

Abstract
Fungal communities play a major role as decomposers in the Earth's ecosystems. Their community-level responses to elevated CO2 (eCO2), one of the major global change factors impacting ecosystems, are not well understood. Using 28S rRNA gene amplicon sequencing and co-occurrence ecological network approaches, we analyzed the response of soil fungal communities in the BioCON (biodiversity, CO2, and N deposition) experimental site in Minnesota, USA, in which a grassland ecosystem has been exposed to eCO2 for 12 years. Long-term eCO2 did not significantly change the overall fungal community structure and species richness, but significantly increased community evenness and diversity. The relative abundances of 119 operational taxonomic units (OTU; ∼27% of the total captured sequences) were changed significantly. Significantly changed OTU under eCO2 were associated with decreased overall relative abundance of Ascomycota, but increased relative abundance of Basidiomycota. Co-occurrence ecological network analysis indicated that eCO2 increased fungal community network complexity, as evidenced by higher intermodular and intramodular connectivity and shorter geodesic distance. In contrast, decreased connections for dominant fungal species were observed in the eCO2 network. Community reassembly of unrelated fungal species into highly connected dense modules was observed. Such changes in the co-occurrence network topology were significantly associated with altered soil and plant properties under eCO2, especially with increased plant biomass and NH4+ availability. This study provided novel insights into how eCO2 shapes soil fungal communities in grassland ecosystems.
INTRODUCTION
Fungi represent a significant portion of the microbial community in the Earth biosphere, with an estimated ca. 1.5 to 5.1 million species in total (1, 2). They play a major role as decomposers in natural ecosystems, by degrading organic matters into reusable nutrition in biogeochemical cycling processes (3). Understanding the fungal diversity, their community structure, and their responses to long-term elevated CO2 (eCO2) in grassland ecosystems is an important issue in ecology and global change biology, but little is known about the impacts of eCO2 on the diversity, composition, structure, and function of soil fungal communities due to the high diversity and uncultivable nature of most (>80%) soil fungi (4).
Past studies have shown that eCO2 significantly increases the plant productivity in grassland ecosystems, resulting in more carbon input to the soil (5–10). As consequences, increased carbon input in turn significantly changed bacterial diversity, composition, and structure and increased the functional potential of bacterial communities for carbon degradation and nutrient cycling, although such effects differed across various ecosystems (6, 11–20). In contrast, fungal biomass and relative abundance of total microbial biomass did not change significantly under eCO2 in this BioCON (biodiversity, CO2, and N deposition) experimental site (6, 21). Previous studies of fungal responses to eCO2 were mainly carried out using approaches such as phospholipid fatty-acid analysis, denaturing gradient gel electrophoresis, extracellular enzyme assays, and clone library analysis (6, 12, 16, 21–23) and mostly focused on mycorrhizal fungi (15, 24–26), which have major influences on plant biodiversity and productivity (27). Those previous studies were focused on fungal carbon degradation, nitrogen cycling, and interactions with plants (26, 28, 29); however, knowledge about fungal community-level responses to eCO2 is still limited, though some efforts have been made recently (12, 30, 31).
Microorganisms, including bacteria, archaea, viruses, fungi, and protists, interact with each other in soil to form complex interactive networks (32). Using ecological network approaches, co-occurrence ecological networks of microbial communities can be constructed and analyzed (33–37). For example, a global ecological network analysis of the human microbiome revealed 3,005 co-occurrence and coexclusion relationships among 197 clades occurring throughout the human microbiome (34). For environmental perturbation impacts on microbial network structures, previous studies showed that eCO2 significantly impacted soil bacterial/archaeal community networks in a grassland ecosystem and that significantly different network structures and increased network complexity were observed in response to eCO2 (36, 37). However, co-occurrence patterns of large soil microorganisms (e.g., fungi and protists) and their responses to eCO2 have not yet been characterized. Therefore, much can be learned by exploring co-occurrence relationships within fungal communities under eCO2 using molecular ecological network analysis (MENA) (38).
In the present study, we aimed to comprehensively survey the fungal community diversity and examine their changes in composition, structure, and co-occurrence patterns in response to eCO2 in a grassland soil ecosystem. The following two hypotheses were tested: (i) stimulated plant biomass and changed soil properties as a result of eCO2 would significantly increase the diversity and alter the structure of soil fungal communities, and (ii) such a changed fungal community structure would lead to more effective decomposition of soil organic matters and increase biologically available nitrogen in the soil for maintaining plant growth. To test these hypotheses, we examined the response of fungal communities to long-term eCO2 in the BioCON experimental site, a 12-year CO2 manipulation in temperate grassland in central Minnesota, USA, by sequencing 28S rRNA gene amplicons and comparing fungal community co-occurrence networks under ambient CO2 (aCO2) and eCO2. Our results indicated that fungal communities responded to long-term eCO2 by community reassembly, while overall community structure and species richness were not significantly changed. Such changes of co-occurrence network topology were significantly associated with soil and plant properties. This study provides novel insights into how eCO2 shapes soil fungal communities in grassland ecosystems, improving our understanding of the effects of eCO2 on soil fungal communities.
MATERIALS AND METHODS
Site description and sample collection.
The study was conducted within the BioCON experimental site located at the Cedar Creek Ecosystem Science Reserve in Minnesota, USA (lat 45.4, long 93.2). The long-term experiment was started in 1997 on a secondary successional grassland on a sandy outwash soil after removing the previous vegetation (9). BioCON is a split-plot arrangement of treatments in a completely randomized design. The main BioCON field experiment has 296 plots (2 m by 2 m) distributed in six 20-meter-diameter circular areas. CO2 treatment is the whole-plot factor and is replicated three times among the six rings. The subplot factors of species number and N treatment were assigned randomly and replicated in individual plots among the six rings. In this study, all 24 plots (12 from aCO2 and 12 from eCO2), with 16 plant species and no additional N supply, were used. A detailed experimental design as well as the location of sampled plots can be found in Fig. S1 in the supplemental material.
All of the 16 plant species used in the present study are native or naturalized to the Cedar Creek Ecosystem Science Reserve and can be classified into four functional groups: (i) four C3 grasses (Agropyron repens, Bromus inermis, Koeleria cristata, and Poa pratensis), (ii) four C4 grasses (Andropogon gerardii, Bouteloua gracilis, Schizachyrium scoparium, and Sorghastrum nutans), (iii) four N-fixing legumes (Amorpha canescens, Lespedeza capitata, Lupinus perennis, and Petalostemum villosum), and (iv) four non N-fixing herbaceous species (Achillea millefolium, Anemone cylindrica, Asclepias tuberosa, and Solidago rigida). Plots were regularly manually weeded to remove unwanted species, although the 16 species plots used in this study require minimal weeding.
Bulk soil samples were obtained in July 2009 under ambient and eCO2 conditions for microbial community analysis, and each sample was composited from five soil cores at a depth of 0 to 15 cm. All samples were immediately transported to the laboratory, frozen and stored at −80°C for DNA extraction, PCR amplification, and 454 pyrosequencing.
DNA extraction, purification, and quantification.
Soil DNA was extracted by freeze-grinding mechanical lysis as described previously (39) and was purified using a low-melting-point agarose gel, followed by phenol extraction for all 24 soil samples collected. DNA quality was assessed by using ratios of 260 to 280 nm and 260 to 230 nm using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE), and final soil DNA concentrations were quantified with PicoGreen (40) using a FLUOstar Optima (BMG Labtech, Jena, Germany).
PCR amplification and 454 pyrosequencing.
A total of 23 samples instead of 24 were subjected to 454 pyrosequencing due to insufficient remaining DNA for one of the samples. Amplification was performed using a fungal 28S rRNA gene primer pair with the forward primer LR3 (ACCCGCTGAACTTAAGC) and the reverse primer LR0R (CCGTGTTTCAAGACGGG), whose products are expected to be ∼625 bp (41). A unique 8-mer barcode was added for each sample at the 5′ end of the forward primer. The barcode primers were synthesized by Invitrogen (Carlsbad, CA) and used for the generation of PCR amplicons. Quadruplicate 20-μl PCRs were performed as follows: 4 μl of Promega GoTaq buffer, 0.5 μl of GoTaq DNA polymerase, 1.5 μl of Roche 25 mM MgCl2, 1 μl of Invitrogen 10 mM deoxynucleoside triphosphate mix, 1 μl of each primer (10 pmol μl−1), 0.2 μl of New England BioLabs bovine serum albumin at 10 mg ml−1, 1 μl of template (1.43 ng DNA/μl), and 9.8 μl of H2O. The cycling conditions were an initial denaturation of 94°C for 3 min, followed by 30 cycles of 94°C for 1 min, 51°C for 40 s, and 72°C for 1 min, and then a final extension at 72°C for 10 min. Replicates were pooled and gel purified using a Qiagen gel purification kit after band excision. The products were further purified using a Qiagen PCR purification kit. After adapter ligation, amplicons were sequenced on an FLX 454 system (454 Life Sciences, Branford, CT) by Macrogen (Seoul, South Korea) using Lib-L kits and processed using the shotgun protocol.
Data analysis.
Raw pyrosequencing reads were extracted from the sff file using the sffinfo tool from Roche 454. Two files, a fasta file containing the sequence and a qual file containing the quality information, were generated and then converted into a fastq file using the python script “faqual2fastq2.py” that comes with the UPARSE pipeline (42). The forward reads were extracted and used for data analysis. Quality filtering, chimera removal, and operational taxonomic unit (OTU) clustering were carried out using the UPARSE pipeline (42), which is a recently developed approach that identifies highly accurate OTU from amplicon sequencing data. Only the reads with perfectly matched barcodes and maximum of two primer mismatches were kept for further analysis. Barcodes and primers were deleted from reads. The remaining reads were then truncated to 250 bp to ensure higher sequence quality. Reads with expected error of >0.5 were discarded, which is the default UPARSE parameter and refers to the average number of errors per read that would be found in the sample of reads. The reads were then dereplicated, sorted, and clustered into candidate OTU with an identity cutoff of 0.97. Chimeric OTU were identified and removed by UCHIME by comparing with the LSU reference sequences downloaded from the Silva database (release_111) (43). Finally, qualified reads were mapped to OTU representative sequences for relative abundance calculation.
Taxonomic assignment for OTU was carried out by RDP classifier using the fungal LSU training data set (41). OTU representative sequences were aligned by the MUSCLE program (44), and a phylogenetic tree was built using FASTTREE (45). Significance tests for different taxonomic groups and OTU were performed by response ratio analysis (46) with a 95% confidence interval. UniFrac principal-coordinate analysis (PCoA) was performed using the online tool Fast Unifrac (47). Species richness, evenness, and diversity indices were calculated by the Mothur package (48), with rarefaction analysis of 1,000 bootstrap random sampling iterations and 0.1% incremental sampling efforts. To avoid potential bias caused by sequencing depth, a random subsampling effort of 6,029 reads per sample was performed.
Co-occurrence ecological network construction and analysis.
Fungal co-occurrence ecological networks were constructed and analyzed using the online MENA pipeline, which implements random matrix theory (RMT) for threshold identification (38). In order to construct highly confident fungal co-occurrence ecological networks for comparative analysis, several different approaches were applied. First, we used an RMT-based approach to identify a proper threshold for pairwise Pearson correlation coefficient values between OTU. The RMT identifies the threshold by observing a transition point of nearest-neighbor spacing distribution of eigenvalues from Gaussian to Poisson distribution, which are two universal extreme distributions (36). The RMT-based approach is a reliable and robust tool for network construction and has been successfully applied to construct various networks, including gene regulatory networks (49–53), functional molecular ecological networks (36), and phylogenetic molecular ecological networks (37). Second, the same cutoff of 0.78 was applied to construct co-occurrence networks for fungal communities at aCO2 and eCO2, with the purpose of comparing between different networks. Since a smaller threshold will result in less reliable and larger networks with more nodes, the same cutoff could effectively eliminate imbalances in network comparisons. Third, only OTU presented in at least six samples were used for Pearson correlation coefficient calculations and zero was filled in for missing values for OTU in paired samples. This made the correlation coefficient between two OTU more statistically reliable. Finally, in order to statistically compare the constructed networks, permutation based null model analysis was developed and applied to statistically evaluate whether the constructed networks are significantly different from random ones as well as that between aCO2 and eCO2 networks. Specifically, the matrices for network construction were permutated 100 times, from which 100 random networks were generated by keeping the number of nodes and links constant. A Student test was then performed to evaluate the significance of differences between experimental network structure and random networks and between control and treatment networks. Ecological networks were visualized by Cytoscape (54).
Linking community structure and network topology with soil and plant properties.
To analyze whether the changed fungal community structure and network topology were correlated with soil and plant properties, Mantel tests that calculate the correlation between two matrices were performed. A total of six soil and plant properties, including soil moisture (ca. 0 to 17 cm), pH, midseason in situ net nitrification, ammonification, and N mineralization rates, and total plant biomass, were collected and analyzed. The Euclidean distance was used to construct dissimilarity matrices for both OTU-based tables (community structure, network topology) and environmental variable(s). For Mantel tests of correlations between network topology and soil and plant properties, the correlation between OTU significance (calculated by OTU relative abundance and soil and plant properties) and node connectivity was examined. (More details can be found in reference 38.)
Nucleotide sequence accession number.
Raw pyrosequencing data associated with this study were deposited in the NCBI database under accession number SRR1057903.
RESULTS
CO2 effects on soil and plant characteristics.
Because soil and plant properties are directly related with belowground microbial community, the CO2 effects on soil moisture, pH, mid-season in situ net nitrification, ammonification, and N mineralization rates, and plant biomass were analyzed. No significant change of midseason net soil nitrification rate was found between aCO2 and eCO2 samples. However, the net soil ammonification rate was significantly (P < 0.05) higher in eCO2 samples than that in aCO2 samples, resulting in moderately significantly (P < 0.1) higher net N mineralization rate (Fig. 1A). The total plant biomass, as expected, also increased significantly (P < 0.05) as a result of eCO2 and higher soil N availability (Fig. 1B). The proportional soil moisture and pH, however, did not change significantly (see Fig. S2 in the supplemental material), suggesting that the increased plant biomass and soil ammonification could be the major factors affecting belowground microbial communities, including fungal communities.
FIG 1.
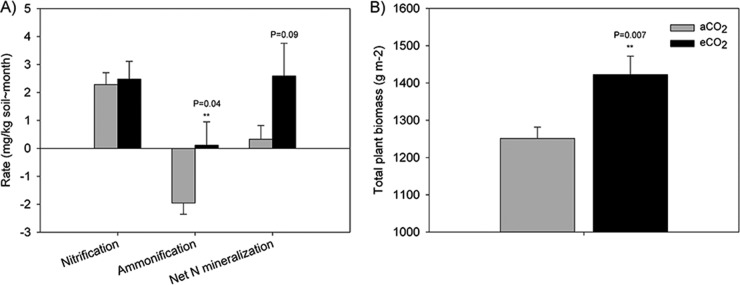
eCO2 effects on soil nitrogen (A) and total plant biomass (B). Soil net nitrification, net ammonification, and net nitrogen mineralization rates were collected and analyzed. The total plant biomass was averaged from the previous 5 years. A significantly increased soil net ammonification rate and total plant biomass were observed.
Sequence summary.
Using 454 pyrosequencing, a total of 402,265 raw sequences of 28S rRNA gene amplicons were obtained with an average length of 477 bp for all 23 samples. A total of 339,048 reads (154,541 for aCO2 samples and 184,507 for eCO2 samples) were then clustered into 1,975 OTU after quality trimming, dereplication, clustering, and chimera removal by the UPARSE pipeline, with an OTU identity cutoff of 97%. Of the identified 1,975 OTU, 407 were found to be singletons. Taxonomic assignment by RDP classifier showed 1,744 OTU covering 97.9% qualified reads were fungal 28S rRNAs, and the remaining 231 were assigned to Eukaryota incertae sedis, but with <50% bootstrap confidence. Of these, 734 OTU belonged to Ascomycota, 326 to Chytridiomycota, 298 to Basidiomycota, 96 to Blastocladiomycota, 53 to Glomeromycota, 41 to Neocallimastigomycota, and 2 to Zygomycota.
Long-term eCO2 did not change the overall fungal community structure, but increased diversity.
The overall community structure between aCO2 and eCO2 samples was not significantly different, as revealed by three different nonparametric multivariate analysis methods (adonis: R = 0.04, P = 0.5; ANOSIM: R = 0.03, P = 0.71; MRPP: δ = 0.55, P = 0.52). However, clear trend of separation of aCO2 samples from eCO2 samples could be observed by UniFrac PCoA that considers the phylogenetic relationship between OTU, indicating potential responses to eCO2 by subgroups of phylogenetically related fungal species (see Fig. S3 in the supplemental material).
To understand how long-term eCO2 affects the fungal community diversity, the species richness and community diversity were analyzed by Chao1 index, Shannon evenness, Shannon diversity, and phylogenetic diversity. Shannon diversity treats each OTU as an independent entity (55), and phylogenetic diversity (56) considers the phylogenetic relationship among different OTU. Owing to the close relationship between diversity indices and sequencing depth, a random subsampling effort of 6,029 reads per sample was carried out by excluding four samples (two aCO2 and two eCO2) with fewer than 3,000 reads. As a result, long-term eCO2 did not significantly change the overall fungal species richness, because 95% confidence intervals were clearly overlapped (see Fig. S4A in the supplemental material). However, the overall phylogenetic diversity (see Fig. S4B in the supplemental material) and taxonomic diversity (measured by Shannon diversity) (see Fig. S4D in the supplemental material) increased significantly, suggesting increased evenness of phylogenetically distant fungal species (see Fig. S4C in the supplemental material).
The composition of fungal community in grassland soil ecosystems.
With a 50% bootstrap confidence cutoff for taxonomy assignment by RDP classifier, the fungal community in this grassland soil was dominated by Ascomycota (81 and 77% of sequences for aCO2 and eCO2, respectively) and Basidiomycota (11 and 14% of sequences for aCO2 and eCO2, respectively), followed by 1% Fungi incertae sedis, 0.25% Chytridiomycota, 0.05% Blastocladiomycota, and 0.03% Glomeromycota at the phylum level. About 7 and 8% sequences in aCO2 and eCO2 samples could not be assigned to any phylum at 50% bootstrap confidence (see Fig. S5A in the supplemental material). At the order level, the most dominant fungal orders were Pleosporales (27.5%), Capnodiales (10.2%), Sordariales (7.5%), Hypocreales (5.4%), Helotiales (4.6%), Agaricales (5.2%), Thelebolales (3.2%), Chaetothyriales (2.6%), Cantharellales (2.3%), Coniochaetales (1.4%), Magnaporthales (1.4%), Xylariales (1.3%), Pezizales (1.3%), and Thelephorales (0.8%) (see Fig. S5B in the supplemental material). These 14 dominant fungal orders accounted for 74.7% of the total 28S rRNA sequences obtained. No significant differences were found for the relative abundances of the above dominant fungal phyla and orders between aCO2 and eCO2 samples.
Of the total 1,975 OTU, the top 20 most abundant OTU accounted for 50.3% and 50.2% of the total sequences for aCO2 and eCO2 samples, respectively. Three OTU (OTU_1, OTU_3, and OTU_6) had ≥5% relative abundance in both aCO2 and eCO2 samples and were assigned to genera Davidiella (70% bootstrap confidence), Corynespora (77% bootstrap confidence), and Didymella (47% bootstrap confidence), respectively. Relative abundance of a total of 119 OTU significantly changed between aCO2 and eCO2 samples. Among these, 28 had ≥0.3% average relative abundance in aCO2 or eCO2 samples, including 18 from Ascomycota, 7 from Basidiomycota, and 3 from Fungi incertae sedis (Fig. 2). Five of these significantly changed OTU belong to the top 20 most abundant OTU. A total of 14 of these 28 OTU were found with significantly increased relative abundance in eCO2 samples, including 7 Ascomycota OTU, 5 Basidiomycota OTU, and 2 incertae sedis fungal OTU. Of the 14 OTU with significantly decreased relative abundance in eCO2, 11 were from Ascomycota, 2 from Basidiomycota, and one from incertae sedis fungus (Fig. 2). Interestingly, these significantly changed OTU under eCO2 were associated with decreased overall relative abundance of Ascomycota (19.3% in aCO2 versus 11.5% in eCO2; analysis of variance [ANOVA], P = 0.01), but increased relative abundance of Basidiomycota (3.5% in aCO2 versus 14.3% in eCO2; ANOVA, P = 0.10). The top three most abundant OTU with significantly increased relative abundance at eCO2 were OTU_10 (Ramaricium, 11% bootstrap confidence), OTU_2 (Lycoperdon, 78% bootstrap confidence), and OTU_11 (Lophiostoma, 59% bootstrap confidence). The top four most abundant OTU with significantly decreased relative abundance were OTU_5 (Alternaria, 100% bootstrap confidence), OTU_8 (Delitschia, 27% bootstrap confidence), OTU_34 (Cudoniella, 19% bootstrap confidence), and OTU_1351 (Thanatephorus, 31% bootstrap confidence). These 28 significantly changed OTU accounted for 24.2 and 19.01% of the total captured sequences in eCO2 and eCO2 samples, respectively, while the total 119 significantly changed OTU accounted for 27.9 and 24.8% of aCO2 and eCO2 samples, respectively (Fig. 2).
FIG 2.

Response ratio (eCO2 versus aCO2) analysis of fungal OTU changes in response to eCO2. Only the top 28 most abundant OTU with relative abundances of ≥0.3% in aCO2 or eCO2 were plotted. Error bar symbols plotted at the right of dashed line indicated increased relative abundances at eCO2, while error bar symbols plotted at the left of dashed line indicated decreased relative abundances at eCO2. The genus information, as well as actual relative abundance along with the standard error, is listed.
The co-occurrence networks of fungal communities and their responses to eCO2.
In order to understand how fungal communities assemble and whether long-term eCO2 affects the fungal community network topology, co-occurrence ecological networks were constructed for aCO2 and eCO2 fungal communities. To ensure the constructed networks are nonrandom biologically meaningful networks, permutation null model analysis was developed and performed by generating 100 networks from the same matrix but randomized. As a result, the constructed networks are significantly different from random networks, as judged by significantly higher geodesic distance, clustering coefficient, and modularity (see Table S1 in the supplemental material). Network comparisons were then carried out at both global level and subnetwork level of selected nodes. The constructed aCO2 fungal network contained 271 nodes (OTU), 647 links, and 19 modules (12 with ≥3 nodes), with an average connectivity of 4.78, an average geodesic distance of 6.0, and a modularity of 0.86, while the eCO2 network had 226 nodes, 600 links, and 13 modules (9 with ≥3 nodes), with an average connectivity of 5.31, average geodesic distance of 5.34, and modularity of 0.80 (see Table S1 and Fig. S6 in the supplemental material). Although the eCO2 network contained fewer nodes and links, it is more complex than the aCO2 network regarding the average connectivity, geodesic distance, and modularity, since the Student t test showed that the average geodesic distance and modularity were significantly smaller in the eCO2 network, suggesting that the nodes in eCO2 network were more intensely connected with each other (see Table S1 in the supplemental material). Both networks were dominated by OTU from Ascomycota, which is also the dominant phylum in the fungal community (see Fig. S6 in the supplemental material). For the 9 modules with ≥3 nodes in the eCO2 network, 92 intermodular connections that linked different modules together were observed. In contrast, only 41 intermodular links were found for the 12 modules in the aCO2 network. Since modules are composed of different fungal OTU/species that have higher connectivity with within module members than outside module members, these modules could be regarded as putative microbial ecological niches (36). Thus, increased intermodular connections might indicate increased relationships between different fungal community “niches,” leading to more collaboration between different fungal subcommunities. In addition, more negative links were found in the eCO2 network than in the aCO2 network (47 in eCO2 versus 37 in aCO2), suggesting that eCO2 may also have increased competition or heterogeneity of fungal species.
In addition to our comparisons of global network topological parameters between aCO2 and eCO2 networks, we also analyzed the effect of eCO2 on subnetworks of fungal communities. Interestingly, 31 nodes that were sparsely distributed in 13 independent modules in the aCO2 network (Fig. 3A) formed five dense modules with high connectivity in the eCO2 network (Fig. 3B). Such interesting community reassembly process was not as obviously observed in the converse manner, i.e., dense aCO2 modules did not separate into sparse individual nodes in eCO2 networks. Of the 31 nodes, 27 were connected to each other in two major modules, and four of the five submodules were connected to another one (Fig. 3B). This was also consistent with the global observation that eCO2 increased the intermodular connections. However, increased connectivity was not found for all the nodes in the eCO2 network. For example, in the aCO2 network, seven OTU with high relative abundances (≥2%) were connected with 37 first neighbors and formed relatively complex subnetworks with 145 links (Fig. 4A). In the eCO2 network, although 5 of the 7 OTU remained as the most abundant OTU in the network, they only connected to 20 neighbors with 31 links (Fig. 4B), resulting in much simpler network structure. The results indicated that long-term eCO2 decreased the connectivity of OTU with high relative abundances but increased the connectivity for OTU with lower relative abundances.
FIG 3.
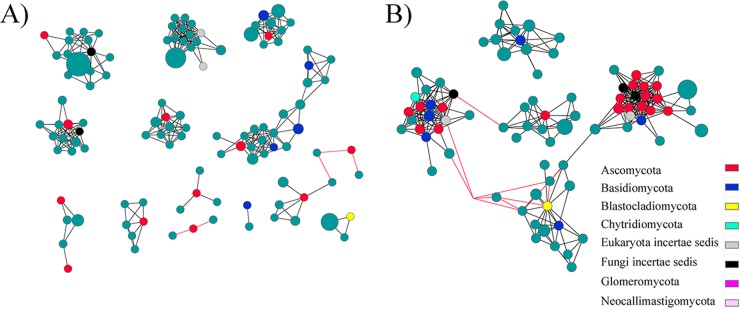
Community reassembly of sparsely distributed OTU in the aCO2 network (A) into highly connected dense modules in the eCO2 network (B). Colored nodes were the OTU involved in community reassembly. Teal nodes were the first neighbor of yellow nodes. Different colors refer to different fungal phyla.
FIG 4.

The subnetwork of top seven most abundant OTU and their first neighbor nodes in the aCO2 (A) and eCO2 (B) networks. Each node represents an OTU, which would be regarded as a fungal species. The size of nodes represents relative abundance of OTU. Each link connects two OTU. Gray links mean positive connections, and red links indicate negative connections. Different colors refer to different fungal phyla. The OTU with top relative abundances are marked with OTU identification numbers.
Linking fungal community structure and network topology with soil and plant properties.
To determine whether the fungal community structure and changed co-occurrence network topology were associated with soil and plant properties, Mantel test was performed. The relationships between community structure and soil moisture (0∼17 cm), pH, mid-season in situ net nitrification, ammonification, and N mineralization rates, and total plant biomass were analyzed (Table 1). Consistent with our dissimilarity testing that the community structure did not differ from each other, no significant (R = −0.259, P = 0.981) correlation was observed between the overall community structure and the overall soil and plant properties, nor with any single soil and plant properties (Table 1).
TABLE 1.
Mantel analysis of the relationships between overall fungal community structure, co-occurrence network topology, and individual soil and plant properties
| Soil/plant property | Community structure |
Network topologya |
||||
|---|---|---|---|---|---|---|
| aCO2 |
eCO2 |
|||||
| rM | P | rM | P | rM | P | |
| Soil moisture (0 to 17 cm) | 0.044 | 0.325 | –0.002 | 0.516 | –0.084 | 1 |
| pH | –0.091 | 0.728 | –0.052 | 0.970 | –0.069 | 0.982 |
| Midseason in situ net nitrification rate | –0.178 | 0.898 | 0.0003 | 0.457 | –0.065 | 0.984 |
| Midseason in situ net ammonification rate | –0.206 | 0.973 | 0.037 | 0.167 | 0.359 | 0.001 |
| Midseason in situ net N mineralization rate | –0.256 | 1 | 0.100 | 0.01 | 0.077 | 0.03 |
| Total plant biomass | 0.022 | 0.392 | –0.021 | 0.272 | 0.063 | 0.07 |
Significant P values are indicated in boldface. rM, correlation based on Mantel test; P, significance (probability) of Mantel test.
For Mantel tests between network topology and soil and plant properties, the trait-based OTU significance measure was used to determine a common group of soil and plant properties important to the network structure (37). Mantel test of network topology and each soil and plant property showed soil ammonification and total plant biomass to be the major factors responsible for changed network topology (Table 1). There was a significant correlation between node connectivity and OTU significance of the selected soil variables based on all nodes (OTU) with P = 0.001 (Table 2). Not all nodes in the network showed significant correlations with soil ammonification rate and plant biomass. Significant correlations mainly occurred for OTU belonging to Ascomycota (P = 0.001), Basidiomycota (P = 0.04) and incertae sedis fungi (P = 0.006). All the four major orders in Ascomycota, including Sordariomycetes (P = 0.001), Dothideomycetes (P = 0.001), Leotiomycetes (P = 0.02), and Lecanoromycetes (P = 0.02), were significantly correlated with soil and plant properties (Table 2). For the aCO2 co-occurrence network, as expected, no significant correlations were found between the node connectivity and OTU significance of the selected soil and plant variables except Sordariomycetes, Fungi incertae sedis, and Blastocladiomycota (Table 2). These results suggest that the changes of the co-occurrence fungal ecological network topology were significantly associated with increased soil ammonification rate and plant biomass under long-term eCO2 and that OTU belonging to Ascomycota were mainly responsible for such changes.
TABLE 2.
Mantel test on network connectivity versus the OTU significances of soil geochemical variablesa
| Phylogeny | aCO2 |
eCO2 |
||||
|---|---|---|---|---|---|---|
| No. of nodes | rM | P | No. of nodes | rM | P | |
| All OTU | 271 | 0.037 | 0.146 | 226 | 0.363 | 0.001 |
| Ascomycota | 135 | 0.052 | 0.133 | 130 | 0.43 | 0.001 |
| Sordariomycetes | 46 | 0.209 | 0.026 | 47 | 0.463 | 0.001 |
| Dothideomycetes | 40 | 0.07 | 0.167 | 42 | 0.471 | 0.001 |
| Leotiomycetes | 16 | –0.012 | 0.394 | 14 | 0.323 | 0.02 |
| Lecanoromycetes | 12 | 0.167 | 0.24 | 14 | 0.499 | 0.02 |
| Basidiomycota | 58 | –0.011 | 0.493 | 34 | 0.217 | 0.038 |
| Eukaryota incertae sedis | 25 | –0.071 | 0.671 | 21 | 0.268 | 0.06 |
| Fungi incertae sedis | 21 | 0.180 | 0.035 | 16 | 0.483 | 0.006 |
| Chytridiomycota | 18 | 0.205 | 0.086 | 11 | 0.036 | 0.424 |
| Blastocladiomycota | 8 | 0.575 | 0.003 | 7 | –0.086 | 0.423 |
| Glomeromycota | 4 | –0.451 | 0.963 | 5 | 0.542 | 0.052 |
Midseason in situ net ammonification and total plant biomass were selected for their significant contribution to network topology differences. rM, correlation based on Mantel test; P, significance (probability) of Mantel test. Significant P values are indicated in boldface.
DISCUSSION
Understanding the response of fungal communities to elevated atmospheric CO2 is important for global change biology. This study comprehensively surveyed soil fungal communities under aCO2 and eCO2 by 454 pyrosequencing of 28S rRNA gene amplicons and RMT-based ecological network analysis. Our results indicated several interesting mechanisms for how fungal communities respond to long-term eCO2. First, long-term eCO2 did not significantly change the overall fungal community structure and species richness, but increased fungal diversity with higher evenness of overall abundance. Second, co-occurrence network analysis suggested that fungal communities respond to long-term eCO2 by community reassembly. Third, such changed co-occurrence network topology was significantly correlated with increased soil ammonification rate and plant biomass, and OTU belonging to Ascomycota were mainly responsible for such changes. These results will provide novel insights on how the ongoing global elevated atmospheric CO2 affects the Earth's fungal community.
Our first hypothesis is that long-term eCO2 would change the fungal community structure and diversity due to changed soil and plant properties. Unexpectedly, we did not see significant changes of overall fungal community structure and species richness between aCO2 and eCO2 samples, as revealed by dissimilarity analysis. However, clear trend of separation could be observed by UniFrac PCoA that considers the phylogenetic relationship among OTU. Both taxonomic and phylogenetic diversity increased as a result of higher species evenness of overall abundance. Although no significant differences were observed at the phylum/order level, relative abundances of 119 OTU (ca. 27% of all captured sequences) were significantly different between aCO2 and eCO2 fungal communities. Interestingly, decreased and increased overall relative abundances of Ascomycota and Basidiomycota in eCO2 samples were observed for the 119 OTU. Compared to the recent study in a forest FACE site by Weber et al. (31), our results were generally consistent that eCO2 had no significant effects on high-level fungal groups when relative abundances for all OTU were considered. Our results were also consistent with a previous study that the fungal richness was not significantly affected by eCO2 (57).
Another objective of the present study was to determine the diversity and composition of fungal communities in the BioCON grassland ecosystem. The grassland soil ecosystem in the BioCON experimental site in Minnesota was dominated by Ascomycota (81% at aCO2 and 77% at eCO2) and Basidiomycota (11% at aCO2 and 14% at eCO2). Compared to the reports by previous studies (31, 58–61), fungal community composition in soil varied greatly across different types of soil ecosystems. Such variations in fungal community composition between different studies might be caused by different coverage of different primer sets or phylogenetic markers (such as ITS versus 28S) (62), but more likely caused by plant species, soil, and/or climate differences (58). Nonetheless, all of these studies suggested Ascomycota and Basidiomycota to be the dominant fungal phyla in soil ecosystems. Notably, we found that only ca. 0.03% of reads (53 OTU) were from Glomeromycota. Glomeromycota is the phylum that most arbuscular mycorrhizal fungi belong to and was previously reported to be dominant in grasslands (63) and widespread among different global ecosystems (64). Since a previous study using the same primer set identified at least 15% Glomeromycota in an Oklahoma tallgrass prairie soil, the low relative abundance of Glomeromycota identified here did not arise from the primer set used for PCR amplification, which was also verified by the NCBI primer tool (60). Since arbuscular mycorrhizal fungi form symbioses with many herbaceous land plants, the low relative abundance of Glomeromycota may result from different plant species composition in these ecosystems because fine roots were not removed prior to DNA extraction and rhizosphere soil and bulk soil were not specifically distinguished during sampling process in either study.
Our second hypothesis is that the changed fungal community would lead to more effective decomposition of soil organic matters and increase biologically available nitrogen in the soil for maintaining plant growth. To test this hypothesis, co-occurrence ecological network analysis was implemented. Ecological network analysis is a systems-level method to identify species interactions/co-occurrence within an ecosystem that cannot be directly observed (65). Similar to the food web network analyses in macroecosystems, microorganisms, including fungi, should also form complex interactions with positive or negative impacts on other species (32). “It would not be surprising to see entire patterns of community organization jumbled as a result of global change” (66). For macroecosystems, many lines of evidence have shown that global change exerts pervasive impacts on various antagonistic and mutualistic interactions among species (67). Similarly, changed co-occurrence network topology is expected for fungal communities. Our comparative analysis of fungal co-occurrence networks verified our hypothesis and indicated that long-term eCO2 affected the fungal community in the following ways. First, eCO2 increased the complexity of co-occurrence patterns within the fungal community, as evidenced by increased intermodular and intramodular connectivity, decreased geodesic distance, and decreased modularity, suggesting more intense interspecies correlations. Second, eCO2 increased negative relationships between fungal species/OTU with lower relative abundances (all <1%, except one), suggesting that increased carbon inputs into soil increased competition between less dominant fungal species. Third, eCO2 decreased the connectivity for abundant OTU. In the aCO2 network, dominant OTU formed relatively complex networks by co-occurring with other less abundant ones, while in the eCO2 network, much fewer connections were observed for the same dominant OTU. Finally, eCO2 promoted fungal community reassembly. At least 31 OTU that were sparsely distributed in different modules in the aCO2 network became connected with each other and formed dense modules in the eCO2 network, suggesting a possible community reassembly process.
Interestingly, the changed fungal network topology under eCO2 was significantly correlated with increased plant biomass and NH4+ availability in the soil. This indicated that the increased plant biomass and NH4+ availability in the soil might be the driving force for the changed network topologies, providing novel insights into how fungal communities respond to eCO2. Fungal communities are well known as decomposers in the ecosystem, by degrading organic matters into biologically available nutrients (26, 68, 69). Under eCO2, both aboveground and belowground plant biomass was stimulated (5–10), providing more organic matters for fungal communities as well as proposing higher demand for biologically available nitrogen (8, 70–72). Statistical testing suggested significant correlations between the changed network topology and increased soil ammonification rate and plant biomass. Such increased network complexity was not only observed in fungal communities. Bacterial communities responded similarly to eCO2, as revealed by both phylogenetic and functional microbial ecological networks (36, 37).
In conclusion, our study suggested that microbial fungal communities mainly responded to long-term eCO2 by community reassembly with overall community structure and species richness unchanged. Such responses were closely related with altered soil and plant properties, especially with increased plant biomass and NH4+ availability in soil, and thus were expected to sustain as long as the plant biomass is stimulated by eCO2. However, studies have shown that the microbial decomposition and plant biomass stimulation by eCO2 were constrained by limited nitrogen availability in natural soil ecosystems (69, 71, 72). Therefore, the described responses of fungal community to eCO2 may be subject to change when a new balance between microbial decomposition, plant biomass and nitrogen availability is reached.
Supplementary Material
ACKNOWLEDGMENTS
This study is supported by the U.S. Department of Agriculture (project 2007-35319-18305) through the NSF-USDA Microbial Observatories Program, by the Department of Energy under contract DE-SC0004601 through the Genomics: GTL Foundational Science, Office of Biological and Environmental Research, and by National Science Foundation grants DEB-0716587 and DEB-0620652, as well as grants DEB-0322057, DEB-0080382 (the Cedar Creek Long Term Ecological Research project), DEB-0218039, DEB-0219104, DEB-0217631, and DEB-0716587 (BioComplexity, LTER and LTREB projects), the DOE Program for Ecosystem Research, and the Minnesota Environment and Natural Resources Trust Fund.
Q.T., Z.H., and Y.D. analyzed the data. Q.T., Z.H., P.B.R., S.E.H., and J.Z. wrote the manuscript. M.Y., K.X., and L.W. performed sampling and lab experiments. All authors read and approved the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04040-14.
REFERENCES
- 1.Hawksworth DL. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105:1422–1432. doi: 10.1017/S0953756201004725. [DOI] [Google Scholar]
- 2.O'Brien HE, Parrent JL, Jackson JA, Moncalvo J-M, Vilgalys R. 2005. Fungal community analysis by large-scale sequencing of environmental samples. Appl Environ Microbiol 71:5544–5550. doi: 10.1128/AEM.71.9.5544-5550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webster J, Weber R. 2007. Introduction to fungi. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 4.Bridge P, Spooner B. 2001. Soil fungi: diversity and detection. Plant Soil 232:147–154. doi: 10.1023/A:1010346305799. [DOI] [Google Scholar]
- 5.Drake JE, Gallet-Budynek A, Hofmockel KS, Bernhardt ES, Billings SA, Jackson RB, Johnsen KS, Lichter J, McCarthy HR, McCormack ML. 2011. Increases in the flux of carbon belowground stimulate nitrogen uptake and sustain the long-term enhancement of forest productivity under elevated CO2. Ecol Lett 14:349–357. doi: 10.1111/j.1461-0248.2011.01593.x. [DOI] [PubMed] [Google Scholar]
- 6.He Z, Xu M, Deng Y, Kang S, Kellogg L, Wu L, Van Nostrand JD, Hobbie SE, Reich PB, Zhou J. 2010. Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2. Ecol Lett 13:564–575. doi: 10.1111/j.1461-0248.2010.01453.x. [DOI] [PubMed] [Google Scholar]
- 7.Langley JA, Megonigal JP. 2010. Ecosystem response to elevated CO2 levels limited by nitrogen-induced plant species shift. Nature 466:96–99. doi: 10.1038/nature09176. [DOI] [PubMed] [Google Scholar]
- 8.Reich PB, Hobbie SE. 2013. Decade-long soil nitrogen constraint on the CO2 fertilization of plant biomass. Nat Clim Change 3:278–282. [Google Scholar]
- 9.Reich PB, Knops J, Tilman D, Craine J, Ellsworth D, Tjoelker M, Lee T, Wedin D, Naeem S, Bahauddin D, Hendrey G, Jose S, Wrage K, Goth J, Bengston W. 2001. Plant diversity enhances ecosystem responses to elevated CO2 and nitrogen deposition. Nature 410:809–810. doi: 10.1038/35071062. [DOI] [PubMed] [Google Scholar]
- 10.Zak DR, Pregitzer KS, Kubiske ME, Burton AJ. 2011. Forest productivity under elevated CO2 and O3: positive feedbacks to soil N cycling sustain decade-long net primary productivity enhancement by CO2. Ecol Lett 14:1220–1226. doi: 10.1111/j.1461-0248.2011.01692.x. [DOI] [PubMed] [Google Scholar]
- 11.Blagodatskaya E, Blagodatsky S, Dorodnikov M, Kuzyakov Y. 2010. Elevated atmospheric CO2 increases microbial growth rates in soil: results of three CO2 enrichment experiments. Global Change Biol 16:836–848. doi: 10.1111/j.1365-2486.2009.02006.x. [DOI] [Google Scholar]
- 12.Castro HF, Classen AT, Austin EE, Norby RJ, Schadt CW. 2010. Soil microbial community responses to multiple experimental climate change drivers. Appl Environ Microbiol 76:999–1007. doi: 10.1128/AEM.02874-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng Y, He Z, Xu M, Qin Y, Van Nostrand JD, Wu L, Roe BA, Wiley G, Hobbie SE, Reich PB, Zhou J. 2012. Elevated carbon dioxide alters the structure of soil microbial communities. Appl Environ Microbiol 78:2991–2995. doi: 10.1128/AEM.06924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drigo B, Kowalchuk GA, Knapp BA, Pijl AS, Boschker HTS, van Veen JA. 2013. Impacts of 3 years of elevated atmospheric CO2 on rhizosphere carbon flow and microbial community dynamics. Global Change Biol 19:621–636. doi: 10.1111/gcb.12045. [DOI] [PubMed] [Google Scholar]
- 15.Drigo B, Pijl AS, Duyts H, Kielak AM, Gamper HA, Houtekamer MJ, Boschker HT, Bodelier PL, Whiteley AS, van Veen JA. 2010. Shifting carbon flow from roots into associated microbial communities in response to elevated atmospheric CO2. Proc Natl Acad Sci U S A 107:10938–10942. doi: 10.1073/pnas.0912421107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drigo B, Van Veen JA, Kowalchuk GA. 2009. Specific rhizosphere bacterial and fungal groups respond differently to elevated atmospheric CO2. ISME J 3:1204–1217. doi: 10.1038/ismej.2009.65. [DOI] [PubMed] [Google Scholar]
- 17.Feng X, Simpson AJ, Schlesinger WH, Simpson MJ. 2010. Altered microbial community structure and organic matter composition under elevated CO2 and N fertilization in the duke forest. Global Change Biol 16:2104–2116. doi: 10.1111/j.1365-2486.2009.02080.x. [DOI] [Google Scholar]
- 18.Hayden HL, Mele PM, Bougoure DS, Allan CY, Norng S, Piceno YM, Brodie EL, DeSantis TZ, Andersen GL, Williams AL, Hovenden MJ. 2012. Changes in the microbial community structure of bacteria, archaea and fungi in response to elevated CO2 and warming in an Australian native grassland soil. Environ Microbiol 14:3081–3096. doi: 10.1111/j.1462-2920.2012.02855.x. [DOI] [PubMed] [Google Scholar]
- 19.He Z, Piceno Y, Deng Y, Xu M, Lu Z, DeSantis T, Andersen G, Hobbie SE, Reich PB, Zhou J. 2012. The phylogenetic composition and structure of soil microbial communities shifts in response to elevated carbon dioxide. ISME J 6:259–272. doi: 10.1038/ismej.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S, Zak D, Van Der Lelie D. 2008. Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10:926–941. doi: 10.1111/j.1462-2920.2007.01512.x. [DOI] [PubMed] [Google Scholar]
- 21.Chung H, Zak DR, Reich PB, Ellsworth DS. 2007. Plant species richness, elevated CO2, and atmospheric nitrogen deposition alter soil microbial community composition and function. Global Change Biol 13:980–989. doi: 10.1111/j.1365-2486.2007.01313.x. [DOI] [Google Scholar]
- 22.Drigo B, Kowalchuk G, Veen J. 2008. Climate change goes underground: effects of elevated atmospheric CO2 on microbial community structure and activities in the rhizosphere. Biol Fertil Soils 44:667–679. doi: 10.1007/s00374-008-0277-3. [DOI] [Google Scholar]
- 23.Parrent JL, Vilgalys R. 2007. Biomass and compositional responses of ectomycorrhizal fungal hyphae to elevated CO2 and nitrogen fertilization. New Phytol 176:164–174. doi: 10.1111/j.1469-8137.2007.02155.x. [DOI] [PubMed] [Google Scholar]
- 24.Alberton O, Kuyper TW, Gorissen A. 2005. Taking mycocentrism seriously: mycorrhizal fungal and plant responses to elevated CO2. New Phytol 167:859–868. doi: 10.1111/j.1469-8137.2005.01458.x. [DOI] [PubMed] [Google Scholar]
- 25.Antoninka A, Reich PB, Johnson NC. 2011. Seven years of carbon dioxide enrichment, nitrogen fertilization and plant diversity influence arbuscular mycorrhizal fungi in a grassland ecosystem. New Phytol 192:200–214. doi: 10.1111/j.1469-8137.2011.03776.x. [DOI] [PubMed] [Google Scholar]
- 26.Cheng L, Booker FL, Tu C, Burkey KO, Zhou L, Shew HD, Rufty TW, Hu S. 2012. Arbuscular mycorrhizal fungi increase organic carbon decomposition under elevated CO2. Science 337:1084–1087. doi: 10.1126/science.1224304. [DOI] [PubMed] [Google Scholar]
- 27.van der Heijden MG, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, Wiemken A, Sanders IR. 1998. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 396:69–72. doi: 10.1038/23932. [DOI] [Google Scholar]
- 28.Phillips RP, Meier IC, Bernhardt ES, Grandy AS, Wickings K, Finzi AC. 2012. Roots and fungi accelerate carbon and nitrogen cycling in forests exposed to elevated CO2. Ecol Lett 15:1042–1049. doi: 10.1111/j.1461-0248.2012.01827.x. [DOI] [PubMed] [Google Scholar]
- 29.Verbruggen E, Veresoglou SD, Anderson IC, Caruso T, Hammer EC, Kohler J, Rillig MC. 2013. Arbuscular mycorrhizal fungi–short-term liability but long-term benefits for soil carbon storage? New Phytol 197:366–368. doi: 10.1111/nph.12079. [DOI] [PubMed] [Google Scholar]
- 30.Edwards IP, Zak DR. 2011. Fungal community composition and function after long-term exposure of northern forests to elevated atmospheric CO2 and tropospheric O3. Global Change Biol 17:2184–2195. doi: 10.1111/j.1365-2486.2010.02376.x. [DOI] [Google Scholar]
- 31.Weber CF, Vilgalys R, Kuske CR. 2013. Changes in fungal community composition in response to elevated atmospheric CO2 and nitrogen fertilization varies with soil horizon. Front Microbiol 4:78. doi: 10.3389/fmicb.2013.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 33.Barberan A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. 2012. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steele JA, Countway PD, Xia L, Vigil PD, Beman JM, Kim DY, Chow C-ET, Sachdeva R, Jones AC, Schwalbach MS, Rose JM, Hewson I, Patel A, Sun F, Caron DA, Fuhrman JA. 2011. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J 5:1414–1425. doi: 10.1038/ismej.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Deng Y, Luo F, He Z, Tu Q, Zhi X. 2010. Functional molecular ecological networks. mBio 1(4):e00169-10. doi: 10.1128/mBio.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J, Deng Y, Luo F, He Z, Yang Y. 2011. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2:e00122-11. doi: 10.1128/mBio.00122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng Y, Jiang Y-H, Yang Y, He Z, Luo F, Zhou J. 2012. Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn SJ, Costa J, Rettig Emanuel J. 1996. PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res 24:2623–2625. doi: 10.1093/nar/24.13.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu K-L, Porras-Alfaro A, Kuske CR, Eichorst SA, Xie G. 2012. Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes. Appl Environ Microbiol 78:1523–1533. doi: 10.1128/AEM.06826-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 43.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned rRNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedges LV, Gurevitch J, Curtis PS. 1999. The meta-analysis of response ratios in experimental ecology. Ecology 80:1150–1156. doi: 10.1890/0012-9658(1999)080[1150:TMAORR]2.0.CO;2. [DOI] [Google Scholar]
- 47.Hamady M, Lozupone C, Knight R. 2009. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin L, Ji Y, Tu Q, Huang R, Teng L, Zeng X, Song H, Wang K, Zhou Q, Li Y. 2013. Microevolution from shock to adaptation revealed strategies improving ethanol tolerance and production in Thermoanaerobacter. Biotechnol Biofuels 6:103. doi: 10.1186/1754-6834-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin L, Song H, Tu Q, Qin Y, Zhou A, Liu W, He Z, Zhou J, Xu J. 2011. The Thermoanaerobacter glycobiome reveals mechanisms of pentose and hexose co-utilization in bacteria. PLoS Genet 7:e1002318. doi: 10.1371/journal.pgen.1002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo F, Yang Y, Zhong J, Gao H, Khan L, Thompson DK, Zhou J. 2007. Constructing gene coexpression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinformatics 8:299. doi: 10.1186/1471-2105-8-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y, Harris DP, Luo F, Wu L, Parsons AB, Palumbo AV, Zhou J. 2008. Characterization of the Shewanella oneidensis Fur gene: roles in iron and acid tolerance response. BMC Genomics 9:S11. doi: 10.1186/1471-2164-9-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou A, He Z, Redding-Johanson AM, Mukhopadhyay A, Hemme CL, Joachimiak MP, Luo F, Deng Y, Bender KS, He Q. 2010. Hydrogen peroxide-induced oxidative stress responses in Desulfovibrio vulgaris Hildenborough. Environ Microbiol 12:2645–2657. doi: 10.1038/ismej.2009.97. [DOI] [PubMed] [Google Scholar]
- 54.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill MO. 1973. Diversity and evenness: a unifying notation and its consequences. Ecology 54:427–432. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 56.Vane-Wright RI, Humphries CJ, Williams PH. 1991. What to protect? Systematics and the agony of choice. Biol Conserv 55:235–254. doi: 10.1016/0006-3207(91)90030-D. [DOI] [Google Scholar]
- 57.Parrent JL, Morris WF, Vilgalys R. 2006. CO2-Enrichment and nutrient availability alter ectomycorrhizal fungal communities. Ecology 87:2278–2287. doi: 10.1890/0012-9658(2006)87[2278:CANAAE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 58.Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F. 2009. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184:449–456. doi: 10.1111/j.1469-8137.2009.03003.x. [DOI] [PubMed] [Google Scholar]
- 59.Jumpponen A, Jones KL, Blair J. 2010. Vertical distribution of fungal communities in tallgrass prairie soil. Mycologia 102:1027–1041. doi: 10.3852/09-316. [DOI] [PubMed] [Google Scholar]
- 60.Penton CR, St Louis D, Cole JR, Luo Y, Wu L, Schuur EA, Zhou J, Tiedje JM. 2013. Fungal diversity in permafrost and tallgrass prairie soils under experimental warming. Appl Environ Microbiol 79:7063–7072. doi: 10.1128/AEM.01702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu L, Ravnskov S, Larsen J, Nicolaisen M. 2012. Linking fungal communities in roots, rhizosphere, and soil to the health status of Pisum sativum. FEMS Microbiol Ecol 82:736–745. doi: 10.1111/j.1574-6941.2012.01445.x. [DOI] [PubMed] [Google Scholar]
- 62.Toju H, Tanabe AS, Yamamoto S, Sato H. 2012. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One 7:e40863. doi: 10.1371/journal.pone.0040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santos-González JC, Finlay RD, Tehler A. 2007. Seasonal dynamics of arbuscular mycorrhizal fungal communities in roots in a seminatural grassland. Appl Environ Microbiol 73:5613–5623. doi: 10.1128/AEM.00262-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.ÖPik M, Moora M, Liira J, Zobel M. 2006. Composition of root-colonizing arbuscular mycorrhizal fungal communities in different ecosystems around the globe. J Ecol 94:778–790. doi: 10.1111/j.1365-2745.2006.01136.x. [DOI] [Google Scholar]
- 65.Fath BD, Scharler UM, Ulanowicz RE, Hannon B. 2007. Ecological network analysis: network construction. Ecol Modeling 208:49–55. doi: 10.1016/j.ecolmodel.2007.04.029. [DOI] [Google Scholar]
- 66.Kareiva PM, Kingsolver JG, Huey RB. 1993. Biotic interactions and global change. Sinauer Associates, Sunderland, MA. [Google Scholar]
- 67.Tylianakis JM, Didham RK, Bascompte J, Wardle DA. 2008. Global change and species interactions in terrestrial ecosystems. Ecol Lett 11:1351–1363. doi: 10.1111/j.1461-0248.2008.01250.x. [DOI] [PubMed] [Google Scholar]
- 68.Chung H, Zak D, Lilleskov E. 2006. Fungal community composition and metabolism under elevated CO2 and O3. Oecologia 147:143–154. doi: 10.1007/s00442-005-0249-3. [DOI] [PubMed] [Google Scholar]
- 69.Hu S, Chapin FS, Firestone MK, Field CB, Chiariello NR. 2001. Nitrogen limitation of microbial decomposition in a grassland under elevated CO2. Nature 409:188–191. doi: 10.1038/35051576. [DOI] [PubMed] [Google Scholar]
- 70.Luo Y, Hui D, Zhang D. 2006. Elevated CO2 stimulates net accumulations of carbon and nitrogen in land ecosystems: a meta-analysis. Ecology 87:53–63. doi: 10.1890/04-1724. [DOI] [PubMed] [Google Scholar]
- 71.Luo Y, Su BO, Currie WS, Dukes JS, Finzi A, Hartwig U, Hungate B, Murtrie REM, Oren RAM, Parton WJ, Pataki DE, Shaw MR, Zak DR, Field CB. 2004. Progressive nitrogen limitation of ecosystem responses to rising atmospheric carbon dioxide. Bioscience 54:731–739. doi: 10.1641/0006-3568(2004)054[0731:PNLOER]2.0.CO;2. [DOI] [Google Scholar]
- 72.Reich PB, Hobbie SE, Lee T, Ellsworth DS, West JB, Tilman D, Knops JM, Naeem S, Trost J. 2006. Nitrogen limitation constrains sustainability of ecosystem response to CO2. Nature 440:922–925. doi: 10.1038/nature04486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.