

Abstract
Protein–peptide interactions, where one partner is a globular protein (domain) and the other is a flexible linear peptide, are key components of cellular processes predominantly in signaling and regulatory networks, hence are prime targets for drug design. To derive the details of the protein–peptide interaction mechanism is often a cumbersome task, though it can be made easier with the availability of specific databases and tools. The Peptide Binding Protein Database (PepBind) is a curated and searchable repository of the structures, sequences and experimental observations of 3100 protein–peptide complexes. The web interface contains a computational tool, protein inter-chain interaction (PICI), for computing several types of weak or strong interactions at the protein–peptide interaction interface and visualizing the identified interactions between residues in Jmol viewer. This initial database release focuses on providing protein–peptide interface information along with structure and sequence information for protein–peptide complexes deposited in the Protein Data Bank (PDB). Structures in PepBind are classified based on their cellular activity. More than 40% of the structures in the database are found to be involved in different regulatory pathways and nearly 20% in the immune system. These data indicate the importance of protein–peptide complexes in the regulation of cellular processes. PepBind is freely accessible at http://pepbind.bicpu.edu.in/.
Keywords: Peptide-binding proteins database, PepBind, Protein–peptide complex, Protein–peptide interface, Protein–peptide interaction tool, Protein inter-chain interaction
Introduction
Functional analyses of proteins involve the exploration of their interactions with other molecules, which plays vital roles in different pathways. Nearly 60% of the interaction pathways such as signal transduction, apoptotic, immune system and other pathways contain domains with bound peptides [1]. These interactions are prevalent in Src homology 2 (SH2) domain, major histocompatibility complex (MHC), antibodies, proteases, calmodulin, PapD chaperone and OppA (oligopeptide permease A) structures, with variable sequence specificity and binding affinity [2]. Protein–peptide interactions require only a small interface and can occur in many interaction networks. Hence, these are attractive drug targets both for small molecules and inhibitory peptides [3–5]. This implies that synthetic peptides can be designed to alter specific interactions in disease or other pathways [1,6,7]. Out of the structures deposited in the Protein Data Bank (PDB) [8], every month around 20 new entries are shown to exhibit interactions with small peptides. As the number of new and interesting protein–peptide complex structures continue to expand, our understanding of these protein–peptide recognition events should improve. To understand and analyze the protein–peptide interaction mechanisms, a reliable database of protein–peptide complexes is necessary. A number of sequence-based protein–peptide interaction databases are available, such as ELM [9], PhosphoELM [10], DOMINO [11], SCANSITE [12], PepBank [13], APD [14], ASPD [15] and BIOPEP [16]. Structural data are also available on protein–peptide complex structures in peptiDB [17] and PepX [18]. While peptiDB is a set of 103 curated PDB files for non-redundant protein–peptide complexes, PepX contains 1431 non-redundant X-ray structures clustered based on their binding interfaces and backbone variations. Previous studies report heterogeneity of domains or proteins to bind multiple peptides (e.g., at least 13 different types of peptides have been reported to bind to SH3 domains [19]). For detailed analysis of interactions of similar proteins with different peptides, an enormous amount of data concerning protein–peptide complex structures are needed. To address this problem, we have created the Peptide Binding Protein Database (PepBind), which contains 3100 available protein structures from the PDB, irrespective of the structure determination methods and similarity in their protein backbone.
Different kinds of interactions have been noted in the stabilization of protein–peptide binding. Analyses of various interacting interfaces between linear peptide and protein domains help us in distinguishing transient and permanent complexes [20–22]. It has been demonstrated that protein-peptide interfaces contain more hydrogen bonds per 100 Å2 solvent accessible surface area (ASA) (i.e., 50% more than protein–protein interactions and 100% more than intrinsically-unstructured regions to protein interactions) [17]. The importance of other interactions such as interactions between nonpolar hydrophobic amino acid residues and ionic interactions in the structure and function of proteins is also well known [23,24]. Knowing the importance of protein–peptide interface hydrogen bonds and other kinds of interactions, we developed and integrated a web-based interaction tool, protein inter-chain interaction (PICI), which calculates all the interface hydrogen bonds along with other interactions (such as disulfide bonds, hydrophobic interactions and ionic interactions) in tertiary structures of protein–peptide complexes and can be visualized with an integrated Jmol [25] viewer. Although a similar tool, Protein Interaction Calculator (PIC) [26], has been available, this tool calculates interface interactions specific for the peptide chain of a protein–peptide complex structure and visualizes them in a single web page along with highlighted interacting residues on sequences. We have also developed a binding prediction server built in PepBind (http://pepbind.bicpu.edu.in/PepBind_prediction_beta.php) to predict the possible protein domains in the PepBind database that may bind the user-defined peptide sequence.
Results
The PepBind database provides researchers with residue and atomic-level information about sequences and structures of protein–peptide complexes and their interfaces, helping in the analysis of protein–peptide interactions by computing various interface interactions and by providing structural information both interactively on screen and in a text format (Figure 1). The PepBind database also maintains a repository of structure coordinate files, PDBML [27] data files and protein–peptide interaction files generated by PICI tool. The database is updated on a regular basis to serve as a resource for structural, functional and protein–peptide interaction studies of peptide-binding proteins. Researchers can also submit protein–peptide complexes to the database, which will be uploaded to PepBind after manual verification.
Figure 1.
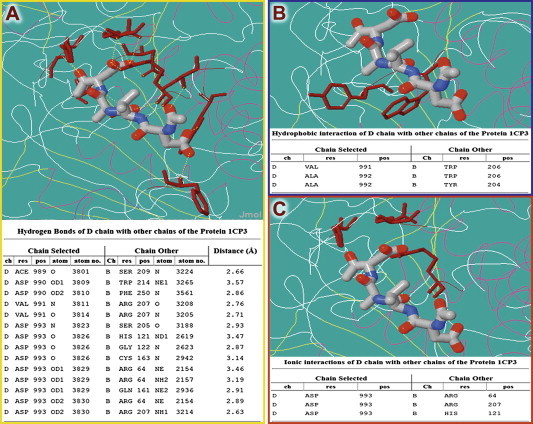
Interactions between the complex of Apopain with the tetrapeptide inhibitor ACE-DVA-ASK (PDB ID: 1CP3) Hydrogen bonds (A), hydrophobic interaction (B) and ionic interactions (C) were identified by PICI server. Interacting residues are colored in brown.
Database statistics
As shown in Table 1, current version of PepBind contains structural information for a total of 3100 protein–peptide complexes. Based on cellular activity, 1745 complexes of all the 3100 proteins (56.3%) are involved in regulatory pathways, along with inhibitory complexes. Our study shows 1278 structures (41.2%) in the database play major roles in hormonal activity, gene regulation, transcription and signal transduction pathways along with transferases. Furthermore, 600 structures (19.3%) in the database are found to function in the immune system. It has been found that 252 proteins (8.1%) are structural, contractile and membrane proteins involved mainly in transport (5.2%) and cell adhesion (1.9%). In addition, 953 (30.7%) structures have protease or other hydrolase activities, while 10.5% structures in the database are associated with proteins involved in other cellular activities.
Table 1.
Contents of the PepBind database
| Cellular activity | No. of complexes (%) | Functional category | No. of complexes (%) |
|---|---|---|---|
| Cell cycle | 90 (2.9) | Structural, contractile and membrane proteins | 252 (8.1) |
| Structural proteins | 126 (4.0) | ||
| Cell adhesion | 59 (1.9) | ||
| Transporta | 163 (5.2) | ||
| Calmodulin (CaM) | 42 (1.3) | Regulatory proteins | 1278 (41.2) |
| Apoptosis | 125 (4.0) | ||
| Signaling | 626 (20.2) | ||
| Hormones | 84 (2.7) | ||
| Transferasesb | 415 (12.7) | ||
| Transcription | 268 (8.6) | ||
| Gene regulation | 38 (1.2) | ||
| Inhibitory complex | 663 (21.4) | Inhibitory complexes | 663 (21.4) |
| MHC | 340 (10.9) | Immune system | 600 (19.3) |
| Immunoglobulin (Ig) | 250 (8.0) | ||
| Antibiotics | 15 (0.5) | ||
| Other immune system proteins | 98 (3.1) | ||
| Proteases | 687 (22.1) | Proteases and other hydrolases | 953 (30.7) |
| Other hydrolases | 266 (8.5) | ||
| Others | 326 (10.5) | Others | 326 (10.5) |
Note: There are totally 3100 protein–peptide complexes in PepBind. Since some proteins are multi-functional, there are overlaps among different categories.
Transporters, channels and pumps;
Transferases along with kinase, phosphomutase, transaldolase and transketolase.
Web interface
The user interface has been developed for browsing through all the contents of the database as a list or by different categories (Figure 2). For the ease of users to search and access data, we have integrated many search tools (Figure 2A) into the web interface. Using the ‘simple search’ function, users can retrieve information about protein–peptide complexes using their PDB ID or protein name. Our ‘keyword search’ tool scans all the fields of all the tables in PepBind for the matched word and returns a list of all protein structures related to the query. Using the ‘advanced search’ function, users can filter search based on peptide length, cellular activity of proteins, structure determination methods (e.g., X-ray diffraction, nuclear magnetic resonance and electron microscopy) and authors contributing to solving protein structure. All these search options with their parameters are joined by ‘AND’ operator for an intensive search. Additionally, to find any protein sequences homologous to the sequence submitted, we provide BLAST searching [28] against PepBind/PDB/SwissProt.
Figure 2.
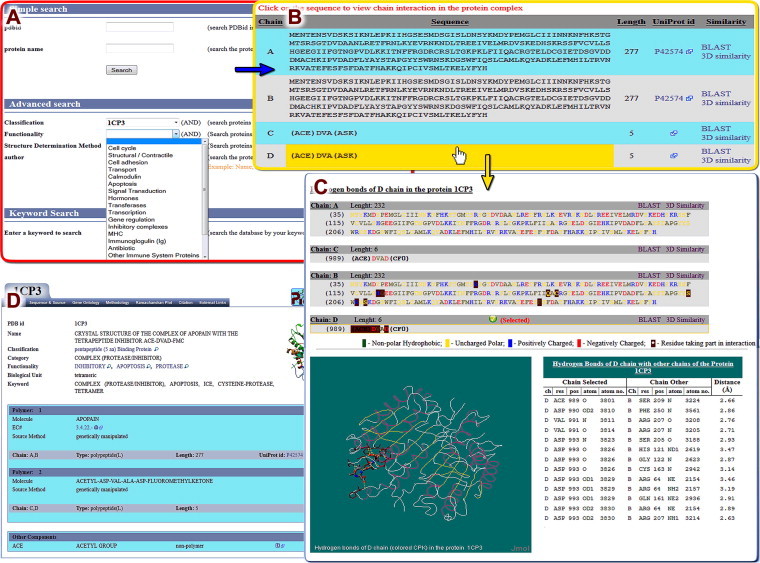
Snapshots of PepBind output A. Search page with search parameters. B. Result summary page showing all the chains with their sequence. C. Jmol showing protein–peptide interface and sequence viewer showing protein chains with identified residues highlighted. D. Detailed result page displaying summary of the protein and other tab options.
The web interface for the output result has been designed to show all the chains present in the protein structure (Figure 2B). Each chain is linked to the PICI web tool for analyzing its interactions with other chains of the protein. This tool shows the interaction details by highlighting the corresponding interacting residues in the displayed sequence along with the Jmol visualization tool for the identified interactions between the residues (Figure 2C). Different tab viewers have been designed for various types of interactions. The protein detail page shows information about protein complex on a single web page under different tabs (Figure 2D), such as summary, sequence and source, gene ontology, methodology, Ramachandran plot, citation and external links. While the ‘sequence and source’ tab displays amino acid sequence in different colors as per their biochemical properties along with source organism data, the ‘Ramachandran plot’ tab shows the Ramachandran plot image developed by the MolProbity [29] server, and the ‘Gene Ontology’ tab shows GO functional annotation [30]. For a structure similarity search, we take advantage of the web service of PDB, which employs the FATCAT algorithm [31] to recognize homologous domains available at PepBind, SCOP [32] and PDP [33].
Discussion
Protein–peptide interactions are the key components of cellular processes such as signal transduction, protein trafficking, defense mechanisms and enzyme regulation. Various databases are available on protein interactions. They can be grouped as protein-small molecule, protein-nucleic acid and protein–protein interaction databases. However, the retrieval of structural and functional information of protein–peptide interactions in biological processes is tedious due to the lack of specific databases to provide such details. The establishment of the PepX database has resolved the difficulty of unavailability of a protein–peptide interaction database, whereby authors have classified the proteins based on backbone variations and binding interfaces. While in PepX, grouping is based solely on 3D similarity, PepBind complements PepX by providing interface information for both the peptide and protein chains of the complexes along with their cellular functions and options for sequence and structure similarity searches. PepBind is integrated with the Jmol viewer to visualize the interface residues along with the interaction files generated by the PICI tool. Furthermore, PepBind provides BLAST search and structure similarity search for protein chains. It also provides a prediction service for binding of user-given peptides to possible protein domains present in the PepBind database.
Links to other related databases and servers for the queried protein are provided for further analysis of the structures. These resources include PDB [8], PDBsum [34], Pfam [35], CASTp [36], OCA Browser (http://bip.weizmann.ac.il/oca/), PSI/KB (http://sbkb.org/kb/), SRS [37], MMDB [38], PQS [39], SCOP [32], CATH [40], Proteopedia [41], Jena Library [42] and UniProt [43].
Currently our interaction tool PICI is capable of analyzing inter-chain interactions like hydrogen bond, disulfide bridge, hydrophobic interaction and ionic interaction. Keeping in view the importance of other weak interactions in stabilizing the protein structure, we plan to improve our tool to study interactions such as aromatic-aromatic interactions [44], cation-pi interactions [45] and aromatic-sulfur interactions [46]. In addition, the current interaction tool capabilities will be extended to user-submitted structures, allowing for examination of interfaces in complexes currently not present in the PepBind.
Methods
Data collection and curation
Files for atomic coordinate (pdb files – version 3.30), sequences (fasta files) and other data (pdbml files – version 4.0) of 3100 protein–peptide complexes in the PDB were downloaded following a thorough manual screening of all the available structures in the PDB. Because PepBind intends to be a comprehensive collection of protein–peptide complexes from the PDB, the database contains all the available protein–peptide complexes, irrespective of their sequence or structure redundancy. Classification of all the collected structure data was done in three steps: (I) an automated program to scan the amino acid sequences and classify them based on length of the bound peptide, (II) manual curation for the cellular activity of the complexes through study of the literature and (III) an automated program to read the data file and group the complexes as per their structure determination methods. Functionality has been analyzed through literature studies and classified as proteins involved in different cellular activities and grouped in 19 categories.
Database schema and implementation
The PepBind database consists of a series of server-side scripts written in the PHP programming language with HTML and JavaScript for user interface functions, which runs on the Apache 2.2 web server, using MySQL 5.1 as a database back-end. Atomic coordinate information from the PDB and other related information from other remote databases and web servers were mined through an automated program and stored in a file repository for further processing. We developed sets of PHP scripts for operating with the available data and process them for easy integration in the database and front-end user interface. The first set of scripts reads the PDBML files [27], extracts the data, and inserts them into the database tables; the second set sorts these data with respect to each attribute and the third set generates web pages with specific information about individual complexes.
Utilities and tools
The PICI tool for depicting potential hydrogen bonds and other interactions between the short peptide and core protein was developed and integrated into PepBind. This tool parses the structure coordinate files, removes the hetero atoms and water molecules, and predicts the interaction based on coordinate distance between atoms of amino acid residues of small peptide and the protein. For structures determined by NMR, the first model in the file is taken for calculation by PICI tool. For the two atoms A(x1, y1, z1) and B(x2, y2, z2), linear distance D is calculated as per the Euclidean distance equation D(A, B) = √{(x1 − x2)2 + (y1 − y2)2 + (z1 − z2)2}.
Various potential interactions are calculated based on standard and published criteria. The hydrogen bond is detected if the distance between oxygen or nitrogen atoms of the peptide and the protein domain is ⩽3.5 Å [47]. Interactions between hydrophobic residues (such as alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan, proline and tyrosine) [48] have been predicted if they fall within 5 Å range. Apart from these interactions, ionic residue (arginine, lysine, histidine, aspartic acid and glutamic acid) pairs falling within 6 Å contribute to ionic interactions. The tool with integrated Jmol viewer shows various interactions between the peptide and the amino acid residues of the interacting protein chains. Moreover, it highlights the positions of interacting amino acid residues on the displayed sequence (Figure 2D). This tool also generates an interaction file for each type of interactions.
A sequence modification tool has been developed and incorporated into the result page, which can read the protein sequence file and color the amino acid sequence (using single letter code) of protein according to their biochemical properties (such as green for non-polar hydrophobic amino acids, yellow for uncharged polar amino acids, blue for positively charged amino acids, red for negatively charged amino acids and black for non standard amino acids). A web-based prediction server has been provided to find the protein domains present in the database that likely bind to the user-given peptide. The sequence search tool present in the web interface allows users to BLAST search the queried sequence in the database using various parameters.
All data related to structure, sequence and interface interactions currently in the PepBind database have been made available for further analysis. These files along with the complete list of the PepBind dataset can be downloaded freely from our database. A reporting tool has been integrated to generate the result in a printer-friendly PDF file.
Authors’ contributions
PPM, RK and MSK conceived and designed the project. AAD collected the data, developed the database, developed the tools and designed the website. OPS developed the BLAST search script. AAD and OPS wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors have no competing interests to declare.
Acknowledgements
This work is supported by the Department of Biotechnology (Grant No. BT/BI/03/015/2002) and Department of Information Technology (Grant No. DIT/R&D/15 (9)2007), Government of India.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

References
- 1.Neduva V., Russell R.B. Peptides mediating interaction networks: new leads at last. Curr Opin Biotechnol. 2006;17:465–471. doi: 10.1016/j.copbio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Stanfield R.L., Wilson I.A. Protein–peptide interactions. Curr Opin Struct Biol. 1995;5:103–113. doi: 10.1016/0959-440x(95)80015-s. [DOI] [PubMed] [Google Scholar]
- 3.Parthasarathi L., Casey F., Stein A., Aloy P., Shields D.C. Approved drug mimics of short peptide ligands from protein interaction motifs. J Chem Inf Model. 2008;48:1943–1948. doi: 10.1021/ci800174c. [DOI] [PubMed] [Google Scholar]
- 4.Zhao L., Chmielewski J. Inhibiting protein-protein interactions using designed molecules. Curr Opin Struct Biol. 2005;15:31–34. doi: 10.1016/j.sbi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Kim J.E., Chen J., Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 6.Petsalaki E., Russell R.B. Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol. 2008;19:344–350. doi: 10.1016/j.copbio.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Vagner J., Qu H., Hruby V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman H.M., Kleywegt G.J., Nakamura H., Markley J.L. The protein data bank at 40: reflecting on the past to prepare for the future. Structure. 2012;20:391–396. doi: 10.1016/j.str.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinkel H., Michael S., Weatheritt R.J., Davey N.E., Van Roey K., Altenberg B. ELM–the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–D251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dinkel H., Chica C., Via A., Gould C.M., Jensen L.J., Gibson T.J. Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceol A., Chatr-aryamontri A., Santonico E., Sacco R., Castagnoli L., Cesareni G. DOMINO: a database of domain-peptide interactions. Nucleic Acids Res. 2007;35:D557–D560. doi: 10.1093/nar/gkl961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obenauer J.C., Cantley L.C., Yaffe M.B. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shtatland T., Guettler D., Kossodo M., Pivovarov M., Weissleder R. PepBank – a database of peptides based on sequence text mining and public peptide data sources. BMC Bioinformatics. 2007;8:280. doi: 10.1186/1471-2105-8-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z., Wang G. APD: the antimicrobial peptide database. Nucleic Acids Res. 2004;32:D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valuev V.P., Afonnikov D.A., Ponomarenko M.P., Milanesi L., Kolchanov N.A. ASPD (Artificially Selected Proteins/Peptides Database): a database of proteins and peptides evolved in vitro. Nucleic Acids Res. 2002;30:200–202. doi: 10.1093/nar/30.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minkiewicz P., Dziuba J., Iwaniak A., Dziuba M., Darewicz M. BIOPEP database and other programs for processing bioactive peptide sequences. J AOAC Int. 2008;91:965–980. [PubMed] [Google Scholar]
- 17.London N., Movshovitz-Attias D., Schueler-Furman O. The structural basis of peptide–protein binding strategies. Structure. 2010;18:188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Vanhee P., Reumers J., Stricher F., Baeten L., Serrano L., Schymkowitz J. PepX: a structural database of non-redundant protein–peptide complexes. Nucleic Acids Res. 2010;38:D545–D551. doi: 10.1093/nar/gkp893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S.S. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J. 2005;390:641–653. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein A., Aloy P. Contextual specificity in peptide-mediated protein interactions. PLoS One. 2008;3:e2524. doi: 10.1371/journal.pone.0002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins J.R., Diboun I., Dessailly B.H., Lees J.G., Orengo C. Transient protein–protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Nooren I.M., Thornton J.M. Structural characterisation and functional significance of transient protein–protein interactions. J Mol Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 23.Gromiha M.M., Selvaraj S. Inter-residue interactions in protein folding and stability. Prog Biophys Mol Biol. 2004;86:235–277. doi: 10.1016/j.pbiomolbio.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Barlow D.J., Thornton J.M. Ion-pairs in proteins. J Mol Biol. 1983;168:867–885. doi: 10.1016/s0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
- 25.Herraez A. Biomolecules in the computer: Jmol to the rescue. Biochem Mol Biol Educ. 2006;34:255–261. doi: 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- 26.Tina K.G., Bhadra R., Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35:W473–W476. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westbrook J., Ito N., Nakamura H., Henrick K., Berman H.M. PDBML: the representation of archival macromolecular structure data in XML. Bioinformatics. 2005;21:988–992. doi: 10.1093/bioinformatics/bti082. [DOI] [PubMed] [Google Scholar]
- 28.Mount DW. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc 2007; 2007: pdb. top17. [DOI] [PubMed]
- 29.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gene Ontology Consortium The Gene Ontology project in 2008. Nucleic Acids Res. 2008;36:D440–D444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye Y., Godzik A. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004;32:W582–W585. doi: 10.1093/nar/gkh430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andreeva A., Howorth D., Chandonia J.M., Brenner S.E., Hubbard T.J., Chothia C. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov N., Shindyalov I. PDP: protein domain parser. Bioinformatics. 2003;19:429–430. doi: 10.1093/bioinformatics/btg006. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski R.A. PDBsum new things. Nucleic Acids Res. 2009;37:D355–D359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zdobnov E.M., Lopez R., Apweiler R., Etzold T. The EBI SRS server–recent developments. Bioinformatics. 2002;18:368–373. doi: 10.1093/bioinformatics/18.2.368. [DOI] [PubMed] [Google Scholar]
- 38.Madej T., Addess K.J., Fong J.H., Geer L.Y., Geer R.C., Lanczycki C.J. MMDB: 3D structures and macromolecular interactions. Nucleic Acids Res. 2012;40:D461–D464. doi: 10.1093/nar/gkr1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henrick K., Thornton J.M. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 40.Knudsen M., Wiuf C. The CATH database. Hum Genomics. 2010;4:207–212. doi: 10.1186/1479-7364-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodis E., Prilusky J., Sussman J.L. Proteopedia: a collaborative, virtual 3D web-resource for protein and biomolecule structure and function. Biochem Mol Biol Educ. 2010;38:341–342. doi: 10.1002/bmb.20431. [DOI] [PubMed] [Google Scholar]
- 42.Reichert J., Suhnel J. The IMB Jena Image Library of Biological Macromolecules: 2002 update. Nucleic Acids Res. 2002;30:253–254. doi: 10.1093/nar/30.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium Uni.Prot. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burley S.K., Petsko G.A. Aromatic–aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 45.Sathyapriya R., Vishveshwara S. Interaction of DNA with clusters of amino acids in proteins. Nucleic Acids Res. 2004;32:4109–4118. doi: 10.1093/nar/gkh733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reid K.S.C., Lindley P.F., Thornton J.M. Sulphur-aromatic interactions in proteins. FEBS Lett. 1985;190:209–213. [Google Scholar]
- 47.Berg J.M., Tymoczko J.L., Stryer L. 5th ed. W H Freeman; New York: 2002. Biochemistry. [Google Scholar]
- 48.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]