

Abstract
Microbial production of propionic acid (PA), an important chemical building block used as a preservative and chemical intermediate, has gained increasing attention for its environmental friendliness over traditional petrochemical processes. In previous studies, we constructed a shuttle vector as a useful tool for engineering Propionibacterium jensenii, a potential candidate for efficient PA synthesis. In this study, we identified the key metabolites for PA synthesis in P. jensenii by examining the influence of metabolic intermediate addition on PA synthesis with glycerol as a carbon source under anaerobic conditions. We also further improved PA production via the overexpression of the identified corresponding enzymes, namely, glycerol dehydrogenase (GDH), malate dehydrogenase (MDH), and fumarate hydratase (FUM). Compared to those in wild-type P. jensenii, the activities of these enzymes in the engineered strains were 2.91- ± 0.17- to 8.12- ± 0.37-fold higher. The transcription levels of the corresponding enzymes in the engineered strains were 2.85- ± 0.19- to 8.07- ± 0.63-fold higher than those in the wild type. The coexpression of GDH and MDH increased the PA titer from 26.95 ± 1.21 g/liter in wild-type P. jensenii to 39.43 ± 1.90 g/liter in the engineered strains. This study identified the key metabolic nodes limiting PA overproduction in P. jensenii and further improved PA titers via the coexpression of GDH and MDH, making the engineered P. jensenii strain a potential industrial producer of PA.
INTRODUCTION
Propionic acid (PA), a valuable C3 platform chemical, has many industrial applications, mainly as a chemical intermediate in the synthesis of cellulose fiber, herbicides, perfumes, and pharmaceuticals (1, 2). It is also an important mold inhibitor, and its ammonia, calcium, sodium, and potassium salts are widely used as preservatives in animal feed and human foods. According to the U.S. Department of Energy, PA is among the top 30 candidate platform chemicals in use (3). Currently, PA is primarily produced via oxo synthesis using a petrochemical route (4). However, the production of biobased PA from renewable resources using Propionibacterium species has attracted increasing attention due to the escalating prices of petroleum resources and concerns about diminishing oil supplies and the serious environmental pollution caused by petroleum refining. The genus Propionibacterium is a class of Gram-positive organisms and is divided into dairy propionibacteria (isolated mainly from dairy products) and cutaneous propionibacteria (typically found on skin and known human pathogens). Propionibacterium acidipropionici (5–13), P. freudenreichii (14), P. thoenii (15), and P. jensenii (16) are dairy propionibacteria and have been used for PA production, and research has focused mainly on process optimization for improved PA titers and productivity.
Although several endogenous plasmids have been found in Propionibacterium (16–20), very little metabolic engineering of Propionibacterium has been carried out. Metabolic engineering in these bacteria progresses slowly because of their thick cell walls, strong restriction-modification systems, and high GC content and a lack of detailed genome information (21). The metabolic engineering of Propionibacterium is of great interest, particularly because dairy propionibacteria have been granted generally recognized as safe status by the U.S. Food and Drug Administration (22). To date, only three studies have undertaken improved PA production via metabolic engineering, namely, acetate kinase gene knockout in P. acidipropionici (11), glycerol dehydrogenase (GDH) overexpression in P. jensenii (16), and phosphoenolpyruvate carboxylase expression in P. freudenreichii (23). Figure 1 shows the metabolic pathway of PA from glycerol in Propionibacterium under anaerobic conditions. Due to its abundance, low price, and high degree of reduction, glycerol is an ideal carbon source for the production of PA by microbial fermentation (24). In general, glycerol is first converted to pyruvate, which is then converted to PA, lactic acid (LA), acetic acid (AA), succinate, propanol, and cell biomass. The PA biosynthesis pathway from glycerol can be divided into two steps, with the catalysis of 10 enzymes. In the first step (Embden-Meyerhof-Parnas pathway), glycerol is converted to dihydroxyacetone by GDH, and dihydroxyacetone is further converted to dihydroxyacetone phosphate and pyruvate. This step is an NADH-forming pathway. The second step (Wood-Werkman cycle) proceeds under anaerobic conditions. Pyruvate is converted to oxaloacetate and then to malate by malate dehydrogenase (MDH). Fumarate hydratase (FUM) catalyzes the conversion of malate to fumarate, which is converted to PA via a series of intermediates, namely, succinate, succinyl coenzyme A (CoA), methylmalonyl CoA, and propionyl CoA (25). This step is an NADH-consuming pathway. Although the synthesis pathway of PA is known, the key metabolic nodes or pathways limiting PA overproduction remain unclear.
FIG 1.

Metabolic pathways for propionic acid, lactic acid, and acetic acid biosynthesis from glycerol in Propionibacterium. Engineered biosynthesis steps by the overexpression of the corresponding genes are indicated by color blocks. The arrows with dotted lines represent multiple reactions. Lactic acid and acetic acid are the main by-products in propionic acid production. EMP, Embden-Meyerhof-Parnas; DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; TCA, tricarboxylic acid; FP, flavoprotein; FPH3, flavoprotein-H3.
In previous studies, we constructed a novel endogenous plasmid, named pZGX01, and a shuttle vector, designated pZGX04, between Propionibacterium and Escherichia coli as tools for engineering P. jensenii to produce PA efficiently (16). The present study aimed to identify the key pathways for PA synthesis in P. jensenii and then overexpress the relevant enzymes to improve the PA titer. Three enzymes, GDH (which converts glycerol to dihydroxyacetone), MDH (which converts oxaloacetate to malate with NADH oxidation), and FUM (which converts malate to fumarate), were identified as the metabolic flux bottlenecks by measurement of the effects of intermediate addition on PA production. The corresponding genes gldA (encoding GDH), mdh (encoding MDH), and fumC (encoding FUM) from Klebsiella pneumoniae were then expressed singly and combinatorially to study their influence on cell growth, by-product formation, and PA production.
MATERIALS AND METHODS
Strains, plasmids, and media.
P. jensenii ATCC 4868 was used as the host for gene expression and PA production. The DNA isolated from K. pneumoniae subsp. pneumoniae ATCC 12657 was used as a template to obtain mdh and fumC. E. coli JM110 (Stratagene, La Jolla, CA) was used as a host for all recombinant plasmid cloning and maintenance. P. jensenii was cultured anaerobically at 32°C in sodium lactate broth medium containing 10 g/liter sodium lactate, 10 g/liter yeast extract, and 10 g/liter Trypticase soy broth. K. pneumoniae and E. coli were grown at 37°C in Luria-Bertani medium containing 10 g/liter NaCl, 10 g/liter peptone, and 5 g/liter yeast extract. When necessary, the medium was supplemented with 10 μg/ml chloramphenicol for P. jensenii culture and 100 μg/ml ampicillin for E. coli culture. The bacterial strains and plasmids used are listed in Table 1, and detailed maps of the constructed plasmids are shown in Fig. 2.
TABLE 1.
Strains and plasmids used in this studya
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli JM110 | dam and dcm mutant; host for vector preparation | Stratagene |
| K. pneumoniae subsp. pneumoniae ATCC 12657 | Genomic DNA for gldA, mdh, and fumC | ATCC |
| P. jensenii ATCC 4868 | Host for gene expression and PA production | ATCC |
| Plasmids | ||
| pZGX04 | Shuttle vector between P. jensenii and E. coli; Ampr Cmr | 13 |
| pZGX04-gldA | gldA-overexpressing plasmid | 13 |
| pZGX04-mdh | mdh-overexpressing plasmid | This work |
| pZGX04-fumC | fumC-overexpressing plasmid | This work |
| pZGX04-gldA-mdh | gldA- and mdh-overexpressing plasmid | This work |
| pZGX04-gldA-fumC | gldA- and fumC-overexpressing plasmid | This work |
| pZGX04-mdh-fumC | mdh- and fumC-overexpressing plasmid | This work |
Ampr, ampicillin selection; Cmr, chloramphenicol selection; ATCC, American Type Culture Collection, Manassas, VA, USA.
FIG 2.

Plasmid maps of the shuttle vector and expression vectors pZGX04, pZGX04-mdh, pZGX04-fumC, pZGX04-gldA-mdh, pZGX04-gldA-fumC, and pZGX04-mdh-fumC. Shuttle vector pZGX04 was constructed by using plasmids pZGX01 and pUC18 and the chloramphenicol resistance gene. The other vectors were constructed based on pZGX04.
Plasmid construction and transformation.
The DNA of K. pneumoniae subsp. pneumoniae ATCC 12657 was extracted with a Uniq-10 column bacterial genomic DNA isolation kit (Sangon, Shanghai, China) and used as a template to amplify mdh and fumC. The oligonucleotides used in the amplification reactions are listed in Table 2. The PCR mixture (50 μl) was prepared from 10 μl 5× PrimerSTAR buffer (TaKaRa, Dalian, China), 4 μl of a 2.5 mM deoxyribonucleotide triphosphate mixture, 2 μl primer mix (10 μM each), 1 μl template DNA, and 0.5 μl DNA polymerase. PCR amplification was performed with a C1000 thermal cycler (Bio-Rad, Hercules, CA) for 30 thermal cycles under the following conditions: an initial denaturation step at 98°C for 15 s, an annealing step at 56°C to 60°C for 15 s, and an extension step at 72°C for 1 to 8.5 min (Table 2). The PCR products were purified by using a TaKaRa MiniBEST agarose gel DNA extraction kit (TaKaRa). The shuttle vector (pZGX04) and expression vector (pZGX04-gldA) used in these experiments were constructed in a previous study (13). The original promoters of pZGX04 and pZGX04-gldA were maintained in the construction of the vectors described below and used to express the target genes.
TABLE 2.
Oligonucleotides and conditions used for PCR in this study
| Primer target (gene) or name | Nucleotide sequence (5′→3′) | Annealing temp (°C) | Extension time (min) |
|---|---|---|---|
| mdh | ATGAAAGTTGCAGTCCTTGGCG | 58 | 1 |
| TTACTTGTTGACGAAGTCTTCGCC | |||
| fumC | ATGACAACGCATCGCAGTGAAAAA | 60 | 1.5 |
| CTAGCGAATAGCCAGGCTGCCG | |||
| pZGX04-orf2 | CGGGGTACCTGGACGATTGAGTACACCGAC | 56 | 8 |
| CCATCGATCATCACGCACCTCCTGACTAC | |||
| pZGX04-orf4 | GACCCACCCTCCCATTCTGTACCC | 60 | 8.5 |
| CATCCTCAGTGCTCGGTGGGG |
The 0.9-kb mdh and 1.4-kb fumC fragments were amplified from K. pneumoniae subsp. pneumoniae ATCC 12657 genomic DNA. We replaced orf2 in pZGX04 with fumC and mdh to construct pZGX04-fumC and pZGX04-mdh, respectively. The original promoter of orf2 in pZGX04 was maintained in the construction of the pZGX04-fumC and pZGX04-mdh vectors and used to express fumC and mdh. Thus, the expression of these genes in the transformants was controlled by the original promoter of orf2 in pZGX04. Vectors pZGX04-mdh-fumC and pZGX04-gldA-fumC were constructed by using fumC in place of orf4 in pZGX04-mdh and pZGX04-gldA, respectively. The construction of pZGX04-gldA-mdh was done according to the same method. The promoter of orf4 was used to express mdh. The constructed vectors were transformed into E. coli JM110, a methylation-deficient strain, and the demethylated vectors were then transformed into P. jensenii ATCC 4868. The preparation of competent cells and transformation into P. jensenii were performed as described previously (16).
Reverse transcription-qPCR (RT-qPCR).
Total RNA was extracted from P. jensenii by using an RNAiso Plus kit (TaKaRa) when the optical density at 600 nm reached 0.6 to 0.7 in sodium lactate broth medium. The total RNA was then treated with a PrimeScript II first-strand cDNA synthesis kit (TaKaRa) to obtain cDNA, which was used for quantitative PCR (qPCR) with a LightCycler 2.0 system (Roche, Basel, Switzerland) and SYBR Premix Ex Taq (TaKaRa).
Cultivation of P. jensenii for PA production.
An inoculum was prepared in 100-ml anaerobic jars containing 100 ml of seed medium with 10 g/liter yeast extract, 5 g/liter Trypticase soy broth, 2.5 g/liter K2HPO4, and 1.5 g/liter KH2PO4 supplemented with 10 μg/ml chloramphenicol. The jars were sealed with butyl rubber caps, the air was evacuated, and the cultures were incubated for 48 h at 32°C. Intermediate addition experiments were carried out with 100-ml anaerobic jars containing 50 ml culture medium (25 g/liter glycerol, 10 g/liter yeast extract, 5 g/liter peptone, 2.5 g/liter K2HPO4, 1.5 g/liter KH2PO4, and 8 mg/liter CoCl2) with 30 g/liter CaCO3 to maintain the pH. Inocula (10%) were transferred to the culture jars for 160 h at 32°C.
Three-liter anaerobic cultures were carried out according to a protocol developed in our previous study (26). The inoculum was prepared in 250-ml anaerobic jars containing 200 ml of seed medium for 48 h at 32°C. Fed-batch cultivations were performed in a 3-liter Bioflo 115 bioreactor (Eppendorf, Hamburg, Germany) containing 2 liters of culture medium under anaerobic conditions with nitrogen gas to remove any traces of oxygen. The medium was autoclaved at 121°C for 21 min. The culture temperature and agitation speed were maintained at 32°C and 200 rpm, respectively. A two-stage pH control strategy was undertaken via the automatic addition of Ca(OH)2 (10% [wt/vol]): the pH was controlled at 5.9 for 0 to 36 h and then shifted to 6.5 after 36 h. Concentrated glycerol (300 g/liter) was fed at a constant rate of 3.33 ml/h between 60 and 132 h of culture.
Analytical methods.
Ten-milliliter aliquots were drawn from the bioreactor every 12 h to determine cell, organic acid, and glycerol concentrations and GDH, MDH, and FUM activities. The influence of sampling volume was negligible, as the sampling volume relative to the culture volume was rather minimal. The change of volume caused by the addition of the Ca(OH)2 solution was removed for calculation. Methods for determining cell, organic acid, and glycerol concentrations and GDH activity were described previously (16).
The samples were centrifuged at 10,000 × g for 5 min, and the cell pellets were washed with 0.5 ml of 50 mM potassium phosphate buffer (pH 7.5). The cell suspension was centrifuged at 10,000 × g at 4°C for 15 min after the cell walls were disrupted by ultrasonication. The supernatant was used to determine the activities of GDH, MDH, and FUM with a UV-2450 temperature-controlled spectrophotometer (Shimadzu, Kyoto, Japan). A control experiment was conducted to eliminate interference, and the linear square of the activity to the amount of extract was >0.99.
MDH activity was measured at 340 nm and at 30°C. The standard assay mixture (1 ml) contained 60 mM sodium phosphate-potassium phosphate buffer (pH 7.5), 0.2 mM NADH, and 0.3 mM oxaloacetate, and the reaction was started by adding the enzyme solution to the mixture. One unit of MDH activity was defined as the amount of enzyme required to consume 1 μM NADH per minute.
FUM activity was determined at 250 nm and at 30°C. The standard assay mixture (1 ml) contained 50 mM potassium phosphate buffer (pH 7.5) and 50 mM l-malate, and the reaction was started by adding the enzyme solution to the mixture (ε = 1.45 mM−1). One unit of FUM activity was defined as the amount of enzyme required to consume 1 μM l-malate per minute. The fed-batch culture parameters studied included YPA/DCW (ratio of the concentration of PA/concentration of dry cell weight [DCW]), YPA/LA (ratio of the concentration of PA/concentration of LA), YPA/AA (ratio of the concentration of PA/concentration of AA), and YPA/glycerol (ratio of the concentration of PA/concentration of glycerol consumed).
Statistical analysis.
Unless otherwise noted, all experiments were performed independently at least three times, and average values with standard errors are reported. Student's t test was performed by using SPSS 12.0 software (SPSS Inc., Chicago, IL, USA). P values of <0.05 were considered statistically significant.
RESULTS
Effects of intermediate addition on PA production by P. jensenii.
Eight intermediates (dihydroxyacetone, phosphoenolpyruvate, pyruvate, oxaloacetate, malate, fumarate, succinate, and acetate) involved in the PA synthesis pathway were chosen, and their influence on PA production was evaluated. The results indicated that the addition of malate, fumarate, and dihydroxyacetone significantly improved PA production, and the effects of the addition of these intermediates on cell growth and LA, AA, and PA formation are shown in Fig. 3. The addition of phosphoenolpyruvate, pyruvate, oxaloacetate, succinate, or acetate at a proper concentration can improve PA production to a certain degree. Especially, the addition of malate, fumarate, and dihydroxyacetone at proper concentrations accelerated cell growth, improved the PA titer, and reduced the formation of main by-products significantly. PA production increased by 57.1% ± 4.3%, 37.3% ± 3.5%, and 31.9% ± 3.1% with the addition of malate, fumarate, and dihydroxyacetone at 42 mM, 20 mM, and 17 mM, respectively. The improved PA titers indicated that malate, fumarate, and dihydroxyacetone are crucial for the synthesis of PA and that the heterologous expression of the three corresponding enzymes in P. jensenii ATCC 4868, MDH, FUM, and GDH, may increase PA production.
FIG 3.

Effects of addition of intermediates with different concentrations on propionic acid production. Intermediate addition experiments were carried out in 100-ml anaerobic jars containing 50 ml culture medium with 30 g/liter CaCO3. Inocula (10%) were transferred to the culture jars for 160 h at 32°C. Black columns, dry cell weight (DCW); red columns, lactic acid (LA); blue columns, acetic acid (AA); purple columns, propionic acid (PA).
Overexpression of fumC and mdh in P. jensenii ATCC 4868.
The lack of information on the genome and codon bias of Propionibacterium was a barrier to genetic engineering. So far, there is no information about the genome of P. jensenii. Although several strains of other species were sequenced, the genes are difficult to manipulate due to the high GC content of the genes. Thus, we aimed to find exogenous genes that can be used in P. jensenii. Some reports declared that attempts to express exogenous genes from some other bacteria failed and that there might be a special codon bias in P. jensenii (18). The genes from Klebsiella pneumoniae were found to be expressed in P. jensenii successfully, so we expressed fumC and mdh genes that were cloned from K. pneumoniae in P. jensenii ATCC 4868, and the expression of these genes in the transformants was controlled by the original promoter of orf2, which came from pZGX01 (16). The two corresponding vectors were transformed into E. coli JM110 for cloning and preservation. Finally, the two demethylated vectors were transformed into P. jensenii ATCC 4868 to construct the engineered strains. The expression of gldA was described in our previous study (16).
Three combinations, mdh and fumC, gldA and fumC, and gldA and mdh, were coexpressed in P. jensenii ATCC 4868. The expression of these genes in the transformants was controlled by the original promoters of orf2 and orf4. orf2 and orf4 are the two open reading frames (ORFs) in wild plasmid pZGX01, which was screened in our previous work, and the analysis revealed that they had relatively high transcription levels (16). Therefore, their corresponding promoters were deduced to have a high efficiency.
The results of RT-qPCR analysis revealed that all three genes could be transcribed to the corresponding mRNAs. As shown in Fig. 4, the changes in the transcription levels of the three genes relative to those of the corresponding genes in the wild type ranged from 2.85- ± 0.19- to 8.07- ± 0.63-fold. The transcription levels of the genes at the position of orf2 were higher than those of the genes at the position of orf4, indicating that the promoter of orf2 had a higher efficiency than the promoter of orf4. These results showed that all vectors in the engineered strains can be successfully transcribed.
FIG 4.
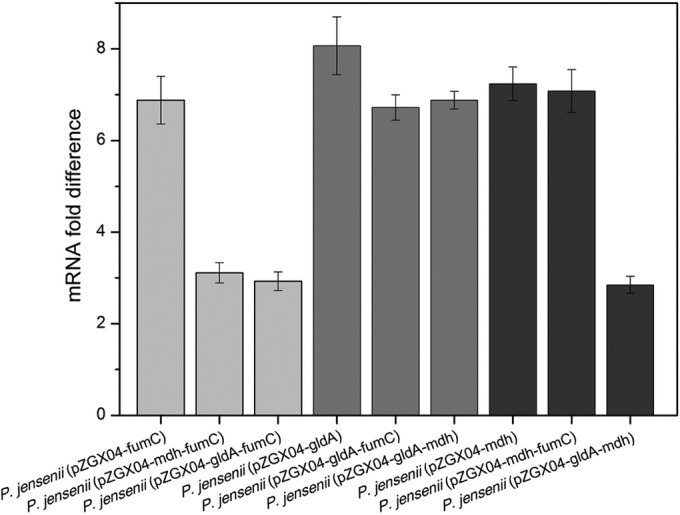
Transcription levels of fumC, gldA, and mdh relative to those of the corresponding genes in Propionibacterium jensenii ATCC 4868. The total RNA extracted from cells was reverse transcribed to cDNA. The obtained cDNA was subjected to quantitative PCR, and fumC, gldA, and mdh in the wild type were set as the guide sample. Light gray bars, fumC; gray bars, gldA; dark gray bars, mdh.
In fed-batch cultivation, samples were obtained after 48 h to analyze GDH, MDH, and FUM activities. Figure 5 shows the specific activities of GDH, MDH, and FUM in wild-type P. jensenii and the engineered strains. Compared with those in the wild type, the specific FUM, GDH, and MDH activities in the engineered strains increased by 2.91- ± 0.17- to 8.12- ± 0.37-fold. This increased activity is attributed to the higher copy number of the genes in the corresponding engineered strains.
FIG 5.
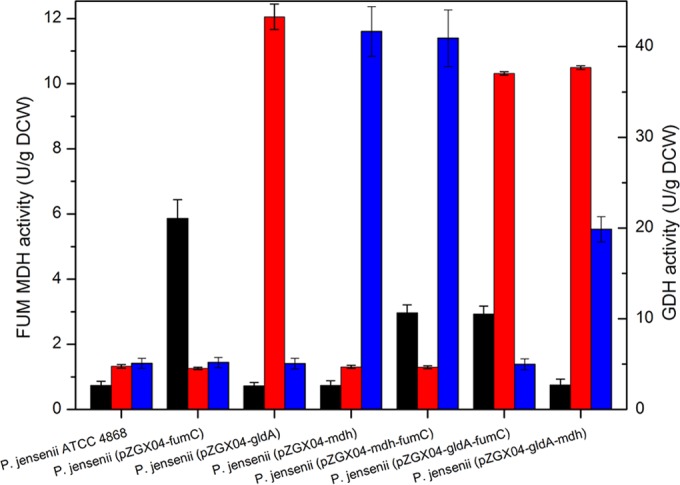
Comparison of specific enzyme activities in wild-type Propionibacterium jensenii ATCC 4868 and transformants overexpressing the gldA, mdh, and fumC genes. In fed-batch cultures of wild-type P. jensenii ATCC 4868 and transformants, samples were obtained after 48 h to analyze GDH, MDH, and FUM activities. Black columns, FUM activity; red columns, GDH activity; blue columns, MDH activity.
Microbial production of PA by engineered P. jensenii strains.
To evaluate the effects of gene overexpression on PA synthesis in P. jensenii, we studied fed-batch cultures of the wild-type and engineered strains in a 3-liter bioreactor under optimized culture conditions (26). The effects of gldA gene overexpression in the engineered P. jensenii(pZGX04-gldA) strain were described in our previous study (16). The results of analysis of the fed-batch culture parameters are summarized in Table 3, and Fig. 6 shows the culture kinetics of the wild-type and engineered strains. The wild-type strain produced PA (26.95 ± 1.21 g/liter) as the main product, with LA (2.86 ± 0.17 g/liter) and AA (2.44 ± 0.14 g/liter) as the two main by-products (Fig. 6A), and DCW reached 3.55 g/liter. The six engineered strains produced more PA and smaller amounts of by-products than did the wild type. However, compared to the wild type, the DCW dropped significantly in the six engineered strains (Fig. 6) under the same cultivation conditions. The lowest DCW decreased to 3.16 ± 0.11 g/liter, compared to 3.55 ± 0.19 g/liter for the wild type. The engineered strains grew poorly, perhaps due to the extra burden of plasmid replication, transcription, and expression.
TABLE 3.
Analysis of fed-batch culture parameters for PA production with different strainsa
| Strain | Mean DCW (g/liter) ± SD | Mean YPA/DCW (g/g) ± SD | Mean YPA/LA (g/g) ± SD | Mean YPA/AA (g/g) ± SD | Mean YPA/glycerol (g/g) ± SD | Mean productivity (g · liter−1 · h−1) ± SD |
|---|---|---|---|---|---|---|
| P. jensenii ATCC 4868 | 3.55 ± 0.19 | 7.59 ± 0.46 | 9.42 ± 0.38 | 11.05 ± 0.48 | 0.44 ± 0.019 | 0.118 ± 0.005 |
| P. jensenii(pZGX04-fumC) | 3.34 ± 0.17 | 10.34 ± 0.52 | 12.28 ± 0.46 | 14.38 ± 0.67 | 0.57 ± 0.022 | 0.151 ± 0.005 |
| P. jensenii(pZGX04-gldA) | 3.42 ± 0.19 | 10.12 ± 0.48 | 11.46 ± 0.53 | 13.26 ± 0.65 | 0.57 ± 0.028 | 0.152 ± 0.006 |
| P. jensenii(pZGX04-mdh) | 3.35 ± 0.16 | 10.77 ± 0.55 | 13.12 ± 0.61 | 15.23 ± 0.49 | 0.59 ± 0.030 | 0.158 ± 0.006 |
| P. jensenii(pZGX04-mdh-fumC) | 3.16 ± 0.11 | 11.66 ± 0.62 | 13.91 ± 0.66 | 15.82 ± 0.69 | 0.60 ± 0.027 | 0.162 ± 0.007 |
| P. jensenii(pZGX04-gldA-fumC) | 3.26 ± 0.12 | 11.51 ± 0.41 | 13.16 ± 0.45 | 14.95 ± 0.54 | 0.62 ± 0.031 | 0.165 ± 0.007 |
| P. jensenii(pZGX04-gldA-mdh) | 3.28 ± 0.13 | 12.02 ± 0.68 | 14.18 ± 0.57 | 15.65 ± 0.57 | 0.65 ± 0.036 | 0.173 ± 0.008 |
YPA/DCW, mass ratio of PA to DCW; YPA/LA, mass ratio of PA to LA; YPA/AA, mass ratio of PA to AA; YPA/glycerol, mass ratio of PA to glycerol.
FIG 6.

Fed-batch culture kinetics of propionic acid production from glycerol with wild-type Propionibacterium jensenii ATCC 4868 and transformants with single and combinatorial expression of the gldA, mdh, and fumC genes. (A) P. jensenii ATCC 4868; (B) P. jensenii(pZGX04-fumC); (C) P. jensenii(pZGX04-mdh); (D) P. jensenii(pZGX04-mdh-fumC); (E) P. jensenii(pZGX04-gldA-fumC); (F) P. jensenii(pZGX04-gldA-mdh). The culture temperature and agitation speed were maintained at 32°C and 200 rpm, respectively. A two-stage pH control strategy was carried out via the automatic addition of Ca(OH)2 (10% [wt/vol]) as follows: the pH was controlled at 5.9 for 0 to 36 h and was shifted to 6.5 after 36 h. Concentrated glycerol (300 g/liter) was fed at a constant rate of 3.33 ml/h between 60 and 132 h of the culture process. Samples were taken from the fermentor every 12 h. △, PA; □, residual glycerol; ■, DCW; ◆, AA; ▲, LA.
Expressed singly, mdh showed a more important role in PA production than those of the other two genes (Fig. 6C). The PA production and productivity of P. jensenii(pZGX04-mdh) reached 36.09 ± 1.53 g/liter and 0.158 ± 0.0069 g · liter−1 · h−1, respectively. The productivity of P. jensenii(pZGX04-mdh) reached 10.77 ± 0.55 g · liter−1 · g−1 DCW, compared to 7.59 ± 0.46 g · liter−1 · g−1 DCW for the wild type. Compared with the single expression of the three genes, the coexpression of two genes in P. jensenii led to higher levels of PA production and productivity. Coexpression of gldA and mdh in P. jensenii(pZGX04-gldA-mdh) resulted in greater production of PA than that of the other engineered strains (Fig. 6F). PA production and productivity reached 39.43 ± 1.90 g/liter and 0.173 ± 0.008 g · liter−1 · h−1, respectively. The productivity of P. jensenii(pZGX04-gldA-mdh) reached 12.02 ± 0.68 g · liter−1 · g−1 DCW, compared to 7.59 ± 0.46 g · liter−1 · g−1 DCW for the wild type.
DISCUSSION
P. jensenii produces PA through the dicarboxylic acid pathway, with LA and AA as by-products (16). Several genomes of Propionibacterium have now been sequenced (3, 25, 27–30). However, genetic engineering of these bacteria has not been thoroughly investigated because of the difficulty in transforming Gram-positive bacteria, the various restriction-modification systems of these bacteria, the high GC content in their genomes, and the lack of cloning tools. To date, several shuttle vectors have been developed by using endogenous plasmids of Propionibacterium (16–20, 31). However, only three reports have focused on improving PA production through metabolic engineering (11, 16, 23).
In a recent study, we constructed a heterologous gene expression vector, pZGX04, and developed a transformation system in P. jensenii (16). pZGX04 is the only reported vector that can be used for gene manipulation in P. jensenii. In the present study, we further improved PA production in P. jensenii ATCC 4868 through metabolic engineering based on our transformation system. By examining the influences of different intermediates on PA yield, we detected three potentially rate-limiting enzymes in the PA biosynthesis pathway from glycerol: FUM, GDH, and MDH.
To verify the results described above and improve PA production, we overexpressed these enzymes in P. jensenii. A native promoter is required to construct the heterologous gene expression vector in Propionibacterium (32). We chose the promoters of orf2 and orf4 in pZGX04 as promoters to express the heterologous genes, as only these native promoters of Propionibacterium were available in pZGX04. The fold changes in mRNA levels of orf2 and orf4 relative to 16S rRNA were 14.47 ± 3.39 and 5.62 ± 1.02, respectively, and the two genes were not essential for pZGX04 (16). Therefore, we chose these positions for exogenous gene insertion. It can be seen that gldA was the most important gene compared to the other two genes, followed by mdh. Therefore, in the coexpression study, we put the more important gene at the position of orf2, which had a higher efficiency of transcription.
gldA was important for PA biosynthesis and cell growth (16). GDH was a key enzyme, and it not only can improve the entire metabolic flux by accelerating the glycerol dissimilation rate but also can generate NADH along with glycerol dissimilation (Fig. 1). As the metabolic pathway from pyruvate to PA is an NADH-consuming process, the overexpression of gldA seemed more important, although the overexpression of mdh improved PA biosynthesis distinctly. More AA will be generated to balance NADH/NAD+ if NADH cannot fulfill the requirements of PA metabolism, as the metabolic pathway from pyruvate to AA is an NADH-generating process (23). Moreover, the generation of NADH by GDH reduced AA accumulation and improved cellular energy metabolism and redox equilibrium, thereby shifting the reaction in favor of cell growth and PA biosynthesis (33).
In theory, the overexpression of fumC and mdh should increase the metabolic flux from pyruvate to PA and reduce it to AA biosynthesis and thereby increase PA production and reduce by-product (LA and AA) production (Fig. 6B and C). The engineered strains consistently had higher YPA/LA and YPA/AA values in glycerol cultures than those of the wild type (Table 3), confirming our hypothesis that the expression of fumC and mdh would increase the production of PA over that of by-products. The overexpression of fumC, gldA, and mdh improved PA production, indicating that the three corresponding enzymes are some of the metabolic bottlenecks in the metabolic pathway. These results certified the results of our analysis of the effects of intermediates on PA production.
Because the single overexpression of these three genes improved PA production significantly, we coexpressed combinations of these genes in P. jensenii to enhance PA production further. The highest level of production reached 39.43 ± 1.90 g/liter with the coexpression of gldA and mdh, compared with 26.95 ± 1.21 g/liter for wild-type P. jensenii (Fig. 6A and F). In theory, GDH overexpression increases the glycerol dissimilation rate, improving NADH generation, and MDH overexpression accelerates the conversion from pyruvate to PA, increasing PA biosynthesis. The cell densities of P. jensenii(pZGX04-gldA-fumC) and P. jensenii(pZGX04-gldA-mdh) were higher than that of P. jensenii(pZGX04-mdh-fumC) due to the generation of NADH by GDH (23). However, compared with the cell density of the wild-type strain, those of all the engineered strains decreased significantly. Figure 6 shows that the engineered strains grew slowly during the cultivation process compared to the wild-type strain. The reason for this finding may be that the replication and expression of the vector increased the burden of cellular metabolism.
The coexpression of fumC and mdh showed the highest YPA/LA and YPA/AA values compared to those for the wild type. Although the DCW in the engineered strains was lower than that in the wild type, YPA/DCW (the mass ratio of PA to DCW) increased substantially. In other words, the rate of productivity per unit cell became higher in the engineered strains. The highest YPA/DCW (the mass ratio of PA to DCW), 58.4% ± 4.4% higher than that of the wild type, was obtained in engineered P. jensenii(pZGX04-gldA-mdh) (Table 3). In general, plasmids in the engineered strains increased the burden of the cells, slowing cell growth and lowering DCW (Fig. 6). It should be noted that coexpression of the three genes has been attempted by using a polycistron under the regulation of the orf2 promoter. Although the three genes can be expressed, the transcriptional levels were rather low. The DCW and PA titers decreased to 2.98 ± 0.10 g/liter and 34.82 ± 1.53 g/liter, respectively. These results confirmed that the replication and expression of genes from the vector caused a metabolic burden to the engineered strain.
Conclusions.
In this study, we identified the metabolic bottlenecks for PA biosynthesis in P. jensenii and improved PA production substantially by overexpressing the corresponding genes fumC and mdh. To our knowledge, this study is the first to attempt the coexpression of two genes to engineer the PA biosynthesis pathway in Propionibacterium to increase PA production. The highest PA titer (39.43 ± 1.90 g/liter) and productivity (0.173 ± 0.008 g · liter−1 · h−1) were obtained with the coexpression of gldA and mdh in P. jensenii in a 3-liter fed-batch culture. The results obtained here provide a potential robust producer of PA and may be helpful in the metabolic engineering of P. jensenii to produce other nutraceuticals and platform chemicals.
REFERENCES
- 1.Hsu ST, Yang ST. 1991. Propionic acid fermentation of lactose by Propionibacterium acidipropionici: effects of pH. Biotechnol Bioeng 38:571–578. doi: 10.1002/bit.260380603. [DOI] [PubMed] [Google Scholar]
- 2.Roberto MC, Mayra T. 2002. Production of propionate by fed-batch fermentation of Propionibacterium acidipropionici using mixed feed of lactate and glucose. Biotechnol Lett 24:427–431. doi: 10.1023/A:1014562504882. [DOI] [Google Scholar]
- 3.Vörös A, Horváth B, Hunyadkürti J, McDowell A, Barnard E, Patrick S, Nagy I. 2012. Complete genome sequences of three Propionibacterium acnes isolates from the type IA(2) cluster. J Bacteriol 194:1621–1622. doi: 10.1128/JB.06758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers P, Chen JS, Zidwick MJ. 2006. Organic acid and solvent production. Part II: propionic and butyric acids and ethanol, p 611–671. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes: a handbook on the biology of bacteria, 3rd ed, vol 1 Symbiotic associations, biotechnology, applied microbiology. Springer, New York, NY. [Google Scholar]
- 5.Coral J, Karp SG, Porto de Souza Vandenberghe L, Parada JL, Pandey A, Soccol CR. 2008. Batch fermentation model of propionic acid production by Propionibacterium acidipropionici in different carbon sources. Appl Biochem Biotechnol 151:333–341. doi: 10.1007/s12010-008-8196-1. [DOI] [PubMed] [Google Scholar]
- 6.Dishisha T, Alvarez MT, Kaul RH. 2012. Batch- and continuous propionic acid production from glycerol using free and immobilized cells of Propionibacterium acidipropionici. Bioresour Technol 118:553–562. doi: 10.1016/j.biortech.2012.05.079. [DOI] [PubMed] [Google Scholar]
- 7.Goswami V, Srivastava AK. 2001. Propionic acid production in an in situ cell retention bioreactor. Appl Microbiol Biotechnol 56:676–680. doi: 10.1007/s002530000582. [DOI] [PubMed] [Google Scholar]
- 8.Himmi EH, Bories A, Boussaid A, Hassani L. 2000. Propionic acid fermentation of glycerol and glucose by Propionibacterium acidipropionici and Propionibacterium freudenfeichii ssp. shermanii. Appl Microbiol Biotechnol 53:435–440. doi: 10.1007/s002530051638. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Zhang YG, Zhang RB, Zhang F, Zhu J. 2011. Glycerol/glucose co-fermentation: one more proficient process to produce propionic acid by Propionibacterium acidipropionici. Curr Microbol 62:152–158. doi: 10.1007/s00284-010-9683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Z, Ma C, Gao C, Xu P. 2012. Efficient utilization of hemicellulose hydrolysate for propionic acid production using Propionibacterium acidipropionici. Bioresour Technol 114:711–714. doi: 10.1016/j.biortech.2012.02.118. [DOI] [PubMed] [Google Scholar]
- 11.Suwannakham S, Huang Y, Yang ST. 2006. Construction and characterization of ack knock-out mutants of Propionibacterium acidipropionici for enhanced propionic acid fermentation. Biotechnol Bioeng 94:383–395. doi: 10.1002/bit.20866. [DOI] [PubMed] [Google Scholar]
- 12.Zhang A, Yang ST. 2009. Propionic acid production from glycerol by metabolically engineered Propionibacterium acidipropionici. Process Biochem 44:1346–1351. doi: 10.1016/j.procbio.2009.07.013. [DOI] [Google Scholar]
- 13.Zhu L, Wei P, Cai J, Zhu X, Wang Z, Huang L, Xu Z. 2012. Improving the productivity of propionic acid with FBB-immobilized cells of an adapted acid-tolerant Propionibacterium acidipropionici. Bioresour Technol 112:248–253. doi: 10.1016/j.biortech.2012.01.055. [DOI] [PubMed] [Google Scholar]
- 14.Feng X, Chen F, Xu H, Wu B, Li H, Li S, Ouyang P. 2011. Green and economical production of propionic acid by Propionibacterium freudenreichii CCTCC M207015 in plant fibrous-bed bioreactor. Bioresour Technol 102:6141–6146. doi: 10.1016/j.biortech.2011.02.087. [DOI] [PubMed] [Google Scholar]
- 15.Boyaval P, Corre C, Madec MN. 1994. Propionic acid production in a membrane bioreactor. Enzyme Microbiol Technol 16:883–886. doi: 10.1016/0141-0229(94)90063-9. [DOI] [Google Scholar]
- 16.Zhuge X, Liu L, Shin HD, Chen RR, Li J, Du G, Chen J. 2013. Development of a Propionibacterium-Escherichia coli shuttle vector as a useful tool for metabolic engineering of Propionibacterium jensenii, an efficient producer of propionic acid. Appl Environ Microbiol 79:4595–4602. doi: 10.1128/AEM.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brede DA, Faye T, Stierli MP, Dasen G, Theiler A, Nes IF, Meile L, Holo H. 2005. Heterologous production of antimicrobial peptides in Propionibacterium freudenreichii. Appl Environ Microbiol 71:8077–8084. doi: 10.1128/AEM.71.12.8077-8084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jore J, Van Luijk N, Luiten R, Van der Werf M, Pouwels P. 2001. Efficient transformation system for Propionibacterium freudenreichii based on a novel vector. Appl Environ Microbiol 67:499–503. doi: 10.1128/AEM.67.2.499-503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiatpapan P, Hashimoto Y, Nakamura H, Piao YZ, Ono H, Yamashita M, Murooka Y. 2000. Characterization of pRGO1, a plasmid from Propionibacterium acidipropionici, and its use for development of a host-vector system in propionibacteria. Appl Environ Microbiol 66:4688–4695. doi: 10.1128/AEM.66.11.4688-4695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehberger TG, Glatz BA. 1990. Characterization of Propionibacterium plasmids. Appl Environ Microbiol 56:864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiatpapan P, Murooka Y. 2002. Genetic manipulation system in propionibacteria. J Biosci Bioeng 93:1–8. doi: 10.1016/S1389-1723(02)80045-7. [DOI] [PubMed] [Google Scholar]
- 22.Ammar EM, Wang Z, Yang ST. 2013. Metabolic engineering of Propionibacterium freudenreichii for n-propanol production. Appl Microbiol Biotechnol 97:4677–4690. doi: 10.1007/s00253-013-4861-6. [DOI] [PubMed] [Google Scholar]
- 23.Ammar EM, Jin Y, Wang Z, Yang ST. 2014. Metabolic engineering of Propionibacterium freudenreichii: effect of expressing phosphoenolpyruvate carboxylase on propionic acid production. Appl Microbiol Biotechnol 98:7761–7772. doi: 10.1007/s00253-014-5836-y. [DOI] [PubMed] [Google Scholar]
- 24.Clomburg J, Gonzalez R. 2013. Anaerobic fermentation of glycerol: a platform for renewable fuels and chemicals. Trends Biotechnol 31:20–28. doi: 10.1016/j.tibtech.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Parizzi LP, Grassi MC, Llerena LA, Carazzolle MF, Queiroz VL, Lunardi I, Zeidler AF, Teixeira PJ, Mieczkowski P, Rincones J, Pereira GA. 2012. The genome sequence of Propionibacterium acidipropionici provides insights into its biotechnological and industrial potential. BMC Genomics 13:562. doi: 10.1186/1471-2164-13-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuge X, Liu L, Shin HD, Li J, Du G, Chen J. 2014. Improved propionic acid production from glycerol with metabolically engineered Propionibacterium jensenii by integrating fed-batch culture with a pH-shift control strategy. Bioresour Technol 152:519–925. doi: 10.1016/j.biortech.2013.11.063. [DOI] [PubMed] [Google Scholar]
- 27.Brüggemann H, Henne A, Hoster F, Liesegang H, Wiezer A, Strittmatter A, Hujer S, Dürre P, Gottschalk G. 2004. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305:671–673. doi: 10.1126/science.1100330. [DOI] [PubMed] [Google Scholar]
- 28.Falentin H, Deutsch SM, Jan G, Loux V, Thierry A, Parayre S, Maillard MB, Dherbécourt J, Cousin FJ, Jardin J, Siguier P, Couloux A, Barbe V, Vacherie B, Wincker P, Gibrat JF, Gaillardin C, Lortal S. 2010. The complete genome of Propionibacterium freudenreichii CIRM-BIA1, a hardy actinobacterium with food and probiotic applications. PLoS One 5:e11748. doi: 10.1371/journal.pone.0011748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horváth B, Hunyadkürti J, Vörös A, Fekete C, Urbán E, Kemény L, Nagy I. 2012. Genome sequence of Propionibacterium acnes type II strain ATCC 11828. J Bacteriol 194:202–203. doi: 10.1128/JB.06388-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meuricea G, Jacoba D, Debordea C, Chailloub S, Rouaulta A, Leverriera P, Jana G, Thierrya A, Maillarda MB, Ameta P, Lalandec M, Zagorecb M, Boyavala P, Dimovaa D. 2004. Whole genome sequencing project of a dairy Propionibacterium freudenreichii subsp. shermanii genome: progress and first bioinformatics analysis. Lait 84:15–24. doi: 10.1051/lait:2003041. [DOI] [Google Scholar]
- 31.Stierli MP. 2002. DNA transformation of propionibacteria based on plasmids pLME106 and pLME108. Ph.D. dissertation. ETHZ, Zurich, Switzerland. [Google Scholar]
- 32.Kiatpapan P, Murooka Y. 2001. Construction of an expression vector for propionobacteria and its use in production of 5-aminolevulinic acid by Propionibacterium freudenreichii. Appl Microbiol Biotechnol 56:144–149. doi: 10.1007/s002530100603. [DOI] [PubMed] [Google Scholar]
- 33.Liu R, Liang L, Jiang M, Ma J, Chen K, Jia H, Wei P, Ouyang P. 2014. Effects of redox potential control on succinic acid production by engineered Escherichia coli under anaerobic conditions. Process Biochem 49:740–744. doi: 10.1016/j.procbio.2014.02.010. [DOI] [Google Scholar]