

ABSTRACT
Receptors expressed on the host cell surface adhere viruses to target cells and serve as determinants of viral tropism. Several viruses bind cell surface glycans to facilitate entry, but the contribution of specific glycan moieties to viral disease is incompletely understood. Reovirus provides a tractable experimental model for studies of viral neuropathogenesis. In newborn mice, serotype 1 (T1) reovirus causes hydrocephalus, whereas serotype 3 (T3) reovirus causes encephalitis. T1 and T3 reoviruses engage distinct glycans, suggesting that glycan-binding capacity contributes to these differences in pathogenesis. Using structure-guided mutagenesis, we engineered a mutant T1 reovirus incapable of binding the T1 reovirus-specific glycan receptor, GM2. The mutant virus induced substantially less hydrocephalus than wild-type virus, an effect phenocopied by wild-type virus infection of GM2-deficient mice. In comparison to wild-type virus, yields of mutant virus were diminished in cultured ependymal cells, the cell type that lines the brain ventricles. These findings suggest that GM2 engagement targets reovirus to ependymal cells in mice and illuminate the function of glycan engagement in reovirus serotype-dependent disease.
IMPORTANCE
Receptor utilization strongly influences viral disease, often dictating host range and target cell selection. Different reovirus serotypes bind to different glycans, but a precise function for these molecules in pathogenesis is unknown. We used type 1 (T1) reovirus deficient in binding the GM2 glycan and mice lacking GM2 to pinpoint a role for glycan engagement in hydrocephalus caused by T1 reovirus. This work indicates that engagement of a specific glycan can lead to infection of specific cells in the host and consequent disease at that site. Since reovirus is being developed as a vaccine vector and oncolytic agent, understanding reovirus-glycan interactions may allow manipulation of reovirus glycan-binding properties for therapeutic applications.
INTRODUCTION
Viruses are capable of binding a variety of cell surface receptors to initiate the process of infection. Many viruses use glycans to facilitate attachment and entry (1–6). Some viruses, such as influenza virus, appear to engage glycans as a primary receptor (5), while others, such as herpes simplex virus (7) and reovirus (1, 8) engage glycans as an initial adhesive event prior to binding a proteinaceous attachment receptor in a process known as adhesion strengthening. Virus-glycan interactions govern cell susceptibility, yet the contribution of individual glycans to viral pathogenesis is not understood for most glycan-binding viruses.
Mammalian reoviruses display serotype-dependent pathology in the murine central nervous system (CNS). Serotype 1 (T1) reovirus spreads via hematogenous routes (9–11) and infects ependymal cells (12, 13), resulting in hydrocephalus (13, 14). Conversely, serotype 3 (T3) reovirus disseminates via neural and hematogenous routes (15–17), infects CNS neurons, and causes lethal encephalitis (9, 18–20). The basis for these serotype-specific differences in neuropathogenesis is not known. However, studies using reassortant strains (i.e., strains containing mixtures of gene segments derived from two parental strains) demonstrate that the viral S1 gene, which encodes attachment protein σ1, dictates serotype-dependent differences in CNS pathology (9, 11, 17, 18). These findings suggest that differences in CNS disease likely are attributable to differential engagement of cell surface receptors.
While T1 and T3 reovirus engage the same known protein receptors, junctional adhesion molecule A (JAM-A) (8) and Nogo receptor 1 (NgR1) (21), the different reovirus serotypes interact with distinct glycans. We previously demonstrated that T1 reovirus binds the GM2 glycan, which is a branched oligosaccharide composed of a glucose and galactose backbone with terminal α2,3-linked sialic acid (Neu5Ac) and β1,4-linked N-acetylgalactosamine (GalNAc) moieties (22). The GM2 glycan is found on glycoproteins and the GM2 ganglioside, and it is possible that T1 reovirus can bind GM2-conjugated glycoproteins, glycolipids, or both. T1 reovirus-GM2 binding is likely physiologic, as soluble GM2 glycan inhibits the capacity of T1 but not T3 reovirus to infect mouse embryonic fibroblasts (MEFs) (22). Conversely, T3 reovirus binds the GM3 glycan (23), which contains α2,3-linked sialic acid, as well as α2,6- and α2,8-linked sialyloligosaccharides (23). Soluble GM3 glycan does not inhibit the capacity of T1 reovirus to infect MEFs (22), indicating that reovirus-glycan interactions are serotype dependent. These observations suggest that differences in glycan utilization by T1 and T3 reoviruses contribute to serotype-specific disease. In support of this idea, sialic acid-binding T3 strains infect neurons more efficiently than do non-sialic acid-binding strains (24), and consequently, sialic acid engagement enhances the capacity of T3 reovirus to cause encephalitis (24). The contribution of T1 reovirus-glycan interactions to the pathogenesis of hydrocephalus is not known.
Reovirus attachment protein σ1 is an elongated, trimeric fiber comprising tail, body, and head domains. Using X-ray crystallography and saturation transfer difference nuclear magnetic resonance (STD-NMR) spectroscopy, we found that the T1 σ1 head domain interacts with the Neu5Ac and GalNAc components of the GM2 glycan (22). Structure-guided mutagenesis revealed that residues Ser370, Gln371, and Val354 are required for functional GM2 engagement in cell culture. Viruses containing the S370P or Q371E mutation in σ1 display an impaired capacity to agglutinate human erythrocytes and infect MEFs, which are most susceptible to GM2-binding T1 strains. Viruses containing these mutations replicate efficiently in L929 (L) cells, which support infection by strains incapable of binding sialic acid (22). Thus, the defect in infectivity in MEFs by strains altered in GM2 binding is linked to an altered capacity to engage glycans.
In this study, we engineered a virus containing both the S370P and Q371E mutations in the σ1 attachment protein of prototype T1 strain type 1 Lang (T1L) for use in studies to determine the function of reovirus-GM2 interactions in disease. STD-NMR experiments demonstrate that in comparison to wild-type T1L σ1, the S370P/Q371E mutant σ1 does not bind GM2. We infected mice with T1L and the S370P/Q371E mutant virus and found that glycan-binding activity does not markedly influence replicative capacity in the brain. However, the degree of hydrocephalus induced by wild-type T1L is substantially greater than that induced by the S370P/Q371E mutant. Concordantly, T1L induces less severe hydrocephalus in mice that do not express GM2. These findings establish GM2 binding as an important determinant of serotype-specific reovirus disease and shed light on mechanisms of virus-induced hydrocephalus.
RESULTS
Molecular characterization of the GM2-binding mutant virus strain.
To engineer a T1 reovirus incapable of engaging GM2, we used site-directed mutagenesis to construct an S370P/Q371E double-residue mutant virus combining two single-residue T1 σ1 mutations that alter glycan-binding capacity (22). This double-residue mutant replicates comparably to wild-type T1L in L cells (see Fig. S1 in the supplemental material), which are equally susceptible to glycan-binding and non-glycan-binding reovirus strains (22, 23). Particle-to-PFU ratios were comparable, averaging 162:1 and 148:1 for wild-type and mutant virus stocks, respectively. Additionally, both wild-type and S370P/Q371E virus strains are neutralized by monoclonal antibody (MAb) 5C6 (see Fig. S2 in the supplemental material), a conformation-specific antibody that binds to a trimeric form of the T1 σ1 head domain (2). Collectively, these data suggest that the engineered mutations do not alter the folding or encapsidation of the T1 σ1 molecule.
T1 reovirus-mediated agglutination of human erythrocytes requires binding to carbohydrates on the erythrocyte surface (25). To test whether the S370P/Q371E mutation impairs glycan-binding capacity, we quantified the hemagglutination titers of wild-type and mutant virus using human erythrocytes. Although the mutant retained some hemagglutination capacity, hemagglutination titers of the S370P/Q371E mutant were 32-fold lower than those of wild-type T1L (Fig. 1A). To determine whether residual hemagglutination capacity reflects residual glycan binding, we treated erythrocytes with Arthrobacter ureafaciens neuraminidase, which removes cell surface sialic acid, or phosphate-buffered saline (PBS) as a control prior to incubation with strain T1L and the S370P/Q371E mutant. T1L-mediated hemagglutination was impaired following neuraminidase treatment, whereas S370P/Q371E was not (Fig. 1B), indicating that the residual hemagglutination capacity of the S370P/Q371E mutant is not attributable to sialylated glycan engagement. As expected, hemagglutination activity of prototype T3 strain type 3 Dearing (T3D) was abolished by neuraminidase treatment of erythrocytes (26). Incubation of wild-type and mutant T1 reovirus strains with T1 σ1-specific MAb 5C6 prevented hemagglutination but had no effect on hemagglutination by strain T3D (Fig. 1B). These findings suggest that T1L, but not the S370P/Q371E mutant, binds sialic acid to agglutinate human erythrocytes.
FIG 1 .

Glycan binding properties of wild-type and σ1 mutant viruses. (A) Purified virions of the strains shown (1011 particles/well) were serially diluted 1:2 in PBS in 96-well U-bottom plates. Human erythrocytes at a concentration of 1% (vol/vol) in PBS were added to virus-containing wells and incubated at 4°C for 3 h. Results are expressed as log2 (HA titer) for three independent experiments. ***, P < 0.001, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison test. (B) Human erythrocytes were washed with PBS, resuspended at a concentration of 1% (vol/vol), and treated with PBS (vehicle control) or 200 mU of A. ureafaciens neuraminidase (NM) at room temperature for 1 h prior to adsorption with 4 HA units of the virus strains shown in 96-well U-bottom plates. Viruses were pretreated with either PBS or T1L σ1 conformation-specific MAb 5C6. Erythrocytes were incubated with virions at 4°C for 3 h. PBS was used as a virus-free control. The data shown are representative of three independent experiments. (C) Wild-type (T1L) and S370P/Q371E σ1 proteins at a final concentration of 16.8 µM each were incubated with the GM2 glycan at a final concentration of 2 mM. Resonances that can be unambiguously assigned to individual protons are labeled and color coded according to the sugar moieties within the GM2 glycan: glucose (Glc), yellow; galactose (Gal), green; neuraminic acid (Neu5Ac), red; and N-acetylgalactosamine (GalNAc), blue.
To determine whether the S370P/Q371E σ1 attachment protein retains any residual GM2-binding activity, we assessed the binding of wild-type T1L and mutant S370P/Q371E σ1 proteins to GM2 by STD-NMR, a technique capable of assessing low-affinity interactions between a large molecule and a small ligand (27), including virus-glycan interactions (28, 29). STD-NMR is based on selective irradiation of protons in the protein and detection of the subsequent magnetization transfer from the protein to the ligand. Protons in the Neu5Ac and GalNAc moieties of GM2 were found to interact with wild-type T1L σ1 (Fig. 1C), reflecting the binding contacts in the crystal structure of the glycan with the T1L σ1 head (22). The S370P/Q371E double-residue mutant σ1 protein did not interact with the GM2 glycan as assessed by STD-NMR (Fig. 1C), indicating that the S370P/Q371E virus is incapable of binding GM2.
To test whether the S370P/Q371E mutant virus displays an altered capacity to infect cells that are maximally susceptible to glycan-binding reovirus strains, we inoculated MEFs, which require binding to GM2 for optimal infection by T1 strains (22), with wild-type T1L and the S370P/Q371E mutant. The S370P/Q371E mutant displayed a diminished capacity to infect these cells compared with T1L at all multiplicities of infection (MOIs) tested (Fig. 2A). To establish whether the differences in infectivity displayed by wild-type and mutant viruses in MEFs result from compromised viral attachment, T1L and the S370P/Q371E mutant were adsorbed to MEFs and L cells at 4°C to prevent internalization, and the percentage of cells bound by virus and the mean fluorescence intensity (MFI) were quantified using flow cytometry. The S370P/Q371E mutant adhered much less efficiently to MEFs than did T1L (Fig. 2B; also, see Fig. S3 in the supplemental material), whereas both strains bound comparably to L cells (see Fig. S4 in the supplemental material). Thus, the S370P/Q371E mutant has a defect in attachment to and infectivity of cells that require glycan engagement for optimal infection.
FIG 2 .

Infectivity and binding of T1L and the S370P/Q371E mutant in MEFs. (A) MEFs were adsorbed with either T1L or the S370P/Q371E mutant at the MOIs shown (in focus-forming units [FFU]/cell as determined by titration on L cells) at room temperature for 1 h. Cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed, and reovirus antigen was detected by indirect immunofluorescence. The percentage of infected cells in three fields of view per well was determined. Results are expressed as the percent infected cells from the combined means of three independent experiments, each performed in triplicate wells. Error bars represent standard errors of the means. *, P < 0.05, **, P < 0.01, and ***, P < 0.001, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. (B) MEFs were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-647-labeled reovirus antiserum. The percentage of cells with bound virus was quantified using flow cytometry. Results are from a representative experiment of three independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test.
Contribution of glycan engagement to reovirus tropism.
To determine whether T1 σ1 GM2-binding capacity influences T1 reovirus infection of the murine brain, wild-type mice were inoculated with either 102 or 108 PFU of T1L or the S370P/Q371E mutant intracranially, and viral titers in brain homogenates were quantified at 4, 8, and 12 days postinoculation by plaque assay. Following inoculation with 102 PFU of virus, T1L produced slightly higher titers in the brain than the S370P/Q371E mutant on days 4 and 8 postinoculation, and the difference reached statistical significance on day 8 (Fig. 3A). These findings suggest that T1 σ1-glycan interactions contribute to optimal viral replication in the murine brain. However, no significant differences in titer were observed following inoculation with 108 PFU of virus (Fig. 3B), suggesting that the replication capacity of T1L and S370P/Q371E in the brain is equalized following high-MOI inoculation.
FIG 3 .

Viral titers in the brain following intracranial inoculation with either T1L or the S370P/Q371E mutant. Newborn mice were inoculated intracranially with either 102 (A) or 108 (B) PFU of either T1L or the S370P/Q371E mutant. At days 4, 8, and 12 postinoculation, mice were euthanized, and brains were excised and homogenized. Viral titers in brain homogenates were determined by plaque assay. Results are expressed as mean viral titers for 4 to 11 mice per virus per time point. Error bars represent standard errors of the means. *, P < 0.05, as determined by two-way ANOVA followed by Bonferroni’s multiple-comparison test.
To determine whether T1L and the S370P/Q371E mutant differ in cell tropism, we inoculated mice in the right cerebral hemisphere with 108 PFU of T1L or the S370P/Q371E mutant, which is a dose that produces comparable titers in the brain (Fig. 3B). At 4 and 12 days postinoculation, mice were euthanized, and brains were excised, sectioned, and stained for reovirus antigen. We did not detect major differences in cells infected by wild-type and mutant virus. However, ependymal denuding was observed in the brain tissues of mice inoculated with wild-type virus (Fig. 4). Less viral antigen was observed in the contralateral hemisphere of mice inoculated with the S370P/Q371E mutant virus compared with mice inoculated with T1L (Fig. 4). We did not detect hydrocephalus in mice inoculated with either virus strain at this time point.
FIG 4 .
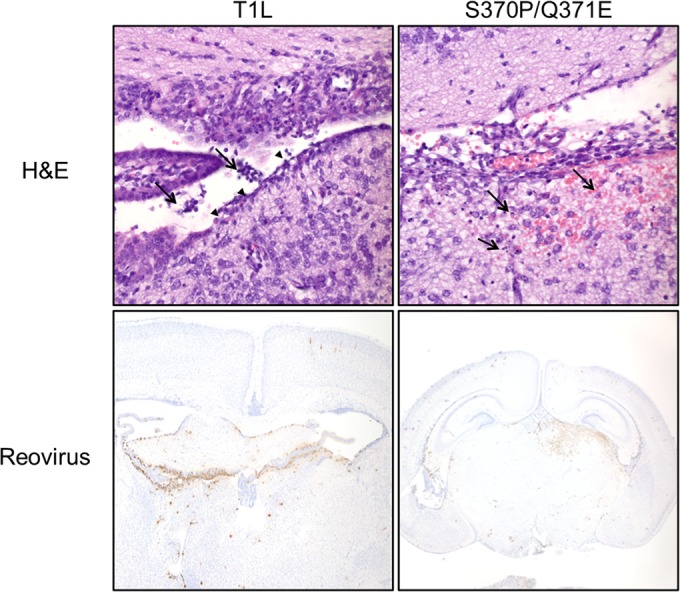
Brain histology 4 days following intracranial inoculation with either T1L or the S370P/Q371E mutant. Newborn mice were inoculated intracranially with 108 PFU of T1L or the S370P/Q371E mutant. Four days postinoculation, mice were euthanized, and brains were excised, fixed, and paraffin embedded. Brain sections were stained with H&E to examine tissue pathology (top). In the section from the T1L-infected brain, arrowheads indicate areas of ependymal cell denuding. Arrows point to sloughed cells in the ventricles, which may contribute to hydrocephalus. In the section from the S370P/Q371E-infected brain, arrows indicate regions of apoptotic neurons and glia with mild hemorrhage. Samples were stained with polyclonal reovirus antiserum to detect viral antigen (bottom). Viral antigen is dispersed in the T1L-infected brain but more localized to the inoculation site in the S370P/Q371E-infected brain.
At 12 days postinoculation, signs of pathological injury were clearly evident (Fig. 5). Histological evaluation of brain tissue from mice inoculated with either T1L or the S370P/Q371E mutant showed greater cell density within the ventricles than tissue from mice inoculated with PBS as a control. However, the cellular infiltrate in T1L-infected animals was greater than in those infected with the S370P/Q371E virus (Fig. 5). Staining with antibodies specific for CD3 and F4/80, which mark T cells and macrophages, respectively, demonstrated that the exudates in brain tissue of mice infected with either virus contained both of these cell types. These observations suggest that T1L induces more infiltrating inflammatory cells than S370P/Q371E in the cerebral ventricles of infected mice.
FIG 5 .

Brain histology 12 days following intracranial inoculation with either T1L or the S370P/Q371E mutant. Newborn mice were inoculated intracranially with PBS or 108 PFU of either T1L or the S370P/Q371E mutant. Twelve days postinoculation, mice were euthanized, and brains were excised, fixed, and paraffin embedded. Coronal brain sections were stained with H&E to examine tissue pathology and anti-CD3 and anti-F4/80 to mark T cells and macrophages, respectively. Boxes indicate portions of the lateral ventricles that are enlarged in the images on the right.
Reovirus-GM2 interactions influence hydrocephalus severity.
To test whether GM2-binding capacity contributes to hydrocephalus induction by T1 reovirus, mice were inoculated intracranially with either T1L or the S370P/Q371E mutant, and the magnitude of hydrocephalus was assessed using magnetic resonance imaging (MRI) scanning 21 days postinoculation (Fig. 6A). Mice infected with T1L developed much more substantial hydrocephalus than those infected with the S370P/Q371E mutant. The mean ventricular volume of T1L-infected animals was 69.2 mm3, whereas that in S370P/Q371E mutant-infected mice was 13.7 mm3 (Fig. 6B). The ventricular volume in S370P/Q371E-infected mice did not differ appreciably from that in mock-infected animals, which was 9.4 mm3. These observations suggest that the capacity to bind GM2 enhances T1 reovirus-induced hydrocephalus.
FIG 6 .
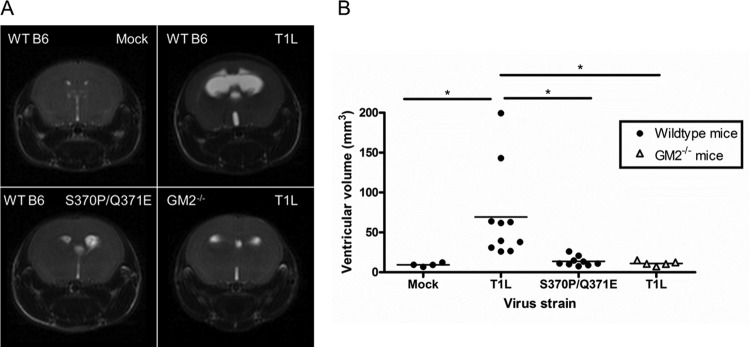
T1 reovirus glycan-binding capacity influences hydrocephalus induction. Wild-type C57BL/6 and GM2−/− mice were inoculated intracranially with PBS or 108 PFU of either T1L or the S370P/Q371E mutant. T2-weighted magnetic resonance images were obtained 21 days postinoculation. (A) Coronal images from representative wild-type mice inoculated with PBS (top left), wild-type mice inoculated with T1L (top right), wild-type mice inoculated with the S370P/Q371E mutant (bottom left), and GM2−/− mice inoculated with T1L (bottom right) are shown. Cerebrospinal fluid appears white, allowing ventricular volume to be quantified. The images shown were obtained from mice with the median ventricular volume for each virus and mouse strain (n = 4 to 10 mice per group). (B) Ventricular volume of reovirus-infected mice. Each symbol represents the ventricular volume from a single mouse. Mean ventricular volume is indicated by a horizontal bar. *, P < 0.05, as quantified by one-way ANOVA followed by Bonferroni’s correction for multiple tests.
To complement studies evaluating hydrocephalus induction by viruses that differ in the capacity to engage GM2, we assessed the magnitude of hydrocephalus using mice that differ in expression of the GM2 glycan. The galgt1 and galgt2 genes encode enzymes required to express the GM2 glycan on gangliosides and glycoproteins, respectively (30). Thus, galgt1 × galgt2 double-knockout mice, herein referred to as GM2−/− mice, lack GM2 glycan expression on both gangliosides and glycoproteins. We inoculated wild-type and GM2−/− mice intracranially with T1L and assessed hydrocephalus using MRI scanning 21 days postinoculation. T1L-mediated hydrocephalus, evidenced by MRI results (Fig. 6A) and quantification of ventricular volume (Fig. 6B), was more severe in wild-type mice than in GM2−/− mice. The mean ventricular volume in T1L-infected GM2−/− mice was 11.14 mm3, approximating that of wild-type mice inoculated with the S370P/Q371E mutant. Taken together, these results demonstrate that disruption of the σ1-GM2 interaction through manipulation of the viral GM2-binding site or host expression of GM2 diminishes the severity of hydrocephalus caused by T1 reovirus.
Glycan binding is required for maximal infection of ependymal cells in vitro.
Damage to ependymal cells caused by infection with T1 reovirus plays a key role in the pathogenesis of hydrocephalus caused by this reovirus serotype (12). To evaluate whether glycan binding activity influences reovirus infection of ependymal cells, we tested T1L and the S370P/Q371E mutant for the capacity to infect BXD-1425EPN ependymoma cells (31). T1L infected these cells much more efficiently than did the S370P/Q371E mutant at all MOIs tested (Fig. 7A). To determine whether the differences in infectivity displayed by wild-type and mutant virus in ependymal cells results from differences in viral attachment, T1L and the S370P/Q371E mutant were adsorbed to BXD-1425EPN cells at 4°C, and the percentage of cells bound by virus was quantified using flow cytometry. Concordant with the infectivity results, T1L adhered more efficiently to these cells than did the S370P/Q371E mutant (Fig. 7B). Incubation of virus with soluble GM2 glycan prior to adsorption diminished the capacity of wild-type but not mutant virus to infect these cells, as did pretreatment of cells with A. ureafaciens neuraminidase (Fig. 7C). Thus, the capacity of T1 reovirus to bind GM2 confers a replication advantage in ependymal cells that is associated with enhanced viral attachment.
FIG 7 .

Infectivity and binding of T1L and the S370P/Q371E mutant in ependymoma cells. (A) BXD-1425EPN cells were adsorbed with either T1L or the S370P/Q371E mutant at the MOIs shown (in FFU/cell as determined by titration on L cells) at room temperature for 1 h. Cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed, and reovirus antigen was detected by indirect immunofluorescence. The percentage of infected cells in three fields of view per well was determined. Results are expressed as the percent infected cells from a representative experiment of two independent experiments, each performed using triplicate wells. Error bars represent standard deviations. *, P < 0.05, **, P < 0.01, and ***, P < 0.001, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. (B) BXD-1425EPN cells were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-488-labeled reovirus antiserum. The percentage of cells with bound virus was quantified using flow cytometry. Results are from a representative experiment of two independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test. (C) T1L and the S370P/Q371E mutant at an MOI of 1 FFU/cell were incubated with the GM2 glycan at a final concentration of 2 mM for 1 h prior to adsorption to BXD-1425EPN cells at room temperature for 1 h. Cells were incubated with PBS or A. ureafaciens neuraminidase for 1 h prior to viral adsorption. Following adsorption, cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed, and reovirus antigen was detected by indirect immunofluorescence. The percentage of infected cells in three fields of view per well was determined. Results are expressed as the percent infected cells from the combined means of three independent experiments, each performed using duplicate wells. Error bars represent standard errors of the means. *, P < 0.05, and **, P < 0.01, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test.
DISCUSSION
An S370P/Q371E mutant virus was selected to establish the function of GM2 engagement in T1 reovirus disease. This double-residue mutant virus replicates with wild-type efficiency in L cells and is neutralized by a T1 σ1 conformation-specific antibody (see Fig. S1 and S2 in the supplemental material), suggesting that the mutations do not adversely affect the structural integrity of σ1. However, the mutant virus displays impaired hemagglutination capacity (Fig. 1A) and infectivity in MEFs relative to wild-type T1L (Fig. 2). Moreover, the S370P/Q371E σ1 protein does not interact with GM2 in solution as assessed by STD-NMR (Fig. 1C). Using this mutant virus, we found that T1 reovirus σ1-GM2 interactions contribute to serotype-specific disease. Wild-type T1L induces more severe hydrocephalus in wild-type mice than does the S370P/Q371E virus. Concordantly, T1L induces less severe hydrocephalus in GM2−/− mice than wild-type mice, mimicking the phenotype of limited hydrocephalus induced by the S370P/Q371E mutant in wild-type mice (Fig. 6). These observations provide strong evidence that GM2-binding capacity is a T1 reovirus virulence determinant for hydrocephalus severity.
Infection of mice with viruses that differ in the capacity to engage GM2 resulted in marked differences in disease severity, yet titers of wild-type and mutant viruses in the brain were comparable. Interestingly, histological analysis suggests that T1L spreads more efficiently to the contralateral hemisphere than does the S370P/Q371E mutant following intracranial inoculation (Fig. 4), suggesting that glycan-binding capacity enhances dissemination within a tissue target.
Previous reports of reovirus-induced hydrocephalus noted cytoplasmic inclusion formation in ependymal cells accompanied by denuding of the ependymal layer (13, 14, 32). Inflammatory exudates appear in the ventricles, peaking between days 10 and 12 postinoculation. Reparative processes also are evident, including proliferation of astroglia and capillaries at sites of ependymal damage (13, 14). Histological evaluation of brain tissue in our study is consistent with these previous reports. Reovirus titers 12 days postinoculation had subsided to levels undetectable by immunohistochemistry. However, we noted increased cell density in the ventricles of mice inoculated with wild-type T1L and to a lesser extent the S370P/Q371E mutant. Immunohistochemistry revealed that T cells and macrophages infiltrated the ventricles of animals infected with either virus (Fig. 5). Concordantly, T1L produced severe hydrocephalus in wild-type mice, but disease was minimal in mice inoculated with the mutant virus incapable of GM2 binding and in mice that do not express the GM2 glycan.
The brain is composed of several cell types, and one possibility is that some cell types, such as ependymal cells, display glycan-dependent susceptibility to T1 reovirus. We tested this hypothesis using an ependymal cell line and found that T1L binds and infects these cells more efficiently than does the S370P/Q371E virus (Fig. 7A and B). Moreover, preincubation of virus with soluble GM2 or cells with neuraminidase decreased infectivity of T1L but not the S370P/Q371E mutant (Fig. 7C). As ependymal cells constitute only a small fraction of total brain tissue, assessment of viral load in whole-brain homogenates by plaque assay may not be sufficiently sensitive to detect differences in replication at such discrete foci of infection, including the ependymal cell layer. Taken together, we think that T1 reovirus causes hydrocephalus by GM2-dependent infection of ependymal cells, which are damaged as a consequence of viral infection or virus-induced inflammation. Such damaged ependymal cells could be sloughed into the ventricles and obstruct cerebrospinal fluid flow or fail to efficiently reabsorb cerebrospinal fluid, resulting in ventricle enlargement.
An alternative possibility, which is not mutually exclusive with the one above, is that the T1L-GM2 interaction somehow interferes with normal brain activities. The neurobiological function of cell surface glycans is not completely understood. Gangliosides modulate several important properties, including leukocyte activation, migration, and differentiation (33–35). T1L cross-linking of GM2 could inhibit physiologic functions dependent on GM2, although we would then expect to see similar alterations in mice lacking GM2, and we did not. However, T1L-GM2 engagement may trigger unknown effects not observed following infection of either wild-type mice with the mutant incapable of GM2 binding or GM2−/− mice with T1L. Many proteins, including sialic acid-binding immunoglobulin-like lectins (siglecs), interact with sialylated glycans (36) and trigger signaling cascades with immunological effects. Since glycans, including GM2, bind siglecs (34), it is possible that the T1L-GM2 interaction interferes with normal GM2-siglec interactions. Alternatively, gangliosides are required for tissue repair, including axon regeneration in the brain (37). Ligands that bind gangliosides, including GD1a-specific antibody, interfere with axon regeneration following injury (38). Thus, it is possible that T1L-GM2 cross-linking somehow impedes the damage repair response in the brain.
Differences in serotype-specific neurologic disease caused by reovirus segregate with the viral S1 gene and are thought to be attributable to differences in receptor engagement. Results from our study, coupled with previous reports (24, 39), support this hypothesis. Our data demonstrate that T1L induces more severe hydrocephalus than does the S370P/Q371E mutant, which is deficient in glycan binding. Additionally, hydrocephalus is less severe following T1L infection of mice lacking the GM2 glycan than in wild-type mice. A function for glycan engagement also is apparent in T3 reovirus pathogenesis. T3 reoviruses differing only in the capacity to engage sialic acid differ in the capacity to infect neurons and cause lethal encephalitis (24). Thus, glycan binding contributes to reovirus serotype-specific tropism and virulence.
T1 reovirus interactions with GM2 likely function in an adhesion-strengthening mechanism, similar to the process used by T3 reovirus to infect cells (1). In this mechanism, binding to an abundantly expressed glycan with low affinity precedes binding to a relatively low-abundance protein receptor with high affinity. The STD-NMR studies of σ1-GM2 binding (Fig. 1C), together with the high doses of GM2 required to block T1 reovirus infection (22) (Fig. 7C), suggest that the virus binds GM2 with relatively low affinity compared with the high-affinity binding of reovirus to JAM-A (8). In further support of this idea, glycan binding is required for T1 reovirus infection of MEFs, which express modest levels of JAM-A, and dispensable for infection of L cells, which express significantly higher JAM-A levels (22). Accordingly, the S370P/Q371E mutant virus displays an impaired capacity to infect MEFs yet is fully replication competent in L cells. The modest differences in viral titer in the ependymal cell line and striking differences in hydrocephalus induction by the wild-type and mutant viruses in our study provides further support for the hypothesis that GM2 functions in adhesion strengthening and is not the sole receptor responsible for disease. Since JAM-A is dispensable for T1L replication in the murine brain (15), we think that serotype-specific glycan-binding preferences dictate neurotropism by facilitating binding to yet-unknown cell-specific protein or nonsialylated glycan receptors.
We were unable to compare wild-type and GM2−/− mice for the capacity to support infection by the reovirus strains used in our study. Unfortunately, GM2−/− mice breed exceedingly poorly, yielding numbers of pups sufficient only for the hydrocephalus experiments. Nonetheless, the limited studies conducted using these mice complement experiments using the S370P/Q371E virus, thereby strengthening the overall conclusions.
Reovirus is being developed as an oncolytic therapeutic due to its preferential replication in transformed cells (40–43) and relative avirulence in adult humans. Reovirus strain T3D (Reolysin) is now undergoing evaluation in clinical trials for the treatment of several types of human cancer (44–46). The specific targeting of reovirus to transformed cells is not completely understood, but cancer cells display increased expression of sialylated glycans (47), suggesting that glycan engagement influences infectivity. GM2 antibodies show promise in killing lung cancer (48) and glioma (49) cells in culture and demonstrate therapeutic efficacy in a mouse model of lung cancer (50). Understanding reovirus-glycan interactions may help target reovirus to cancer cells to initiate preferential lysis. In support of this idea, a T3 reovirus that lacks the JAM-A-binding σ1 head domain yet retains the glycan-binding region in the body domain of this serotype is less toxic than the native virus but retains oncolytic potential (51). Moreover, human glioblastoma cells grown under standard culture conditions require JAM-A binding for reovirus infection, but in spheroid cultures of these cells, which more closely resemble tumors in humans, reovirus infection is independent of JAM-A expression (52), suggesting that other factors, such as sialylated glycans, mediate reovirus attachment. T1 reovirus has not been tested for oncolytic capacity, but altered glycan expression by certain tumor cells (47, 53) provides sufficient rationale to initiate such experiments. Another possible approach would be to engineer an oncolytic T3 reovirus that contains the T1 GM2-binding site. Such a virus might display increased infection of tumors that express GM2. Thus, understanding the molecular basis of reovirus-glycan interactions has enhanced knowledge of mechanisms used by reovirus to cause hydrocephalus and may improve the design of oncolytic vectors.
MATERIALS AND METHODS
Cells.
Spinner-adapted murine L cells (54), MEFs (22, 55), and BHK-T7 cells (56) were maintained as described previously. BXD-1425EPN cells (31) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented to contain 10% fetal bovine serum, 100 U/ml of penicillin, and 100 µg/ml streptomycin.
Viruses.
Viruses were generated using plasmid-based reverse genetics (56–58). Purified virions were prepared as described previously (16, 59). To generate mutant viruses, codons for residues S370 and Q371 in the T1L S1 gene plasmid were altered by QuikChange (Stratagene) site-directed mutagenesis. S1 gene sequences were confirmed using the OneStep reverse transcription-PCR (RT-PCR) kit (Qiagen), gene-specific primers, and viral double-stranded RNA (dsRNA) extracted from infected L cells (Trizol, Invitrogen). Genotypes were confirmed by electrophoresis of viral particles (60). Particle concentrations were determined using the following conversion: 1 absorbance unit at 260 nm (AU260) = 2.1 × 1012 particles. Viral titers were quantified by plaque assay (61) or fluorescent focus assay (8).
Antibodies.
Polyclonal immunoglobulin G (IgG) raised against T1L and T3D was used to stain for reovirus antigen (62). Alexa-488- and Alexa-647-conjugated goat anti-rabbit antisera (Invitrogen) were used as secondary antibodies. Neutralizing MAb 5C6 specific for T1L σ1 (63) was used in neutralization assays as described previously (22). CD3 and F4/80 antibodies (Vanderbilt University Histology Core) were used as markers for T cells and macrophages, respectively.
Infectivity studies.
MEFs were adsorbed at room temperature for 1 h with virus at various MOIs determined using L cells. The inoculum was removed, and cells were washed twice with PBS and incubated at 37°C for 20 h. Cells were fixed in methanol and visualized by indirect immunofluorescence (22).
T1L σ1 protein expression and purification.
Construct σ1long, comprising the three most C-terminal β-spirals of T1L σ1 and the entire head domain (amino acids 261 to 470), was purified as described previously (22) and used in STD-NMR experiments. Mutations were generated using the GeneArt site-directed mutagenesis PLUS system (Invitrogen) along with AccuPrime Pfx DNA polymerase (Invitrogen) and One Shot MAX Efficiency DH5α-T1R competent cells (Invitrogen).
Viral replication assays.
Viral plaque assays were performed using L cells (61). Viral yield was calculated using the following formula: log10 yieldtx = log10(PFU/ml)tx − log10(PFU/ml)t0, where t is the time postinoculation.
Conformation-specific antibody neutralization.
Virus strains were incubated with 10 µg/ml of MAb 5C6 or a mouse IgG2α isotype control at room temperature for 1 h. Cells seeded in confluent monolayers in 24-well plates (Corning) were inoculated with the virus-antibody mixture at room temperature for 1 h. Cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed, and infectivity was determined by flow cytometry using Alexa-647-labeled reovirus polyclonal antiserum.
Hemagglutination assay.
Purified reovirus virions (1011 particles) were distributed into 96-well U-bottom microtiter plates (Costar) and serially diluted 2-fold in 0.05 ml of PBS. Human type O erythrocytes (Vanderbilt University Blood Bank) were washed twice with PBS and resuspended at a concentration of 1% (vol/vol). Erythrocytes (0.05 ml) were added to wells containing virus particles and incubated at 4°C for 3 h. A partial or complete shield of erythrocytes on the well bottom was interpreted as a positive hemagglutination result; a smooth, round button of erythrocytes was interpreted as a negative result. Hemagglutination (HA) titer is expressed as 1011 particles divided by the number of particles/HA unit. One HA unit equals the number of particles sufficient to produce hemagglutination.
Hemagglutination inhibition assay.
Human erythrocytes were treated with A. ureafaciens neuraminidase (M.P. Biomedicals, LLC) at room temperature for 1 h, incubated with 4 HA units of reovirus, incubated at 4°C for 3 h, and scored for agglutination.
STD-NMR spectroscopy.
NMR spectra were recorded at 283 K using 3-mm tubes and a Bruker AVIII-600 spectrometer equipped with a room temperature probe head and processed with TOPSPIN 3.0 (Bruker). Samples contained 2 mM GM2 glycan (Elicityl), 20 mM potassium phosphate (pH 7.4), and 150 mM NaCl with and without 16.8 µM T1L σ1 or the mutant protein. Samples were prepared in D2O, and no additional water suppression was used to preserve the anomeric proton signals. A sample without protein also was used for spectral assignment. The off- and on-resonance irradiation frequencies were set to −30 ppm and 7.3 ppm, respectively. A selective pulse irradiation power of 57 Hz and saturation time of 2 s were used, and the total relaxation delay was 3 s. A 50-ms continuous-wave spin-lock pulse with a strength of 3.2 kHz was employed to suppress residual protein signals. Spectra were referenced using HDO as an internal standard (64) and multiplied with a Gaussian window function prior to Fourier transformation.
Virus attachment by flow cytometry.
Cells were adsorbed with reovirus strains at 4°C for 1 h to prevent internalization, washed twice with PBS, and stained with Alexa-647 or Alexa-488 labeled reovirus polyclonal antiserum. Labeling was performed using the AlexaFluor antibody labeling kit (Molecular Probes) according to the manufacturer’s instructions. The percentage of cells bound by virus was quantified using an LSR-II flow cytometer (BD Biosciences). The data were analyzed with FlowJo software (Tree Star).
Infection of mice.
C57BL/6 mice were obtained from the Jackson Laboratory to establish a breeding colony at Vanderbilt University. GM2−/− mice lacking functional copies of galgt1 and galgt2 on a C57BL/6 background (30) were generated by The Center for Functional Glycomics (The Scripps Research Institute) and provided by Dapeng Zhou (MD Anderson Cancer Center).
Two- to three-day-old mice were inoculated intracranially in the right cerebral hemisphere with reovirus diluted in 5 µl PBS using a Hamilton syringe and 30-gauge needle. For analysis of viral replication, mice were euthanized at various intervals postinoculation, and organs were excised, collected in 1 ml of PBS, and frozen and thawed twice prior to homogenization using a TissueLyser (Qiagen). Viral titer was quantified by plaque assay using L cells. For histopathological and immunohistochemical analyses, mice were euthanized at various intervals following inoculation, and organs were excised and fixed overnight in 10% formalin. Fixed organs were paraffin embedded, and 6-µm sections were prepared. Sections were evaluated for histological damage following hematoxylin-and-eosin (H&E) staining. Reovirus proteins were detected using reovirus polyclonal antiserum. T cells and macrophages were detected using CD3- and F4/80-specific antibodies, respectively.
All animal husbandry and experimental procedures were performed in accordance with U.S. Public Health Service policy and approved by the Vanderbilt University School of Medicine Institutional Animal Care and Use Committee, All animal work was performed under Vanderbilt animal protocol M/05/198.
Magnetic resonance imaging.
Mice were inoculated with 108 PFU of reovirus. Twenty-one days postinoculation, mice were anesthetized via inhalation of 2% isoflurane and 98% oxygen. Animals were secured in a prone position with the head in a 25-mm-inner-diameter radiofrequency (RF) coil and placed in a Varian 7T horizontal-bore magnetic resonance imaging system (Varian Inc.) to collect imaging data. Respiration rate and internal body temperature were continuously monitored. A constant body temperature of 37°C was maintained using heated airflow.
For each animal, multislice scout images were collected in all three imaging planes (axial, sagittal, and coronal) using a gradient echo sequence with a repetition time (TR) of 75 ms, an echo time (TE) of 5 ms, a slice thickness of 2 mm, and a flip angle of 35°. An average of four acquisitions were obtained for each animal. Additional parameters include a 50-mm by 50-mm field of view (FOV) and a 128-by-128 data matrix.
Following localization of the brain, T2-weighted fast-spin echo images were collected over 12 to 20 imaging slices for all three imaging planes (axial, coronal, and sagittal), with a 20-mm by 20-mm FOV, a slice thickness of 0.75 mm, and a 128-by-128 data matrix. Additional parameters include a TR of 5 s, an echo train length of 16, echo spacing of 8 ms, and a TE of 64 ms.
Ventricular quantification.
Ventricular volume was quantified using MatLab 2013a (The MathWorks, Inc.). A region of interest (ROI) encompassing the entire brain was manually drawn for each slice, and a signal intensity threshold 1.25 times the mean signal intensity in a manually draw region of cortical gray matter was used to segment voxels within the ROI corresponding to a ventricle. The total ventricular volume was calculated as the sum of the number of voxels within the segmented ventricular region multiplied by the volume of each voxel. The appearance of both eyes in the coronal plane of analysis was used as a marker to standardize regions for ventricular volume quantification. Eight consecutive images following the landmark image were used for volumetric determination.
Statistical analysis.
Statistical analyses were performed using Prism (GraphPad). P values of less than 0.05 were considered to be statistically significant. Descriptions of the specific tests are found in the figure legends. Viral replication assays, hemagglutination assays, infectivity assays comprising more than two virus strains, and animal studies were analyzed by analysis of variance (ANOVA) followed by Bonferroni’s correction for multiple tests. Student’s t tests were used for analysis of infectivity and binding experiments in which only two strains were compared.
SUPPLEMENTAL MATERIAL
Replication and binding of wild-type T1L and mutant S370P/Q371E in L cells. L cells were adsorbed with the strains shown at an MOI of 0.1 PFU/cell at room temperature for 1 h. Cells were washed with PBS, and fresh medium was added. At 0, 24, and 48 h postadsorption, cells were frozen and thawed twice, and viral titer in cell lysates was quantified by plaque assay. Results are expressed as the mean log10 viral yield from three independent experiments, each performed in triplicate. Error bars represent standard errors of the means. Differences are not statistically significant, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. Download
Multiple-residue T1 σ1 mutant virus is neutralized by MAb 5C6. Reovirus strain T1L, the S370P/Q371E mutant, and strain T3D were incubated with T1L σ1 conformation-specific MAb 5C6 at room temperature for 1 h. The virus-antibody mixture was adsorbed to L cells at room temperature for 1 h, cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed and stained with Alexa-647-labeled polyclonal reovirus antiserum. The percentage of infected cells was quantified using flow cytometry. The data shown are from a representative experiment of two independent experiments, each performed in duplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. Download
Binding of T1L and the S370P/Q371E mutant to MEFs. MEFs were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-647-labeled reovirus antiserum. The mean fluorescence intensity (MFI) was quantified using flow cytometry. Results are from a representative experiment of three independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test. Download
Binding of T1L and the S370P/Q371E mutant to L cells. L cells were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-647-labeled reovirus antiserum. The percentage of cells with bound virus (A) and the mean fluorescence intensity (MFI) (B) were quantified using flow cytometry. Results are from a representative experiment of three independent experiments, each performed in triplicate. Error bars represent standard deviations. Differences are not statistically significant, as determined by two-tailed Student’s t test. Download
Binding of T1L and the S370P/Q371E mutant to BXD-1425 EPN cells. BXD-1425EPN cells were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-488-labeled reovirus antiserum. The mean fluorescence intensity (MFI) of bound virus was quantified using flow cytometry. Results are from a representative experiment of two independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test. Download
ACKNOWLEDGMENTS
We are grateful to Melanie Dietrich and Alexandra Thor (Interfaculty Institute of Biochemistry, University of Tübingen) for assistance with the expression and purification of the σ1 proteins. We acknowledge the staff in the Vanderbilt University Center for Small Animal Imaging, Flow Cytometry Shared Resource, and Translational Pathology Shared Resource for technical support. We thank William S. Bush (Case Western University) for help with statistical analysis and Remco Sprangers (Max Planck Institut for Developmental Biology, Tübingen) for assistance in recording the NMR data. We thank James Paulson (The Scripps Research Institute) and Dapeng Zhou (MD Anderson Cancer Center) for establishing and providing the GM2−/− mice, respectively.
This work was supported by U.S. Public Health Service award R01 AI076983 and the Elizabeth B. Lamb Center for Pediatric Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation Stencel-Baerenwald J, Reiss K, Blaum BS, Colvin D, Li X, Abel T, Boyd K, Stehle T, Dermody TS. 2015. Glycan engagement dictates hydrocephalus induction by serotype 1 reovirus. mBio 6(2):e02356-14. doi:10.1128/mBio.02356-14
REFERENCES
- 1.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 2.Chappell JD, Duong JL, Wright BW, Dermody TS. 2000. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J Virol 74:8472–8479. doi: 10.1128/JVI.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. 1997. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein σ1. J Virol 71:1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22:4346–4355. doi: 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. 1983. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304:76–78. doi: 10.1038/304076a0. [DOI] [PubMed] [Google Scholar]
- 6.Neu U, Bauer J, Stehle T. 2011. Viruses and sialic acids: rules of engagement. Curr Opin Struct Biol 21:610–618. doi: 10.1016/j.sbi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haywood AM. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J Virol 68:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 9.Weiner HL, Drayna D, Averill DR Jr., Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci U. S. A. 74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J Infect Dis 141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 11.Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- 12.Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421. doi: 10.1126/science.6276976. [DOI] [PubMed] [Google Scholar]
- 13.Kilham L, Margolis G. 1969. Hydrocephalus in hamsters, ferrets, rats, and mice following inoculations with reovirus type 1. Lab Invest 22:183–188. [PubMed] [Google Scholar]
- 14.Margolis G, Kilham L. 1969. Hydrocephalus in hamsters, ferrets, rats, and mice following inoculations with reovirus type I. II. Pathologic studies. Lab Invest 21:189–198. [PubMed] [Google Scholar]
- 15.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus σ1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J Virol 85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison LA, Sidman RL, Fields BN. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci U S A 88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiner HL, Greene MI, Fields BN. 1980. Delayed hypersensitivity in mice infected with reovirus. I. Identification of host and viral gene products responsible for the immune response. J Immunol 125:278–282. [PubMed] [Google Scholar]
- 19.Pruijssers AJ, Hengel H, Abel TW, Dermody TS. 2013. Apoptosis induction influences reovirus replication and virulence in newborn mice. J Virol 87:12980–12989. doi: 10.1128/JVI.01931-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenson AB, Rabin ER, Phillips CA, Melnick JL. 1965. Reovirus encephalitis in newborn mice: an electron microscopic and virus assay study. Am J Pathol 47:223–239. [PMC free article] [PubMed] [Google Scholar]
- 21.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor “NgR1” mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional co-receptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermody TS. 2012. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J Virol 86:13164–13173. doi: 10.1128/JVI.01822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerner AM, Cherry JD, Finland M. 1963. Haemagglutination with reoviruses. Virology 19:58–65. doi: 10.1016/0042-6822(63)90024-2. [DOI] [PubMed] [Google Scholar]
- 26.Gentsch JR, Pacitti AF. 1987. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology 161:245–248. doi: 10.1016/0042-6822(87)90192-9. [DOI] [PubMed] [Google Scholar]
- 27.Meyer B, Peters T. 2003. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed Engl 42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 28.Haselhorst T, Fiebig T, Dyason JC, Fleming FE, Blanchard H, Coulson BS, von Itzstein M. 2011. Recognition of the GM3 ganglioside glycan by rhesus rotavirus particles. Angew Chem Int Ed Engl 50:1055–1058. doi: 10.1002/anie.201004116. [DOI] [PubMed] [Google Scholar]
- 29.Neu U, Allen SA, Blaum BS, Liu Y, Frank M, Palma AS, Ströh LJ, Feizi T, Peters T, Atwood WJ, Stehle T. 2013. A structure-guided mutation in the major capsid protein retargets BK polyomavirus. PLoS Pathog 9:e1003688. doi: 10.1371/journal.ppat.1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singhal N, Xu R, Martin PT. 2012. Distinct contributions of Galgt1 and Galgt2 to carbohydrate expression and function at the mouse neuromuscular junction. Mol Cell Neurosci 51:112–126. doi: 10.1016/j.mcn.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu L, Baxter PA, Voicu H, Gurusiddappa S, Zhao Y, Adesina A, Man TK, Shu Q, Zhang YJ, Zhao XM, Su JM, Perlaky L, Dauser R, Chintagumpala M, Lau CC, Blaney SM, Rao PH, Leung HC, Li XN. 2010. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol 12:580–594. doi: 10.1093/neuonc/nop056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masters C, Alpers M, Kakulas B. 1977. Pathogenesis of reovirus type 1 hydrocephalus in mice: significance of aqueductal changes. Arch Neurol 34:18–28. doi: 10.1001/archneur.1977.00500130038008. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto K, Takada K, Furukawa K, Furukawa K, Kusunoki S. 2008. Roles of complex gangliosides in the development of experimental autoimmune encephalomyelitis. Glycobiology 18:408–413. doi: 10.1093/glycob/cwn017. [DOI] [PubMed] [Google Scholar]
- 34.Rapoport E, Mikhalyov I, Zhang J, Crocker P, Bovin N. 2003. Ganglioside binding pattern of CD33-related siglecs. Bioorg Med Chem Lett 13:675–678. doi: 10.1016/S0960-894X(02)00998-8. [DOI] [PubMed] [Google Scholar]
- 35.Yu RK, Tsai YT, Ariga T, Yanagisawa M. 2011. Structures, biosynthesis, and functions of gangliosides—an overview. J Oleo Sci 60:537–544. doi: 10.5650/jos.60.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varki A. 2007. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 37.Schnaar RL. 2010. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett 584:1741–1747. doi: 10.1016/j.febslet.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lehmann HC, Lopez PHH, Zhang G, Ngyuen T, Zhang J, Kieseier BC, Mori S, Sheikh KA. 2007. Passive immunization with anti-ganglioside antibodies directly inhibits axon regeneration in an animal model. J Neurosci 27:27–34. doi: 10.1523/JNEUROSCI.4017-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest 111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashiro G, Loh PC, Yau JT. 1977. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch Virol 54:307–315. doi: 10.1007/BF01314776. [DOI] [PubMed] [Google Scholar]
- 41.Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. 2004. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci U. S. A. 101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. 1998. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coffey MC, Strong JE, Forsyth PA, Lee PW. 1998. Reovirus therapy of tumors with activated Ras pathway. Science 282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 44.Gollamudi R, Ghalib MH, Desai KK, Chaudhary I, Wong B, Einstein M, Coffey M, Gill GM, Mettinger K, Mariadason JM, Mani S, Goel S. 2010. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs 28:641–649. doi: 10.1007/s10637-009-9279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vidal L, Pandha HS, Yap TA, White CL, Twigger K, Vile RG, Melcher A, Coffey M, Harrington KJ, DeBono JS. 2008. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res 14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- 46.Sahin E, Egger ME, McMasters KM, Zhou HS. 2013. Development of oncolytic reovirus for cancer therapy. J Cancer Ther 4:1100–1115. doi: 10.4236/jct.2013.46127. [DOI] [Google Scholar]
- 47.Harduin-Lepers A, Krzewinski-Recchi MA, Colomb F, Foulquier F, Groux-Degroote S, Delannoy P. 2012. Sialyltransferases functions in cancers. Front Biosci (Elite Ed) 4:499–515. doi: 10.2741/396. [DOI] [PubMed] [Google Scholar]
- 48.Livingston PO, Hood C, Krug LM, Warren N, Kris MG, Brezicka T, Ragupathi G. 2005. Selection of GM2, fucosyl GM1, globo H and polysialic acid as targets on small cell lung cancers for antibody mediated immunotherapy. Cancer Immunol Immunother 54:1018–1025. doi: 10.1007/s00262-005-0663-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjerkvig R, Engebraaten O, Laerum OD, Fredman P, Svennerholm L, Vrionis FD, Wikstrand CJ, Bigner DD. 1991. Anti-GM2 monoclonal antibodies induce necrosis in GM2-rich cultures of a human glioma cell line. Cancer Res 51:4643–4648. [PubMed] [Google Scholar]
- 50.Yamada T, Bando H, Takeuchi S, Kita K, Li Q, Wang W, Akinaga S, Nishioka Y, Sone S, Yano S. 2011. Genetically engineered humanized anti-ganglioside GM2 antibody against multiple organ metastasis produced by GM2-expressing small-cell lung cancer cells. Cancer Sci 102:2157–2163. doi: 10.1111/j.1349-7006.2011.02093.x. [DOI] [PubMed] [Google Scholar]
- 51.Kim M, Garant KA, zur Nieden NI, Alain T, Loken SD, Urbanski SJ, Forsyth PA, Rancourt DE, Lee PW, Johnston RN. 2011. Attenuated reovirus displays oncolysis with reduced host toxicity. Br J Cancer 104:290–299. doi: 10.1038/sj.bjc.6606053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van den Wollenberg DJ, Dautzenberg IJ, van den Hengel SK, Cramer SJ, de Groot RJ, Hoeben RC. 2012. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS One 7:e48064. doi: 10.1371/journal.pone.0048064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raval G, Biswas S, Rayman P, Biswas K, Sa G, Ghosh S, Thornton M, Hilston C, Das T, Bukowski R, Finke J, Tannenbaum CS. 2007. TNF-alpha induction of GM2 expression on renal cell carcinomas promotes T cell dysfunction. J Immunol 178:6642–6652. doi: 10.4049/jimmunol.178.10.6642. [DOI] [PubMed] [Google Scholar]
- 54.Boehme KW, Hammer K, Tollefson WC, Konopka-Anstadt JL, Kobayashi T, Dermody TS. 2013. Nonstructural protein sigma1s mediates reovirus-induced cell cycle arrest and apoptosis. J Virol 87:12967–12979. doi: 10.1128/JVI.02080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Danthi P, Pruijssers AJ, Berger AK, Holm GH, Zinkel SS, Dermody TS. 2010. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog 6:e1000980. doi: 10.1371/journal.ppat.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boehme KW, Ikizler M, Kobayashi T, Dermody TS. 2011. Reverse genetics for mammalian reovirus. Methods 55:109–113. doi: 10.1016/j.ymeth.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 60.Shatkin AJ, Sipe JD, Loh P. 1968. Separation of 10 reovirus genome segments by polyacrylamide gel electrophoresis. J Virol 2:986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Virgin HW IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wetzel JD, Wilson GJ, Baer GS, Dunnigan LR, Wright JP, Tang DS, Dermody TS. 1997. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J Virol 71:1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Virgin HW IV, Mann MA, Fields BN, Tyler KL. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1995. 1H, 13C and 15-N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Replication and binding of wild-type T1L and mutant S370P/Q371E in L cells. L cells were adsorbed with the strains shown at an MOI of 0.1 PFU/cell at room temperature for 1 h. Cells were washed with PBS, and fresh medium was added. At 0, 24, and 48 h postadsorption, cells were frozen and thawed twice, and viral titer in cell lysates was quantified by plaque assay. Results are expressed as the mean log10 viral yield from three independent experiments, each performed in triplicate. Error bars represent standard errors of the means. Differences are not statistically significant, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. Download
Multiple-residue T1 σ1 mutant virus is neutralized by MAb 5C6. Reovirus strain T1L, the S370P/Q371E mutant, and strain T3D were incubated with T1L σ1 conformation-specific MAb 5C6 at room temperature for 1 h. The virus-antibody mixture was adsorbed to L cells at room temperature for 1 h, cells were washed twice with PBS, and fresh medium was added. After incubation at 37°C for 20 h, cells were fixed and stained with Alexa-647-labeled polyclonal reovirus antiserum. The percentage of infected cells was quantified using flow cytometry. The data shown are from a representative experiment of two independent experiments, each performed in duplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-way ANOVA followed by Bonferroni’s multiple comparison test. Download
Binding of T1L and the S370P/Q371E mutant to MEFs. MEFs were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-647-labeled reovirus antiserum. The mean fluorescence intensity (MFI) was quantified using flow cytometry. Results are from a representative experiment of three independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test. Download
Binding of T1L and the S370P/Q371E mutant to L cells. L cells were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-647-labeled reovirus antiserum. The percentage of cells with bound virus (A) and the mean fluorescence intensity (MFI) (B) were quantified using flow cytometry. Results are from a representative experiment of three independent experiments, each performed in triplicate. Error bars represent standard deviations. Differences are not statistically significant, as determined by two-tailed Student’s t test. Download
Binding of T1L and the S370P/Q371E mutant to BXD-1425 EPN cells. BXD-1425EPN cells were adsorbed with the virus strains shown at an MOI of 5 × 104 particles per cell at 4°C for 1 h. Cells were washed twice with PBS and stained with Alexa-488-labeled reovirus antiserum. The mean fluorescence intensity (MFI) of bound virus was quantified using flow cytometry. Results are from a representative experiment of two independent experiments, each performed in triplicate. Error bars represent standard deviations. ***, P < 0.001, as determined by two-tailed Student’s t test. Download