


Abstract
Although the RNase H-dependent mechanism of inhibition of gene expression by chemically modified antisense oligonucleotides (ASOs) has been well characterized, little is known about the interactions between ASOs and intracellular proteins that may alter cellular localization and/or potency of ASOs. Here, we report the identification of 56 intracellular ASO-binding proteins using multi-step affinity selection approaches. Many of the tested proteins had no significant effect on ASO activity; however, some proteins, including La/SSB, NPM1, ANXA2, VARS and PC4, appeared to enhance ASO activities, likely through mechanisms related to subcellular distribution. VARS and ANXA2 co-localized with ASOs in endocytic organelles, and reduction in the level of VARS altered lysosome/ASO localization patterns, implying that these proteins may facilitate ASO release from the endocytic pathway. Depletion of La and NPM1 reduced nuclear ASO levels, suggesting potential roles in ASO nuclear accumulation. On the other hand, Ku70 and Ku80 proteins inhibited ASO activity, most likely by competition with RNase H1 for ASO/RNA duplex binding. Our results demonstrate that phosphorothioate-modified ASOs bind a set of cellular proteins that affect ASO activity via different mechanisms.
INTRODUCTION
RNase H1 is a conserved endonuclease that selectively degrades the RNA strand within a DNA/RNA duplex (1). Synthetic antisense oligonucleotides (ASOs) that harness this property to direct sequence-specific cleavage of the RNAs are used for both biological research and biomedical purposes (2,3). The mechanism of ASO-directed RNase H1 cleavage of target RNAs has been well characterized, and the crystal structure of RNase H1 has been solved (1,4). Evidence clearly shows that ASOs introduced into mouse or human cells trigger substrate RNA degradation through RNase H1, but not RNase H2 (5). Recently we showed that the mitochondrial protein P32 associates with RNase H1 to increase RNase H1 activity, most likely by increasing the turnover rate of the enzyme (6). It also appears that the level of RNase H1 protein is a limiting factor in terms of ASO potency, as reducing RNase H1 decreased and over-expressing RNase H1 increased ASO-directed RNA cleavage (5).
The chemical composition of one class of ASOs currently used as therapeutic agents has 10 deoxyribonucleotides flanked on either side by five 2′-O-methoxyethyl (MOE) modified nucleotides with nucleotides linked via phosphorothioate (PS) backbones; this configuration is often referred to as a ‘gapmer’. This chemical composition ensures efficient cellular uptake and stability against nuclease digestion (7). Internalization of ASOs has been shown to be largely through endocytosis (8,9). At low concentrations (<1 μM), ASOs may enter cells predominately through receptor-mediated endocytosis, whereas at high concentrations, fluid phase endocytosis plays a major role (10). It has been proposed that two pathways may exist for ASO uptake and accumulation in cells—a non-productive pathway in which internalized ASOs are not functionally active and a productive pathway through which ASOs can hybridize with RNAs and trigger RNase H1 cleavage (9,11,12).
When ASOs are incubated with cells in culture in the absence of transfection reagents (free uptake), internalized ASOs predominantly accumulate in lysosomes; and GW/P-body localization of PS-ASOs in Huh-7 cells was also reported previously (13). Lysosome-localized ASOs are considered to be inactive (8,9,14). However, ASOs can escape from the membrane-enclosed endocytosis particles to the cytosol during intracellular trafficking (11,12), and the released ASOs thus have the potential to reach the target RNAs. The productive pathway of ASO uptake may involve AP2M1, since reduction of AP2M1, but not clathrin or caveolin, reduces ASO activity upon free uptake (9). Although the molecular mechanism of the productive pathway has not been defined, previous studies showed that nuclear localization of ASOs positively correlates with the antisense activity (15). This is evidenced by the observations that upon transfection, ASOs accumulate in the nucleus, and ASO activity is significantly increased as compared with ASO free uptake, by which ASOs are barely detectable in the nucleus (15). However, upon free uptake, ASOs can still be active even without significant nuclear accumulation (9,13). In addition, ASOs were shown to shuttle between the cytoplasm and nucleus, and nuclear import is most likely mediated by an active transport pathway (16,17). These results suggest that in the case of free uptake, the portion of ASOs in the productive pathway is small relative to the ASOs accumulated in endosomes and lysosomes, whereas upon transfection, the fraction of ASOs entering the productive pathway, or ASOs released to the cytosol, is increased.
In addition to the many properties of the targeted RNAs, such as RNA structure, sequence, and RNA-binding proteins that can affect ASO accessibility to the target RNA (18), the binding of intracellular proteins to ASOs may also influence antisense activity at many steps, e.g. release of ASOs from endocytosis pathways, proper subcellular localization, interaction of ASOs with RNA substrates and recruitment of RNase H1 to the ASO/RNA duplex. Previous studies have demonstrated that PS-ASOs bind more proteins, including NCL1, hnRNP A1 and albumin, than ASOs containing phosphodiester (PO) backbone (19–22). Some proteins may compete with RNase H1 for binding to the mRNA–ASO duplex. Indeed, it was shown very recently that hspA8 and hnRNP F could bind to an ASO/RNA duplex and compete with RNase H1, leading to inhibition of RNase H1 cleavage (23). In addition, we recently demonstrated that the chaperone protein TCP1 complex interacts with PS-ASOs and can increase ASO activity (22). The beta subunit of the TCP1 complex co-localizes with ASOs in nuclear PS bodies upon transfection. Several TCP1 proteins also co-localize with ASOs in cytoplasmic endosomes and lysosomes in the case of free uptake, suggesting a potential role of TCP1 proteins in ASO release from the endocytosis pathway and/or in nuclear retention (22). Additionally, we also found that paraspeckle proteins, including P54nrb, PSF and PSPC1, bind to ASOs and inhibit ASO activity (24). These paraspeckle proteins co-localize with ASOs in different nuclear structures, such as nuclear paraspeckles, ASO-induced nuclear filaments and perinucleolar cap structures (24). The chemical modification at the 2′ moiety also affects protein binding. For example, ASOs with more hydrophobic 2′ modifications (e.g. constrained ethyl) tend to bind more proteins and more avidly than those with more hydrophilic 2′ modifications (e.g. 2′-MOE) [(22,24) and our unpublished data]. In addition, some proteins bind primarily to single-stranded ASOs (ssASOs), whereas others, such as P54nrb and PSF proteins, bind the ASO/RNA duplex (24). These results suggest that a number of cellular proteins interact with ASOs and affect the antisense activity positively or negatively. However, information about intracellular ASO-binding proteins and their effects on ASO activity and localization is still limited.
To better understand the interactions between intracellular proteins and ASOs, we sought to identify the major ASO-binding proteins using multiple-step affinity selection approaches. Here we report the identification of 56 intracellular ASO-binding proteins. To our knowledge, this is the first systematic study investigating the binding of ASOs to intracellular proteins and the effects of ASO-binding proteins on ASO activity and/or localization. Although PS-ASOs interact with a number of proteins, a relatively small subset of intracellular proteins appears to be the major binders of PS-ASOs as determined using our competition-mediated approach. Certain proteins, including La, NPM1, valyl-tRNA synthetase (VARS), PC4 and ANXA2, moderately increased ASO activity, likely through different mechanisms, such as by altering ASO subcellular distribution. ANXA2 and VARS were found to co-localize with ASOs in the cytoplasm, similar to previously reported results with TCP1 proteins. On the other hand, Ku70/Ku80 complex proteins were found to inhibit ASO activity, most likely due to competition with RNase H1 for binding to ASO/RNA duplex. Thus, intracellular proteins can bind to and affect ASO activity and/or subcellular localization.
MATERIALS AND METHODS
Antibodies, siRNAs, ASOs and quantitative real time PCR (qRT-PCR) primer probe sets are listed in Supplementary data.
Cell culture and transfection of plasmids, siRNAs and ASOs
HEK293 cells stably over-expressing Flag-tagged human RNase H1 were grown as described previously (6). HeLa cells were grown as described elsewhere. One day before transfection, cells were re-seeded at ∼50–70% confluency. Transfection of plasmids was performed using Effectene (Qiagen) based on manufacturer's instructions. At 36 h after plasmid transfection, cells were collected for western analysis or were reseeded at ∼50% confluency, grown overnight, and transfected with ASOs. Transfection of siRNAs and ASOs was performed as described previously (25). Briefly, HeLa cells grown to ∼70% confluency in complete Dulbecco's modified Eagle's medium without antibiotics were transfected with 3–5-nM siRNA using Lipofectamine RNAiMax (Life Technologies). Eight or 24 h post siRNA transfection, cells were reseeded in 6-well or 96-well plates at 50% confluency and incubated for overnight. Cells were then transfected with indicated concentration of ASOs using Lipofectamine 2000 (Life Technologies), and, after 4 h, RNA or protein was prepared. For ASO activity assay upon free uptake, HEK293 cells treated with siRNAs for 24 h were washed and reseed at ∼40% confluency. ASOs were added 8 h later directly to the medium without transfection reagent. After 16 h of ASO incubation, RNA was prepared from cells and mRNA levels were determined by qRT-PCR.
Affinity selection of ASO or ASO/RNA duplex-binding proteins
Affinity selection of ASO-binding proteins was performed as described recently (22,24). Briefly, 100-μl Neutravidin beads (Thermo Scientific) were incubated with 100 μl of 200-μM biotinylated ASO ISIS386652 at 4°C for 2–3 h in W-100 buffer [50-mM Tris–HCl (pH 7.5), 100-mM KCl, 5-mM ethylenediaminetetraacetic acid (EDTA), 0.1% NP-40 and 0.05% sodium dodecyl sulphate (SDS)]. The beads were then blocked for 30 min at 4°C in 300-μl blocking buffer [1-mg/ml bovine serum albumin (BSA), 0.2-mg/ml glycogen and 0.2-mg/ml yeast tRNA in W-100]. After three washes with W-100, ASO-coated beads were incubated for 2–4 h at 4°C with 3-mg HeLa cell extract prepared in buffer A [25-mM Tris-HCl (pH 8.0), 5-mM MgCl2, 150-mM KCl, 10% glycerol, 0.5-mM PMSF, 5-mM β-mercaptoethanol and one tablet of Protease Inhibitor Cocktail/50 ml (Roche)]. After three washes with 500-μl W-200 buffer [50-mM Tris-HCl (pH 7.5), 200-mM KCl, 5-mM EDTA, 0.05% SDS, 0.1% NP-40], the beads were transferred to a 1-ml column and washed seven times with W-200, 1 ml each time. Bound proteins were eluted with 200 μl of 50-μM ASO116847 in W-100 by incubation at room temperature for 30 min with gentle rotation. The eluted ASO–protein complexes were precipitated and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE). Alternatively, to identify binding proteins that can bind ASO/RNA duplex, the eluted ASO–protein complexes were diluted with 200-μl W-100 supplemented with 100 units of RNaseOUT (Life Technologies) and further incubated at 4°C for 2 h with 40-μl Neutravidin beads pre-incubated with 40 μl of 200-μM biotinylated oligonucleotides complementary (XL180) or non-complementary (XL181) to ASO116847. After washing seven times with W-200 supplemented with 20-unit/ml RNaseOUT, the beads were treated with 100-μl Tris-EDTA buffer containing 5-units/μl RNase I at 30°C for 30 min, and the eluted proteins were precipitated and separated on 4–12% SDS-PAGE. Sliver staining was carried out using ProteoSilver Plus Silver Stain Kit (Sigma) based on manufacturer's instructions. The interesting protein bands were excised and identified by mass spectrometry by Alphalyse Inc. Briefly, the samples were reduced and alkylated with iodoacetamide, followed by trypsin digestion. The digested peptides were concentrated and analyzed on a Bruker Autoflex Speed MALDI TOF/TOF instrument. MALDI MS/MS was conducted on 15 peptides for peptide fragmentation analysis. The MS and MS/MS spectra were combined and used to search database with the Mascot software. Alternatively, the isolated proteins were analyzed by western blot.
Immunoprecipitation of RNase H1-associated proteins
Whole cell lysates were prepared from HEK293 cells expressing Flag-tagged RNase H1 (6) using IP buffer (Pierce) supplemented with proteinase inhibitor and 5-mM EDTA. Aliquots of cell lysate (∼2-mg protein) were incubated at 4°C for 1 h with 100 μl of 200-μM ASO116847, or with 100 μl of 200 μM of pre-annealed ASO116847/XL279 duplex, or without ASO. Next, 50-μl anti-FLAG beads were added, and immunoprecipitation was performed at 4°C for 2 h. After seven washes with wash buffer (50-mM Tris-HCl (pH 7.5), 150-mM NaCl, 5-mM EDTA, 0.1% NP-40, 0.05% SDS), co-precipitated proteins were eluted by competition using 100 μl of 1-mg/ml 3×FLAG peptide in wash buffer. The eluted proteins were precipitated and analyzed by western blot.
RNA preparation and qRT-PCR
Total RNA was prepared from HeLa cells using the RNeasy mini kit (Qiagen) or Tri-Reagent (Sigma). Northern hybridization and qRT-PCR using TaqMan primer probe sets were conducted as described previously (25). qRT-PCR reactions were performed in triplicate, and average values were calculated and normalized to the amount of total RNA measured using the Ribogreen approach. The P-values were calculated based on unpaired t-test (n = 3), with P < 0.05 considered to be significant. The sequences of primer probe sets for different RNAs are listed in Supplementary data.
Subcellular fractionation
Untreated HeLa cells or cells treated with PS-ASOs for indicated times were harvested, washed with phosphate buffered saline (PBS) and pelleted. Cytoplasmic and nuclear fractions were prepared using Qproteome Nuclear Protein kit (Qiagen) based on the manufacturer's instructions. Cytoplasmic and nuclear proteins were separated on 4–12% SDS-PAGE and analyzed by western blot.
Immunofluorescence staining
Cells were grown in glass-bottomed culture dishes (MatTek) for 16 h and were transfected or not transfected with 50-nM fluorescent-labeled ASOs for 4–8 h. For free uptake, cells were incubated with 2-μM fluorescent-labeled ASOs for 16–24 h. Immunofluorescence staining was performed as described (22), with minor revisions. Briefly, cells were fixed with 4% paraformaldehyde for 30 min at room temperature and were permeabilized for 5 min using 0.1% Triton X-100 in PBS. Following three washes with PBS, cells were treated with blocking buffer (1-mg/ml BSA in PBS) at room temperature for 30 min and were incubated with primary antibodies (1:100–1:200) in blocking buffer at room temperature for 2–4 h, or at 4°C overnight. After three 5-min washes with wash buffer (0.1% NP-40 in PBS), cells were incubated with secondary antibodies (1:200) in blocking buffer at room temperature for 1–2 h and washed three times (5 min each) with wash buffer. For double staining, two antibodies were used together. Anti-fade reagent containing DAPI (Life Technologies) was added, and slides were covered with cover slips. Images were taken using a confocal microscope (Olympus, FV-1000) and were analyzed with Fluoview Ver. 2.0b Viewer (Olympus).
Western analysis
Equal portions of ASO-binding proteins isolated by affinity selection or an equal amount of proteins from whole cell extracts (∼20 μg) were separated on 4–12% SDS-PAGE and transferred to a membrane. Blocking, antibody incubation and detection of proteins with enhanced chemiluminescence were performed as described previously (26).
RESULTS
Identification of ASO-binding and ASO/RNA duplex-binding proteins
Intracellular ASO-binding proteins were isolated from HeLa cell extracts using a biotinylated 5-10-5 PS/MOE gapmer ASO ISIS386652. To reduce non-specific protein binding, the bound proteins were eluted from the beads by competition using ASO ISIS116847, an ASO with the same sequence and chemical composition as ISIS386652 but lacking biotin (Figure 1A). The isolated ASO–protein pool was either separated by SDS-PAGE for silver staining (Figure 1B, lane 1) or was used as starting material to isolate proteins that bind the ASO/RNA duplex. The ASO/RNA duplex-binding proteins are particularly interesting since these proteins might be expected to affect the activity of RNase H1, which recognizes the ASO/RNA duplex and cleaves the target RNA. This was done using a biotinylated oligonucleotide (XL180) complementary to the elution ASO ISIS116847, or using a biotinylated oligonucleotide (XL181) that does not base-pair with the ASO and served as a negative control. Both XL180 and XL181 contain 20 nucleotides modified with 2′-O-methyl and an unmodified linker sequence. We have shown that 2′-O-methyl-modified oligonucleotides are more stable to nucleases including RNase H1 than unmodified RNA and create duplexes that behave similarly to RNA–ASO duplexes with regard to binding of proteins including RNase H1 [(27,28) and our unpublished data]. The duplex-binding proteins were released from the beads using RNase I digestion, separated by SDS-PAGE, and visualized using silver staining (Figure 1B, lanes 2).
Figure 1.
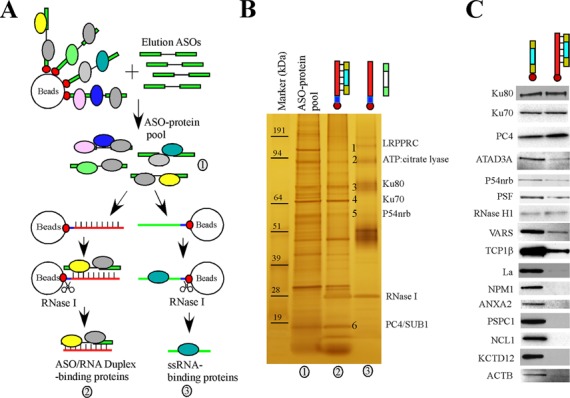
Identification of ASO-binding proteins by affinity selection. (A) Schematic of the affinity selection approaches. The ASO-binding proteins are indicated using colored circles. The biotin tag is indicated by the red dots at the ends of the gapmer ASOs. (B) A representative silver-stained 4–12% SDS-PAGE gel used to resolve affinity-selected proteins. Certain protein bands identified by mass spectrometry are numbered and indicated. Proteins were isolated by competition with ASO (lane 1), ASO/RNA-like duplex (lane 2) or a non-complementary RNA-like 2′-O-methyl oligonucleotide (lane 3). The marker was pre-stained protein Benchmark (Life Technologies). (C) Western analyses of proteins isolated with either ssASO or ASO/RNA-like duplex. Isolated proteins were separated on a 4–12% SDS-PAGE, transferred to a membrane, and probed for indicated proteins by immunoblotting.
As exemplified in Figure 1B, multiple proteins were isolated with the duplex and the proteins of interest were excised and identified by mass spectrometry. The affinity selection was performed multiple times and the interesting proteins were selected mainly based on whether they were consistently observed to bind to ASO or ASO/RNA-like duplex, or whether the proteins exhibited different binding preference to PS-ASOs with different 2′-modifications, which can affect protein binding [(22,24) and our unpublished data]. Collectively, 56 proteins were identified (Table 1) that bind to either the ssASO or the ASO/RNA-like duplex (Figure 1B and data not shown). Our system enabled identification of reasonably abundant proteins that bind ASOs or ASO–RNA duplexes with moderate to high affinity. The identified proteins included 30 DNA- or RNA-binding proteins and 11 proteins of the chaperone family. The RNA-binding class of proteins included hnRNP proteins and the nuclear paraspeckle proteins P54nrb, PSF and PSPC1, which we showed recently to co-localize with ASOs and inhibit ASO activity (24). Consistent with previous findings (20), the ASO-binding protein NCL1 was co-selected with ASOs in our study. Some abundant chaperone proteins were also isolated. It is unlikely that these proteins were identified due to contamination, since we previously showed that TCP1 proteins co-localize with ASOs in nuclear PS bodies and in cytoplasmic endocytosis particles and enhance ASO activity (22). It was also previously reported that heat shock protein hspA8 binds to an ASO/RNA duplex and inhibits RNase H1-mediated cleavage (23).
Table 1. List of the identified ASO-binding proteins.
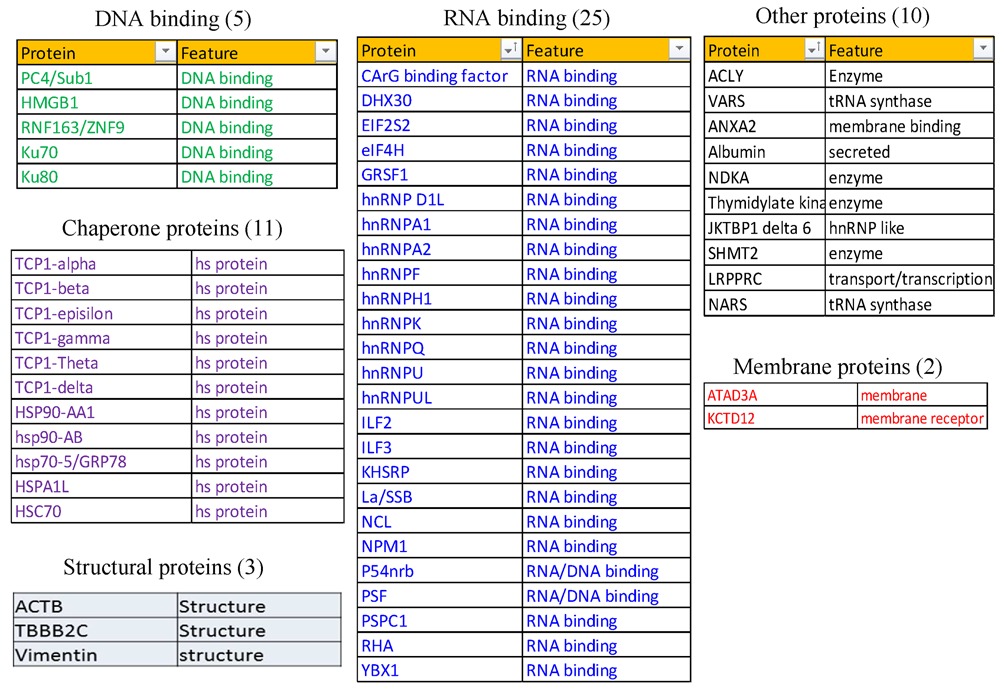
To confirm that the proteins identified by mass spectrometry are able to bind ssASO or the ASO/RNA-like duplex, isolated proteins were subjected to western analyses (Figure 1C). Consistent with silver staining results, the binding of Ku80, Ku70, PC4/Sub1 and P54nrb with either the ssASO or the ASO/RNA-like duplex was confirmed. Several of these proteins were previously shown to bind double-stranded DNA (29–31). Certain proteins, such as PSF, ATAD3A, VARS and TCP1β, were also found to be able to bind the ASO/RNA-like duplex, to different extents. However, some proteins, including La, NPM1, PSPC1, NCL1, ANXA2 and KCTD12, appeared to bind primarily to ssASO. That the binding of these proteins is relevant was supported by our finding that competition with PO oligonucleotides carrying 2′-O-methyl modification did not significantly elute Ku70, TCP1 and P54nrb proteins from the captured PS-ASO [(22,24) and data not shown]. Together, these results confirmed the binding of these proteins to PS-ASOs and indicate that these proteins can have different affinities to ssASO or ASO/RNA-like duplex.
La or NPM1 enhances ASO activity
Protein binding to ASOs may impact the ability of the ASO to inhibit gene expression. We recently showed that reduction in the levels of three different subunits of the TCP1 complex reduced antisense activity, whereas reduction of paraspeckle proteins P54nrb, PSF and PSPC1 increased ASO activity (22,24). Using similar approaches, we analyzed the effects of reducing the levels of La and NPM1 on ASO activity. La and NPM1 are abundant nuclear RNA-binding proteins that function in maturation of polymerase III transcripts and ribosome biogenesis, respectively (32,33). HeLa cells were treated for 24 h with siRNAs specific to La or NPM1. As controls, cells were treated with an siRNA targeting the mRNA encoding another ASO-binding protein NCL1, which was previously shown to have no effect on RNaseH-mediated ASO activity (22), or with an inactive siRNA with sequence complementary to U16 snoRNA (34). As expected, the levels of the targeted mRNAs and proteins were dramatically and specifically reduced by the corresponding siRNAs as determined by qRT-PCR (Figure 2A) and western analyses (Figure 2B), respectively. To ascertain the effects of protein reduction on ASO activity, control and siRNA-treated cells were transfected for 4 h with ASOs specific to four different RNAs, and levels of the ASO-targeted RNAs were analyzed by qRT-PCR (Figure 2C–E). In cells depleted of La or NPM1, the antisense activity of an ASO targeting PTEN mRNA was reduced, as evidenced by less reduction of the PTEN mRNA especially at lower doses (<10 nM) of ASO (Figure 2C) when compared with mock-transfected control cells (UTC). Consistent with our previous studies (22), treatment with NCL1- or U16-targeted siRNAs had no significant effect on the activity of the PTEN ASO. Similar observations were made for an ASO targeting drosha mRNA (Figure 2D): reduced ASO activity was observed in cells depleted of La or NPM1 but not in NCL1- or U16-siRNA-treated cells. Although the protein effects on ASO activity were modest, the difference was statistically significant. Decreased activity was also found in cells treated with La- and NPM1-siRNAs for ASOs targeting NCL1 mRNA (Figure 2E) and U16 snoRNA (Figure 2F), suggesting that La and NPM1 proteins enhance ASO activity independently of ASO sequence or targeted RNA.
Figure 2.
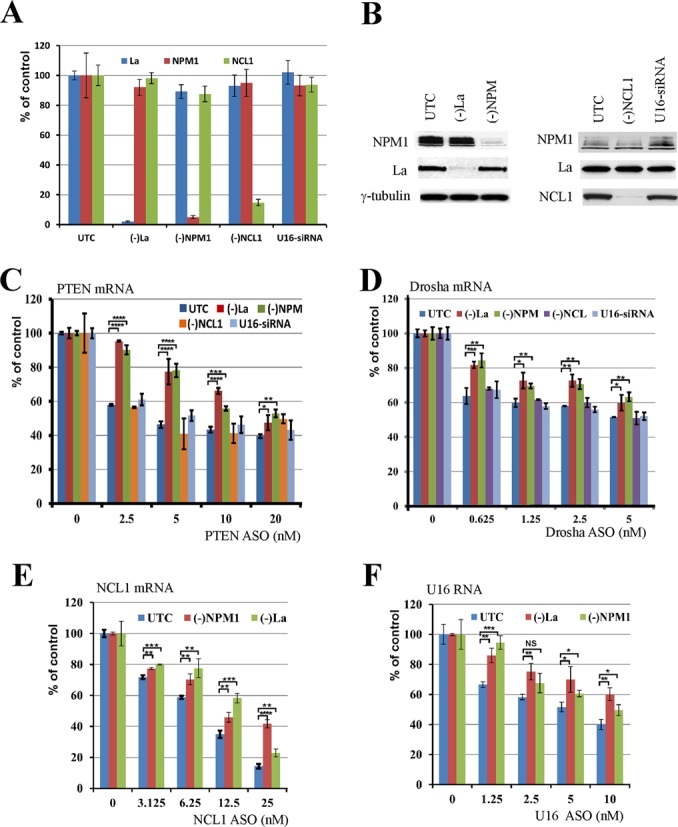
La and NPM1 enhance ASO activity. (A) Levels of mRNAs encoding La and NPM1 were reduced by siRNA treatment for 48 h, as determined by qRT-PCR analyses. Cells treated with NCL1 siRNA or a functionally inactive U16 siRNA were used as controls, and mRNA levels were determined by qRT-PCR. (B) Reduction of La and NPM1 protein levels was confirmed by western analysis. Whole cell lysates from mock-transfected (UTC) or siRNA-treated cells were subjected to western immunoblotting. γ-tubulin served as a loading control. (C–F) Depletion of La or NPM1 protein reduced the activity of ASOs targeting PTEN, drosha, NCL1 and U16 RNAs, respectively. Control cells or cells treated with siRNAs were transfected with indicated concentrations of ASOs for 4 h, and RNA was prepared and subjected to qRT-PCR analyses using primer probe sets specific to each ASO-targeted RNA. Where shown, error bars are standard deviations of three independent experiments. P-values were calculated based on unpaired t-test (n = 3), and the significance is indicated above the bars. ‘NS’: not significant. ‘*’: 0.01<P<0.05; ‘**’: 0.001<P<0.01; ‘***’: 0.0001<P<0.001; and ‘****’: 0.00001<P<0.0001.
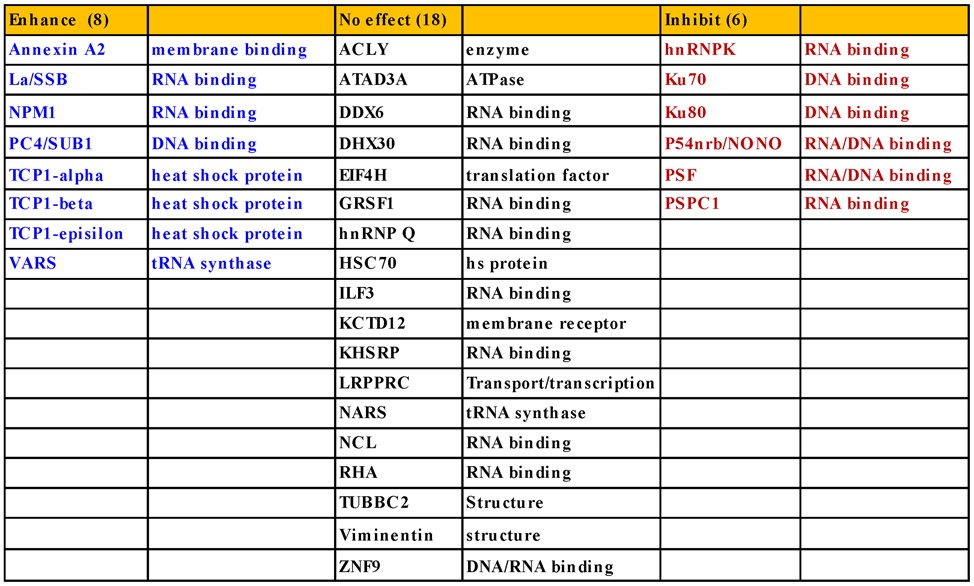
Since reductions in levels of La and NPM1 had only modest effects on ASO activity, the experiments were repeated multiple times. Similar results were observed in each replicate [data not shown and (22)], suggesting that the experimental system is reliable. Although siRNA treatment can cause unexpected effects (35), the reduced ASO activity in La- and NPM1-siRNA-treated cells appears to be specific, since treatments with NCL1-siRNA and U16-siRNA did not have significant effects on ASO-directed target reduction (Figure 2C and D). We performed similar siRNA-mediated reduction experiments for an additional 18 proteins identified in the ASO-binding assay and found no significant effect on ASO activity (Table 2). For example, siRNA-mediated reduction of ASO-binding proteins ACLY, eIF4H, KCTD12, ATAD3A and GRSF1 did not alter the activity of ASOs targeting different RNAs (Supplementary Figures S1–S4).
Table 2. Effects of proteins on ASO activity.
To further confirm that decreased ASO activity was caused by reductions in levels of La or NPM1 proteins and not experimental artifacts, La protein was reduced in HeLa cells either by treatment with an siRNA or with a La-specific ASO with a sequence different from the siRNA (Figure 3A). The siRNA and the ASO cause mRNA degradation through different pathways (i.e. RISC and RNase H1 pathways, respectively). The activity of the PTEN ASO was reduced in cells depleted of La protein, regardless of the methods used (Figure 3B). Similarly, reduction in levels of NPM1 using either siRNA or ASO treatment decreased the PTEN ASO activity (Figure 3C and D). Finally, we transfected HeLa cells with plasmids designed to over-express La or NPM1 proteins (Figure 3E) and found that over-expression of either protein led to moderately increased ASO activity, as compared with control cells transfected with an empty vector (Figure 3F and G). Together, these results confirm that La and NPM1 enhance ASO antisense activity.
Figure 3.
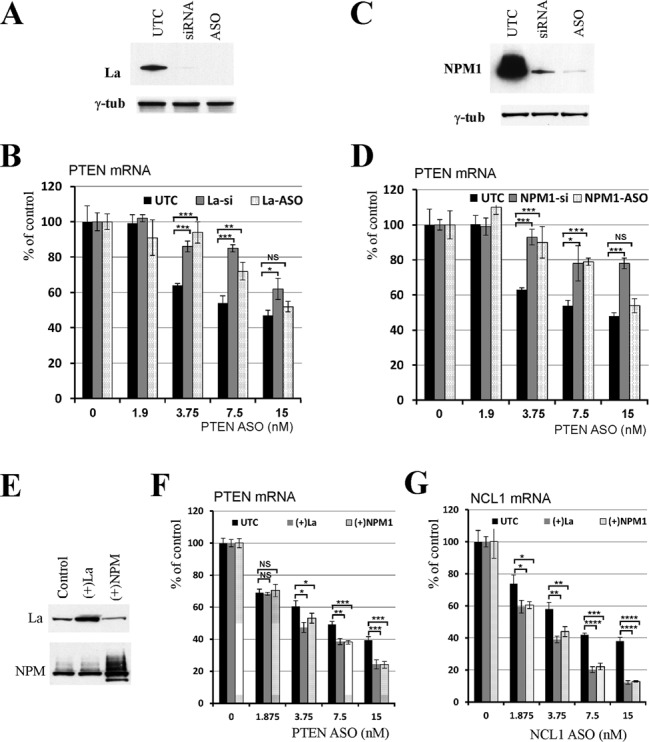
The effects on ASO activity were specific to depletion of La or NPM1. (A) Western analysis for La protein in cells treated with either siRNA or an ASO targeting La mRNA. γ-tubulin served as a loading control. UTC indicates mock-transfected cells. (B) Reduction of La levels by either siRNA or ASO caused similar effects on the activity of an ASO targeting PTEN mRNA. ASO treatment and qRT-PCR analysis were performed as described in Figure 2. (C) NPM1 protein was reduced by either an siRNA or an ASO targeting NPM1 mRNA as determined by western. γ-tubulin was used as a loading control. (D) Reduction of NPM1 protein by either siRNA or ASO decreased the activity of an ASO targeting PTEN mRNA. (E) La or NPM1 was transiently over-expressed in HeLa cells. After 48 h, protein levels were detected by western. (F, G) Over-expression of La or NPM1 protein increased the activity of ASOs targeting PTEN or NCL1 mRNAs, respectively. Where shown, the error bars are standard deviations of three independent experiments. P-values were calculated based on unpaired t-test (n = 3), and the significance is indicated above the bars. ‘NS’: not significant. ‘*’: 0.01<P<0.05; ‘**’: 0.001<P<0.01; ‘***’: 0.0001<P<0.001; and ‘****’: 0.00001<P<0.0001.
The effects of depleting La and NPM1 proteins on ASO activity do not appear to be due to alterations in the normal functions of these proteins. La is required for the maturation of polymerase III-transcribed RNAs (32); however, under the experimental conditions we used for depletion of La, the steady-state levels of polymerase III-transcribed tRNAtyr and 7SL RNA were not affected (Supplementary Figure S5A). NPM1 is involved in ribosome biogenesis and nuclear transport of RPL5 protein (36,37), and steady-state rRNA levels and the nuclear/cytoplasmic distribution of RPL5 protein were not affected by NPM1 depletion under our experiment conditions (Supplementary Figure S5B and C). Altogether, our results indicate that our experimental system is capable to identify the moderate changes in ASO activities induced by intracellular proteins and that La and NPM1 can enhance ASO activity.
Identification of additional proteins that can enhance ASO activity
Using the experimental systems described above and previously (22), we examined the effects of other ASO-binding proteins on ASO activity, as summarized in Table 2. The effects of reducing each of these proteins on ASO activity were repeated multiple times to confirm the observed phenotypes [data not shown and (22)]. Representative results are shown throughout this study. Collectively, 32 proteins have been characterized thus far. Eighteen of the tested ASO-binding proteins do not appear to impact ASO activity, as described above. However, eight proteins enhanced ASO activity. In addition to La and NPM1, and the three TCP1 subunits that we reported recently (22), ANXA2, VARS and PC4/Sub1 were also found to enhance ASO activity. ANXA2 is a phospholipid binding protein implicated in multiple cellular processes including exocytosis, endocytosis, membrane reorganization, vesicular trafficking and regulation of gene expression (38). Depletion of ANXA2 by siRNA treatment reduced the activities of tested ASOs targeting PTEN, NCL1 and U16 RNAs (Figure 4A–D). As a control, reduction of LRPPRC by siRNA treatment had no effect on ASO activity, as we reported previously (22,24). Similarly, reduction of VARS by two different siRNAs caused similar reduction of ASO activities (Figure 4E–H), suggesting that VARS can also enhance ASO activity in cells. Although VARS functions in protein synthesis, the observed effects on ASO activity do not appear to be due to impaired translation, since reduction of the asparaginyl-tRNA synthetase NARS, another ASO-binding protein, by two different siRNAs had no significant effect (Supplementary Figure S6). In addition, depletion of the transcription co-factor PC4/Sub1, which binds both ssASO and the ASO/RNA duplex (Figure 1C), also moderately reduced ASO activity (Supplementary Figure S7). Currently it is not clear how PC4/Sub1 protein enhances ASO activity, as no co-localization of this protein with ASOs was observed and reduction of PC4 did not significantly affect ASO subcellular distribution (data not shown). However, this protein may affect ASO activity in ways other than affecting ASO distribution.
Figure 4.
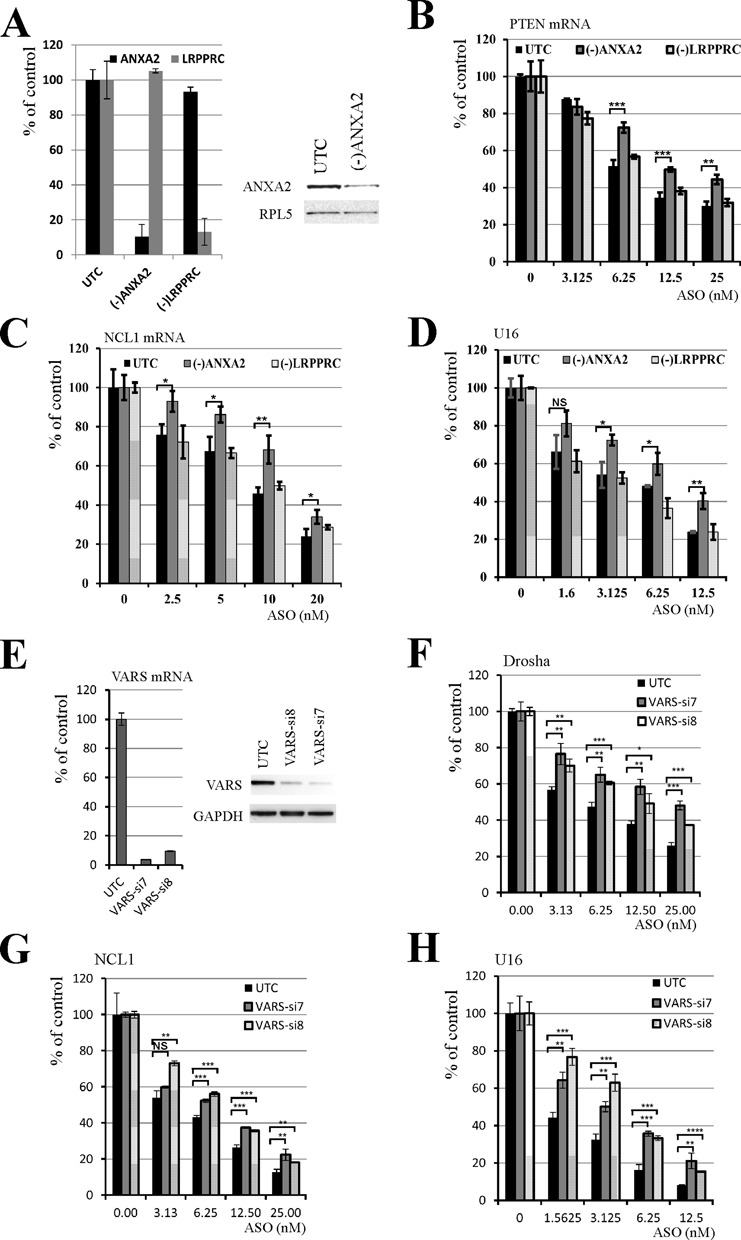
ANXA2 and VARS enhance ASO activity. (A) HeLa cells were either mock transfected (UTC) or treated with ANXA2 siRNA or a control LRPPRC siRNA for 32 h, and total RNA was prepared from an aliquot of cells and subjected to qRT-PCR analysis for ANXA2 or LRPPRC mRNA (left panel). Whole cell lysates prepared from an aliquot of cells were subjected to western analysis for ANXA2 (right panel). RPL5 served as a loading control. (B–D) Reduction of ANXA2, but not LRPPRC, decreased the activity of ASOs targeting (B) PTEN, (C) NCL1 or (D) U16 RNAs. Control or siRNA-treated cells were transfected with ASOs targeting indicated mRNAs, and the antisense activity was determined by qRT-PCR. (E) VARS expression in HeLa cells was reduced by treatment with two different siRNAs. VARS mRNA (left) and protein (right) levels were detected by qRT-PCR or western analysis, respectively. GAPDH protein served as a loading control. (F–H) Reduction of VARS protein decreased the antisense activity of ASOs targeting (F) drosha, (G) NCL1 and (H) U16 RNAs, as determined by qRT-PCR analyses. Where shown, error bars are standard deviations of three independent experiments. P-values were calculated based on unpaired t-test (n = 3), and the significance is indicated above the bars. ‘NS’: not significant. ‘*’: 0.01<P<0.05; ‘**’: 0.001<P<0.01; ‘***’: 0.0001<P<0.001; and ‘****’: 0.00001<P<0.0001.
Ku70/Ku80 proteins inhibit ASO activity
In addition to the proteins that enhance ASO activity, some proteins may inhibit ASO activity, as we reported recently for the paraspeckle proteins P54nrb and PSF (24). Indeed, we found in the current study that Ku70 and Ku80 proteins, which bind ssASOs and ASO/RNA duplexes as was the case for P54nrb and PSF (Figure 1C), also inhibited ASO activity. Ku70 and Ku80 proteins interact with each other to form a heterodimer that binds dsDNA break ends and is involved in DNA repair (29). Levels of Ku70 or Ku80 protein were reduced using specific siRNAs; depletion of either protein led to a reduction in levels of the other (Figure 5A), consistent with previous findings (39). Compared with mock-transfected control cells, depletion of Ku70/Ku80 caused moderately greater reduction of PTEN (Figure 5B) and NCL1 mRNAs (Figure 5C) targeted by specific ASOs. As a negative control, siRNA-mediated reduction of LRPPRC had no effect on ASO activity.
Figure 5.
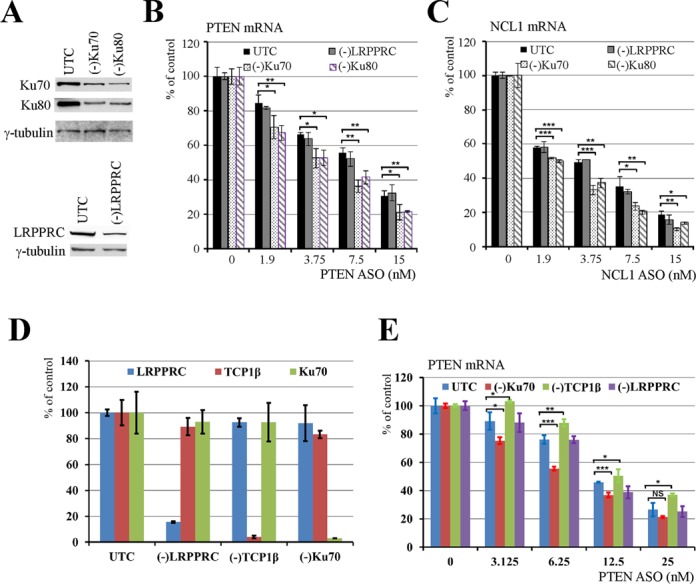
Ku70 and Ku80 inhibit ASO activity. (A) siRNA mediated reduction of Ku70 or Ku80 proteins. siRNA targeting LRPPRC was used as a negative control. Whole cell lysates from control cells or cells treated with siRNAs for 36 h were subjected to western analyses for Ku70 or Ku80 protein. γ-tubulin served as a loading control. (B, C) Reduction of Ku70 or Ku80 protein, but not LRPPRC, increased the activity of ASOs targeting (B) PTEN and (C) NCL1 mRNAs as detected by qRT-PCR. (D) Reduction of Ku70, TCP1β or LRPPRC mRNAs by siRNA treatment was demonstrated by qRT-PCR. (E) qRT-PCR analysis for ASO-directed PTEN mRNA reduction in different test cells. Where shown, the error bars are standard deviations of three independent experiments. P-values were calculated based on unpaired t-test (n = 3), and the significance is indicated above the bars. ‘NS’: not significant. ‘*’: 0.01<P<0.05; ‘**’: 0.001<P<0.01; ‘***’: 0.0001<P<0.001; and ‘****’: 0.00001<P<0.0001.
Since the effect of reducing Ku70/Ku80 on ASO activity was modest, we confirmed this observation by analyzing effects of inhibiting expression of three different proteins in the same experiment to minimize experimental variation. These proteins included Ku70, LRPPRC and TCP1β. Treatment with siRNAs resulted in specific reductions in mRNA levels (Figure 5D). Consistent with our previous observations (22), reduction of TCP1β moderately reduced ASO activity, whereas depletion of Ku70 caused the opposite effect (Figure 5E). As expected, reduction of LRPPRC had no significant effect on ASO activity. Together, our results indicate that Ku70/Ku80 complex proteins negatively affect ASO activity.
The effects of proteins on ASO activity are not unique to HeLa cells
As the identification and characterization of the ASO-binding proteins described above were performed using HeLa cells, next, we determined if these proteins could also affect ASO activity in other cell types. To this end, La, Ku70 and ANXA2 were reduced by siRNA treatment in HEK293 cells (Supplementary Figure S8A), and the effect on ASO activity was analyzed using the NCL1 ASO (Supplementary Figure S8B–D), as described above. The qRT-PCR results indicate that reduction of La or ANXA2 decreased, whereas reduction of Ku70 increased, ASO activity, as compared with the effects in control cells treated with a luciferase siRNA. These results suggest that the identified ASO-binding proteins can also affect ASO activity in other cell types.
Since ASO uptake and intracellular trafficking may utilize different pathways between transfection and free uptake, we then investigated if these proteins affect ASO activity upon free uptake. For this purpose, La, Ku70 or ANXA2-reduced HEK293 cells were washed, reseeded and incubated for 16 h with the NCL1 ASO at different concentrations without transfection reagent, as described in the Materials and Methods section. Total RNA was prepared from cells and the levels of NCL1 mRNA were measured by qRT-PCR. The results showed that, similar to transfection, the ASO activity was reduced in La- or ANXA2-depleted cells, but increased in Ku70-reduced cells (Supplementary Figure S8E). Together, these results indicate that the observed protein effects on ASO activity are not unique to a single cell type, and not unique to transfection delivery of ASOs.
The effects of proteins on ASO activity do not appear to be due to direct interactions with RNase H1
Since ASO-mediated RNA reduction results from cleavage by RNase H1 (5), it is possible that the proteins identified or the treatments with different siRNAs or ASOs may affect the level of RNase H1. This was not the case, as RNase H1 protein levels were comparable in cells treated with various siRNAs (Supplementary Figure S9A). In addition, the levels of RNase H1 and levels of ASO-binding proteins were not affected by transfection of cells with ASOs (Supplementary Figure S9B).
Next, we analyzed whether any of the proteins that impact ASO activity interact with RNase H1, directly or bridged by ASOs or the duplex. For this purpose, cell lysates were prepared from HEK293 cells over-expressing Flag-tagged RNase H1 and were incubated with an ASO or ASO/RNA-like duplex. RNase H1 and associated proteins were co-immunoprecipitated using anti-Flag beads. The co-isolated proteins were eluted by competition using Flag peptides, precipitated and analyzed by western (Supplementary Figure S9C). The Flag-tagged RNase H1 was efficiently isolated with or without ASOs. However, none of the tested ASO-binding proteins, including La, NPM1, PC4, ANXA2, VARS, Ku70 or Ku80, was co-selected with RNase H1 in the presence or absence of ASOs or the ASO/RNA-like duplex. Thus, the tested proteins do not have stable physical interactions with RNase H1, either directly or mediated by ASOs or ASO/RNA-like duplexes, although we cannot exclude the possibility that some proteins may transiently interact with RNase H1 in cells, but not detected in our experiments.
Ku80 and P54nrb compete with RNase H1 for ASO/RNA duplex binding
Since no physical interaction was observed between the ASO-binding proteins and RNase H1, these proteins may affect ASO activity through other indirect mechanisms. For example, proteins like Ku70/Ku80 and P54nrb that inhibit antisense activity may bind to the ASO/RNA duplex to reduce the accessibility of RNase H1to the substrate. To evaluate this possibility, levels of P54nrb and Ku80 were reduced in HeLa cells by siRNA treatment, and affinity selection was performed using an ASO/RNA-like duplex formed with ISIS386652 and XL279. The co-isolated proteins were determined by western analysis (Figure 6A). As expected, moderately less P54nrb and Ku80 were co-isolated with the duplex from cell lysates depleted of the corresponding proteins as compared with amounts from control cells. However, the level of co-isolated RNase H1 was moderately increased upon depletion of P54nrb or Ku80, suggesting that P54nrb and Ku80 compete with RNase H1 for binding to the duplex. This view is further supported by the observation that simultaneous depletion of P54nrb and Ku80 further increased the binding of RNase H1 to the duplex (Figure 6B and C).
Figure 6.
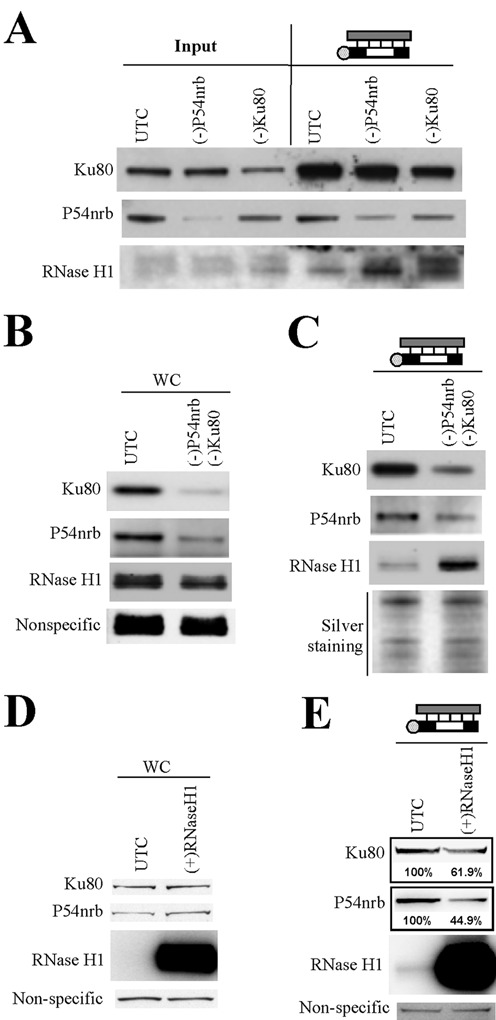
Ku80 and P54nrb compete with RNase H1 for binding to ASO/RNA duplex. (A) Reduction of P54nrb or Ku80 protein levels by siRNA treatment increased the binding of RNase H1 protein to the ASO/RNA-like duplex, as determined by affinity selection using an ASO/RNA-like duplex, followed by western analyses. (B) Simultaneous treatment with siRNAs targeting P54nrb and Ku80 reduced levels of both proteins, as shown by western analysis. (C) Simultaneous reduction of P54nrb and Ku80 led to a significant increase in the binding of RNase H1 to the ASO/RNA-like duplex. Silver staining of an aliquot of the affinity selected proteins analyzed on a separate SDS-PAGE is shown as a loading control. (D) The protein levels of RNase H1, Ku80 and P54nrb in control cells (UTC) or cells over-expressing RNase H1 were evaluated by western blot. (E) Over-expression of RNase H1 led to reduced binding of P54nrb and Ku80 proteins to the ASO/RNA-like duplex, as determined by affinity selection followed by western analyses. The numbers below the lanes indicate the estimated protein level relative to control.
Next, we determined whether over-expression of RNase H1 reduced the binding of P54nrb or Ku80 proteins to the RNA/ASO duplex. Cell lysates were prepared from control HEK293 cells and HEK293 cells over-expressing Flag-RNase H1. The over-expression of RNase H1 was confirmed by western analysis, and no significant changes in the levels of Ku80 or P54nrb were detected (Figure 6D). The endogenous RNase H1 was not detected due to its low abundance. Next, affinity selection was performed using the duplex, and co-isolated proteins were analyzed by western blot. The level of co-isolated RNase H1 was dramatically increased in cells that over-expressed RNase H1 as compared with that in control cells (Figure 6E). Moreover, reduced binding of P54nrb (∼55% reduction) and Ku80 (∼38% reduction) to the duplex was observed upon RNase H1 over-expression. Together, these results indicate that P54nrb and Ku80 compete with RNase H1 for binding to the ASO/RNA duplex and suggest that competition with RNase H1 may contribute to the inhibitory effects of P54nrb and Ku80/Ku70 proteins on ASO activity. Thus, the data on these proteins and the data on the hspA8 and hnRNP F proteins described earlier identify a general mechanism affecting RNase H1-mediated ASO activity and the type of proteins likely to be involved.
La and NPM1 affect ASO nuclear accumulation
Next, we analyzed the potential mechanisms by which some ASO-binding proteins enhance ASO activity. Previous studies have shown that ASO activity positively correlates with ASO nuclear localization upon transfection (15). We thus examined the effects of reduction of certain identified proteins on ASO subcellular distribution using confocal microscopy. Control cells or cells depleted of different ASO-binding proteins were transfected with Cy3-labeled ASOs for 4 h, and ASO localization was determined in live cells by confocal imaging. No significant difference in the nuclear levels of ASOs was detected in cells individually depleted of Ku70, Ku80, PC4, VARS or ANXA2 as compared with control cells (data not shown). However, upon depletion of NPM1, reduced nuclear ASO accumulation was observed in many cells, as compared with that in control cells, in which ASOs strongly accumulated in nuclei (Figure 7A). Note that the reduced nuclear ASO accumulation was not due to the presence of siRNAs, as siRNA-mediated depletion of NCL1 in the same way did not reduce ASO nuclear accumulation.
Figure 7.
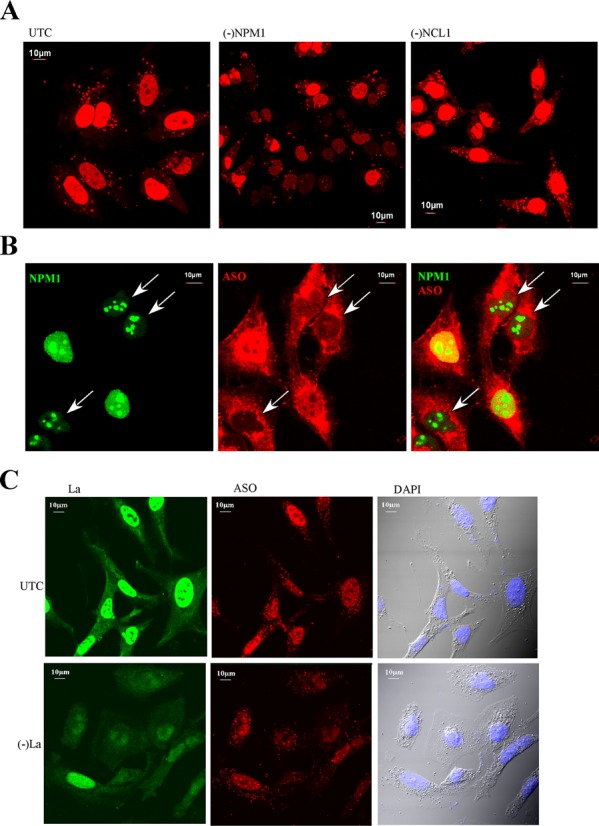
Reduction of NPM1 or La can moderately reduce nuclear ASO levels. (A) Control cells or cells depleted of NPM1 or NCL1 were transfected with 50-nM cy3-labeled ASOs (red) for 5 h, and images of live cells were taken. In many NPM1-siRNA-treated cells nuclear ASO levels were reduced as compared with control cells or NCL1-siRNA-treated cells. (B) NPM1-siRNA-treated cells and control cells were mixed, reseeded and transfected with 50-nM cy3-labeled ASO. After 5 h, cells were fixed and stained for NPM1 protein. The arrows indicate cells in which NPM1 levels are reduced. (C) Reduction of La protein (green) by treatment with La-siRNA moderately reduced the nuclear ASO level, as compared with the levels in control cells. Scale bars: 10 μm.
To rule out the possibility that the difference in nuclear ASO levels in control and NPM1-depleted cells resulted from variation in ASO transfection, control cells and NPM1-depleted cells were mixed and reseeded in the same dish, followed by transfection of Cy-3 labeled ASOs. Cells were fixed and stained for NPM1 protein. More intense ASO nuclear signal was observed in cells that exhibited strong NPM1 staining than in cells showing weaker NPM1 staining (Figure 7B). These results indicate that reduction of NPM1 protein decreased ASO nuclear accumulation. Similarly, reduction of La protein also caused a significant reduction of ASO nuclear accumulation (Figure 7C). Together, these results suggest that La and NPM1 proteins may enhance ASO activity by facilitating nuclear accumulation of ASOs.
VARS is recruited to Lamp1-containing organelles in the presence of ASOs
In similar co-staining experiments, no significant co-localization with ASOs transfected into cells was detected for La, NPM1, Ku70, Ku80 or PC4 proteins; ASOs and these proteins were evenly distributed in the nucleoplasm (data not shown). Similarly, no significant co-localization of transfected ASOs with VARS was detected in either cytoplasm or nucleus (data not shown). However, upon ASO free uptake, VARS appeared to be enriched in some cytoplasmic structures that co-localized with ASOs (Figure 8A, right panels). In cells without ASO, VARS was evenly distributed in the cytoplasm (Figure 8A, upper left panel). To determine whether these VARS-enriched structures were related to endocytosis organelles, ASO-incubated cells were co-stained for VARS and Lamp1, a lysosomal marker that is also present in some late endosomes (40). Consistently, in cells incubated with ASOs in the absence of transfection reagent, we observed aggregation of VARS in cytoplasmic structures that co-localized with Lamp1 (Figure 8B). By examining ∼300 cells, we found that 86.4% VARS-stained structures co-localized with Lamp1 and ASO. These results suggest that in the presence of ASOs VARS can localize to lysosomes/late endosomes.
Figure 8.
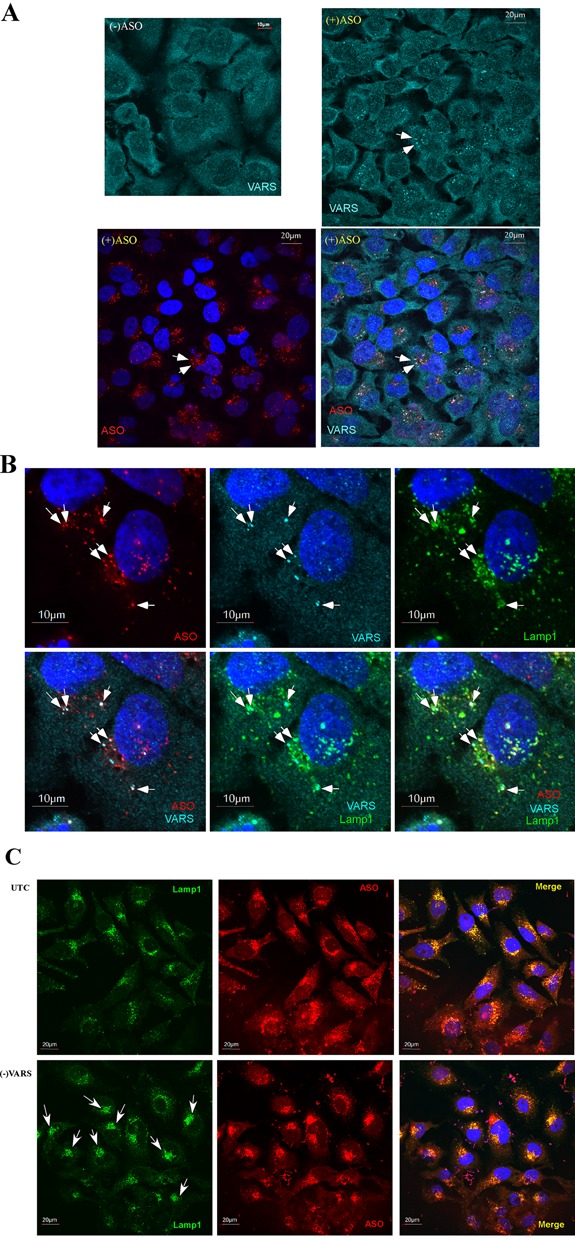
VARS can co-localize with ASOs in Lamp1-stained structures upon free uptake and can affect lysosome pattern. (A) VARS staining (cyan) in control cells [(-)ASO] or cells incubated with 2-μM cy3-labeled ASOs (red) for 24 h. Upon ASO incubation, VARS appears in aggregated dot structures that co-localize with ASOs (exemplified by arrows). Scale bars: 20 μm. (B) VARS/ASO-containing structures co-localized with Lamp1, as indicated by arrows. HeLa cells incubated with 2-μM Cy3-labeled ASOs for 24 h were fixed and co-stained for VARS and Lamp1. Scale bars: 10 μm. (C) Reduction of VARS led to more condensed ASO/lysosome localization in perinuclear structures, as indicated by arrows. Control cells or cells treated with VARS-siRNA for 24 h were incubated with 2-μM ASO for an additional 24 h, then fixed and stained for Lamp1. Scale bars: 20 μm.
We next determined whether VARS depletion affected ASO subcellular localization. HeLa cells treated with an siRNA targeting VARS or control cells were incubated with 2-μM Cy3-labeled ASO for 24 h, fixed and stained for Lamp1. Consistent with previous studies, ASOs localized in perinuclear structures that co-localized with Lamp1 in the cytoplasm of control cells (Figure 8C, upper panel), suggesting a dominant localization of ASOs in lysosomes/late endosomes upon free uptake. However, in VARS-depleted cells, ASOs localized in smaller, but condensed perinuclear structures (Figure 8C, lower panel; Supplementary Figure S10A). Similarly, Lamp1 also appeared to be present in condensed perinuclear regions that co-localized with ASOs in VARS-depleted cells, but in control cells much of the Lamp1 appeared in dot-like structures scattered throughout the cytoplasm. By analyzing about 200 cells in different images, we found that the condensed lysosome structure was observed in 42.7 ± 6.6% of VARS reduced cells, and only in ∼17.6 ± 7.9% of control cells. The condensed Lamp1 staining pattern in VARS-depleted cells was not driven by the uptake of ASOs, since a similar pattern of Lamp1 staining was observed in cells depleted of VARS using two different siRNAs but not incubated with ASOs (Supplementary Figure S10B). This effect seems to be specific to VARS reduction, since depletion of NARS did not alter the Lamp1 pattern (data not shown). Together, these results suggest that VARS may affect lysosome structure or localization and also affects ASO localization into lysosomes/endosomes upon ASO free uptake.
ANXA2 co-localizes with ASOs in late endosomes
In cells transfected with ASOs, ANXA2 co-localized with ASOs in distinct cytoplasmic structures containing Lamp1 (Supplementary Figure S10C). Since ANXA2 is required for endocytosis and for the biogenesis of late endosomes (38), this protein may be involved in ASO trafficking through the endocytosis pathway. To better understand this possibility, HeLa cells were incubated with ASOs by free uptake; under these conditions, ASOs mainly enter cells through endocytosis pathways (12). Interestingly, ANXA2 protein was observed in distinct cytoplasmic loci in the presence of ASO, whereas in cells not treated with ASO an even staining pattern was observed (Figure 9A), suggesting that ASO treatment enriched ANXA2 in these cytoplasmic loci. Indeed, ANXA2-stained loci were not obvious 2 h after ASO treatment, although at this time significant amount of ASOs was already detected in cells (Supplementary Figure S10D). However, 4 h after ASO incubation, ANXA2 was observed to co-localize with ASOs in distinct structures. These cytoplasmic ANXA2-containing structures co-localized with ASOs and also with Lamp1 (Figure 9B), indicating lysosomal and/or late endosomal localization. However, fewer ANXA2-stained loci were observed than Lamp1-stained structures, suggesting ANXA2 was only enriched in a subset of Lamp1-containing organelles. To determine whether the ANXA2-stained structures were late endosomes, we co-stained for Rab7, a marker of late endosomes. Indeed, upon ASO incubation, majority of ANXA2-stained structures co-localized with Rab7 (Figure 9C). Statistical analyses showed that 74.7 ± 5.3% ANXA2-stained structures co-localized with RAB7, whereas 98 ± 1.8% ANXA2-stained structures co-localized with LAMP1, indicating that some ANXA2 co-localized with ASOs in lysosomes. Early endosome enrichment of ANXA2 was not detected upon ASO incubation as shown by co-staining for the early endosome marker EEA1 (Supplementary Figure S11). Together, these results indicate that ASO and ANXA2 are localized to late endosomes and lysosomes, implying that ANXA2 functions in ASO intracellular trafficking.
Figure 9.
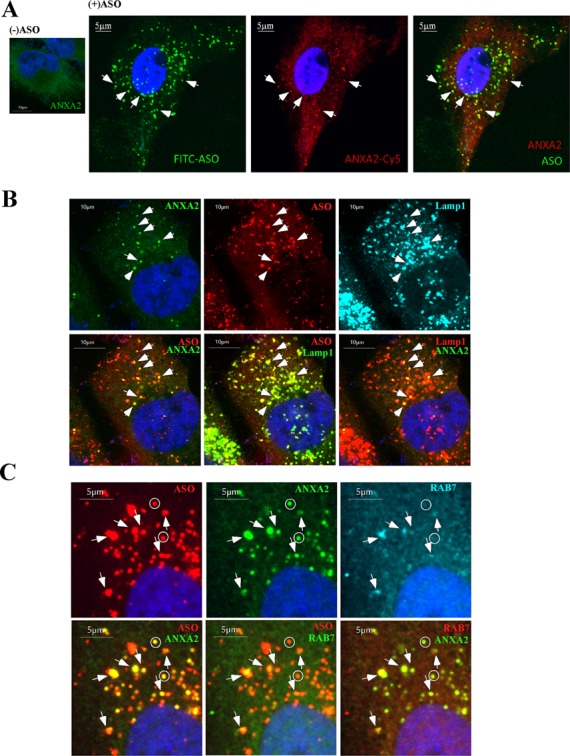
ANXA2 can co-localize with ASOs in late endosomes/lysosomes upon free uptake. (A) ANXA2 staining in control cells (left panel) or cells incubated with 2-μM FITC-labeled ASOs for 24 h (right panels). ANXA2-stained structures that co-localize with ASOs are exemplified by arrows. Scale bars: 5 μm. (B) Co-staining of ANXA2 and Lamp1 in cells incubated with 2-μM Cy3-labeled ASOs for 24 h. Arrows indicate the co-localization of ASO, ANXA2 and Lamp1. Scale bars: 10 μm. (C) Co-staining of ANXA2 and Rab7 in cells incubated with 2-μM Cy3-labeled ASOs for 24 h. Co-localization of ASO/ANXA2/Rab7 is exemplified by arrows; whereas some ANXA2/ASO containing structures that do not co-localize with Rab7 are indicated by circles. Scale bars: 5 μm.
DISCUSSION
PS ASOs bind proteins with higher affinity than do PO ASOs (20). To reduce non-specific binding and to enhance the probability of identification of proteins that are likely to impact ASO activity or localization, we developed a multi-step affinity selection assay to isolate PS ASO-binding proteins and proteins that bind ASO/RNA duplex. Using these approaches, we identified 56 intracellular proteins that bound to PS-ASOs. As our goal and the approach were to identify intracellular ASO-binding proteins, the set of proteins we identified does not include many membrane proteins, which may also play roles in ASO uptake and trafficking. Thirty-two proteins were characterized for effects on ASO activity and localization. Among these proteins, 14 exhibited effects on ASO activity. That most characterized ASO-binding proteins had no significant impact on ASO activity was not unexpected, since ASOs can bind many different proteins and the effects of reducing levels of a particular protein might be compensated by other proteins. In addition, we cannot exclude the possibility that some isolated proteins may not bind ASOs in cells as there are many differences between live cells and cell homogenates.
Effects of the characterized proteins on ASO activity were modest, with approximately doubling or halving of potencies in most cases. These changes were specific as evidenced by the following observations: (i) similar results were observed using different approaches (siRNA or RNase H-dependent ASOs for inhibition of protein expression) and using different siRNAs to target the same mRNAs, ruling out potential off-target effects; (ii) reduction of 18 ASO-binding proteins did not cause significant change in ASO activity, indicating the experimental system we used for the ASO activity assay is suitable for detecting these moderate changes; and (iii) over-expression of La or NPM1 led to opposite effects on ASO activity compared to protein depletion, confirming these observed phenotypes are specific to the tested proteins. In addition, the effects on ASO activity appeared not to be due to secondary effects related to depletion of the proteins. The ASO activity assay was performed at relatively early time points after protein depletion to reduce the potential secondary effects due to inhibition of the endogenous activities of the depleted proteins. For example, under the experimental conditions we used, reduction of La protein, which is required for the maturation of polymerase III-transcribed RNAs (e.g. tRNAs and 7SL RNA), did not cause detectable changes in the level of evaluated polymerase III transcripts. In addition, reduction of NPM1, which is involved in nucleocytoplasmic shuttling of RPL5 protein, did not change the subcellular distribution of RPL5 under our experimental conditions. Furthermore, the effects of reducing VARS on ASO activity were not due to impaired translation related to a deficiency in the charged tRNA, as reduction of NARS, another tRNA synthetase, had no detectable effect on ASO activity. It is very unlikely that the effects of proteins on ASO activity were derived from altered stability of PS-ASOs with 2′-MOE or other 2′-modifications by reduction of these proteins, since these chemically modified ASOs are very stable, with a half-life of 2–4 weeks in vivo (41). These results all argue that the influence of these proteins on ASO activity is specific, although indirect effects for certain protein cannot be completely excluded.
The moderate effect of each protein on ASO activity was not unexpected. ASOs can bind a variety of proteins, and each protein may associate with a small portion of cellular ASOs. In addition, due to the relatively high capacity of numerous ASO-binding proteins, reduction of one particular protein may lead to binding of ASOs by other cellular proteins. Thus, the effect of reduction of an individual protein could be partially compensated by many other proteins, resulting in moderate phenotypes.
One mechanism through which cellular proteins reduce ASO activity is by competition with RNase H1 for binding sites on the ASO–RNA duplex. Such competition has been observed for hnRNP F and hspA8, which were previously shown to inhibit ASO-directed RNase H cleavage (23). A similar mechanism was also observed in this study. We showed that P54nrb and Ku80 directly compete with RNase H1. We consider this as an important observation and already have reported that such mechanisms may complicate interpretation of results from ASO experiments (23). Equally importantly, our results emphasize the complexity of ASO–RNA–protein interactions and show that, once again, due to the low cellular concentration of RNase H1 (2), abundant proteins may have a competitive advantage. That RNase H1 clearly has an affinity for the RNA/ASO duplex that is similar to Ku70/Ku80 is demonstrated by the fact that over-expression of RNase H1 overcame the effect of Ku70/Ku80 for binding and on antisense activity. The screening process to identify optimal sites for ASO activity in specific RNAs would be facilitated if preferred binding sequences for proteins that compete with RNase H1 were bettered elucidated.
Proteins may enhance ASO activity through multiple mechanisms. Previously we showed that TCP1 complex proteins enhance ASO activity (22). These proteins can be recruited to cytoplasmic structures related to endocytosis, raising the possibility that these proteins may facilitate escape of ASOs from endosomes or lysosomes. Here, we showed that VARS, which enhanced ASO activity, was enriched in lysosome/endosome-related structures upon ASO incubation, suggesting that it may be involved in the release of ASOs from endocytosis structures into the cytosol. VARS is required for aminoacyl-tRNA synthesis (42), and a role in cellular uptake has not been documented. It is possible that this protein is recruited on the endosome/lysosomes when these structures are loaded with ASOs, which may cause some topological changes of the membrane structures. It is also possible that VARS may be directed by ASOs to localize in these organelles. Further investigation is needed to evaluate these possibilities. However, the effect of this protein on ASO activity appears to be specific as reduction of this protein using two different siRNAs caused similar effect on ASO activity, whereas reduction of another aminoacyl-tRNA synthase, NARS, had no detectable effects. Moreover, reduction of VARS, but not reduction of NARS, led to strong perinuclear condensation of lysosomes, suggesting that this protein may be involved, directly or indirectly, in the distribution or localization of lysosomes. Thus, the increased ASO activity upon VARS depletion may stem from altered ASO release from endosomes or lysosomes.
In addition to VARS, we also found that ANXA2 was enriched in late endosomes and lysosomes upon ASO incubation. This is not surprising, since ANXA2 is known to be involved in endocytosis and exocytosis (38). ANXA2 is a phospholipid-binding protein that is required for the biogenesis of late endosomes from early endosomes (43). Given that this protein binds negatively charged phospholipids of the membrane and that ASO release from liposomes may occur through a flip-flop mechanism (44), binding of ANXA2 to the membrane of endosomes or lysosomes may change the local conformation of the lipid bilayer to facilitate ASO release and subsequently increased ASO activity. In addition, it is also likely that ANXA2 may be involved in ASO trafficking within the endocytosis pathways, such as from early endosome to late endosomes.
Nuclear localization of ASOs is positively correlated with ASO activity after transfection (15). Thus, altering the ASO concentration in the nucleus may alter also activity. We tested the ASO localization upon free uptake in cells depleted of various proteins, no significant nuclear localization of ASOs was observed under this situation, since upon free uptake, the nuclear level of ASOs is too low to be convincingly detected by confocal imaging. Thus, judging if the nuclear ASO level is affected by reduction of these different proteins awaits more sensitive detection approaches. However, upon transfection, alteration in the nuclear ASO levels appears to be the mechanism through which La and NPM1 proteins enhance ASO activity. Reduction of La or NPM1 decreased nuclear ASO levels, but did not completely prevent nuclear localization of ASOs. A role of NPM1 in nucleocytoplasmic shuttling has already been demonstrated for some ribosomal proteins. Thus it is possible that NPM1 may also be involved in ASO trafficking, directly or indirectly via other shuttling proteins. In addition, a role of La in nuclear export has also been reported (45); it is therefore likely that this protein may contribute to ASO shuttling. However, other possibilities also exist. For example, it has been reported that La has non-canonical helicase activity (46) and can accelerate duplex formation and enhance duplex dissociation (47). Thus, La may facilitate ASO binding to mRNA substrate or unwinding of the ASO/RNA duplex after RNase H1 cleavage.
Further understanding how additional ASO-binding proteins impact ASO activity, especially the roles of these proteins on ASO intracellular trafficking/distribution, awaits further investigation. We are currently investigating the structure–activity relationships of ASO binding to various types of proteins. This information should enable us to design more effective ASOs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We wish to thank Walt Lima and Timothy Vickers for stimulating discussions.
FUNDING
ISIS Pharmaceuticals. Funding for open access charge: ISIS Pharmaceuticals.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lima W., Wu H., Crooke S.T. The RNase H Mechanism. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Applications. 2nd edn. Boca Raton, FL: CRC Press; 2008. pp. 47–74. [Google Scholar]
- 2.Crooke S.T., Vickers T.A., Lima W.F., Wu H.-J. Mechanisms of Antisense Drug Action, an Introduction. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Applications. 2nd edn. Boca Raton, FL: CRC Press; 2008. pp. 3–46. [Google Scholar]
- 3.Watts J.K., Corey D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nowotny M., Gaidamakov S.A., Crouch R.J., Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 5.Wu H., Lima W.F., Zhang H., Fan A., Sun H., Crooke S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 6.Wu H., Sun H., Liang X., Lima W.F., Crooke S.T. Human RNase H1 is associated with protein P32 and is involved in mitochondrial pre-rRNA processing. PLoS One. 2013;8:e71006. doi: 10.1371/journal.pone.0071006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett C.F., Swayze E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 8.Marcusson E.G., Bhat B., Manoharan M., Bennett C.F., Dean N.M. Phosphorothioate oligodeoxyribonucleotides dissociate from cationic lipids before entering the nucleus. Nucleic Acids Res. 1998;26:2016–2023. doi: 10.1093/nar/26.8.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koller E., Vincent T.M., Chappell A., De S., Manoharan M., Bennett C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beltinger C., Saragovi H.U., Smith R.M., LeSauteur L., Shah N., DeDionisio L., Christensen L., Raible A., Jarett L., Gewirtz A.M. Binding, uptake, and intracellular trafficking of phosphorothioate-modified oligodeoxynucleotides. J. Clin. Invest. 1995;95:1814–1823. doi: 10.1172/JCI117860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juliano R.L., Ming X., Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug. Chem. 2012;23:147–157. doi: 10.1021/bc200377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juliano R.L., Carver K., Cao C., Ming X. Receptors, endocytosis, and trafficking: the biological basis of targeted delivery of antisense and siRNA oligonucleotides. J. Drug Targ. 2013;21:27–43. doi: 10.3109/1061186X.2012.740674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein C.A., Hansen J.B., Lai J., Wu S., Voskresenskiy A., Hog A., Worm J., Hedtjarn M., Souleimanian N., Miller P., et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010;38:e3. doi: 10.1093/nar/gkp841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dias N., Stein C.A. Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- 15.Bennett C.F., Chiang M.Y., Chan H., Shoemaker J.E., Mirabelli C.K. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Mol. Pharmacol. 1992;41:1023–1033. [PubMed] [Google Scholar]
- 16.Lorenz P., Misteli T., Baker B.F., Bennett C.F., Spector D.L. Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000;28:582–592. doi: 10.1093/nar/28.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartig R., Shoeman R.L., Janetzko A., Grub S., Traub P. Active nuclear import of single-stranded oligonucleotides and their complexes with non-karyophilic macromolecules. Biol. Cell. 1998;90:407–426. [PubMed] [Google Scholar]
- 18.Lima W.F., Vickers T.A., Nichols J., Li C., Crooke S.T. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One. 2014;9:e101752. doi: 10.1371/journal.pone.0101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown D.A., Kang S.H., Gryaznov S.M., DeDionisio L., Heidenreich O., Sullivan S., Xu X., Nerenberg M.I. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994;269:26801–26805. [PubMed] [Google Scholar]
- 20.Weidner D.A., Valdez B.C., Henning D., Greenberg S., Busch H. Phosphorothioate oligonucleotides bind in a non sequence-specific manner to the nucleolar protein C23/nucleolin. FEBS Lett. 1995;366:146–150. doi: 10.1016/0014-5793(95)00517-d. [DOI] [PubMed] [Google Scholar]
- 21.Abdul-Manan N., Williams K.R. hnRNP A1 binds promiscuously to oligoribonucleotides: utilization of random and homo-oligonucleotides to discriminate sequence from base-specific binding. Nucleic Acids Res. 1996;24:4063–4070. doi: 10.1093/nar/24.20.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang X.H., Shen W., Sun H., Prakash T.P., Crooke S.T. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014;42:7819–7832. doi: 10.1093/nar/gku484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vickers T.A., Crooke S.T. Antisense oligonucleotides capable of promoting specific target mRNA reduction via competing RNase H1-dependent and independent mechanisms. PLoS One. 2014;9:e108625. doi: 10.1371/journal.pone.0108625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen W., Liang X.H., Crooke S.T. Phosphorothioate oligonucleotides can displace NEAT1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014;42:8648–8662. doi: 10.1093/nar/gku579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang X.H., Crooke S.T. RNA helicase A is not required for RISC activity. Biochim. Biophys. Acta. 2013;1829:1092–1101. doi: 10.1016/j.bbagrm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Liang X.H., Crooke S.T. Depletion of key protein components of the RISC pathway impairs pre-ribosomal RNA processing. Nucleic Acids Res. 2011;39:4875–4889. doi: 10.1093/nar/gkr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swayze E.E., Bhat B. The medicinal Chemistry of Oligonucleotides. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Applications. 2nd edn. Boca Raton, FL: CRC Press; 2008. pp. 143–182. [Google Scholar]
- 28.Yu Y.T., Shu M.D., Steitz J.A. A new method for detecting sites of 2′-O-methylation in RNA molecules. RNA. 1997;3:324–331. [PMC free article] [PubMed] [Google Scholar]
- 29.Jin S., Weaver D.T. Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO J. 1997;16:6874–6885. doi: 10.1093/emboj/16.22.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conesa C., Acker J. Sub1/PC4 a chromatin associated protein with multiple functions in transcription. RNA Biol. 2010;7:287–290. doi: 10.4161/rna.7.3.11491. [DOI] [PubMed] [Google Scholar]
- 31.Li S., Kuhne W.W., Kulharya A., Hudson F.Z., Ha K., Cao Z., Dynan W.S. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009;37:6746–6753. doi: 10.1093/nar/gkp741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolin S.L., Cedervall T. The La protein. Annu. Rev. Biochem. 2002;71:375–403. doi: 10.1146/annurev.biochem.71.090501.150003. [DOI] [PubMed] [Google Scholar]
- 33.Okuwaki M. The structure and functions of NPM1/Nucleophosmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 2008;143:441–448. doi: 10.1093/jb/mvm222. [DOI] [PubMed] [Google Scholar]
- 34.Liang X.H., Vickers T.A., Guo S., Crooke S.T. Efficient and specific knockdown of small non-coding RNAs in mammalian cells and in mice. Nucleic Acids Res. 2011;39:e13. doi: 10.1093/nar/gkq1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang X.H., Hart C.E., Crooke S.T. Transfection of siRNAs can alter miRNA levels and trigger non-specific protein degradation in mammalian cells. Biochim. Biophys. Acta. 2013;1829:455–468. doi: 10.1016/j.bbagrm.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Maggi L.B. Jr., Kuchenruether M., Dadey D.Y., Schwope R.M., Grisendi S., Townsend R.R., Pandolfi P.P., Weber J.D. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol. Cell. Biol. 2008;28:7050–7065. doi: 10.1128/MCB.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Y., Maggi L.B. Jr, Brady S.N., Apicelli A.J., Dai M.S., Lu H., Weber J.D. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol. Cell. Biol. 2006;26:3798–3809. doi: 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bharadwaj A., Bydoun M., Holloway R., Waisman D. Annexin A2 heterotetramer: structure and function. Int. J. Mol. Sci. 2013;14:6259–6305. doi: 10.3390/ijms14036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song J.Y., Lim J.W., Kim H., Morio T., Kim K.H. Oxidative stress induces nuclear loss of DNA repair proteins Ku70 and Ku80 and apoptosis in pancreatic acinar AR42J cells. J. Biol. Chem. 2003;278:36676–36687. doi: 10.1074/jbc.M303692200. [DOI] [PubMed] [Google Scholar]
- 40.Manzoni M., Monti E., Bresciani R., Bozzato A., Barlati S., Bassi M.T., Borsani G. Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett. 2004;567:219–224. doi: 10.1016/j.febslet.2004.04.080. [DOI] [PubMed] [Google Scholar]
- 41.Geary R.S., Yu R.Z., Siwkoswki A., Leivin A.A. Pharmacokinetic/Phar,acodynamic Properties of Phosphorothioate 2′-O-(2′Methoxyethyl)-Modified Antisense Oligonucleotides in Animals and Man. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Applications. Boca Raton, FL: CRC Press; 2008. pp. 305–326. [Google Scholar]
- 42.Yaniv M., Gros F. Studies on valyl-tRNA synthetase and tRNA from Escherichia coli. 3. Valyl-tRNA synthetases from thermosensitive mutants of Escherichia coli. J. Mol. Biol. 1969;44:31–45. doi: 10.1016/0022-2836(69)90403-3. [DOI] [PubMed] [Google Scholar]
- 43.Morel E., Gruenberg J. Annexin A2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J. Biol. Chem. 2009;284:1604–1611. doi: 10.1074/jbc.M806499200. [DOI] [PubMed] [Google Scholar]
- 44.Zelphati O., Szoka F.C. Jr. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bayfield M.A., Kaiser T.E., Intine R.V., Maraia R.J. Conservation of a masked nuclear export activity of La proteins and its effects on tRNA maturation. Mol. Cell. Biol. 2007;27:3303–3312. doi: 10.1128/MCB.00026-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachmann M., Pfeifer K., Schroder H.C., Muller W.E. Characterization of the autoantigen La as a nucleic acid-dependent ATPase/dATPase with melting properties. Cell. 1990;60:85–93. doi: 10.1016/0092-8674(90)90718-t. [DOI] [PubMed] [Google Scholar]
- 47.Naeeni A.R., Conte M.R., Bayfield M.A. RNA chaperone activity of human La protein is mediated by variant RNA recognition motif. J. Biol. Chem. 2012;287:5472–5482. doi: 10.1074/jbc.M111.276071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.