis (CatRNAHis, PDB code 3WC1) (36) consisting of the acceptor stem and TΨC region as the search models. The two components were then searched simultaneously to produce a partial solution. To obtain the complete complex structure, a second round of MR was carried out with the partial solution and residues 8–49 of human tRNAGly(CCC) (the D- and anticodon regions of PDB code 4KR2) (34) as the search models. The initial phases for both the protein and a complete tRNA molecule were evident once the solution was found. However, the insertion domain (Arg172-Glu319) was found to undergo large conformational changes and this region was manually rebuilt with the program Coot (37). The rebuilt model was fed to phenix.refine and multiple cycles of refinement were alternated with model rebuilding (38). Titration-Liberation-Screw (TLS) refinement was carried out at the late stage of the refinement using a total of 22 TLS groups. The final R-factor was 20.73% (Rfree = 26.17%) (Table 1). The Ramachandran plot of the final models had 96.05%, 3.46%, 0.49% of the residues in the most favorable, generously allowed and disallowed region as indicated by the program Molprobity (39). All the figures about structures were created with Pymol (www.pymol.org) and the domain architecture was prepared by DOG (40).
Table 1. Data collection and refinement statistics.
| Data collection | |
|---|---|
| Space group | C 2221 |
| Cell dimensions (Å) | |
| a, b, c (Å) | 84.061, 159.703, 123.988 |
| α, β, γ (°) | 90 90 90 |
| Resolution (Å) | 50-2.55 (2.64-2.55)a |
| Rmergeb | 0.148 (0.700) |
| I/σ(I) | 13.86 (2.61) |
| Completeness (%) | 98.8 (98.8) |
| Redundancy | 5.4 (5.1) |
| Refinement | |
| Resolution (Å) | 29.47- 2.55 (2.64 - 2.55) |
| No. reflections | 27018 (2329) |
| Rworkc/Rfreed | 0.207/0.262 |
| No. atoms | |
| Protein | 3217 |
| tRNA | 1654 |
| Ligand | 23 (AMP) |
| 11 (Histidine) | |
| Water molecules | 76 |
| B-factors (Å2) | |
| protein | 62.69 |
| tRNA | 94.13 |
| Ligand | 47.37 (AMP) |
| 128.39 (Histidine) | |
| Water molecules | 61.38 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (º) | 0.80 |
aValues in parentheses are for the highest-resolution shell. bRmerge = Σ|(I -< I >)|/σ(I), where I is the observed intensity. cRwork = Σhkl∥Fo| - |Fc∥/ Σhkl|Fo|, calculated from working data set. dRfree is calculated from 5.0% of data randomly chosen and not included in refinement.
Aminoacylation assay
The histidylation assay mixture contained 50 mM Tris-HCl (pH 7.7), 100 mM KCl, 10 mM MgCl2, 8 mM DTT, 2.5 mM ATP, 20 μM L-[14C] -histidine (PerkinElmer, 320 mCi/mmol) and 5 μM annealed tRNAHis(GUG). 50 nM HisRS or mutants were added to initiate the reactions. All the activity assays were performed with 5′-GMP tRNAs due to their higher activities as the substrates than that of the 5′-GTP tRNAs. The reaction was carried out at 60°C, and aliquots were removed at the designated time points, spotted onto 5% trichloroacetic acid (TCA)-soaked filter pads, and washed twice with 5% cold TCA as well as 95% ethanol. The filter pads were dried and the radioactivity was measured by scintillation counting.
The aminoacylation assays for hSerRS/hGlyRS were carried out under similar conditions to those of HisRS assays. hSerRS assays contained 50 mM Tris-HCl (pH 7.7), 100 mM KCl, 10 mM MgCl2, 8 mM DTT, 2.5 mM ATP, 20 μM cold serine, 1 μM L-[3H]-serine (PerkinElmer, 22 Ci/mmol), 10 μM annealed tRNASer(UGA), whereas that of hGlyRS contained 150 mM HEPES (pH 7.5), 20 mM KCl, 4 mM MgCl2, 2 mM DTT, 3 mM ATP, 20 μM cold glycine, 1 μM L-[3H]-glycine (PerkinElmer, 48.7 Ci/mmol) and 5 μM annealed tRNAGly(CCC). 200 nM hSerRS and 50 nM hGlyRS were added last, and all the reactions were performed at room temperature.
RESULTS
Overview of the complex
The cocrystal structure is determined at 2.55 Å resolution. The space group of the crystal belongs to C2221, with a completeness of 97.86% and a Wilson B-factor of 48.7 Å2. Gel filtration chromatography indicates that TtHisRS exists as a single species with an approximate mass of 80 kDa, suggesting a dimer in solution. Continuous electron density is observed for the polypeptide from Thr2 to Gly421, except for the disorder from Gly53 to Lys63. This region is located on a loop connecting η1 and β3. Ala57-Arg62 of this region is also unstructured in the apo TtHisRS structure, but it becomes fully ordered in the histidine- or HAM-bound structures (30). The tRNA model is almost intact except for nucleotide A38 and the map shows clear electron density for the triphosphate group of G-1. The final model contains one AMP (coming from AMPPNP), one histidine and 76 water molecules (Table 1).
The protein structure in the complex is similar to that of the apoprotein. The N-terminal catalytic domain (Met1-Asp171 and Leu229-Gly320) is formed by six- stranded antiparallel β-sheet surrounded by three long α-helices. This domain contains the necessary functional elements for HAM synthesis as well as tRNAHis aminoacylation (41). In addition, the extra domain (Arg172-Phe228) splits the catalytic domain. The C-terminal domain (Leu331-Gly421) displays the typical α/β fold shared by the Class IIa enzymes (Figure 1A and B).
Figure 1.
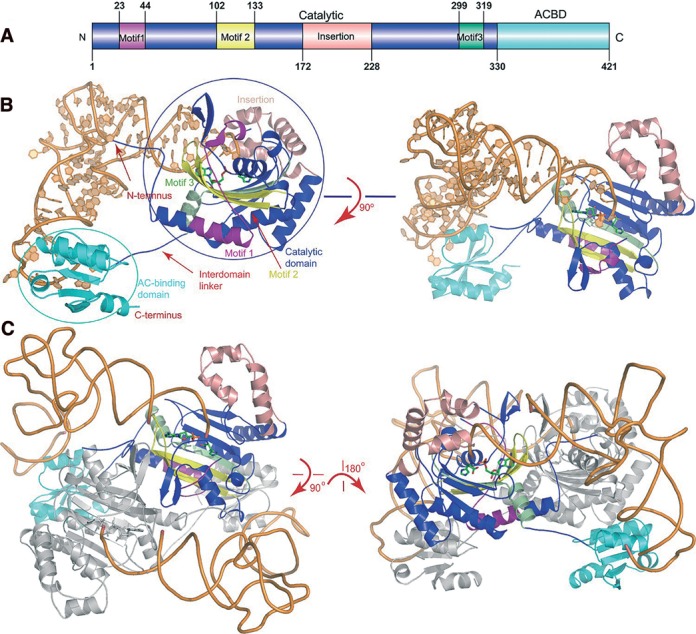
The overall view of the quaternary complexes. (A) The TtHisRS domain architecture. The catalytic, anticodon binding domains (ACBD) as well as the insertion domains are colored blue, cyan and salmon, respectively. The catalytic domain (colored blue) contains three signature motifs 1–3 (colored purple, yellow and pale green, respectively). (B) The side view and the top views of the complex in ribbon rendition. The active site AMP and histidine are shown as sticks while RNA is in orange. (C) The dimeric complex. Only one monomer is colored as described in 1A and the other molecule is colored gray.
HisRS is an α2 homodimeric enzyme and the molecular dimer axis coincides with the crystallographic 2-fold axis. tRNAHiss bind to the protein dimer in a symmetrical fashion, interacting with both subunits (Figure 1C). However, the majority of the contacts come from subunit one, and is with the active site and the anticodon binding domain, burying a surface area of 3706.4 Å2. The contacts with subunit two are mainly between G-1 and the 70′DRG72′ tripeptide (the primes indicate the residues from another subunit), and between the D-stem end (G10, A26 and C27) and the Gly94′-Pro99′ fragment, resulting in an additional buried surface area of 779.9 Å2.
Recognition of acceptor stem
The most interesting feature of tRNAHis is the presence of the G-1 nucleotide. Identification of the elements responsible for the specific recognition of the critical G-1:C73 base pair holds the key to the histidylation mechanism. The interactions at the 5′ end of tRNA are mainly on G-1 and G1, whereas at the 3′ end, contacts are on nucleotides C68-A76. Surprisingly, most of these interactions are nonspecific and electrostatic interactions between positively charged residues and the backbone phosphates of tRNA (Supplementary Table S3). tRNAHis used for crystallization begins with a guanosine triphosphate and three salt bridges are formed between the β-, γ-phosphate oxygens and Arg7 as well as Arg122. Arg122 is absolutely conserved and Arg7 is usually replaced by a similar lysine residue in other organisms (Supplementary Figure S1). These interactions are not biologically relevant because the natural tRNAHis bears a 5′ GMP. Besides recognizing the γ-phosphate oxygen, Arg7 also forms a salt bridge with C68 backbone phosphate. The most critical interactions may come from the two salt bridges with Arg115 and an additional one from Arg122, between their guanidino groups and the oxygen atoms of G-1 α-phosphate. Furthermore, Arg115 donates a hydrogen bond to the O6 and N7 atoms of G-1, respectively, through its main chain nitrogen, and the former allows the enzyme to specifically recognize G-1 instead of A-1. Aside from the interactions with subunit one, the subunit forming the majority of protein-RNA contacts, G-1 also accepts four more hydrogen bonds and two salt bridges from the 71′RGGR74′ tetrapeptide across the subunit. However, two hydrogen bonds on the base ring involve Arg71′, whose side chain is not well defined in the electron density map. In addition, Arg71′ NH1 atom is relatively far from its hydrogen bonding partner N9 of G-1 (3.48 Å), therefore the contributions from these interactions may be rather weak (Figure 2A).
Figure 2.
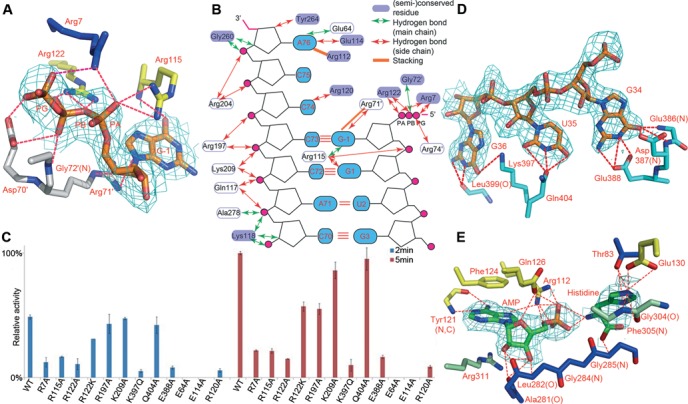
Substrate recognition by TtHisRS. All the 2Fo-Fc maps were contoured at 1σ. (A) The specific interactions of TtHisRS with G-1 in the acceptor stem. Note that G-1 is in the triphosphate form. The color scheme is the same as in Figure 1A and subunit two is in gray. (B) The diagram illustrating the interactions of TtHisRS with the acceptor stem. (C) Time course of the relative aminoacylation activities of HisRS mutants that are involved in tRNA recognition. Two sets of data are shown, representing the measurements at 2- (blue) and 5-min time points (red), respectively. The activity of WT TtHisRS at the 5 min time point was normalized to 100% and the readings at time point zero were used as blanks. Error bars represent SD calculated from two measurements. (D) The recognition of the anticodon loop bases GUG. (E) The interactions with AMPPNP and histidine substrates at the active site.
Other than the contacts on G-1, G1 also makes three hydrogen bonds with Arg115 side chain through its purine ring, as well as a salt bridge through its backbone phosphate (Figure 2B). We mutated the arginines to alanines individually and discovered that the both the R7A and R115A mutations reduce enzymatic activities by ∼80%, while the R122A mutant is only 15% as active as the WT, attesting to the significance of G-1 interactions (Figure 2C and Supplementary Figure S2).
Contacts on the 3′ end are also mainly nonspecific, and the key residues responsible for the recognition are concentrated in three regions. Gln117, Lys118, Arg120 and Glu114 from the conserved motif 2, interact with C70-C72, C74 and A76 (Figure 2B). Gln117 donates two hydrogen bonds to A71 and C72 backbone oxygens using its NE2 atom, while Lys118 hydrogen bonds to C70 and A71 using its main chain nitrogen. The invariant Arg120 is responsible for the specific recognition of C74 while the semiconserved Glu114 makes specific contacts with A76 purine ring. The second region responsible for C72-C75 recognition (Arg197, Arg204 and Lys209) is located in the α5 and η4 helices within the insertion domain, and these contacts are mostly salt bridges between the side chains of positively charged residues and tRNA backbone phosphates. Among these, the NZ atom of the highly conserved Lys209 electrostatically interacts with C72 phosphate oxygen, whereas Arg197 and Arg204 make a total of nine salt bridges with C73-A76. However, neither arginine is conserved. The R197A mutant retains more than half of the WT activity while the activity of K209A is comparable to that of WT. The third region involves residues Gly260, Tyr264 and Ala278, and is between the insertion domain and motif 3. Gly260 and Ala278 form nonspecific hydrogen bonds using their main chain nitrogens. The 3′ terminal A76 alone makes seven hydrogen bonds, three of which are donated from the 6-exocyclic amino group to the carboxylate group of Glu114, and to the main chain carbonyl oxygen of Glu64.
Consequently, the acceptor stem is clamped between the insertion domain, and the Arg115/Arg122 arginine pair. Surprisingly, most interactions on the G-1:C73 base pair are neither specific nor from conserved residues. In contrast, G1, C74 and A76 each form multiple base-specific hydrogen bonds. We introduced mutations to these residues to test their importance (except for residues with main chain contacts). We found that while Glu64 and Glu114 are absolutely essential for aminoacylation, the rest residues are dispensable or play minor roles in substrate recognition (Figure 2C). Therefore the contribution to aminoacylation from these nonspecific interactions is negligible. Nonetheless, these nonspecific interactions together with the contacts on the essential identity determinants, help to adjust and place the acceptor end of tRNA.
Recognition of anticodon loop
The anticodon binding domain in aaRSs is responsible for recognizing the anticodon loop. In our cocrystal structure, it forms the most specific hydrogen bonds with the anticodon trinucleotide. This domain approaches the anticodon loop from the major groove (Figure 1B and C). Bases of U33 to G37 are splayed out, and the GUG triplet forms extensive interaction network with the enzyme (Figure 2D). The residues for the anticodon recognition mainly cluster in the α12-β15 region, and all the interactions are base-specific. Particularly, the wobble base G34 forms six hydrogen bonds with residues Glu386-Glu388 through the base ring, two of which are with the main chain nitrogens of Glu386 and Asp387 respectively. U35 forms three hydrogen bond contacts with the side chain of Gln404. G36 forms two hydrogen bonds with the main chain carbonyl oxygen of Lys399 and one with the side chain of Lys397. Activity tests show that the two most severe mutations are K397Q and E388A, which dropped the activities by 9- and 5-fold, respectively, whereas the Q404A activity is on a similar level to that of WT (Figure 2C). The weak contribution to aminoacylation from the C-terminal domain is similar to its Escherichia coli counterpart, in which the catalytic domain of the enzyme alone retains ∼0.6% aminoacylation efficiency of that of the full-length enzyme (41).
In contrast to the anticodon loop, the region outside of the anticodon is largely flexible. Despite the relative high resolution of the crystal structure, the tRNA molecule shows poor electron density in this region except for the 34GUGG37 fragment, and A38 is completely disordered. We also obtained another form of cocrystal structure at a lower resolution, which contains a TtHisRS dimer bound by two tRNAs in the asymmetric unit (Tian et al., unpublished). The dimeric complex displays even larger flexibility in the anticodon loop and only nucleotides G34-G37 are visible.
Active site
Although AMPPNP was added during the crystallization process, only the AMP moiety is found present in the final refined structure for reasons that are not clear to us. Additionally, the histidine molecule is not fully covered by the map and its main chain density is not evident either in the 2Fo-Fc or Fo-Fc map (Figure 2E and Supplementary Figure S3). On the other hand, the imidazole ring occupies a similar position to the tRNA-free structures (PDB code 1ADJ and 1ADY), forming two hydrogen bonds with the terminal carboxylate group of the motif 2 residue Glu130. In addition, Thr83 also forms two more hydrogen bonds with the ring. These contacts hold the side chain of histidine from the side and top, and Gly304 and Phe305 form the bottom of the imidazole-binding pocket, adding two more hydrogen bonds (Figure 2E). However, the main chain of histidine is in a different orientation from previous histidine-bound structure. Particularly, the amino group points to the A76 ribose as well as the AMP phosphate, and makes two hydrogen bonds with the latter; the carbonyl oxygen is directed towards histidine-2 peptide (30) and are involved in two hydrogen bonds with the main chain of the Gly284Gly285 dipeptide. The orientation of histidine may not represent its genuine intermediate state. The nonproductive stalemate due to the existence of the inhibitor AMPPNP may have promoted histidine to flip, in favor of the hydrogen bonds as well as reducing the repulsion with the AMP phosphate group. However, A76 is in a functional position for histidine acceptance with its 3′ OH within a hydrogen bonding distance of the O3P of AMP, and AMP in our structure adopts a very similar conformation to HAM in 1ADJ (Supplementary Figure S4). Furthermore, a tetraglycine motif is present from positions 283 to 286 and forms part of the histidine-binding motif. The last two glycines are absolutely conserved while the first two are usually small residues (Supplementary Figure S1). Slightest size increases on the side chain by the G-to-A mutation render the enzyme inactive: the activity of G285A is undetectable and G286A loses its activity by more than 30-fold (Figure 3C).
Figure 3.
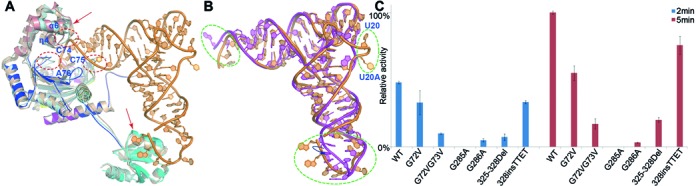
Structural changes upon the formation of complex. (A) Structure comparison of the enzyme in the apo form (PDB code 1H4V, colored wheat), in complex with HAM (PDB code 1ADJ, colored cyan), and in complex with tRNA (the same color scheme as in 1A). The tRNA is depicted in orange. The regions of possible steric clashes between the tRNA-free enzyme and tRNA are indicated by the open circles. The arrows show relative large conformational changes occurring in the insertion and the anticodon binding domain. (B) RNA structural changes during catalysis. tRNAHis is superimposed onto the canonical, free tRNAphe (PDB code 1TN1). tRNAphe and tRNAHis are shown in magenta and orange, respectively. The open circles indicate large structural differences between two tRNA molecules and the blue arrow shows the rotation direction of the anticodon loop. (C) Activity assays of residues involved in the binding pockets of G-1 and histidine, in addition to the interdomain linker.
Most of the residues that contact with AMP are conserved, which have been described previously in great details (30,31), and will not be discussed here (Figure 2E).
Conformational changes of HisRS upon RNA binding
Upon the binding of tRNA, TtHisRS as well as tRNA undergo some local structural changes. Compared to the protein bound with HAM, the insertion domain exhibits the largest movements (Figure 3A and Supplementary Figure S5). The insertion domain would pose clashes with C74 and C75 backbone phosphates and has to open up the active site to allow tRNA to enter (Figure 3A). The entire domain moves as a rigid body without changing its overall shape. The largest translation occurs at the η4-α6 loop (Asp207-Glu211). In the extreme case, Arg212 Cα moves 9.9 Å relative to the apo structure and 5.2 Å relative to the HAM-bound HisRS structure (Supplementary Figure S6). The conformational changes in this α-helical insertion allow it to clamp onto the accepter stem of tRNA through charged interactions. In addition, the histidine-1 motif Val258-Asp262 and the Gln117-Arg120 fragment also show noticeable shifts. The histidine-1 motif closes upon histidine binding (30). In the tRNA complex, this loop reopens to some extent and occupies an intermediate position between the apo- and histidine-bound TtHisRS in order to avoid steric hindrance with A76 phosphate (Figure 3A and Supplementary Figure S4). In the anticodon binding region, Ala379-Arg398 also displays local readjustments from apo TtHisRS due to the interactions with the anticodon loop, but these adjustments are relatively small. On the other hand, the ordering loop (L78-M86) is quite similar to those observed in the apoprotein or histidine-bound structure (Supplementary Figure S4).
In terms of tRNA, nucleotides U35-G37 experience unwinding compared to yeast tRNAPhe (PDB code 1TN1), to avoid the hindrance from α11 (Figure 3B). The only contacts with the enzyme are through the anticodon and the remainder contributes little to aminoacylation. As a result, the entire anticodon loop (residues 30–43) shows weak density apart from the anticodon triplet, and the averaged B-factor of this region is well above that of the whole tRNA (146.5 Å2 vs. 94.1 Å2). The substantial flexibility of tRNAHis is consistent with the difficulty of running molecular replacement during the structure determination process, in which the tRNA model had to be split in halves to generate a reasonable solution. The poorly ordered model in this region also suggests that the majority of the anticodon loop plays a nonessential role in aminoacylation. Besides the disorder in the anticodon loop, bases 16 and 17 display weak densities as well (Figure 3B).
Role of the interdomain linker
HisRSs contain a 9∼14-residue interdomain hinge, which links the two otherwise stand-alone domains. The N- and C-terminal domains within the same subunit barely make any direct contacts with or without tRNA. Judging from several bacterial HisRS structures, this loop appears to be relatively tense and little changes occur to this region after the binding of tRNA substrate. One would speculate that this loop bears much tension in order to communicate between the two domains. Bacterial HisRSs usually possess shorter linker sequences whereas eukaryotic HisRSs have an insertion (a TTET motif in vertebrates) at the end of the linkers (Supplementary Figure S1). To assess the importance of the linker length, we intentionally shortened (by removing the 325EEKG328 tetrapeptide) or extended (by inserting the TTET sequence after Gly328) this loop and tested activities. We discovered that the insertion mutant retains ∼ 3/4 activity of that of WT and the deletion mutant only 1/5 (Figure 3C). We also noticed that the deletion of the EEKG motif is highly detrimental to protein stability because the deletion mutant failed to form a peak on the size exclusion column, indicating some folding problems. Thus, the hinge joining the two domains is important for the crosstalk between the two domains. Structural alterations resulting from the anticodon binding domain are coupled to those of active site, caused by substrate binding via this unique linker. Through evolution, the length of the linker has attained an optimal value. While too little distance causes serious clashes between the N- and C-terminal domains, too much distance reduces the communication efficiencies between them. The two domains act as rigid units, and the linker length may affect directly their relative positions and orientations, which in turn affect the interactions with the tRNA substrate and hence aminoacylation.
The binding pocket of G-1
The binding pocket of G-1 is composed of two important glycine residues Gly72′Gly73′ across the subunit. These two glycines are very conserved across species and the accommodation of G-1 may originate from the flexibility of the dipeptide located on a loop as well as the small size for glycine (Figure 4A, B and Supplementary Figure S7). To explore the tolerance capacity of the pocket, we reduced its size by creating the G72V single- as well as the G72VG73V double mutants, and the later has proved to reduce the enzymatic activities by more than 4/5 (Figure 3C). Structural comparison with other class IIa synthetases in complex with their cognate tRNAs reveals that such a pocket is probably only unique to HisRS. We therefore created models of tRNAHis in complex with other class IIa enyzmes by superimposing the catalytic domains of hGlyRS (34), hSerRS (42) or EcThrRS (43), respectively, onto that of tRNA-bound TtHisRS. In our models, G-1 of tRNAHis clashes into Glu323′Asn324′ as well as the motif 2 residues around Glu365 in EcThrRS (PDB code 1QF6), Gly233′Pro234′ as well as the motif 2 residues around Ile280 in hGlyRS (PDB code 4KR2), and Lys253′-Ser262′ as well as the motif 2 residues around Val305 in hSerRS (Figure 4C–E). It is therefore reasonable to predict that these tRNA with G-1 would make poor substrates for their enzymes. Our aminoacylation assays also lent support to these theoretical models, by showing that hSerRS or hGlyRS only retained ∼1/3 activity for their G-1-containing tRNA substrates, compared to their WT tRNA substrates, respectively (Figure 4F).
Figure 4.
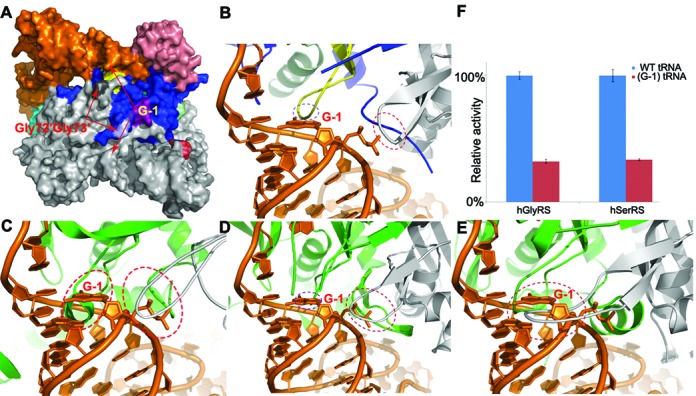
The comparison of the putative G-1 binding pockets. (A) The surface representation of HisRS-tRNA complex dimer. The color scheme is the same as in Figure 1A. Both the Gly72′Gly73′ dipeptides (colored in light gray and light blue) and the G-1 bases (in orange and gray) of the dimer are indicated by arrows, respectively. (B)–(E) The close-up views of G-1 environment when tRNAHis is in complex with TtHisRS (B), in the EcThrRS-tRNA model (C), in the hGlyRS-tRNA model (D) and in the hSerRS-tRNA model (E). (F) The activity assays of hGlyRS and hSerRS using their cognate WT tRNA substrates, or tRNAs with an artificially implanted G-1. The ovals show the areas that possibly have significant steric conflicts between the enzyme and G-1-containing tRNAs, as well as the same area in HisRS, which is structurally compatible.
DISCUSSION
The substrate recognition of tRNAHis is an outstanding problem that has remained unclear for many years. In this work, we determined the crystal structure from TtHisRS-tRNA complex and studied the structural basis of histidylation. Our crystal structure is in good agreement with the modeling studies carried out by Åberg et al. (30) but with much more information on the structural details. Structure-based mutagenesis was subsequently conducted and mutants were tested for activities. G(-1)MP-containing tRNAHis was used in the activity assays due to its reportedly higher activity than the GTP-form of tRNAHis. Our pilot experiments show that appropriate enzyme concentrations for initial velocity measurements range from 35 to 70 nM (Supplementary Figure S8). Hence we fixed the concentration of the enzyme at 50 nM throughout the assays. A few conclusions can be drawn from our crystallographic and biochemical studies.
First, the recognition of tRNAHis is principally a type of backbone recognition, in which the -1:73 base-pairing geometry plays an important role. The enzyme makes very few specific contacts with tRNA except for the anticodon triplet. The interactions on the acceptor stem are mainly on the 3′ end while those on the 5′ end are concentrated on G-1 and G1. Most charged electrostatic interactions between conserved Arg/Lys residues and the backbone phosphates bear little consequences on aminoacylation. Additionally, many polar interactions are formed by the main chains of the protein. The only specific recognition on G-1 arises from the hydrogen bond through O6, allowing it to be possibly distinguished from A-1 by the enzyme. Interestingly, we identified two semiconserved residues Glu64 and Glu114 that cause the most activity loss. Glu64 is located at the tip of a disordered loop that may contact with the minor groove of the acceptor stem and its only direct contact with the tRNA molecule is a possible hydrogen bond with the N6 atom of A76. The density of neither hydrogen-bonding partner is clearly resolved, similar to the case of Glu114. Glu114 is located to an adjacent loop that contacts with the major groove. The two glutamate residues are spatially close and may form some interactions (Supplementary Figure S9). The reasons for the significant effects on histidylation from these two residues are not clear, and whether this phenomenon is TtHisRS-specific awaits further investigation.
Second, the conformational changes for protein upon the formation of the complex are not dramatic, with detectable rearrangements occurring in the insertion domain. There are also several local structural changes induced by tRNA binding that lead to the ordering or disordering of several flexible loops. However, these conformational changes are relatively small, compared to those incurred by the binding of histidine or synthesis of HAM. Consequently, we propose that the synthesis histidyl-adenylate is likely to be the rate-limiting step in histidylation, to solicit the large conformational changes needed for the first step. Only minor rearrangements ensue in the insertion domain as well as in other regions in order to complete the reaction.
In contrast, the anticodon of tRNA becomes highly mobile upon the formation of complex. In the structure of CatRNAHis in complex with its processing enzyme Thg1, the anticodon loop is intact and its B-factor is on a similar level to that of entire tRNA (36). Therefore, the flexibility of the anticodon loop is a consequence of the induced fit brought about by the binding of tRNAHis. The high flexibility of anticodon loop from both cocrystal structures suggests that this region is dispensable for aminoacylation, and offers an explanation for the observation of mini- or microhelices or even transplanted yeast tRNAAsp being fair substrates for HisRS (22). It is reasonable to deduce that the heavy reliance of HisRS on G-1 as the critical identity determinant is a safety mechanism, compensating for the weak contribution of the anticodon to aminoacylation.
Third, the accommodation of the extra base G at the -1 position appears to be related to the enlarged substrate-binding pocket of HisRS for tRNAHis at the very 5′ extremity. The conserved Gly72′Gly73′ dipeptide from across the subunit allows G-1 to fit, while the steric hindrance from hGlyRS, hSerRS and EcThrRS excludes the entry of their cognate tRNAs with an implanted G-1 base. This hypothesis is confirmed by the aminoacylation activities of hSerRS and hGlyRS for G-1 tRNAs, which reduced 2/3 activities of the enzymes.
Caulobacter crescentus HisRS (CcHisRS) is reported to have a G-1-less tRNAHis. The sequence alignment indicates that a pair of aspartates substitute the conserved G72′G73′ dipeptide and thus blocks the binding site of G-1 (Supplementary Table S3). The covariation of both the enzyme/tRNA allows for the unusual histidylation system lacking G-1 recognition, but the activity of CcHisRS is considerably lower than HisRSs from other organisms, due to the loss of interactions with G-1. To compensate for this loss of substrate binding, CcHisRS has to rely more on the anticodon recognition (28).
ACCESSION CODES
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 4RDX.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Shanghai Synchrotron Radiation Facility (SSRF) for access to beamline 17U (BL17U).
FUNDING
National Sciences Foundation of China [31100579]; Guangdong Innovative Research Team Program [201001Y0104687244]. Funding for open access charge: National Sciences Foundation of China [31100579]; Guangdong Innovative Research Team Program [201001Y0104687244].
Conflict of interest statement. None declared.
REFERENCES
- 1.Cusack S. Eleven down and nine to go. Nat. Struct. Biol. 1995;2:824–831. doi: 10.1038/nsb1095-824. [DOI] [PubMed] [Google Scholar]
- 2.Arnez J.G., Moras D. Structural and functional considerations of the aminoacylation reaction. Trends Biochem. Sci. 1997;22:211–216. doi: 10.1016/s0968-0004(97)01052-9. [DOI] [PubMed] [Google Scholar]
- 3.Eriani G., Delarue M., Poch O., Gangloff J., Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 4.Cusack S., Hartlein M., Leberman R. Sequence, structural and evolutionary relationships between class 2 aminoacyl-tRNA synthetases. Nucleic Acids Res. 1991;19:3489–3498. doi: 10.1093/nar/19.13.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ribas de Pouplana L., Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104:191–193. doi: 10.1016/s0092-8674(01)00204-5. [DOI] [PubMed] [Google Scholar]
- 6.Frugier M., Moulinier L., Giege R. A domain in the N-terminal extension of class IIb eukaryotic aminoacyl-tRNA synthetases is important for tRNA binding. EMBO J. 2000;19:2371–2380. doi: 10.1093/emboj/19.10.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berthet-Colominas C., Seignovert L., Hartlein M., Grotli M., Cusack S., Leberman R. The crystal structure of asparaginyl-tRNA synthetase from Thermus thermophilus and its complexes with ATP and asparaginyl-adenylate: the mechanism of discrimination between asparagine and aspartic acid. EMBO J. 1998;17:2947–2960. doi: 10.1093/emboj/17.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Commans S., Plateau P., Blanquet S., Dardel F. Solution structure of the anticodon-binding domain of Escherichia coli lysyl-tRNA synthetase and studies of its interaction with tRNA(Lys) J. Mol. Biol. 1995;253:100–113. doi: 10.1006/jmbi.1995.0539. [DOI] [PubMed] [Google Scholar]
- 9.Delarue M., Moras D. The aminoacyl-tRNA synthetase family: modules at work. Bioessays. 1993;15:675–687. doi: 10.1002/bies.950151007. [DOI] [PubMed] [Google Scholar]
- 10.Ibba M., Francklyn C., Cusack S. The aminoacyl-tRNA synthetases. Georgetown, TX: Landes Bioscience; 2005. [Google Scholar]
- 11.Sprinzl M., Steegborn C., Hübel F., Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1996;24:68–72. doi: 10.1093/nar/24.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooley L., Appel B., Söll D. Post-transcriptional nucleotide addition is responsible for the formation of the 5′ terminus of histidine tRNA. Proc. Natl. Acad. Sci. U.S.A. 1982;79:6475–6479. doi: 10.1073/pnas.79.21.6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackman J.E., Phizicky E.M. tRNAHis guanylyltransferase adds G-1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA. 2006;12:1007–1014. doi: 10.1261/rna.54706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkard U., Willis I., Söll D. Processing of histidine transfer RNA precursors. Abnormal cleavage site for RNase P. J. Biol. Chem. 1988;263:2447–2451. [PubMed] [Google Scholar]
- 15.Orellana O., Cooley L., Soll D. The additional guanylate at the 5′ terminus of Escherichia coli tRNAHis is the result of unusual processing by RNase P. Mol. Cell. Biol. 1986;6:525–529. doi: 10.1128/mcb.6.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crothers D., Seno T., Söll D. Is there a discriminator site in transfer RNA. Proc. Natl. Acad. Sci. U.S.A. 1972;69:3063–3067. doi: 10.1073/pnas.69.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Himeno H., Hasegawa T., Ueda T., Watanabe K., Miura K.-i., Shimizu M. Role of the extra GC pair at the end of the acceptor stem of tRNAHis in aminoacylation. Nucleic Acids Res. 1989;17:7855–7863. doi: 10.1093/nar/17.19.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nameki N., Asahara H., Shimizu M., Okada N., Himeno H. Identity elements of Saccharomyces cerevisiae tRNAHis. Nucleic Acids Res. 1995;23:389–394. doi: 10.1093/nar/23.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan W., Francklyn C. Cytosine 73 is a discriminator nucleotide in vivo for histidyl-tRNA in Escherichia coli. J. Biol. Chem. 1994;269:10022–10027. [PubMed] [Google Scholar]
- 20.Rosen A.E., Musier-Forsyth K. Recognition of G-1: C73 atomic groups by Escherichia coli histidyl-tRNA synthetase. J. Am. Chem. Soc. 2004;126:64–65. doi: 10.1021/ja0381609. [DOI] [PubMed] [Google Scholar]
- 21.Fromant M., Plateau P., Blanquet S. Function of the extra 5′-phosphate carried by histidine tRNA. Biochemistry. 2000;39:4062–4067. doi: 10.1021/bi9923297. [DOI] [PubMed] [Google Scholar]
- 22.Rudinger J., Florentz C., Giegé R. Histidylation by yeast HisRS of tRNA or tRNA-like structure relies on residues–1 and 73 but is dependent on the RNA context. Nucleic Acids Res. 1994;22:5031–5037. doi: 10.1093/nar/22.23.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nameki N., Tadaki T., Muto A., Himeno H. Amino acid acceptor identity switch of Escherichia coli tmRNA from alanine to histidine in vitro. J. Mol. Biol. 1999;289:1–7. doi: 10.1006/jmbi.1999.2754. [DOI] [PubMed] [Google Scholar]
- 24.Francklyn C., Schimmel P. Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc. Natl. Acad. Sci. U.S.A. 1990;87:8655–8659. doi: 10.1073/pnas.87.21.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salomon R., Littauer U. Enzymatic acylation of histidine to mengovirus RNA. Nature. 1974;249:32–34. doi: 10.1038/249032a0. [DOI] [PubMed] [Google Scholar]
- 26.Francklyn C., Shi J.-P., Schimmel P. Overlapping nucleotide determinants for specific aminoacylation of RNA microhelices. Science. 1992;255:1121–1125. doi: 10.1126/science.1546312. [DOI] [PubMed] [Google Scholar]
- 27.Rosen A.E., Brooks B.S., Guth E., Francklyn C.S., Musier-Forsyth K. Evolutionary conservation of a functionally important backbone phosphate group critical for aminoacylation of histidine tRNAs. RNA. 2006;12:1315–1322. doi: 10.1261/rna.78606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan J., Gogakos T., Babina A.M., Söll D., Randau L. Change of tRNA identity leads to a divergent orthogonal histidyl-tRNA synthetase/tRNAHis pair. Nucleic Acids Res. 2011;39:2286–2293. doi: 10.1093/nar/gkq1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnez J., Harris D., Mitschler A., Rees B., Francklyn C., Moras D. Crystal structure of histidyl-tRNA synthetase from Escherichia coli complexed with histidyl-adenylate. EMBO J. 1995;14:4143–4155. doi: 10.1002/j.1460-2075.1995.tb00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Åberg A., Yaremchuk A., Tukalo M., Rasmussen B., Cusack S. Crystal structure analysis of the activation of histidine by Thermus thermophilus histidyl-tRNA synthetase. Biochemistry. 1997;36:3084–3094. doi: 10.1021/bi9618373. [DOI] [PubMed] [Google Scholar]
- 31.Yaremchuk A., Tukalo M., Grotli M., Cusack S. A succession of substrate induced conformational changes ensures the amino acid specificity of Thermus thermophilus prolyl-tRNA synthetase: comparison with histidyl-tRNA synthetase. J. Mol. Biol. 2001;309:989–1002. doi: 10.1006/jmbi.2001.4712. [DOI] [PubMed] [Google Scholar]
- 32.Merritt E.A., Arakaki T.L., Gillespie J.R., Larson E.T., Kelley A., Mueller N., Napuli A.J., Kim J., Zhang L., Verlinde C.L. Crystal structures of trypanosomal histidyl-tRNA synthetase illuminate differences between eukaryotic and prokaryotic homologs. J. Mol. Biol. 2010;397:481–494. doi: 10.1016/j.jmb.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z., Wei Z., Zhou J.J., Ye F., Lo W.-S., Wang F., Lau C.-F., Wu J., Nangle L.A., Chiang K.P. Internally deleted human tRNA synthetase suggests evolutionary pressure for repurposing. Structure. 2012;20:1470–1477. doi: 10.1016/j.str.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin X., Hao Z., Tian Q., Zhang Z., Zhou C., Xie W. Cocrystal structures of glycyl-tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. J. Biol. Chem. 2014;289:20359–20369. doi: 10.1074/jbc.M114.557249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura A., Nemoto T., Heinemann I.U., Yamashita K., Sonoda T., Komoda K., Tanaka I., Soll D., Yao M. Structural basis of reverse nucleotide polymerization. Proc. Natl. Acad. Sci. U.S.A. 2013;110:20970–20975. doi: 10.1073/pnas.1321312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–273. doi: 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
- 41.Augustine J., Francklyn C. Design of an active fragment of a class II aminoacyl-tRNA synthetase and its significance for synthetase evolution. Biochemistry. 1997;36:3473–3482. doi: 10.1021/bi962395y. [DOI] [PubMed] [Google Scholar]
- 42.Xu X., Shi Y., Yang X.L. Crystal structure of human Seryl-tRNA synthetase and Ser-SA complex reveals a molecular lever specific to higher eukaryotes. Structure. 2013;21:2078–2086. doi: 10.1016/j.str.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sankaranarayanan R., Dock-Bregeon A.C., Romby P., Caillet J., Springer M., Rees B., Ehresmann C., Ehresmann B., Moras D. The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.