

Abstract
As a class of cis-regulatory elements, enhancers were first identified as the genomic regions that are able to markedly increase the transcription of genes nearly 30 years ago. Enhancers can regulate gene expression in a cell-type specific and developmental stage specific manner. Although experimental technologies have been developed to identify enhancers genome-wide, the design principle of the regulatory elements and the way they rewire the transcriptional regulatory network tempo-spatially are far from clear. At present, developing predictive methods for enhancers, particularly for the cell-type specific activity of enhancers, is central to computational biology. In this review, we survey the current computational approaches for active enhancer prediction and discuss future directions.
Keywords: Enhancer identification, Active enhancer recognition, Histone modification mark, Transcription factor
Introduction
Gene transcription is regulated by a series of accurately orchestrated interactions between transcription factors (TFs) and cis-regulatory DNA elements, e.g., promoters and enhancers [1]. Enhancers are often found in non-coding regions of a genome and generally distal to their target promoters. The first characterized enhancer was a DNA segment that markedly increased the transcription of the β-globin gene in a transgenic assay in the SV40 tumor virus genome about 30 years ago [2]. Nonetheless, global identification of enhancers and their activities remains challenging, since enhancers can activate transcription regardless of their location or orientation [3]. The development of computational enhancer recognition approaches has been greatly facilitated by the massive amount of genomic data available owing to the rapid advances in sequencing technologies in recent years. Early algorithms were developed largely based on evolutionary constraints with the assumption that highly conserved non-coding regions should have functional potential [4]. However, conservation by itself is not sufficient to confer cell-type specific enhancer activities, suggesting that additional (e.g., epigenetic) information is required for accurate prediction. Genome-wide maps of chromatin marks have been used to show that active enhancers are likely to be associated with certain characteristic chromatin signatures, e.g., monomethylation of histone H3 at lysine residue 4 (H3K4me1) [5]. But, Bonn et al. reported that H3K4me1 is distributed similarly between mesodermally active and inactive enhancers, indicating that the placement of H3K4me1 is not cell type specific during embryonic development [6]. Hitherto, to the best of our knowledge, there is no evidence that active enhancers should necessarily exhibit the same single or a combination of epigenetic marks across all the cell types [7]. Therefore, it is necessary to select optimal combinations of epigenetic marks to predict when and where an enhancer is active [8–11]. In this review, we first survey the most commonly adopted strategies in enhancer recognition and then discuss potential future directions.
The principle of enhancer recognition
Enhancers may be characterized by quantitative measures, termed features, associated with the underlying DNA sequences. In principle, an enhancer recognition algorithm utilizes informative and discriminative features as input to discriminate enhancers from non-enhancers, ideally from other non-enhancer cis-regulatory elements. Algorithms and features are both important. We therefore will discuss them separately.
Features can be briefly classified into three categories, namely comparative genomic features, TF binding related genetic features and epigenetic features (Figure 1). Comparative genomic features mainly refer to the conservation scores calculated by comparing the genome sequences of different species. The predictive power of comparative genomic features stems from the fact that functional genome regions (e.g., enhancers) are subjected to negative selection [12,13]. TF binding related genetic features use quantitative scores presumably representing the TF binding affinity at the DNA sequence of interest. The DNA binding sites of a given TF are usually determined by the DNA nucleotide sequence and the binding affinity between the TF and the DNA sequence [14–16]. It is believed that TFs are the actual operators for enhancer regulatory activities [17], which may explain why TF binding related genetic features are predictive. Direct measurement of the binding affinity between a TF and DNA sequence is not easy. However, the binding affinity can be inferred indirectly, either by experimentally measuring frequency of TF binding events, such as chromatin immunoprecipitation (ChIP) [18], or by calculating the similarity of the DNA sequences with a known TF binding motif [19,20]. The epigenetic feature mainly includes the level of histone modifications and of DNA methylation. Recent experimental evidence supports the association of several histone modifications with enhancer activity. The histone modification levels thus have served as features to predict active enhancers in humans [21,22]. Researchers also attempt to seek optimum combinations of these features for whole-genome prediction of active enhancers [5,9–11] (Table 1). Obviously, not all the aforementioned features are equally important for active enhancer prediction. The level of some dominant features showed strong correlation with enhancer activity [5,23], although the nature of the relationship between the features and enhancer states is poorly understood. Further development of superior predictive methods can not only help us to reveal such structure, but also help to improve sensitivity and specificity of the predictions.
Figure 1.
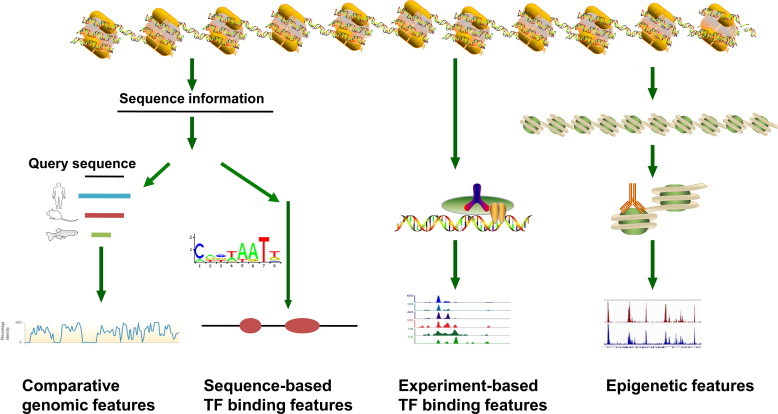
Features used in enhancer prediction algorithms The comparative genomic features are usually generated from comparison between DNA sequences in closely-related species. TF binding features result from two sources, one from known TF binding motifs and the other from ChIP experiments. Epigenetic features can be measured by various technologies. See the main text for more details.
Table 1.
Features of computational methods for enhancer prediction
| Feature | Method | Ref |
|---|---|---|
| Comparative genomic features | Aparicio’s method | [4] |
| Visel’s method (2008) | [30] | |
| Chen’s method | [8] | |
| Yip’s method | [50] | |
| Sequence-based TF binding related features | Narlikar’s method | [65] |
| Chen’s method | [8] | |
| Lee’s method | [44] | |
| Yip’s method | [50] | |
| Experiment-based TF binding related features | Visel’s method (2009) | [46] |
| Zinzen’s method | [67] | |
| May’s method | [48] | |
| Chen’s method | [8] | |
| Epigenetic features | Heintzman’s method | [5] |
| Won’s method | [11] | |
| Firpi’s method | [10] | |
| SEGWAY | [69] | |
| Kharchenko’s method | [60] | |
| He’s method | [23] | |
| Ernst’s method | [61] | |
| ChromaGenSVM | [9] | |
| Yip’s method | [50] | |
| Chen’s method | [8] | |
| Bonn’s method | [6] | |
Note: More than one type of features were employed to build enhancer recognition model in some studies. For example, Chen et al. used all four types of features to develop active enhancer recognition model [8].
Algorithms for enhancer recognition can be roughly divided into two groups. One group comprises probabilistic graphical models which describe the generative process of specific signals, such as Bayesian networks (BNs) [24] and hidden Markov models (HMMs) [25]. The other group employs discriminative filters and includes thresholds or classification boundaries in the features. This group mainly includes support vector machines (SVMs) [26] and artificial neutral networks (ANNs) [27].
The features used in enhancer recognition
Comparative genomic features
Comparative genomic features comprise conservation scores calculated from multi-species genome sequence alignment. With the completion of more vertebrate genome sequencing projects, many methods have been developed to discern slowly evolving genome regions. For example, by comparing point substitutions, insertions and deletions between humans, mice and rats, Cooper et al. comprehensively annotated slowly evolving regions in the human genome [28]. Phastcon score, another example representing the evolutionary conservation of genomic regions [29], has been employed to predict putative enhancer location [8]. In early systematic recognition of potential enhancers in fugu [4], a pair-wise identity score of Hoxb-4 between mouse and fugu was used to detect conserved sequence blocks, followed by transgenic mouse assays to measure their enhancer activities. Likewise, ultraconserved non-coding elements between humans, mice and rats were also found to be highly enriched in enhancer regions [30]. However, conservation per se is not sufficient to deduce enhancer activity in any given cell type. Moreover, several enhancers with little conservation were found carrying identical regulatory patterns in different species [31–33]. Therefore, additional information is required to predict enhancer activity in a given cell type.
Transcriptional factor binding related genetic features
Transcriptional factor binding related genetic features can be roughly classified into two groups. One group includes quantitative scores of similarity to a known TF binding motif, representing the TF binding affinity to the DNA segments (sequence-based TF binding related genetic features). The other group includes experimental measurements of TF binding frequency, which also presumably represents TF binding affinity (experiment-based TF binding related genetic features).
The sequence-based TF binding related genetic features comprise individual TF binding and the enrichment of modular combinations of TF binding. Measuring TF binding affinities is not an easy task experimentally; however, it can be approached from the nucleotide preferences at each sequence position [20], e.g., position weight matrix (PWM). PWM describes the probability of observing the respective nucleotides A, C, G, and T in each position of a sequence motif. It has been found that there is a strong correlation between PWM scores and the TF binding affinity [15,16,20]. PWMs for known TFs have been cataloged in databases [34,35]. These matrices enable people to assign a quantitative score to any sequence to evaluate the binding affinity of the specific TF at that sequence (Figure 1). In vertebrates, functional TF binding sites are usually clustered into a modular structure, which motivates researchers to seek cis-regulatory modules (CRMs) as the advanced predictive features for cis-regulatory element recognition [36,37]. The CRM features are often calculated as the likelihood of the CRM in a given genome context [38]. For example, MSCAN value measures the statistical significance of the appearance of potential combinatorial TF binding sites [39]. All the TF binding sites are represented by PWM scores and MSCAN returns the significance of the CRM. A similar strategy is adopted in MCAST [40].
To further improve the performance, additional phylogenetic footprinting is employed to align interested orthologous DNA sequences to define a conserved region and then the significance of the CRM is calculated in the regions. For example, EEL approach was used to scan a given pair of orthologous sequences to identify conserved TF binding sites, and, then EEL scores were calculated by considering both distances and differences in the angles between adjacent binding sites [41]. Another example, MorphMS, implemented a pair-HMM statistical alignment between two species [42]. A first order Markov network with three states (match, deletion and insertion) was implemented and emits two strings, one for each species. The string emitted in the match state was chosen by another probabilistic process, which models the arrangement of binding sites and non-binding (“background”) sites by PWM. Then, two log likelihood ratio (LLR) scores were reported. The two scores (LLR1 and LLR2) compare the likelihood of a sequence under the MORPH model to the likelihood of the sequences under null models. The null model used in LLR1 only considers background PWM, while the null model for LLR2 assumes that the two orthologous sequences were generated independently.
Besides the similar strategy used in MorphMS, another algorithm EMMA incorporates gains and losses at binding site, a process that is believed to be an important part of CRM evolution [43]. However, the computational cost increases exponentially with the number of TFs considered. One alternate choice for this type of sequence features is k-mer profile, which is the frequency of all possible k-mer (putative motifs with length of k) in a given sequence region [44]. The profile measures how likely the k-mers in one enhancer would be found in another independently-generated sequence. Using such k-mer features, Leung and Eisen developed a profile similarity between pairs of sequences to detect novel enhancers [45]. However, the search space is growing exponentially with k.
The sequence-based TF binding related genetic features alone are not sufficient for active enhancer recognition. First, most of the features are conserved TF binding sites, while many enhancer elements are not conserved. For example, in Drosophila, the cone-specific Pax2 enhancer carries barely-conserved TF binding sites, which have been shown to possess similar enhancer functions in transgenic assays [31]. Similarly, a large proportion of a 40 kb region in the Phox2b locus showed regulatory activity by transgenic assay in zebrafish, while only 29–61% identified regulatory sequences were conserved [32]. Second, in any given tissue, only a subset of enhancers is active. This tissue-specific activity may result from a tissue-specific combination of binding TFs or from regulation at the epigenetic level.
TF binding in given tissues or cell types can be experimentally measured, which gives experiment-based TF binding related genetic features. For example, data from chromatin immunoprecipitation followed by massively parallel DNA sequencing (ChIP-seq) technology precisely provide binding loci for the TFs under the given conditions [18]. Visel et al. mapped the genome-wide occupancy of p300 in three cell lines by ChIP-seq. Using transgenic mouse assay, they show that p300 binding sites are predictive for enhancer activity in the cell types examined [46]. Similarly, CREBBP-bound enhancers also show environment-dependent activity in neurons [47], or in transgenic mouse enhancer assays [48]. Recently, ENCODE project has generated high-throughput sequencing (ChIP-seq or ChIP-chip) data sets for 119 distinct transcription factors over five main cell lines [49]. These experimental results have been used for enhancer recognition [50].
Epigenetic features
Epigenetic features consist of chromatin structure, histone modifications, DNA-methylation levels and non-coding RNAs. In this review, we mainly focus on the first two types of epigenetic features, since other features have been reviewed elsewhere (such as [51]). Chromatin structure controls DNA accessibility of TFs to enhancer or other regulatory elements. DNA accessibility can be inferred as DNase I hypersensitivity [52,53] or by Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) technology [54]. The regions detected by DNase I or FAIRE are associated with all known classes of active DNA regulatory elements, including enhancers [55]. For example, Wiench et al. found that CpG methylation at glucocorticoid receptor (GR) -associated DNase I hypersensitive sites was a cell type-specific event and suggested that these sites could be a unique class of active enhancers [56]. Comparing DNase I-seq and FAIRE-seq data in seven human cell types indicated that data from these two assays were not fully overlapping [57]. DNase I tended to find the regions around transcriptional start sites, while FAIRE was more sensitive in detecting distal regulatory elements. Notably, neither DNase I nor FAIRE hypersensitive sites detected in one cell type are sufficient to demonstrate their enhancer state, as many other regulatory element sites, such as repressors or insulators, are also DNase I or FAIRE hypersensitive [57]. Therefore, DNase I or FAIRE hypersensitivity data should be regarded as a necessary but not sufficient input for active enhancer prediction.
In addition to DNA accessibility, the presence of characteristic histone modifications is another sign for the activity of enhancers, e.g., elevated H3K4 monomethylation (H3K4me1) levels and depleted H3K4 trimethylation (H3K4me3) levels have been correlated with enhancer activity [5]. Further experiments showed that active enhancers marked by H3K4me1 in ES cells are also flanked by H3K27 acetylation (H3K27ac), while regions marked by H3K27 trimethylation (H3K27me3) are associated with early developmental genes which are poised in ES cells [58,59]. In another study, however, Bonn et al. found that H3K4me1 was distributed similarly between mesodermally active and inactive enhancers, indicating that the placement of H3K4me1 is not completely cell type specific during embryonic development [6]. Instead, they found a conditional link between the presence of H3K79me3, H3K27ac marks and enhancer activity.
Although the histone modification patterns mentioned above showed promising potential for enhancer activity prediction in certain cell types, the general pattern of histone modifications for prediction still remains elusive. In human CD4+ T cells, 39 histone modification types have been mapped and several histone mark combinations showed correlation with enhancers, yet no single mark is associated with more than 40% of enhancers [7]. Integrating more epigenetic marks may render a more reliable, robust and precise model to capture active enhancers. Several attempts have been made [5,9–11]. One such attempt employed 10-fold cross-validation for all possible combinations of six histone modification marks to predict p300 binding sites, and found that enrichment of H3K4me1 and depletion of H3K4me3 is the most predictive combination for p300 binding [5]. Many more sophisticated computational technologies have also been applied to search for optimal combinations for active enhancers. For example, Won et al. coupled HMM with simulated annealing to search for the most informative combination of histone modification marks [11]. In Drosophila, Kharchenko and coworkers found that active enhancers lack H3K4me3 and are enriched for H3K4me1, H3K27ac and H3K18ac [60]. Similarly, ChromHMM labeled active enhancers with the H3K4me1, H3K4me2 and H3K27ac signature [61]. In a vast collection of epigenetic marks (20 histone methylations and 18 histone acetylations), genetic algorithms indicated that the most predictive histone modification signals within enhancers are H3K4me1 and H3K4me3 [9]. A similar pattern was also extracted from nearly 40 ENCODE histone modifications by using fisher discriminate analysis [10].
The features we discussed above can also be roughly classified into two classes, based on the prediction power for enhancer activity. One class of features represents the potential of a locus to be an enhancer, e.g., comparative genomic features or sequence-based TF binding related genetic features, because the features describe the static DNA sequence characteristics which are shared by almost all cells in an organism. The other class of features, e.g., experiment-based TF binding related genetic features or epigenetic features, further indicates enhancer activity of the loci in a given tissue or cell type. These features are the actual measurement of cellular or molecular activities that had already been associated with enhancer activity in living cells. For example, when Visel and colleagues compared the evolutionary conservation score and p300 binding sites, they found that only 47% (246 out of 528) of conserved enhancer candidates were active in a transgenic mouse assay, whereas 87.7% of p300 binding sites were reproducibly active in the same transgenic assay [46]. Another study employed chromatin signatures of H3K4me1, H3K4me3 and H3K27ac to recognize active enhancers in 19 mouse cell lines. By comparing predicted enhancers with 726 experimentally validated enhancers, they found that 82% of predicted enhancers were correctly identified [62]. Androgen receptor binds primarily to active enhancers in human prostate cancer cells [63]. Interestingly, He et al. found that the H3K4me2 signal was detected in the known androgen receptor binding sites [23]. At present, although some features showed strong preference in the putative enhancer regions, and some other features showed association with enhancer activity, the relationship between features and enhancer activity is complicated, and sophistic models are still essential to achieve sensitive and specific active enhancer prediction.
Model building
The general process of enhancer prediction is summarized in Figure 2, and the commonly used methods are listed in Table 2. The simplest method to differentiate active enhancers from background is to look for the presence of characteristic features. For example, p300 ChIP-seq data were used to determine p300-enriched regions, which were considered as putative active enhancers. Of the 122 tested p300 binding elements in mouse, 107 (87.7%) showed reproducible enhancer activity [46]. Heintzman et al. exhaustively searched all combinations of six different histone modification marks, and identified the optimal combinations of H3K4me1 and H3K4me3 [5]. Despite the fine performance of this simple model, the best predictive power in one dataset does not guarantee its performance in another. Moreover, an ever increasing number of features would challenge these simple methods. This is not only because of the inter-correlations between the features, but also because of the difficulties in interpreting the relative importance of each feature. A class of computational technology, named feature selection, has been applied to solve such problems [64]. For example, Narlikar et al. built a linear regression model to identify active enhancers in heart based on 727 sequence features including 721 TF binding related genetic features [65]. The LASSO linear regression method was then applied to find features relevant to enhancer activity and 45 features were assigned nonzero weights. The accuracy of 92% was achieved in distinguishing heart enhancers from a large pool of random noncoding sequences.
Figure 2.
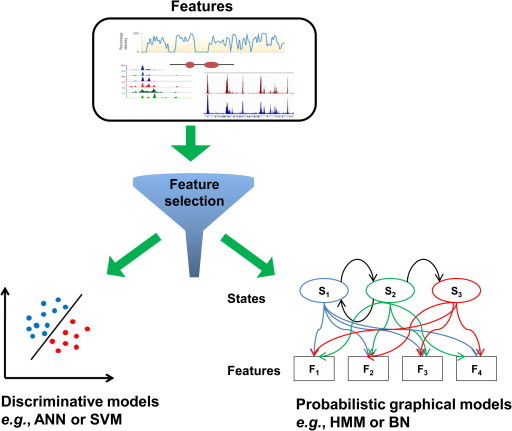
Flow scheme of model building To improve model interpretability and reduce overfitting, sophisticated computational strategies implement feature selection algorithm to select a subset of relevant features for model building. Then, appropriate classification model is employed to differentiate active enhancers from non-enhancers. Generally, there are two major classification models. The first is the discriminative models which find the optimal classification border in the feature space (lower left panel). The other one is the probabilistic graphical models that try to model the joint distribution of states and associated features with graph (lower right panel). ANN, artificial neutral network; BN, Bayesian network; HMM, hidden Markov model; SVM, support vector machine.
Table 2.
Model building strategies and performance of enhancer prediction methods
| Category | Method | Operational model | Positive predictive value (%) | Note | Ref |
|---|---|---|---|---|---|
| Discriminative model | Heintzman’s method | Thresholds of histone modification profiles | 39.5 | Mapped to distal p300 binding sites in HeLa cells | [5] |
| Visel’s method (2009) | Thresholds of p300 binding profiles | 87.7 | With reproducible enhancer activity in transgenic mouse | [46] | |
| Narlikar’s method | Linear regression | 62 | With reproducible enhancer activity in vivo in mouse and zebrafish | [65] | |
| Zinzen’s method | Support vector machine | 71.4 | With reproducible enhancer activity in transgenic Drosophila | [67] | |
| Firpi’s method | Time-delay neural network | 66.3 | Overlapped with p300 binding sites, Dnase I hypersensitivity sites or TRAP220 binding sites in HeLa cells | [10] | |
| Lee’s method | Support vector machine | 74.5 | Overlapped with Dnase I hypersensitive enhancers in embryonic mouse whole brain cells | [44] | |
| ChromaGenSVM | Support vector machine | 57 | Overlapped with p300 binding sites, Dnase I hypersensitivity sites or TRAP220 binding sites in HeLa cells | [9] | |
| Probabilistic graphical model | Won’s method | Hidden Markov model | 54.8 | Overlapped with p300 binding sites, Dnase I hypersensitivity sites or TRAP220 binding sites in HeLa cells | [11] |
| Bonn’s method | Bayesian network | 78 | Overlapped with previously identified TF binding sites in Drosophila | [6] | |
| Other | Chen’s method | Multinomial logistic | 83 | Overlapped with at least one TF peak from 7 mouse embryonic stem cell ChIP-seq datasets | [8] |
| Yip’s method | Random forest | 67 | With enhancer activity in vivo in mouse and medaka fish (28/42) | [50] | |
Note: The performance shown here is the reported performance compared to experimental results. The positive predictive value (percentage) was calculated as follows: positive predictive value = true positive/(true positive + false positive).
Recently, more sophisticated methods have been implemented to find the optimal classification border in the feature space. Typical methods include ANNs and SVMs. A neural network is a parallel system, capable of resolving paradigms that linear computing cannot [27]. A case concerning enhancer recognition is a time-delay neural network (TDNN) which combines 39 histone modifications [10]. In an independent test, 66.3% of the putative regions identified by this model overlapped with experimentally supported enhancers [10]. A SVM performs classification by seeking a hyperplane in high dimensional labeled feature space that optimally separates the data into two categories regarding the classification labels [66]. A SVM model has been applied to ChIP-seq data of five different TFs and 77% of all known muscle-specific enhancers in Drosophila have been correctly predicted [67]. In addition, using ChromaGenSVM, which was based on five histone modification marks, 57.0% of identified potential enhancers overlapped with experimentally supported enhancers in the pilot ENCODE region in HeLa cells [9]. In fact, SVM models are closely related to ANNs. SVMs are alternative training methods for multi-layer perception classifiers, in which the weight of the network is found by solving a quadratic programming problem with linear constrains, rather than by solving an unconstrained minimization problem in ANNs [26]. A comparison in HeLa cells between ChromaGenSVM and TDNN showed that ChromaGenSVM recovered 70.2% of the p300-bound putative active enhancers, while TDNN achieved a precision of 84.0% [9]. However, due to different feature sets used by these two models, these data do not necessarily indicate that SVM is more effective than ANN for active enhancer recognition.
Another type of approaches try to model the joint distribution of states and associated features with graph, generally termed as probabilistic graphical models. The naïve Bayes classifier (NBc) is the simplest one of this type [68]. For enhancer recognition, NBc learns the conditional probability of each feature related to enhancer activity from a training data. For example, a NBc on 6-mer features has been trained to detect active enhancers in the mouse genome [44]. However, compared with a SVM model with the same feature set [area under receiver operating characteristic curve (AUC) > 0.9], the NBc preformed significantly less accurately in discriminating active enhancers from random sequences (AUC < 0.79). HMM is another example in probabilistic graphical models. The current model of the genome is a linear combination of stated DNA sequences, e.g., ‘promoter’, ‘enhancer’ or ‘coding region’. By assuming that the state of any locus is only dependent on its nearest neighbor, HMM provides a natural solution for the task of segmenting the stated DNA sequences [25]. For example, Kharchenko and coworkers used a HMM to identify the prevalent combinatorial pattern of 18 histone modifications and captured the overall complexity of chromatin profiles observed in Drosophila S2 and BG3 cells with 9 states [60]. They found that enhancer regions are always enriched with H3K4me1, H3K27ac and H3K18ac. A similar strategy was implemented in ChromHMM, which mapped 15 chromatin states in nine human cell lines [61]. BN represents another probabilistic graphical model that allows effective representation of the joint probability distribution over feature set [24]. BN provides a powerful framework for modeling the complicated hidden relationships that explain the observed chromatin patterns in a genome. For instance, SEGWAY used BN techniques to simultaneously segment and cluster 1% of the human genome with 31 ENCODE signal tracks including histone modifications, TF binding and open chromatin, and revealed active enhancer associated patterns at nucleosomal resolution [69]. BN has also been applied to predict active enhancers in Drosophila [6], and the trained BN identified a conditional link between the H3K79me3 and H3K27ac marks and enhancer activity. This BN model achieved better performance (AUC = 0.82), compared to the aforementioned NBc model.
Conclusion and outlook
Enhancers are regulatory DNA elements that can activate transcription largely independent of their location or orientation. Often, enhancers regulate gene expression in a tissue-specific manner and play important roles in cell differentiation [17]. In this review, we have described the general computational strategies for enhancer prediction. It has been suggested that H3K4me1 and p300 binding signatures are the most predictive features for active enhancer recognition [5,46], however, this notion may be disputed by new data. For example, a recent study found that H3K79me3 and H3K27ac, instead of H3K4me1, are predictive for cell type specific enhancer activity during embryonic development [6]. Recently, a more complicated picture, which involves nuclear organization, chromatin structure and non-coding RNAs, is emerging for enhancer activation. Accumulating data suggested that the insulators are critical in the regulation of enhancer–promoter interaction which is believed to be accomplished by long-range inter- or intra-chromosomal chromatin interactions [70].
From the perspective of computational biology, the field of enhancer research is now moving toward the modeling of 3D chromatin structure in nuclei, to reveal the principle of enhancer-promoter interactions. Polymer models are valuable tools in 3D chromatin structure study, e.g., the dynamic random loop model [71] and the fractal globular model [72]. To understand enhancers in the context of gene regulatory networks, it is necessary to integrate data from ultra- heterogeneous data sources in this “big data” era. For example, enhancer transcribed RNAs (eRNAs) were recently found prevalent at enhancer loci [47]. Some of such non-coding RNAs even act like enhancers [73]. Therefore, the integration of RNA-seq data is essential for a model which aims to understand eRNA associated enhancer activity.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgements
We apologize to many authors whose important works could not be cited owing to space limitations or our ignorance. This work was supported by grants from the National Natural Science Foundation of China (NSFC, Grant No. 31271398 and 91131012) and 100 Talents Project to ZZ, NSFC (Grant No. 91019016) and National Basic Research Program of China (NBRPC, Grant No. 2012CB316503) to MQZ.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

References
- 1.Carey M., Smale S.T., Peterson C.L., editors. Transcriptional regulation in eukaryotes: concepts, strategies, and techniques. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- 2.Banerji J., Rusconi S., Schaffner W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 3.Li G., Ruan X., Auerbach Raymond K., Sandhu Kuljeet S., Zheng M., Wang P. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aparicio S., Morrison A., Gould A., Gilthorpe J., Chaudhuri C., Rigby P. Detecting conserved regulatory elements with the model genome of the Japanese puffer fish, Fugu rubripes. Proc Natl Acad Sci U S A. 1995;92:1684–1688. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 6.Bonn S., Zinzen R.P., Girardot C., Gustafson E.H., Perez-Gonzalez A., Delhomme N. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44:148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z.B., Zang C.Z., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C.Y., Morris Q., Mitchell J. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 2012;13:152. doi: 10.1186/1471-2164-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez M., Miranda-Saavedra D. Genome-wide enhancer prediction from epigenetic signatures using genetic algorithm-optimized support vector machines. Nucleic Acids Res. 2012;40:e77. doi: 10.1093/nar/gks149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Firpi H.A., Ucar D., Tan K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics. 2010;26:1579–1586. doi: 10.1093/bioinformatics/btq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Won K.J., Chepelev I., Ren B., Wang W. Prediction of regulatory elements in mammalian genomes using chromatin signatures. BMC Bioinformatics. 2008;9:547. doi: 10.1186/1471-2105-9-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nobrega M.A., Ovcharenko I., Afzal V., Rubin E.M. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 13.Pennacchio L.A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 14.Stormo G.D., Fields D.S. Specificity, free energy and information content in protein–DNA interactions. Trends Biochem Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 15.Stormo G.D., Zhao Y. Determining the specificity of protein–DNA interactions. Nat Rev Genet. 2010;11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y., Stormo G.D. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat Biotechnol. 2011;29:480–483. doi: 10.1038/nbt.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ong C.T., Corces V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collas P. The current state of chromatin immunoprecipitation. Mol Biotechnol. 2010;45:87–100. doi: 10.1007/s12033-009-9239-8. [DOI] [PubMed] [Google Scholar]
- 19.Stormo GD. An introduction to recognizing functional domains. Curr Protoc Bioinformatics 2002. http://dx.doi.org/10.1002/0471250953.bi0201s00. [DOI] [PubMed]
- 20.Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 21.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z.B. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Jin C.Y., Zang C.Z., Wei G., Cui K.R., Peng W.Q., Zhao K.J. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He H.H., Meyer C.A., Shin H., Bailey S.T., Wei G., Wang Q. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen T.D., Jensen F.V., editors. Bayesian networks and decision graphs. Springer-Verlag; New York: 2007. [Google Scholar]
- 25.Rabiner L.R. A tutorial on hidden markov models and selected applications in speech recognition. Proc IEEE. 1989;77:257–286. [Google Scholar]
- 26.Vapnik V., Golowich S.E., Smola A., editors. Support vector method for function approximation, regression estimation, and signal processing. MIT Press; Cambridge, MA: 1997. [Google Scholar]
- 27.Haykin S.O., editor. Neural networks: a comprehensive foundation. Prentice Hall; Upper Saddle River: 1998. [Google Scholar]
- 28.Cooper G.M., Brudno M., Stone E.A., Dubchak I., Batzoglou S., Sidow A. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 2004;14:539–548. doi: 10.1101/gr.2034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Visel A., Prabhakar S., Akiyama J.A., Shoukry M., Lewis K.D., Holt A. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson C.I., Evans N.C., Barolo S. Structural rules and complex regulatory circuitry constrain expression of a notch- and EGFR-regulated eye enhancer. Dev Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGaughey D.M., Vinton R.M., Huynh J., Al-Saif A., Beer M.A., McCallion A.S. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. 2008;18:252–260. doi: 10.1101/gr.6929408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hare E.E., Peterson B.K., Iyer V.N., Meier R., Eisen M.B. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008;4:e1000106. doi: 10.1371/journal.pgen.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matys V. TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2009;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman B.P., Nibu Y., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schones D., Smith A., Zhang M. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. doi: 10.1186/1471-2105-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wasserman W.W., Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 39.Johansson Ö., Alkema W., Wasserman W.W., Lagergren J. Identification of functional clusters of transcription factor binding motifs in genome sequences: the MSCAN algorithm. Bioinformatics. 2003;19:i169–i176. doi: 10.1093/bioinformatics/btg1021. [DOI] [PubMed] [Google Scholar]
- 40.Bailey T.L., Noble W.S. Searching for statistically significant regulatory modules. Bioinformatics. 2003;19:ii16–ii25. doi: 10.1093/bioinformatics/btg1054. [DOI] [PubMed] [Google Scholar]
- 41.Hallikas O., Palin K., Sinjushina N., Rautiainen R., Partanen J., Ukkonen E. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 42.Sinha S., He X. MORPH: probabilistic alignment combined with hidden markov models of cis-regulatory modules. PLoS Comput Biol. 2007;3:e216. doi: 10.1371/journal.pcbi.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He X., Ling X., Sinha S. Alignment and prediction of cis-regulatory modules based on a probabilistic model of evolution. PLoS Comput Biol. 2009;5:e1000299. doi: 10.1371/journal.pcbi.1000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee D., Karchin R., Beer M.A. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011;21:2167–2180. doi: 10.1101/gr.121905.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leung G., Eisen M.B. Identifying cis-regulatory sequences by word profile similarity. PLoS One. 2009;4:e6901. doi: 10.1371/journal.pone.0006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim T.K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.May D., Blow M.J., Kaplan T., McCulley D.J., Jensen B.C., Akiyama J.A. Large-scale discovery of enhancers from human heart tissue. Nat Genet. 2012;44:89–93. doi: 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yip K.Y., Cheng C., Bhardwaj N., Brown J.B., Leng J., Kundaje A. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orom U.A., Shiekhattar R. Long non-coding RNAs and enhancers. Curr Opin Genet Dev. 2011;21:194–198. doi: 10.1016/j.gde.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keene M.A., Corces V., Lowenhaupt K., Elgin S.C. DNase I hypersensitive sites in Drosophila chromatin occur at the 5′ ends of regions of transcription. Proc Natl Acad Sci U S A. 1981;78:143–146. doi: 10.1073/pnas.78.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGhee J.D., Wood W.I., Dolan M., Engel J.D., Felsenfeld G. A 200 base pair region at the 5′ end of the chicken adult β-globin gene is accessible to nuclease digestion. Cell. 1981;27:45–55. doi: 10.1016/0092-8674(81)90359-7. [DOI] [PubMed] [Google Scholar]
- 54.Giresi P.G., Kim J., McDaniell R.M., Iyer V.R., Lieb J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xi H., Shulha H.P., Lin J.M., Vales T.R., Fu Y., Bodine D.M. Identification and characterization of cell type – specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 2007;3:e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiench M., John S., Baek S., Johnson T.A., Sung M.-H., Escobar T. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song L., Zhang Z.C., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.K. Open chromatin defined by Dnase I and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kharchenko P.V., Alekseyenko A.A., Schwartz Y.B., Minoda A., Riddle N.C., Ernst J. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia L., Kim J., Shen H., Clark P.E., Tilley W.D., Coetzee G.A. Androgen receptor activity at the prostate specific antigen locus: steroidal and non-steroidal mechanisms. Mol Cancer Res. 2003;1:385–392. [PubMed] [Google Scholar]
- 64.Saeys Y., Inza I., Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23:2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 65.Narlikar L., Sakabe N.J., Blanski A.A., Arimura F.E., Westlund J.M., Nobrega M.A. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vapnik V., editor. The nature of statistical learning theory. Springer; New York: 1995. [Google Scholar]
- 67.Zinzen R.P., Girardot C., Gagneur J., Braun M., Furlong E.E.M. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 68.Domingos P., Pazzani M. On the optimality of the simple bayesian classifier under zero-one loss. Mach Learn. 1997;29:103–130. [Google Scholar]
- 69.Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., Noble W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mateos-Langerak J., Bohn M., de Leeuw W., Giromus O., Manders E.M.M., Verschure P.J. Spatially confined folding of chromatin in the interphase nucleus. Proc Natl Acad Sci U S A. 2009;106:3812–3817. doi: 10.1073/pnas.0809501106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mirny L.A. The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 2011;19:37–51. doi: 10.1007/s10577-010-9177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ørom U.A., Derrien T., Beringer M., Gumireddy K., Gardini A., Bussotti G. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]