

Abstract
Aims
The NLRP3 inflammasome is activated in the ischaemic heart promoting caspase-1 activation, inflammation, and cell death. Ischaemic injury establishes both a priming signal (transcription of inflammasome components) and a trigger (NLRP3 activation). Whether NLRP3 activation, without priming, induces cardiac dysfunction and/or failure is unknown. The aim of this study was to assess the independent and complementary roles of the priming and the triggering signals in the heart, in the absence of ischaemia or myocardial injury.
Methods and results
We used mice with mutant NLRP3 (constitutively active), NLRP3-A350V, under the control of tamoxifen-driven expression of the Cre recombinase (Nlrp3-A350V/CreT mice). The mice were treated for 10 days with tamoxifen before measuring the activity of caspase-1, the effector enzyme in the inflammasome. Tamoxifen treatment induced the inflammasome in the spleen but not in the heart, despite expression of the mutant NLRP3-A350V. The components of the inflammasome were significantly less expressed in the heart compared with the spleen. Subclinical low-dose lipopolysaccharide (LPS; 2 mg/kg) in Nlrp3-A350V/CreT mice induced the expression of the components of the inflammasome (priming), measured using real-time PCR and western blot, leading to the formation of an active inflammasome (caspase-1 activation) in the heart and LV systolic dysfunction while low-dose LPS was insufficient to induce LV systolic dysfunction in wild-type mice (all P < 0.01 for mutant vs. wild-type mice).
Conclusion
The signalling pathway governing the inflammasome formation in the heart requires a priming signal in order for an active NLRP3 to induce caspase-1 activation and LV dysfunction.
Keywords: NLRP3, Inflammasome, Priming, Cryopyrinopathy, Cardiomyopathy, Caspase-1
1. Introduction
Heart failure is the common final pathway in a variety of clinical conditions in which the cardiac function is impaired.1 A close interplay between inflammation and heart failure exists.2,3 IL-1β and IL-18 have been shown to act as endogenous cardio-depressant factors and to be involved in the pathophysiology of heart disease.4–6
The inflammasome is a large macromolecular complex that controls the activation of caspase-1 and the maturation and release of IL-1β and IL-18.7 NLRP3 (cryopyrin/NALP3/CIAS1) is an intracellular danger sensor that upon activation oligomerizes and initiates the assembly of the inflammasome by engaging the apoptosis speck-like protein containing caspase recruitment domain (ASC) and pro-caspase-1.7
Caspase-1 is a key mediator of myocardial damage in ischaemic heart injury and failure.8–12 Inhibition of the NLPR3-initiated inflammasome during experimental myocardial infarction minimizes the activation of caspase-1 in the heart and improves left ventricular remodelling and function.8–10
During physiological conditions, NLRP3 rests in an inactive state. Several pathogen- and damage-associated molecular patterns (PAMPS and DAMPS) have been shown to activate NLRP3.7 The formation of the inflammasome is regulated at the level of the priming and the triggering.13 The priming is an active process leading to the activation of nuclear factor κB (NF-κB) and the transcription of inflammasome components.13 The triggering is represented by the activation of NLRP3, leading to final inflammasome assembly.13 Thus, activation of NLRP3 may be insufficient to induce caspase-1 activation or produce IL-1β or IL-18 if the inflammasome components are not sufficiently expressed, and the priming would then represent the limiting step. Prevention of NLRP3 activation (trigger) during acute myocardial infarction (AMI) is sufficient to inhibit the inflammasome function and the adverse cardiac remodelling, but it is not known whether, without an inciting injury to the heart, NLRP3 activation alone is sufficient to induce cardiac dysfunction. Mutations of Nlrp3 have been identified in a group of autosomal dominant diseases that are collectively known as inflammasomopathies or cryopyrin-associated periodic syndromes (CAPS).14,15 The diseases are driven by the spontaneous oligomerization of NLRP3 and massive production of IL-1β. Knock-in mouse models that express mutated forms of Nlrp3 reproduce the auto-inflammatory disease.15–17 These knock-in mice represent the ideal model to investigate whether the activation of NLRP3 alone induces cardiac dysfunction in the absence of tissue injury and/or of a priming signal. An accurate definition of the hierarchic signalling cascade in LV dysfunction is necessary to develop novel targeted treatment strategies.
2. Methods
2.1. Animals and treatments
The animal experiments were conducted following the guidelines on the humane use and care of laboratory animals for biomedical research published by National Institutes of Health (No. 85-23, revised 2011). The Institutional Animal Care and Use Committee of Virginia Commonwealth University approved the study. The Nlrp3-A350V/NeoR mice were generated as previously described, inserting a floxed, inverted, neomycin cassette in intron 2 of the Nlrp3 gene that prevents its expression.16 The A350V substitution corresponds to the missense mutation of the alanine 352 often found in patients affected by Muckle–Wells syndrome (MWS). Homozygous mice were crossed with mice carrying Tg(CAG-(cre/Esr1*)5Amc) (Jackson Laboratories, Bar Harbor, ME, USA), the transgene for Cre recombinase under a tamoxifen-inducible promoter. Thus, the resulting progeny, Nlrp3-A350V/CreT, heterozygous for the mutated Nlrp3 gene, expresses just the wild-type allele, because the floxed inverted neomycin cassette prevents the expression of the mutant. After tamoxifen treatment, the cassette is removed, and the mutated gene expressed. This mouse model has been previously characterized and showed increased levels of IL-1β and related cytokines.18 Background-matched C57B6 mice (Jackson laboratories) and Tg(CAG-(cre/Esr1*)5Amc) treated with or without tamoxifen and Nlrp3-A350V/CreT mice without tamoxifen were used as controls. The mice were randomly assigned to the treatment groups. Tamoxifen (5 mg/kg in an ethanol:sunflower oil solution 1:9) was injected intraperitoneally (i.p.) daily for 3 days and then once every 3 days until Day 9. The mice were followed up to 30 days. Subgroups of mice Nlrp3-A350V/CreT were treated with tamoxifen for 10 days to induce recombination of the mutant gene, and then, 24 h after last tamoxifen dose, they were stimulated with a subclinical dose of lipopolysaccharide (LPS; 2 mg/kg in normal saline, i.p.), referred as low-dose LPS, to provide the priming signal for the expression of the inflammasome components in the heart. The normal saline was used as control vehicle. Nlrp3-A350V/NeoR (without the Cre) were used as controls. An additional group of wt mice was used to investigate the effects on cardiac function and caspase-1 activation in wt mice, and to assess the cardiac effects of this low-dose LPS in the absence of the mutant NLRP3 (constitutively active).
2.2. Doppler echocardiography
Echocardiography was performed using the 30-MHz probe-equipped Vevo 770 Imaging System (Visual Sonic, Toronto, Ontario, Canada) as previously described.19 M-mode echocardiography was used to measure the left ventricular end-diastolic and end-systolic diameters (LVEDD and LVESD), anterior wall (AW) and posterior wall (PW) systolic and diastolic thickness (AWDT and AWST; PWDT and PWDT), and fractional shortening (LVFS) under light anaesthesia (pentobarbital, 30–50 mg/kg). LVFS was calculated using the formula (LVEDD-LVESD)/LVEDD. The systolic function was measured starting 1 day before tamoxifen treatment, and the measures were repeated at Days 0 (day of injection), 5, and 10. The LV systolic function in the groups of mice primed with LPS was measured 6 h after challenge.
2.3. Left ventricular catheterization
The pressure probe catheter (Millar Instruments, Houston, TX, USA) was inserted retrogradely in the left ventricle through the left carotid artery in deeply sedated mice (pentobarbital, 50–70 mg i.p.) to measure the LV end-diastolic pressure (LVEDP), the LV peak systolic pressure (LVPSP), +dP/dt, and −dP/dt.6
The measurements were recorded and analysed with the Labchart Pro software (Millar).
2.4. Samples collection
The mice were euthanized (pentobarbital, 200 mg/kg i.p.), the hearts excided, the atria removed, and the ventricles snap-frozen and stored at −80°C. Spleens were also collected as control tissue that is enriched with myeloid cells that express higher components of the inflammasome.20
2.5. Detection of genomic recombination following tamoxifen treatment
DNA was isolated from hearts and spleens. Briefly, DNA was extracted with phenol–chloroform following protease K digestion. PCR was performed with two primers that anneal to the sides of the neomycin cassette and produce three different products: (i) a wild-type fragment of 180 bp, (ii) a fragment derived from the silenced mutant cryopyrin of 2208 bp, and (iii) a fragment of 288 bp correspondent to the reactivated mutant cryopyrin allele following neomycin cassette excision. The presence of fragment 3 is evidence of mutant cryopyrin activation. The PCR products were analysed by agarose electrophoresis using a DNA ladder.
2.6. Measurement of caspase-1 tissue activity
Caspase-1 activity was measured in protein extracts as previously described.8 Briefly, proteins were extracted from frozen hearts and spleens using RIPA buffer (Sigma Aldrich, St Louis, MO, USA) and were diluted in caspase assay buffer (31% sucrose, 3.1% HEPES, and 0.31% CHAPS; Sigma Aldrich). The activity was measured using a fluorometric substrate YVAD-AFC (Enzo Life Sciences, Farmingdale, NY, USA), after subtraction of background signal in the presence of the caspase-1 inhibitor YVAD-CHO (Enzo Life Sciences). Fluorescence was read using a Glomax fluorimeter (Promega Corporation, Fitchburg, WI, USA) and a UV filter (em/ex 360/410–450).
2.7. Western blotting
Protein samples were resolved by SDS electrophoresis and were analysed by western blot to determine the protein levels of the inflammasome components. Primary antibodies for NLRP3 (Santa Cruz Biotechnology, Dallas, TX, USA), ASC, and caspase-1 (Sigma Aldrich) were used in combination with peroxidase-conjugated secondary antibodies and chemiluminescence to detect the protein amount. A primary antibody against β-actin was used to correct for protein loading. The protein bands were quantified using Image J (National Institute of Health).
2.8. TUNEL assay
Two subgroups of mice were treated with LPS (2 mg/kg) or normal saline for 6 h, and the hearts were collected, perfused with PBS, and fixed in 4% formalin for 48 h. After fixation, the hearts were embedded in paraffin, sliced in 5 micron sections, and analysed with the TUNEL assay (EMD Millipore, Billerica, MA, USA) to evaluate DNA fragmentation as marker of cell death.
2.9. Real-time PCR
RNA was extracted from hearts and spleens using affinity columns (RNeasy extraction Kit, Qiagen, Valencia, CA, USA) and converted to cDNA using the High Capacity cDNA Reverse Transcription kit (Life Technologies, Grand Island, NY, USA). Real-time PCR was performed using SYBR Green PCR master mix (Life Technologies) with a Roche 480 Thermocycler (Roche, Indianapolis, IN, USA). Gene expression was carried for the following mRNAs: Nlrp3, asc, pro-caspase-1, and pro-IL-1β. TATA Box protein mRNA had the same expression in the spleen and the heart and was used as a reference gene.
2.10. Statistical analysis
The statistical analysis was completed using the SPSS 19.0 software (IBM, Armonk, NY, USA). Values are presented as mean and standard error of mean. The Student t or ANOVA test was used to compare data between two or more groups, respectively. Statistical significance was set at P < 0.05 (two-tailed).
3. Results
3.1. Induction of Nlrp3-A350V in the heart
Nlrp3-A350V/CreT knock-in mice (KI) had a normal appearance (weight and behaviour) and cardiac phenotype as assessed by transthoracic echocardiography and gross pathology, and no differences were noted compared with the wild-type mice (Table 1).
Table 1.
Baseline LV function in wild-type (wt) and Knock-In Nlrp3-A350V/CreT mice (KI)
| wt (n = 7) | KI (n = 6) | |
|---|---|---|
| LVEDD | 3.46 ± 0.05 | 3.51 ± 0.09 |
| LVESD | 2.21 ± 0.07 | 2.19 ± 0.14 |
| LVFS | 36.0 ± 1.3 | 38.1 ± 2.7 |
| LVEF | 66.6 ± 1.8 | 68.8 ± 3.1 |
| LVPSP | 67.1 ± 3.8 | 65.4 ± 3.1 |
| LVEDP | 3.28 ± 0.51 | 3.00 ± 0.50 |
| HR | 403 ± 54 | 334 ± 26 |
| +dP/dt | 3672 ± 396 | 3775 ± 583 |
| −dP/dt | −2440 ± 398 | −1925 ± 288 |
LVEDD, left ventricular (LV) end-diastolic diameter; LVESD, LV end-systolic diameter; LVFS, LV fractional shortening; LVEF, LV ejection fraction; LVPSP, LV peak systolic pressure; LVEDP, LV end-diastolic pressure; HR, heart rate.
Treatment with tamoxifen for 10 days effectively activated the Cre recombinase, leading to excision of the neomycin cassette as shown by PCR analysis (Figure 1). We used RT–PCR to determine NLRP3 expression in the spleen and heart. The expression of Nlrp3 in wt and mutant mice treated with tamoxifen is given in Figure 1. NLRP3 mRNA in the heart of wt mice was significantly lower than that in the spleen. Tamoxifen treatment increased NLRP3 mRNA levels in the heart of Nlrp3-A350V/CreT to levels similar to those observed in the spleen.
Figure 1.

Tamoxifen treatment in wild-type (wt) and the Knock-In (KI) Nlrp3-A350V/CreT mice induces genomic recombination of the transgenic sequence. (A) PCR was used to detect the neomycin cassette in the Nlrp3 gene of KI mice with and without tamoxifen treatment (10 days). (B) mRNA quantification of NLRP3 in the spleen and heart of wt and KI Nlrp3-A350V/CreT mice treated with tamoxifen for 10 days (n = 4 per group); the mRNA was normalized on TATA Box Protein (TBP). Results are reported as fold change compared with spleen wt mRNA expression.
3.2. Caspase-1 activity in the heart following expression of Nlrp3/A350V
Compared with wt mice, Nlrp3-A350V/CreT mice had a five-fold higher caspase-1 activity in the spleen, but not in the heart (Figure 2), suggesting that the tamoxifen-driven transcription of NLRP3-A350V is sufficient to activate the inflammasome in myeloid organs, i.e. spleen, but not in the heart.
Figure 2.

Catalytic activity and mRNA quantification of caspase-1 in spleen and heart of wt and Nlrp3-A350V/CreT mice. (A) Enzymatic activity of caspase-1 reported as relative fluorescence produced compared with controls (n = 5 per group). Values are expressed as mean ± SEM. *P < 0.04 vs. wt spleen. (B) Pro-caspase-1 mRNA quantified and reported as relative amount compared with the spleen of wt mice (n = 5 per group). Values are expressed as mean ± SEM. #P = 0.01 vs. spleen wt or KI.
To determine whether the observed differences reflected the changes in constitutive expression of caspase-1, we measured pro-caspase-1 mRNA and found a several-fold lower expression of pro-caspase-1 mRNA in the heart compared with the spleen in both Nlrp3-A350V/CreT and wt mice, without any significant effect of tamoxifen (Figure 2). These data show that NLRP3-A350V expression is insufficient to lead to inflammasome formation in the heart, possibly due to the lack of constitutive expression of inflammasome components in the heart to induce caspase-1 activity.
3.3. Induction of the Nlrp3/A350V does not affect heart function
LV dimensions and function were measured before and after 5 and 10 days of tamoxifen treatment in Nlrp3-A350V/CreT and wt mice. As an additional control, we included CreT mice (without the Nlrp3-A350V), to determine effects of Cre expression alone. Tamoxifen had no significant effects on cardiac dimensions and function in all the mice strain (Figure 3). A subgroup of Nlrp3-A350V/CreT mice was followed for 30 days, and no changes were detected in cardiac dimensions and function (data not shown). These data show that NLRP3-A350V does not induce measurable changes in cardiac dimensions or function.
Figure 3.

Echocardiographic parameters in wt, CreT, and Nlrp3-A350V/CreT mice 10 days following tamoxifen treatment compared with untreated wt mice. (A) LVFS (expressed as %), (B) LVEDD (expressed in mm), and (C) LV mass (expressed in mg). All parameters were expressed as mean ± SEM (n = 6 per group).
3.4. Low-dose LPS induces cardiac dysfunction in Nlrp3-A350V/CreT mutant mice
Low dose of LPS (2 mg/kg) was used to measure the effects of the priming signal in the Nlrp3-A350V/CreT mice. The mRNA and protein analysis were performed on the hearts of Nlrp3-A350V/CreT mice 6 hours following the LPS injection, showing that LPS induced priming leading to increased expression of the inflammasome components in the heart (Figure 4).
Figure 4.

Priming effects of low-dose LPS administration on the heart of Nlrp3-A350V/CreT mice. (A) mRNA levels of the inflammasome components are reported as change compared with the untreated control and expressed as mean ± SEM (n = 5 per group). *P < 0.01. (B) Representative western blot images of the inflammasome proteins NLRP3, pro-caspase-1 and active caspase-1, and ASC. β-Actin was used as loading control. (C) Densitometric quantification of the proteins analysed by western blot mean ± SEM. *P < 0.05.
Moreover, in the Nlrp3-A350V/CreT mice with constitutively active NLRP3 treatment with the low-dose LPS induced a significant three-fold increase in caspase-1 tissue activity (P < 0.05, Figure 5), indicating that when in combination with constitutively active NLRP3, priming is sufficient to activate caspase-1 without the need for further triggering. Despite a measurable effect on the expression of the NLRP3 inflammasome components, treatment with low-dose LPS did not induce caspase-1 activation in the heart of the Nlrp3-A350V lacking CreT (inactive mutant) or of the wt mice indicating that low-dose LPS is insufficient to induce caspase-1 activity in the Nlrp3-A350 mice and in wt mice due to the absence of an active NLRP3 protein (Figure 5).
Figure 5.
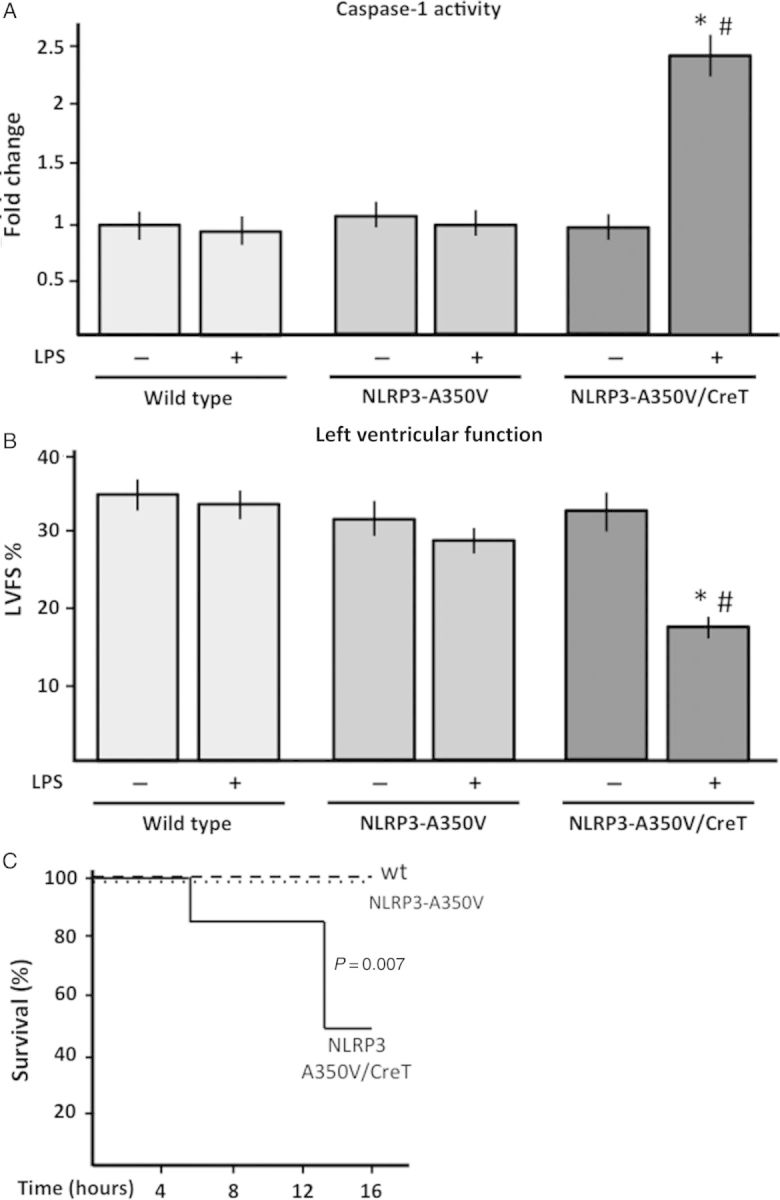
Effects of low-dose LPS in wild-type, Nlrp3-A350V, and KI Nlrp3-A350V/CreT mice. (A) Caspase-1 activity in the heart of wild type, Nlrp3-A350V, and Nlrp3-A350V/CreT treated with tamoxifen and then challenged with low-dose LPS or the vehicle solution for 6 h. Caspase-1 activity was measured and reported as change relative to untreated controls and expressed as mean ± SEM (n = 6–7 per group). *P < 0.05 vs. Nlrp3-A350V/CreT without LPS; #P < 0.05 vs. wt or Nlrp3-A350V with LPS. (B) The LVFS was measured in tamoxifen-treated wild-type, Nlrp3-A350V, and Nlrp3-A350V/CreT mice challenged with low-dose LPS or the vehicle to measure the effects of the simultaneous presence of priming and active NLRP3 on heart function. The values are expressed as mean ± SEM (n = 6–7 per group). *P < 0.05 vs. Nlrp3-A350V/CreT without LPS; #P < 0.05 vs. wt or Nlrp3-A350V with LPS (C) Survival curve of wt (n = 12), Nlrp3-A350V (n = 3), and Nlrp3-A350V/CreT (n = 6) mice treated with tamoxifen for 10 days and then challenged with a single injection of low-dose (subclinical) LPS, P = 0.007.
In the Nlrp3-A350V/CreT mutant mouse, and not in the Nlrp3-A350 mice, or wt mice, low-dose LPS led to significant LV systolic dysfunction, as measured by a reduction in LVFS at 6 h (−34%, P = 0.03; Figure 5), associated with a 26% reduction in +dP/dt (P = 0.05). Administration of vehicle alone had no effects in any of the groups of mice (Figure 5).
To evaluate the effects of the subclinical, low-dose LPS (2 mg/kg) on the expression of the inflammasome components in wild-type mice, mRNA and protein analysis were performed on heart samples collected in mice treated with tamoxifen for 10 days and then challenged with LPS. LPS increased the expression of NLRP3 mRNA and protein, increased the mRNA of pro-IL-1β, and induced a trend in increase in the pro-caspase-1 protein and mRNA although did not reach significance (Figure 6). ASC protein and mRNA were not changed. No active, cleaved, caspase-1 was detected.
Figure 6.

Protein and mRNA analysis of the inflammasome components in the heart of wild-type mice with or without LPS low dose. (A) NLRP3, pro-caspase-1, and ASC protein expression were analysed by western blot and (B) were quantified through densitometric analysis. Results are reported as change in protein levels compared with the untreated control and expressed as mean ± SEM (n = 5 per group). *P = 0.01. (C) mRNA levels of the inflammasome components are reported as change compared with the untreated control and expressed as mean ± SEM (n = 5–7 per group). *P < 0.01.
Within 16 h, low-dose LPS led to death in 50% of the Nlrp3-A350V/CreT mutant mice (P = 0.007 vs. wt mice; Figure 5). Cell death in the heart, measured with the TUNEL assay for DNA fragmentation performed 6 h following LPS, showed no induction in cell death in any of the groups (data not shown).
4. Discussion
The formation of the NLRP3 inflammasome is a critical, finely regulated step in the process of amplification of the inflammatory response following cell or tissue injury.7,8,13 Here we show that priming and triggering are two equally important mechanisms regulating the NLRP3 inflammasome formation in the heart, by demonstrating that triggering alone is insufficient to induce cardiac dysfunction in mice in the absence of priming, thus defining the hierarchical signalling cascade in the events leading to LV dysfunction following systemic or local inflammation (Figure 7).
Figure 7.

Effects of NLRP3 activation or the combination with priming on cardiac function. The induction of the mutant, active, NLRP3 in the absence of priming is insufficient to induce left ventricular dysfunction. The combination of the induction of the mutant NLRP3 with a priming signal induces caspase-1 activation (indicative of inflammasome activation) and LV dysfunction.
Research in the past decade has highlighted the central role of the NLRP3 inflammasome in a variety of diseases ranging from rare genetically determined disorders to very common degenerative diseases (such as osteoarthritis or gout).15–17 Several unrelated stimuli produced under stress or following tissue injury activate NLRP3 leading to inflammasome formation through mechanisms involving either activation of the membrane purinergic receptors P2X7 or intracellular signalling generated by lysosomal destabilization or production of reactive oxygen species.8,9,13 The mechanisms of inflammasome activation are, however, differentially regulated at the cell and tissue levels. The different inflammasome components are indeed differentially expressed, with some cells/tissues expressing them constitutively and others expressing them only when induced by priming.20–22 In fact, NLRP3 expression in the heart of wt mice was lower than that in the spleen, in accordance to a previous observation showing that NLRP3 mRNA in the heart is consistently lower in human and mouse samples.20 During AMI, cell debris and the release of ATP in the myocardium provide a powerful stimulus for NLRP3 inflammasome activation by rendering both the priming, through activation of the Toll-like receptors (TLRs), and the triggering through the P2X7 receptor.7,8,23,24 In the absence of an acute tissue injury, however, the presence of stimuli for NLRP3 inflammation may be insufficient to induce inflammasome activation. Tissue resident cells may not need to respond to injurious/infectious agents as promptly as circulating monocytes or other cells of the innate immune response.22,23
In this study, we used a mouse model that recapitulates CAPS, a disease caused by the presence of an autosomal dominant mutations in the Nlrp3 gene, prone to spontaneously form the NLRP3 inflammasome.16,17 Patients with CAPS have profound systemic inflammation, characterized by recurrent fever, rash, joint and bone deformities, and, in severe cases, premature death, yet highly responsive to IL-1β blockade.14,15 This mouse model, which relies on tamoxifen-dependent DNA recombination, has a normal phenotype until the expression of the constitutively active mutant NLRP3 is induced by tamoxifen, guided by its native promoter (not overexpression) accordingly to our experimental needs.16,17 This model is unique in that it presents the possibility to induce the inflammasome in the absence of tissue injury, permitting us to study the role of NLRP3 activation at the tissue level, in this case the heart, without the additional confounding signalling induced by the associated tissue injury (i.e. during AMI). In this mouse model, the induction of the mutant NLRP3 is sufficient to activate the inflammasome (caspase-1) in the spleen but failed to activate the inflammasome in the heart, highlighting the need for additional signals to activate the NLRP3 inflammasome. This allows us to understand the hierarchy of events needed to form the inflammasome in the heart. Indeed, while mutant NLRP3-A350V mRNA was equally expressed in the heart and spleen, other components of the NLRP3 inflammasome were significantly less expressed in the heart, reflecting that the priming signal is necessary for the formation of the NLRP3 inflammasome in the heart. Myeloid Differentiation Factor 88 (MyD88) is considered to be an important signalling node for the priming of the inflammasome.25 Accordingly, following AMI, the inhibition of MyD88, limiting priming, prevented adverse cardiac remodelling and ameliorated left ventricular function.26
LPS priming in the spleen is dispensable for the inflammasome formation in the mice with mutant NLRP3 (constitutively active), showing an inflammatory phenotype following 10 days of tamoxifen treatment. In these same mice, we observed no measurable caspase-1 activation in the heart or changes in left ventricular dimensions or function. These findings are in agreement with the lack of clinical reports of cardiac dysfunction in patients with CAPS.27
In wild-type mice, low dose of LPS failed to induce any appreciable LV systolic dysfunction, which is indeed generally seen only when higher doses of LPS induce multi-organ injury and failure.28 This is in agreement with the findings that the majority of patients with systemic inflammation or sepsis do not have cardiac dysfunction despite having intense systemic inflammation; on the other hand, some patients who develop cardiac dysfunction also show signs of myocardial injury, such as troponin I increase, which serves as focal trigger for the inflammasome activation in the heart.24 The LPS treatment did not induce cell death, suggesting that the dysfunction is independent from cardiomyocytes loss.
The low-dose LPS, on the other hand, induced the expression of NLRP3 inflammasome components in the heart of the NLRP3-A350V/CreT mice, leading to the formation of the NLRP3 inflammasome and activation of caspase-1 in the heart even in the absence of myocardial injury, showing that despite the lack of a cardiac phenotype, Nlrp3-A350V/CreT mice are exquisitely sensitive to this low-dose LPS, leading to severe impairment in cardiac function (which was not seen in wild-type mice). Low-dose LPS was also lethal in 50% of the mutant mice, and not in wt mice, likely due to a concomitant reduction in systemic vascular resistance and a decrease in cardiac output rather than the expected compensatory increase seen with vasodilation. Whether the excess mortality with low-dose LPS is directly linked to inflammation in the heart or the failure of other organs or multi-organ failure is unexplored in this study. In this study, we indeed limited our analysis to the cardiac function of the mutant NLRP3-A350V/CreT mouse and used a low-dose LPS as a research tool rather than a model of disease. A previous report showed that mice that express the NLRP3-A350V mutation during foetal development present systemic inflammation affecting several organs and leading to death few days after birth, indicating that these mice are indeed prone to multi-organ failure.16 Further studies would be needed to explore the contribution of myocardial dysfunction to premature mortality in sepsis-like preclinical models.
The literature on the role of the inflammasome and its components in AMI has highlighted that the inflammasome has different effects depending on the cell type.8–10,29,30 In Leucocytes and in resident fibroblasts, the inflammasome formation induces release of IL-1β and IL-18, whereas in cardiomyocytes, it seems more involved in the induction of cell death. In the experimental setting proposed in this study, we did not observe increase in TUNEL positivity, although longer time may be needed for the mutant NLRP3/A350V to induce cell death without tissue injury. The lack of a role of the mutant NLRP3 in the unprimed heart in our study is in line with the lack of a significant role of ASC or NLRP3 observed in the isolated heart exposed to ex vivo acute ischaemia.10,31
This study, as any experimental animal study using genetically modified mice strains, has several other limitations worth noting. First and foremost, although the genetic mutation was derived from patients with CAPS and the mutation in the mouse recapitulates the disease, there are differences between the human and mouse CAPS.15 We believe, however, that in this study the genetic mutant is used as an experimental tool rather than for a translational study. Second, the mutation in NLRP3 was systemic and not restricted to one cell line or tissue; therefore, the ability to identify cardiomyocyte-specific effects is rather limited, yet we believe that cardiomyocytes may behave in the same way of all other cardiac resident cells and that the differences between the heart and the spleen reflect the difference between systemic intravascular defense lines and the organ-level defense line.21 Further studies with cell line-restricted mutations or bone marrow chimeras may be very informative in this regard. Third, the use of a low dose of LPS in this study may be viewed as an additional limitation since LPS provides complex signalling beyond simple TLR agonism and priming, and high-dose LPS, in particular, leads to multi-organ failure also associated with LV systolic dysfunction. Nevertheless, we viewed the use of low-dose LPS as a tool to demonstrate that increased expression of the inflammasome components (priming) is sufficient to induce LV systolic dysfunction in the NLRP3 mutant mouse (because constitutively active) in the absence of myocardial injury or sepsis-like picture. We cannot exclude, however, that the NLRP3 mutant mouse may be more sensitive to LPS due to impaired clearance or increased TLR expression. Fourth, this study did not address the role of IL-1β or IL-18 in LV systolic dysfunction. A recent study from our group has shown that the phenotype of CAPS is mediated by the two cytokines IL-1β and IL-18 and also in part independent of the cytokines,32 whether this applies also to LV systolic dysfunction remains unexplored. Previous studies from our group have shown that both systemic administration of IL-1β and IL-18 are able to induce LV systolic dysfunction.5,6,33
In conclusion, the use of genetically modified mice in this study reveals the independent and complementary roles of both the priming and triggering of the NLRP3 inflammasome in the heart. With the recognized role of inflammation in heart disease, the NLRP3 inflammasome occupies a central role in the local and systemic response to tissue injury. Identifying the mechanisms regulating the formation of the inflammasome in the heart may be helpful to reduce the severity of dysfunction or remodelling due to ischaemic or non-ischaemic injury. TLRs and MyD88 activity correlates with worse remodelling, and the function following AMI and NLRP3 and the axis TLR-MyD88 are active also during doxorubicin-induced cardiomyopathy.34–41 A novel small molecule inhibitor of NLRP3 has been recently developed and has been shown efficacious in an experimental model of AMI in the mouse.42 Interventions aimed at targeting the priming through inhibition of the axis TLRs-MyD88-NFκB may prove to be equally effective in reducing the inflammasome effects in the heart following myocardial injury. Moreover, treatments aimed at blocking the main product of the NLRP3 inflammasome, IL-1β, have shown promising results in preclinical animal studies and pilot clinical trials.43 Identifying additional proximal ‘check points' in this process could thus provide new potential targets for therapeutic interventions.
Conflict of interest: none declared.
Funding
This work was supported by the American Heart Association [Post-Doctoral Grant to S.T. and E.M.; Scientist Development Grants to F.N.S. and A.A], by funds from the VCU Pauley Heart Center [to A.A.], and by National Institute of Health [institutional grant KL2TR000057 to B.W.V.T.].
References
- 1.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 3.Satoh M, Minami Y, Takahashi Y, Nakamura M. Immune modulation: role of the inflammatory cytokine cascade in the failing human heart. Curr Heart Fail Rep. 2008;5:69–74. doi: 10.1007/s11897-008-0012-2. [DOI] [PubMed] [Google Scholar]
- 4.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1β induces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 6.Toldo S, Mezzaroma E, O'Brien L, Marchetti C, Seropian IM, Voelkel NF, Van Tassell BW, Dinarello CA, Abbate A. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–H1031. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 10.Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–174. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 11.Merkle S, Frantz S, Schön MP, Bauersachs J, Buitrago M, Frost RJ, Schmitteckert EM, Lohse MJ, Engelhardt S. A role for caspase-1 in heart failure. Circ Res. 2007;100:645–653. doi: 10.1161/01.RES.0000260203.55077.61. [DOI] [PubMed] [Google Scholar]
- 12.Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, Wright JR, Jr, Macdonald JA, Lees-Miller JP, Roach D, Semeniuk LM, Duff HJ. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp Physiol. 2013;98:462–472. doi: 10.1113/expphysiol.2012.068338. [DOI] [PubMed] [Google Scholar]
- 13.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman HM, Brydges SD. Genetic and molecular basis of inflammasome-mediated disease. J Biol Chem. 2011;286:10889–10896. doi: 10.1074/jbc.R110.135491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25:469–484. doi: 10.1016/j.smim.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, Soroosh P, Watford WT, O'Shea JJ, Kastner DL, Hoffman HM. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonar SL, Brydges SD, Mueller JL, McGeough MD, Pena C, Chen D, Grimston SK, Hickman-Brecks CL, Ravindran S, McAlinden A, Novack DV, Kastner DL, Civitelli R, Hoffman HM, Mbalaviele G. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PLoS ONE. 2012;7:e35979. doi: 10.1371/journal.pone.0035979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, Brydges SD. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol. 2012;189:2707–2711. doi: 10.4049/jimmunol.1101737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seropian IM, Abbate A, Toldo S, Harrington J, Smithson L, Ockaili R, Mezzaroma E, Damilano F, Hirsch E, Van Tassell BW. Pharmacologic inhibition of phosphoinositide 3-kinase gamma (PI3Kγ) promotes infarct resorption and prevents adverse cardiac remodeling after myocardial infarction in mice. J Cardiovasc Pharmacol. 2010;56:651–658. doi: 10.1097/FJC.0b013e3181f9a905. [DOI] [PubMed] [Google Scholar]
- 20.Lech M, Avila-Ferrufino A, Skuginna V, Susanti EH, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int Immunol. 2010;22:717–728. doi: 10.1093/intimm/dxq058. [DOI] [PubMed] [Google Scholar]
- 21.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, Yang XF. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. 2009;22:311–322. doi: 10.1177/039463200902200208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi M. NLRP3 Inflammasome as a Novel Player in Myocardial Infarction. Int Heart J. 2014;55:101–105. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol. 2007;193:323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- 26.Van Tassell BW, Seropian IM, Toldo S, Salloum FN, Smithson L, Varma A, Hoke NN, Gelwix C, Chau V, Abbate A. Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J Cardiovasc Pharmacol. 2010;55:385–390. doi: 10.1097/FJC.0b013e3181d3da24. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman HM, Wanderer AA. Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep. 2010;10:229–235. doi: 10.1007/s11882-010-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fallach R, Shainberg A, Avlas O, Fainblut M, Chepurko Y, Porat E, Hochhauser E. Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction following septic shock or myocardial ischemia. J Mol Cell Cardiol. 2010;48:1236–1244. doi: 10.1016/j.yjmcc.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 29.Toldo S, Mezzaroma E, Mauro AG, Salloum FN, Van Tassell BW, Abbate AS. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2014.5989. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.ver Elst KM, Spapen HD, Nguyen DN, Garbar C, Huyghens LP, Gorus FK. Cardiac troponins I and T are biological markers of left ventricular dysfunction in septic shock. Clin Chem. 2000;46:650–657. [PubMed] [Google Scholar]
- 31.Zuurbier CJ, Jong WM, Eerbeek O, Koeman A, Pulskens WP, Butter LM, Leemans JC, Hollmann MW. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. PLoS ONE. 2012;7:e40643. doi: 10.1371/journal.pone.0040643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123:4695–4705. doi: 10.1172/JCI71543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, Shah K, Canada J, Voelkel NF, Dinarello CA, Abbate A. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE. 2012;7:e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu C, Ren D, Wang X, Ha T, Liu L, Lee EJ, Hu J, Kalbfleisch J, Gao X, Kao R, Williams D, Li C. Toll-like receptor 3 plays a role in myocardial infarction and ischemia/reperfusion injury. Biochim Biophys Acta. 2014;1842:22–31. doi: 10.1016/j.bbadis.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, Takahashi H, Arimoto T, Maeda K, Yamakawa M, Takeuchi O, Akira S, Takeishi Y, Kubota I. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 36.Riad A, Jäger S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, Pauschinger M, Schultheiss HP, Tschöpe C. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- 37.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 38.Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–C769. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11:1008–1016. doi: 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation. 2004;110:2869–2874. doi: 10.1161/01.CIR.0000146889.46519.27. [DOI] [PubMed] [Google Scholar]
- 41.Krysko DV, Kaczmarek A, Krysko O, Heyndrickx L, Woznicki J, Bogaert P, Cauwels A, Takahashi N, Magez S, Bachert C, Vandenabeele P. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011;18:1316–1325. doi: 10.1038/cdd.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63:316–322. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]