

ABSTRACT
The opportunistic pathogen Pseudomonas aeruginosa can infect almost any site in the body but most often targets epithelial cell-lined tissues such as the airways, skin, and the cornea of the eye. A common predisposing factor is cystic fibrosis (CF), caused by defects in the cystic fibrosis transmembrane-conductance regulator (CFTR). Previously, we showed that when P. aeruginosa enters epithelial cells it replicates intracellularly and occupies plasma membrane blebs. This phenotype is dependent on the type 3 secretion system (T3SS) effector ExoS, shown by others to induce host cell apoptosis. Here, we examined mechanisms for P. aeruginosa-induced bleb formation, focusing on its relationship to apoptosis and the CFTR. The data showed that P. aeruginosa-induced blebbing in epithelial cells is independent of actin contraction and is inhibited by hyperosmotic media (400 to 600 mOsM), distinguishing bacterially induced blebs from apoptotic blebs. Cells with defective CFTR displayed enhanced bleb formation upon infection, as demonstrated using bronchial epithelial cells from a patient with cystic fibrosis and a CFTR inhibitor, CFTR(Inh)-172. The defect was found to be correctable either by incubation in hyperosmotic media or by complementation with CFTR (pGFP-CFTR), suggesting that the osmoregulatory function of CFTR counters P. aeruginosa-induced bleb-niche formation. Accordingly, and despite their reduced capacity for bacterial internalization, CFTR-deficient cells showed greater bacterial occupation of blebs and enhanced intracellular replication. Together, these data suggest that P. aeruginosa bleb niches are distinct from apoptotic blebs, are driven by osmotic forces countered by CFTR, and could provide a novel mechanism for bacterial persistence in the host.
IMPORTANCE
Pseudomonas aeruginosa is an opportunistic pathogen problematic in hospitalized patients and those with cystic fibrosis (CF). Previously, we showed that P. aeruginosa can enter epithelial cells and replicate within them and traffics to the membrane blebs that it induces. This “bleb-niche” formation requires ExoS, previously shown to cause apoptosis. Here, we show that the driving force for bleb-niche formation is osmotic pressure, differentiating P. aeruginosa-induced blebs from apoptotic blebs. Either CFTR inhibition or CFTR mutation (as seen in people with CF) causes P. aeruginosa to make more bleb niches and provides an osmotic driving force for blebbing. CFTR inhibition also enhances bacterial occupation of blebs and intracellular replication. Since CFTR is targeted for removal from the plasma membrane when P. aeruginosa invades a healthy cell, these findings could relate to pathogenesis in both CF and healthy patient populations.
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic bacterium that can infect almost any part of the human body but typically targets surface-exposed epithelial cells such as in the airways, skin, and eye. P. aeruginosa is particularly devastating in cystic fibrosis (CF), a common hereditary disease that significantly decreases the life span of patients as a result of chronic lung infections characterized by a progressive destructive bronchitis and bronchiolitis (1). P. aeruginosa tends to dominate the CF airways, being present in 80% of CF patients over the age of 18 (2). The cystic fibrosis transmembrane-conductance regulator (CFTR), mutated in patients with CF, has been shown to be involved in P. aeruginosa virulence (reviewed in reference 3).
P. aeruginosa can enter epithelial cells during lung and eye infections in vivo (4–8). However, epithelial cells isolated from CF patients are known to phagocytose fewer P. aeruginosa bacterial cells (9, 10). The mechanism by which bacterial internalization is inhibited in CF cells does not involve the reduced conductance capacity of the chloride (Cl−) channel; instead, P. aeruginosa internalization is mediated by binding to lipid rafts (11).
Our published data show that, after P. aeruginosa enters cultured epithelial cells, a subset of infected cells display plasma membrane blebs to which bacteria traffic, while others show bacteria colocalizing with acidic vacuoles (6, 12, 13). These membrane blebs physically separate internalized bacteria from the remainder of the cytoplasm, allowing bacteria to swim rapidly within them. This phenomenon is not limited to cultured cells. Indeed, published images show that bleb-niche formation also occurs in corneal epithelial cells within excised whole mouse eyes (7).
We previously showed that the ExoS type 3 secretion system (T3SS) effector is required for bleb-niche formation. Mutants in exoS are unable to form blebs while also demonstrating reduced intracellular replication, hinting at a relationship between the two phenomena (6, 12). Mechanisms involved in bleb formation remain unknown, except that our previous studies using annexin V staining of infected cells produced results suggesting activation of programmed cell death in blebbing cells (6).
Plasma membrane blebbing can be associated with multiple types of cell death, including apoptosis and necrosis, which can both cause annexin V staining (14). Apoptotic bleb outgrowth is driven by the cortical tension produced as a result of actin contraction, which occurs when myosin II actively slides actin stress fibers against each other at the neck of the bleb (15–20) along the actin cortex that lies under the plasma membrane (21). Following actin cortex separation from the plasma membrane at the bleb neck, the actin contraction in apoptotic blebs allows unfolding of membrane reservoirs and/or flow of membrane through the bleb neck (18, 20). In contrast, necrotic blebbing differs from the more fully understood apoptotic blebbing in that the blebs are larger and more transparent and the bleb formation is independent of actin contraction (22).
Here, we explored host cell factors involved in bleb formation induced by infection with P. aeruginosa, the role of CFTR, and the consequences of blebbing for survival of intracellular P. aeruginosa in CF cells. Since ExoS has been shown to induce apoptosis in cells following transfection or after injection through the T3SS needle (23–25), we hypothesized that the formation of blebs induced by P. aeruginosa involves the host cell apoptotic machinery.
RESULTS
P. aeruginosa-induced blebbing is independent of actin contraction.
Since actin contraction is required for apoptotic blebbing, we examined its role in P. aeruginosa-induced blebbing using human corneal epithelial cells. Infection-induced blebs were compared to apoptotic blebs generated using tumor necrosis factor alpha (TNF-α) and actinomycin D. Following a 4-h infection or 4-h induction of apoptosis, corneal epithelial cells were incubated either with 5 µM latrunculin A to depolymerize the actin cytoskeleton or with dimethyl sulfoxide (DMSO) as a control for an additional 3 h. This latrunculin treatment was sufficient to remove actin stress fibers in the absence of either bacteria or apoptotic induction (see Fig. S1 in the supplemental material). Treatment with latrunculin completely blocked apoptotic blebbing, without having an effect on blebbing induced by P. aeruginosa (Fig. 1a and b). Further, P. aeruginosa-induced blebs appeared morphologically distinct from apoptotic blebs, the latter being much smaller and more irregularly shaped than the large, ballooning P. aeruginosa-induced blebs (Fig. 1a).
FIG 1 .

Role of actin contraction in P. aeruginosa-induced blebbing. (a) Representative images of blebbing (arrows) induced in human corneal epithelial cells following treatment with TNF-α and actinomycin D to induce apoptosis (upper panels) or infection with P. aeruginosa (MOI = 100) in gentamicin-containing media to kill extracellular bacteria (lower panels). Following 4 h of exposure to either the apoptotic cocktail or bacteria, 5 µM latrunculin A (or the DMSO control) was added for an additional 3 h prior to imaging. (b) Quantification of blebbing frequency in the experiment represented in panel a. Latrunculin inhibited apoptotic blebbing (P = 0.001, Student’s t test) but not infection-induced blebbing (P = 0.5, Student’s t test). NS, not significant. (c) Corneal epithelial cells were pretreated with either the r-enantiomer or the s-enantiomer of blebbistatin (40 µM) prior to inoculation with P. aeruginosa (MOI = 100), and blebbing frequency was quantified at 7 h postinoculation. ***, P < 0.001 (ANOVA with Dunnett’s multiple group comparison post hoc analysis comparing all groups to the untreated group). (d) Western blot analysis of phosphorylated myosin light chain 20 in corneal cells during the course of infection. (e) Rhodamine-phalloidin staining of corneal epithelial cells at various time points before and after infection with P. aeruginosa. All images were taken using the same exposure settings. A representative blebbing cell (the bleb had collapsed after fixation) shown in DIC (right) was free of stress fibers at 6 h postinfection.
To verify results with latrunculin, infected corneal epithelial cells were treated with blebbistatin, which inhibits several types of blebbing, including apoptotic and certain migratory blebs (26–28). Rather than blocking P. aeruginosa-induced blebbing, s-blebbistatin treatment caused significantly more bacterially induced blebs to form (Fig. 1c; see also Fig. S2 in the supplemental material). Nevertheless, the enhanced blebbing caused by s-blebbistatin did not result in a greater number of bacteria occupying blebs (see Fig. S3). Further supporting the idea that infection-induced blebbing occurs independently of actin contraction, myosin light chain peptide 20 (MLC20) was actively dephosphorylated as a result of infection with P. aeruginosa (Fig. 1d). MLC20 in its phosphorylated state is required for myosin-induced actin contraction. Thus, the basal level of phosphorylated MLC20 is an indicator of the presence of active myosin involved in actin contraction. The basal phosphorylated MLC20 levels declined over the course of the infection and became essentially undetectable between 2 and 3 h after bacterial inoculation (Fig. 1d). Likewise, phalloidin staining revealed a basal level of actin stress fibers present in uninfected cells that increased at 2 h postinoculation, decreased by 3 h, and then disappeared in cells harboring blebs at 6 h postinoculation (Fig. 1e).
P. aeruginosa-induced blebbing is osmotically driven.
Having found that blebs induced in response to P. aeruginosa infection form independently of actin contraction, we explored alternatives for potential driving forces. Hydrostatic pressure can play an integral role in bleb formation (22) and is involved in the formation of necrotic blebs, which rely on movement of sodium and water for bleb biogenesis (29). Therefore, we examined the effect of medium osmolarity on P. aeruginosa-induced bleb-niche formation. Bacteria were allowed to internalize before gentamicin and hyperosmotic media were added (Fig. 2a). Blebbing in the corneal cells diminished as the osmolarity of the media increased to up to 600 mOsM (Fig. 2c and d). Infection-induced blebbing could be almost completely eliminated by adding 600 mOsM mannitol to the media at 4 h after bacteria entered epithelial cells (Fig. 2d). Treating apoptotic cells with the same hyperosmotic media (Fig. 2b) did not reduce the number of apoptotic blebs. Indeed, the number of cells with apoptotic blebs slightly increased with increasing osmolarity (Fig. 2c and e).
FIG 2 .
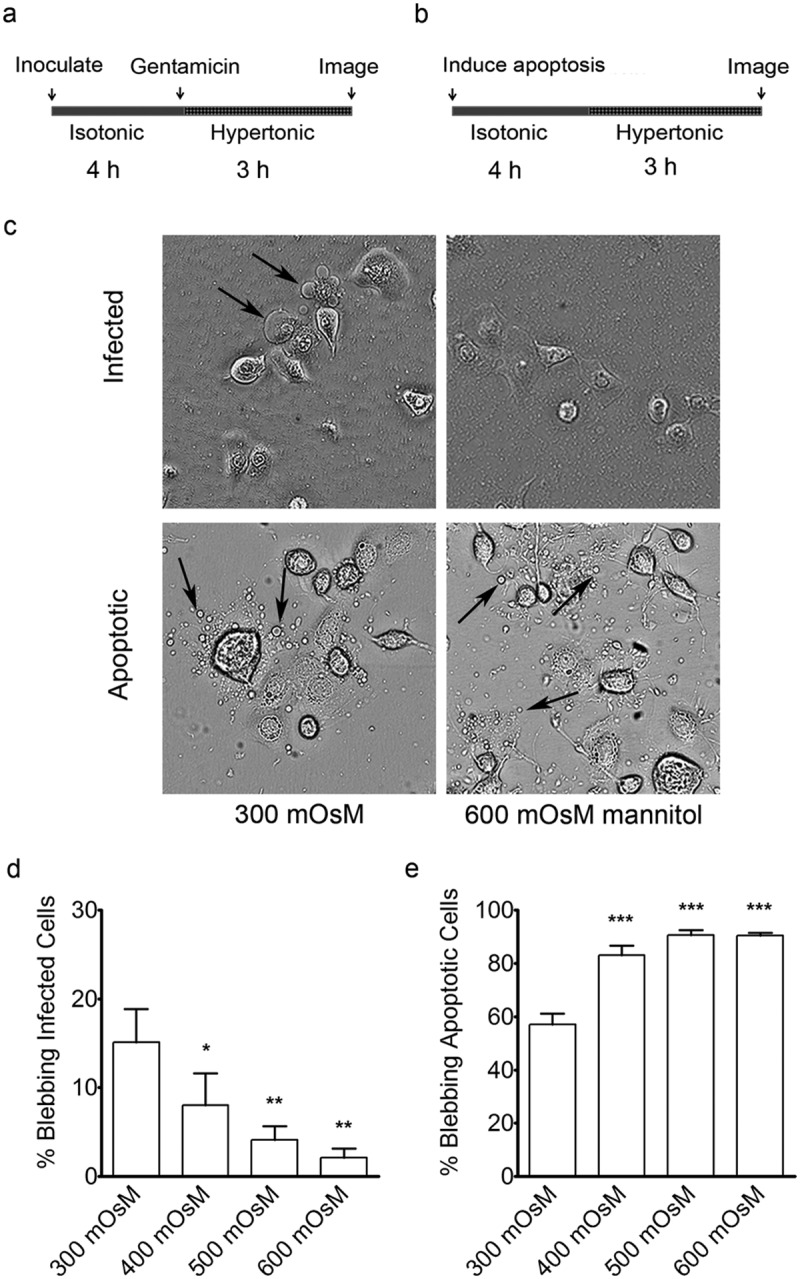
Effects of osmolarity on P. aeruginosa-induced blebs in human corneal epithelial cells. (a) Schematic of the experimental outline for infection prior to treatment with hyperosmotic media. Four hours post-inoculation, after blebs had formed, hyperosmotic media was added to the cells for an additional 3 h (using mannitol to achieve a final osmolarity of 400, 500, or 600 mOsM). (b) Schematic showing a similar experimental outline for induction of apoptosis without infection prior to osmotic treatment. (c) Representative images showing the effects of the hyperosmotic media (600 mOsM) on blebbing (see arrows). (d) Blebbing frequency in human corneal epithelial cells following infection in iso-osmolar media (15% ± 4%) compared to that in increasingly hyperosmolar media (2% ± 1% in 600 mOsM). (e) Blebbing frequency following induction of apoptosis in iso-osmolar media (57% ± 4%) compared to that in increasingly hyperosmolar media (91% ± 1% in 600 mOsM), *, P < 0.05; **, P < 0.01; ***, P < 0.001 (ANOVA with Dunnett’s multiple-comparison post hoc analysis).
Next, we performed long-term imaging experiments to investigate the behavior of the blebs in hypo-osmotic media after using hyperosmotic media to prevent the induction of infection-induced blebs. Bleb size was modulated within seconds of diluting the medium from 600 mOsM down to a 1:1 mixture of medium and water (i.e., 50% medium) and lower, including 25%, 10%, and 0% media (the latter is pure water) (see Movie S1 in the supplemental material). Blebs that had been repressed in the hyperosmolar media, and thus arrested, were derepressed and bulged out almost instantaneously. These imaging experiments also revealed what appeared to be intracellular vacuoles depositing bacteria directly into the newly formed blebs (see Movie S2), suggesting that bleb formation may occur prior to bacterial entrance into the bleb.
CFTR deficiency enhances P. aeruginosa-induced bleb-niche formation.
The CFTR chloride channel regulates fluid-mediated changes in cell volume in response to osmotic pressure (30–33). Given the previously established roles of CFTR in P. aeruginosa interactions with the host, we hypothesized that the chloride-conductance capacity of CFTR may contribute to the osmotically driven bleb formation. Corneal epithelial cells treated with CFTR(Inh)-172 (20 µM) prior to inoculation showed increased bacterially induced blebbing compared to DMSO-treated controls (Fig. 3a). Blebs were not found in uninfected cells treated with inhibitor or DMSO alone.
FIG 3 .

Blebbing frequency and size in epithelial cells following infection with Pseudomonas aeruginosa. (a) Corneal epithelial cells pretreated with 20 µM CFTR(Inh)-172 increased the blebbing frequency from 20% ± 3% (DMSO, n = 3 days) to 50% ± 4% (Inh-172, n = 3 days) (P < 0.001, Student’s t test). Treatment with inhibitor without infection did not induce blebbing. (b) Live imaging of NuLi-1 and CuFi-1 bronchial epithelial cells between 6 and 7 h post-inoculation in the presence of gentamicin (blebs are artificially outlined in black). (c) Box and whiskers plot showing maximum, minimum, and median bleb areas following infection. The median area was 440 µm2 (n = 164 blebs) in CF cells versus 138 µm2 (n = 139 blebs) in non-CF cells, P < 0.0001 (Mann-Whitney U test). (d) The number of cells having blebs was greater in CF cells (26% ± 5%, n = 3 days) than in non-CF cells (15% ± 2%, n = 3 days) (P < 0.05, Student’s t test). (e) Treatment of non-CF bronchial epithelial cells with CFTR(Inh)-172 resulted in an increase in blebbing frequency from 22% ± 3% (DMSO, n = 3 days) to 39% ± 8% (Inh-172, n = 3 days) (P < 0.05, Student’s t test); treating CF cells with the inhibitor had no effect on blebbing (P = 0.6, Student’s t test). NS, not significant. Data are expressed as weighted means ± weighted standard deviations.
Given that blebs are niches for intracellular bacteria and that CFTR mutation predisposes the host to infection of the airways, the results described above prompted us to examine the impact of CF on blebbing in airway cells. After 6 h, CF-derived bronchial epithelial cells (CuFi-1; CF) displayed enhanced bleb-niche formation and size compared to non-CF bronchial cells (NuLi-1; non-CF) (Fig. 3b to d). Similarly to the corneal epithelial cells, NuLi-1 cells treated with CFTR(Inh)-172 (20 µM) displayed an increase in P. aeruginosa-induced blebbing compared to controls, to a level similar in magnitude to that observed in CF versus non-CF cells (Fig. 3e). Treatment of CF cells with the CFTR inhibitor had no effect on blebbing (Fig. 3e), suggesting that the enhancement of bacterially induced blebs by this compound in non-CF cells was not due to off-target effects of the inhibitor.
Over-expression of CFTR prevents P. aeruginosa-induced blebbing.
Since both mutation of CFTR and a CFTR inhibitor enhanced the ability of bacteria to induce blebbing, we investigated the effects of CFTR complementation on the blebbing phenotype. CF cells were transfected with pGFP-CFTR, a plasmid encoding wild-type CFTR under the control of the cytomegalovirus (CMV) promoter. Both green fluorescent protein-positive (GFP+) and GFP− cells were present in each transfected dish, so each dish served as its own infection control. Following infection, GFP− CF cells formed large-membrane blebs whereas none of the GFP+ CF cells formed blebs (see Fig. S4 in the supplemental material). A pEGFP plasmid was also used as a transfection control for the effects of overexpression of GFP on blebbing. Unlike the cells expressing GFP-CFTR, cells expressing GFP alone retained the ability to bleb, even when the GFP was expressed at much higher levels (see Fig. S4). Since the GFP-CFTR fusion construct traffics and folds similarly to native CFTR (34), these data show that overexpression of CFTR prevents P. aeruginosa-induced blebbing in CF cells.
Blebbing in CFTR-deficient cells is rescued by small increases in extracellular osmolarity.
The enhanced bacterially induced blebbing in the CFTR inhibitor-treated non-CF bronchial cells suggested involvement of Cl− channel conductance in bleb formation. Chloride ion conductance is integral to the maintenance of osmotic pressure, and we hypothesized that the osmoregulatory function of CFTR is involved in bleb formation. As in non-CF cells, blebbing in CF cells was almost completely eliminated by increasing the medium osmolarity to 600 mOsM at 3 h after bacteria entered epithelial cells, using either NaCl or mannitol to modulate the medium osmolarity (Fig. 4a). We therefore tested the ability of hyperosmotic media to correct for the enhanced blebbing caused by the CFTR inhibitor in normal cells. Only 400 mOsM was required to eliminate enhanced bleb formation in CFTR inhibitor-treated cells (Fig. 4b). Similarly, increasing osmolarity to 500 mOsM eliminated the enhanced blebbing observed in CF compared to non-CF cells (Fig. 4c).
FIG 4 .
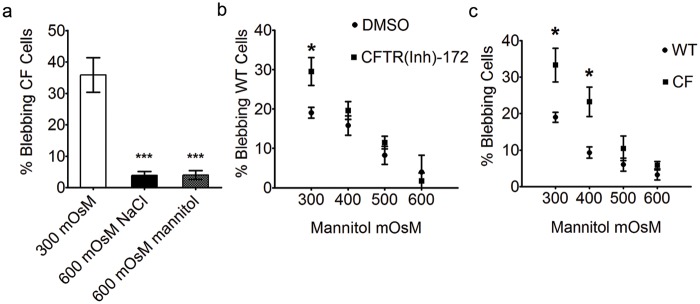
Reversal of blebbing in CFTR-deficient cells by incubation in hyperosmotic media. (a) P. aeruginosa-induced blebbing in CF bronchial epithelial cells decreased from 36% ± 6% in isosmotic media to 4% ± 1%, by increasing media osmolarity to 600 mOsM with either NaCl or mannitol after 3 h (***, P < 0.001, ANOVA with Dunnett’s multiple-comparison post hoc analysis). (b and c) The percentage of blebbing cells was also recorded in PAO1-infected non-CF cells treated with CFTR(Inh)-172 (>244 cells per data point) versus DMSO-treated (>498 cells per data point) (b) and CF (>552 cells per data point) versus non-CF (>462 cells per data point) (c) bronchial cells following treatment with 400, 500, and 600 mOsM media at 3 h postinoculation (*, P < 0.05; Student’s t test comparing CF to non-CF cells under each condition). Data are expressed as means ± standard deviations and represent the results from three independent experiments. WT, wild type.
CF cells show enhanced P. aeruginosa occupation of bleb niches and enhanced intracellular growth.
Microscopy and gentamicin exclusion intracellular-survival assays were used to determine the degree of bacterial internalization and intracellular growth in non-CF and CF bronchial epithelial cells. The P. aeruginosa-induced bleb niches in non-CF bronchial cells frequently contained fewer bacteria than those induced in CF cells (Fig. 5a and b), suggesting that bacteria may have an enhanced survival niche in CFTR-deficient cells. We observed a maximum of 6 bacteria occupying an individual bleb in a non-CF cell and up to 25 bacteria occupying a CF cell bleb at 6 h postinoculation (Fig. 5c). The greater numbers of bacteria occupying individual blebs and the increase in number of blebbing cells resulted in a 2.3-fold population-wide increase in total bacterial numbers within blebs in CF cells (63 ± 5 per 100 cells) compared to non-CF cells (27 ± 2 per 100 cells), as determined by live microscopy. While the number of bacteria occupying individual blebs was often higher in CF cells, as shown in Fig. 5c, the percentages of blebs harboring bacteria were similar in non-CF cells (34% ± 9%, weighted mean ± weighted standard deviation [SD]) and CF cells (23% ± 19%) (P = 0.34, Student’s t test), suggesting that the rate of bacterial trafficking to blebs is independent of the presence of CFTR.
FIG 5 .

Increased P. aeruginosa occupation of bleb niches and survival in CF versus non-CF cells. Bleb occupation was determined using confocal microscopy and P. aeruginosa expressing GFP. (a and b) Non-CF cell blebs (a) contained fewer bacteria on average than CF cell blebs (b). (c) A maximum of 6 bacteria were found in non-CF cell blebs (50 blebs analyzed), while up to 25 bacteria occupied a CF cell bleb (84 blebs analyzed) at 6 h post-inoculation. (d) Gentamicin exclusion assays comparing P. aeruginosa internalization in non-CF and CF bronchial epithelial cells using either the wild type (WT) or the exsA mutant (MOI = 5). CF cells showed a significant reduction in internalization of both the WT (P < 0.01, Student’s t test) and the exsA mutant (P < 0.05, Student’s t test). (e) Intracellular replication of P. aeruginosa and its T3SS mutant over the 3-h period between 4 and 7 h post-inoculation using the gentamicin exclusion assay. Intracellular growth was greater in CF cells than in non-CF cells (P < 0.05, Student’s t test), whereas levels of exsA mutant replication did not differ in CF versus non-CF cells (P = 0.6, Student’s t test). NS, not significant. Data are expressed as means ± SEM. Data represent a composite of the results of three independent experiments.
As previously published (7, 8), internalization of P. aeruginosa by CF cells was reduced compared to that seen with non-CF cells (Fig. 5d). Since the T3SS encodes effectors that can inhibit phagocytosis, we also examined a T3SS mutant (exsA). The exsA mutant had a reduced capacity to invade CF cells similar to that seen with wild-type PAO1, showing that CF does not change the impact of the T3SS on internalization.
We next explored the impact of CF on the fate of wild-type PAO1 following internalization by bronchial epithelial cells by comparing the intracellular population at 4 h (baseline internalized population) to the population at 7 h (post-internalization population). The results showed that the population in CF cells increased 6.9-fold ± 1.9-fold during this 3-h interval compared to only 2.7-fold ± 0.8-fold in non-CF cells (Fig. 5e). In contrast, the levels of intracellular replication by the PAO1 T3SS mutant in non-CF and CF cells were similar (Fig. 5e). Thus, the CF cells favored more efficient intracellular replication for wild-type PAO1 but not for a T3SS mutant.
Blebbing cells in a P. aeruginosa-infected population lack acidic vacuoles for both CF and normal cells.
Previously, we demonstrated that internalized P. aeruginosa T3SS mutants that do not thrive intracellularly colocalize with acidic vacuoles that stain with LysoTracker dye (13) and reside within vacuoles positive for the lysosomal marker LAMP3 (12). Thus, we tested the hypothesis that differences in intracellular survival between non-CF and CF cells are due to differential acidification of vacuoles. Instead, we found that uninfected CF cells displayed LysoTracker dye retention and vacuolar morphology similar to those seen with non-CF cells, and all cells in both populations stained with the dye (Fig. 6a). As shown in Fig. 6a and b, after 6 h of infection with PAO1, similar numbers of CF cells and non-CF cells failed to retain the LysoTracker stain, indicating that they were devoid of acidic vacuoles. Interestingly, approximately 80% of the blebbing cells in both the CF and non-CF cell populations did not retain the dye whereas most nonblebbing cells did. Thus, CFTR deficiency did not affect the bacterially induced changes in vacuolar acidification that we previously reported (13), nor did it impact the overall numbers of acidic vacuoles in uninfected cells.
FIG 6 .

Non-CF and CF blebbing cells lack acidic vacuoles. (a) Representative images of LysoTracker staining in non-CF and CF bronchial epithelial cells before and after infection with PAO1-GFP (MOI = 10). (b) Similar numbers of non-CF and CF cells stained with LysoTracker (56% ± 7% of non-CF cells and 52% ± 7% of CF cells; P = 0.5, Student’s t test). NS, not significant. Few blebbing cells retained the stain compared to nonblebbing cells (22% ± 13% of blebbing versus 88% ± 7% of nonblebbing non-CF cells [P < 0.01, Student’s t test]; 12% ± 11% of blebbing versus 80% ± 7% of non-blebbing CF cells [P < 0.01, Student’s t test]). Data are expressed as means ± standard deviations and were compiled from the results of three independent experiments.
Inhibition of bleb-niche formation reduces intracellular P. aeruginosa replication.
Since differences between non-CF and CF cells in intracellular killing of bacteria could not be explained by differences in vacuolar acidification, we next explored if enhanced blebbing in CF cells following infection was involved. Intracellular survival assays showed that bleb inhibition with 600 mOsM mannitol resulted in a 75% decrease in total intracellular bacterial numbers at the end of a 6-h assay (3 h of incubation followed by 3 h of gentamicin treatment) in CF cells (Fig. 7a). Similar results were observed with other cell types, i.e., human corneal epithelial cells (85% ± 4% reduction; same assay) and RAW 264.7 macrophages (65% ± 25% reduction; modified 5-h assay) (see Fig. S5 in the supplemental material).
FIG 7 .

Treatment of infected CF cells with hyperosmolar media reduces intracellular replication of P. aeruginosa. (a) Bacteria were allowed to internalize into CF cells before the media osmolarity was adjusted as described in the Fig. 5 legend. CFU counts taken at 6 h postinoculation showed a 75% loss in the total number of intracellular bacteria (***, P < 0.001 [ANOVA with Dunnett’s multiple-comparison post hoc analysis]; 3 to 6 independent experiments per condition). (b) Intracellular replication rates between 4 h and 7 h postinoculation in the same assay (gentamicin and hyperosmotic media were added simultaneously at 3 h post-inoculation). Wild-type bacteria replicated 4.6-fold ± 0.6-fold in iso-osmolar media, while replication diminished to 1.8-fold ± 0.3-fold in hyperosmolar media (P < 0.01, Student’s t test). Levels of T3SS mutant bacterial replication were similar in iso-osmolar media (0.99-fold ± 0.16-fold growth) and hyperosmolar media (1.08-fold ± 0.16-fold growth) (P = 0.6, Student’s t test). NS, not significant. Data are presented as means ± SEM. (c) Number of viable (propidium iodide-negative) epithelial cells following treatment with 600 mOsM NaCl or mannitol (P = NS, ANOVA with Dunnett’s multiple-comparison post hoc analysis). Data are expressed as means ± standard deviations of the results from three independent experiments. (d) Effect of hyperosmotic treatments on extracellular bacterial replication.
The hyperosmolar treatment of CF cells also decreased the bacterial replication rate over 3 h (Fig. 7b) to levels similar to those seen with the T3SS mutant (which does not form blebs due to lack of ExoS). Accordingly, 600 mOsM mannitol had no effect on intracellular replication by the T3SS mutant (Fig. 7b). Moreover, we found that the number of CF cells that lacked acidic vacuoles after exposure to 600 mOsM media was similar to that seen with cells in isosmotic media (data not shown). Controls confirmed that mannitol treatment had no impact on epithelial cell viability (Fig. 7c) or replication of extracellular bacteria (Fig. 7d). Taken together, these results point toward the increased number of bleb niches as a potential contributor to increased bacterial replication in CF cells versus non-CF cells.
DISCUSSION
Apoptotic blebbing, in addition to blebbing associated with certain types of cell migration and during cytokinesis, is a multi-step process requiring delamination, a separation of the plasma membrane from the underlying actin cortex, and actin contraction powered by the activity of myosin II (15, 16, 19, 20, 22, 26–28). Here we show that P. aeruginosa-induced bleb formation is independent of actin contraction and is instead sensitive to osmotic pressure, which is not a feature of apoptotic blebs. Thus, P. aeruginosa-induced blebs are more similar to blebs found in necrotic or pyroptotic cells, whose formation is independent of actomyosin contraction (22) and is instead driven by a net gain of sodium and water (29, 35). Notably, the annexin V staining that we observed previously (6) can label cells not undergoing apoptosis if the membrane becomes porous, allowing the phosphatidylserine present on the inner leaflet of the plasma membrane to be labeled (36). However, pyroptotic and necrotic cells are rapidly lysed whereas P. aeruginosa-infected cells remain intact and adherent to the tissue culture dish for over 8 h post-inoculation. This suggests that P. aeruginosa may actively modify the host cell death response in a manner similar to that seen with some other pathogens (reviewed in reference 37). Determining the relationship between blebbing and cell death, if it exists, will be a significant effort requiring a separate dedicated study.
We hypothesized that the chloride channel CFTR may play a role in generating the osmotic pressure required for P. aeruginosa-induced bleb formation. Many glands and epithelial mucosa secrete fluid following activation of the CFTR chloride channel, which plays a critical role in regulating cellular osmolarity (reviewed in reference 38). Our data suggest that loss of the CFTR osmoregulatory function is involved in bleb-niche formation as a specific inhibitor of chloride conductance through the CFTR channel-enhanced formation of P. aeruginosa-induced membrane bleb niches in non-CF bronchial epithelial cells. Moreover, hyperosmotic media corrected for the bleb-inducing effect of the inhibitor, without an impact on actin contraction-mediated (apoptotic) blebbing. We propose a model for P. aeruginosa-induced blebbing (Fig. 8) that contrasts it to actin contraction-mediated blebbing (e.g., apoptotic blebbing), in that loss of actin stress fibers occurs following infection both in normal epithelial cells (see Fig. 1e) and in CF epithelial cells (see Fig. S6 in the supplemental material). Airway cells from CF patients are edematous (swollen), and their volume is greater than that of airway cells from non-CF patients (39). Indeed, cells with defective CFTR are unable to regulate their cell volume following osmotic stress (30–33), indicating that the cell volume is dysregulated as a direct consequence of CFTR deficiency. The greater existing stress on the plasma membrane in CF cells even before the application of external forces, as a result of the abnormally enhanced cell volume, may then make it more susceptible to blebbing upon P. aeruginosa infection (Fig. 8). Overexpressing GFP-CFTR in CF cells completely blocked bleb-niche formation, strongly supporting the idea that loss of CFTR contributes to bleb formation.
FIG 8 .

Model of bleb initiation in apoptotic versus P. aeruginosa-infected non-CF and CF cells. (Top panel) Before bleb induction. (Bottom panel) After bleb induction. Apoptotic bleb initiation (left) requires delamination followed by actin contraction at the bleb neck. When P. aeruginosa infects non-CF cells (middle), CFTR is removed from the plasma membrane (9, 10, 40), which our data suggest provides an osmotic driving force for bleb outgrowth. We also report a loss of actin stress fibers in non-CF cells, which may facilitate osmotically driven bleb formation (middle). Infection of CF cells (right) also results in loss of actin stress fibers, as well as in a higher frequency of blebbing and larger blebs, which are then occupied by greater numbers of bacteria. Since CF cells lack CFTR even prior to infection, we hypothesize that edematous CF cells are osmotically primed for bleb formation following infection.
Intriguingly, P. aeruginosa infection has been shown to result in the removal of CFTR from the host cell membrane (9, 10, 40). The mechanism for this phenomenon involves targeting of the CFTR channel by Cif (CFTR inhibitory factor), a protein that forms part of P. aeruginosa outer membrane vesicles (41). Thus, it is feasible that removal of CFTR from the membrane of non-CF cells by Cif could be involved in providing the osmotic force for bleb formation (Fig. 8). Indeed, this likely explains why blebbing occurs in normal CFTR-expressing cells and why blebbing can be prevented by over-expressing CFTR.
CFTR has previously been shown to mediate P. aeruginosa internalization by epithelial cells (9, 42), attributed to lipid raft association (11). The current study showed that CFTR is also involved in controlling survival/replication of bacteria after internalization by an epithelial cell, with the mechanism involving its osmoregulatory function. While CFTR of epithelial cells has not previously been studied for impact on growth of internalized bacteria, CFTR-deficient macrophages are known to be defective in their ability to kill intracellular P. aeruginosa (43–47). Several studies have suggested that lysosomal acidification may be impaired in CF cells (43, 44, 48–51), but others have also refuted the idea (52–54), leaving the matter unresolved. Our data collected using epithelial cells rather than macrophages suggest that bacterial survival within CF and non-CF cells does not directly involve differences in vacuole acidification. Indeed, both non-CF and CF epithelial cells contained acidified vacuoles, with loss of acidification occurring similarly in the two cell types following infection, as we previously reported for corneal epithelial cells (13). Further, while more than 60% of the intracellular bacterial replication in CF cells was prevented by osmotically blocking bleb-niche formation, there was no impact on the number of cells harboring acidic vacuoles. Since LysoTracker was used to detect acidic vacuoles, it remains possible that there are minor undetectable differences between CF and non-CF epithelial cells in vacuolar pH levels before and after P. aeruginosa infection. Defects in bacterial trafficking to acidified lysosomes, rather than differences in acidification, would be another potential mechanism for making cells more supportive of intracellular bacteria. However, our data do not support this hypothesis for CF cells. T3SS mutants of P. aeruginosa are unable to form bleb niches and instead traffic to acidified lysosomes (12, 13). Given those data, if CFTR contributed to bacterial trafficking to lysosomes, we would have found differences in intracellular bacterial viability for exsA (T3SS) mutants in non-CF versus CF cells, which we did not. Moreover, while there were more blebs in CF cells, rates of bacterial trafficking to the blebs were similar for CF and non-CF cells.
We propose that enhanced bleb formation contributes to the explanation of why CF epithelial cells are more supportive of intracellular bacteria than normal epithelial cells. While difficult to prove directly, this idea is supported by data showing that hyperosmotic medium shrinks blebs and also reduces intracellular survival. Further, our other data rule out obvious alternatives. While it remains possible that hyperosmotic medium somehow more directly impacts the viability of intracellular bacteria, we found that the medium itself had no effect on bacterial viability, and there was no impact on intracellular replication when exsA (T3SS) mutant bacteria rather than wild-type bacteria were used to infect cells, making that an unlikely alternative.
Interestingly, current treatments for CF include 7% hypertonic saline solution and dry powder mannitol (55). These therapies are thought to work by enhancing fluid secretion by CF airway epithelial cells to improve mucus hydration. The 600 mOsM NaCl concentrations used in cultured cells in this study would roughly correspond to a 1% saline solution. Since 600 mOsM media significantly reduced the intracellular replication rate in CF cells, the mechanism(s) by which these treatments work to reduce disease could include limiting intracellular P. aeruginosa replication.
In conclusion, the data presented in this report show that CFTR-defective epithelial cells are more supportive of intracellular bacterial replication and are also more susceptible to bacterially induced blebbing. The data also support the idea that these two findings are related, i.e., that enhanced P. aeruginosa-induced bleb-niche formation in CFTR-deficient cells contributes to providing a supportive environment for intracellular survival. Further research will be needed to determine the contribution of bleb formation to lung infections involving P. aeruginosa in vivo. Since blebs collapse with fixation, visualization requires live tissue. While methods have not been developed to allow subcellular visualization of vital lung tissue without fixation, they are available for eye tissue research, and we have used them to show that blebs containing bacteria exist within P. aeruginosa-infected epithelium of mouse eyes (7). The T3SS is required for bleb formation (6, 12), it influences the progression of acute infection in the lung (56–58) and in the eye (59, 60), and it is expressed by environmental strains of P. aeruginosa that initially colonize the CF lung (61, 62). If the T3SS of P. aeruginosa initially colonizing the lung of a CF patient conspires with defective CFTR to favor P. aeruginosa persistence in airway epithelial cell blebs, where they are protected from non-cell-permeative antibiotics and antimicrobial peptides, while avoiding lysosomes in which they could be processed for immunological recognition, this could contribute to niche establishment in the lung. While this sequence of events could also occur in macrophages (contributing to the significance of our findings), the consequences of intracellular survival in epithelial cells that less readily turn over within the lung tissue could be of equal or even greater significance.
MATERIALS AND METHODS
Cell culture.
Telomerase-immortalized human corneal epithelial cells were maintained in KGM (Lonza) media, plated on traditional tissue culture-treated plastics, and used at passages 30 to 39. Telomerase-immortalized human bronchial epithelial cells derived from a healthy patient (NuLi-1 cells; ATCC CRL-4011) or from a CF patient expressing the ΔF508/ΔF508 CFTR (CuFi-1 cells; ATCC CRL-4013) were grown and assayed on collagen IV (BD BioCoat)-coated cell culture dishes and passaged in bronchial epithelial growth media (BEGM) (Lonza). Assays were performed using cells at passages 1 to 15. RAW 264.7 macrophages were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), and survival assays were performed on collagen-coated 96-well plates as described above.
Bacterial strains.
Pseudomonas aeruginosa strain PAO1 and its isogenic exsA (T3SS) mutant (PAO1 exsA::Ω) were used. Both strains were complemented with GFP on the pSMC2 plasmid. For some assays, PAO1 expressing mCherry on the pUCP18 plasmid was used. Bacteria were grown overnight on carbenicillin-containing Trypticase soy agar (TSA) plates (400 µg/ml) to form a lawn prior to dilution in cell culture media for infection assays.
Antibodies and chemicals.
The antibody against pMLC20 was purchased from AbCam (ab2480). CFTR (Inh)-172 was purchased from Sigma and stored as frozen aliquots of 20 mM in DMSO prior to being used at a final concentration of 20 µM. Blebbistatin enantiomers were purchased from Santa Cruz Biotechnology, Inc., reconstituted in DMSO to a stock of 40 mM, and used at a final concentration of 40 µM. Latrunculin A was purchased from Sigma; stocks were made up in DMSO to 2 mM and used at a final concentration of 5 µM. All inhibitor assays included DMSO in media used at a final concentration of 0.1%. Mannitol and NaCl were also purchased from Sigma. TNF-α was purchased from Shenandoah Biotechnology Inc., stored as a stock of 100 µg/ml in double-distilled water (ddH2O) with 0.1% bovine serum albumin (BSA), and used at a final concentration of 30 ng/ml. Actinomycin D was purchased from Sigma; stocks were made at 5 mg/ml in DMSO and used at a final concentration of 5 µg/ml. All drugs were divided into aliquots and stored in −20°C or −80°C according to the manufacturer’s instructions.
Infection.
As previously described, inoculation of corneal epithelial cells with P. aeruginosa results in heterogeneous phagocytosis events in which some cells take up bacteria before others. However, following a 3-h incubation period with a multiplicity of infection (MOI) of 100, all of the cells have internalized the bacteria. This 3-h internalization period is followed by incubation for at least 1 h in the presence of the non-membrane-permeative antibiotic gentamicin, which is bactericidal only to the extracellular bacteria. Following this procedure, the infected cells begin to form blebs. Given our experience and the reproducibility of this experimental procedure, we therefore designed our assays with both corneal and CF bronchial epithelial cells to work within this time frame.
Induction of apoptosis.
Telomerase-immortalized human corneal epithelial cells were incubated in KGM media with 30 ng/ml TNF-α and 5 µg/ml actinomycin D for 7 h before imaging was performed in order to induce bleb formation. This treatment has been previously shown to induce apoptosis in human corneal fibroblasts (63) and HeLa cells (64).
Epithelial cell transfection.
CuFi-1 cells were transfected with a pGFP-CFTR plasmid created from pS65T-GFP-C1 (34) using a 10-µl-tip Neon transfection system (Invitrogen), according to the manufacturer’s protocol and the following optimized electroporation settings: pulse voltage, 1,350 V; pulse width, 20 ms; and pulse number, 1. Cells were incubated in BEGM culture media in 37°C and 5% CO2 for 48 h before assays were performed.
Assays of bacterially induced bleb-niche formation.
Telomerase-immortalized epithelial cells grown in 35-mm-diameter dishes were infected with 3 × 107 CFU (MOI = 100) of P. aeruginosa PAO1 or its exsA mutant (expressing GFP or mCherry). At 3 h postinoculation, cells were extensively washed in phosphate-buffered saline (PBS), and 200 µg/ml gentamicin was added to kill extracellular bacteria. Cells were imaged at 6 to 8 h postinoculation and analyzed for bleb-niche formation by bright-field or confocal differential inference contrast (DIC) microscopy. The effect of medium osmolarity on bleb-niche formation was examined with and without 20 µM CFTR(Inh)-172. At the 20 µM concentration used in our assays, CFTR(Inh)-172 does not affect cell viability for up to 24 h (65). Cells were pretreated with the inhibitor 30 min prior to inoculation with bacteria, and the inhibitor was maintained at each wash step of the assay.
Microscopy.
Confocal microscopy was performed on a FluoView FV1000 laser scanning confocal microscope (Olympus, PA) equipped with a 60x magnification water-immersion objective, 100-W halogen illumination, and a multiline 488-nm-wavelength argon laser. Fluorescent light and transmitted light were collected simultaneously in z stacks with 1-µm increments along the z axis. For imaging of phalloidin-stained actin, the pinhole was opened almost completely and single z sections were captured. For imaging of transfected cells, z stacks were captured, a single slice of the red and DIC channels was presented, and the entire z stack from the green channel was compiled into a maximum-intensity projection before making the composite image. Wide-field microscopy was performed on an Olympus IX-70 inverted microscope for bright-field and phase-contrast imaging of live blebbing cells in real time (30 frames per second [fps]) with a QImaging QI Click charge-coupled-device (CCD) camera and Volocity software.
Internalization and intracellular replication assays.
Gentamicin exclusion assays were used to measure bacterial invasion and intracellular survival. Cells were plated at a density of 5 × 104 cells per well in collagen IV-coated 96-well plates and inoculated with 1.5 × 106 CFU GFP-expressing PAO1 cells (MOI = 5). Cells were thoroughly washed with PBS at 3 h postinoculation before addition of gentamicin (200 µg/ml) and washed again prior to cell lysis and were lysed with Triton X-100 (0.1% [vol/vol]) for 15 min at room temperature and then plated on MacConkey agar at 4 h and 7 h postinoculation. A modified version of this assay was used on RAW 264.7 macrophage cells because gentamicin is rapidly engulfed by these cells, killing both intracellular and extracellular bacteria. The modified assay consisted of a 1-h infection followed by a PBS wash, including gentamicin in the PBS, and an additional 10-min incubation in DMEM containing gentamicin, followed by another PBS wash before incubation in gentamicin-free media for the remainder of the survival assay. CFU counts were taken at 2 h and 5 h after macrophage inoculation as described above.
Staining of acidic vacuoles.
At 30 min prior to imaging, cells were incubated in a 50 nM solution of LysoTracker Red DND99 (Invitrogen) at 37°C (15 min) and then washed with PBS before addition of culture media for live-cell imaging. LysoTracker is selectively retained within acidic compartments. A low concentration of LysoTracker was used to avoid saturating the cells and to adequately distinguish between the cells that were able to retain the stain and those that were not.
Phalloidin staining.
Following treatments, cells were fixed in 4% paraformaldehyde, quenched in ammonium chloride, permeabilized in 1% Triton X-100, and incubated in PBS containing rhodamine-phalloidin (Invitrogen) in the presence of 1% BSA according to the manufacturer’s instructions prior to mounting.
Hyperosmotic assays.
PAO1 was allowed to internalize into cells (under the conditions described above) for 3 h before treatment with gentamicin (200 µg/ml) in either isosmotic or hyperosmotic media. A 300 mOsM medium was considered isosmotic; NaCl or mannitol was added to increase osmolarity to 600 mOsM. To achieve 600 mOsM NaCl, 0.009 g NaCl was added per ml of BEGM media. For 600 mOsM mannitol, 0.0546 g per ml of media was used.
Extracellular bacterial replication assays.
Two milliliters of cell culture media (BEGM) (equivalent to an MOI of 100) was inoculated as described for the blebbing assays, the reaction mixture was shaken at 37°C, and CFU counts were taken every hour.
Statistics.
Data are expressed as means ± SD or standard errors of the means (SEM) as indicated. Weighted means and weighted SD were used to present data from blebbing frequency assays in order to combine data from independent assays collected from three or more days, each day having a different number of cells analyzed. For survival assays, error propagation was used to calculate the SD resulting from taking the average of averages (including the results of technical and biological replicates in the mean and error calculations). SEM was reported for survival assays in order to take into account sample size (defined as the number of days an assay was repeated; a minimum of n = 3 days was used). Student’s t test was used for most assays to calculate the significance of the differences between the results determined for the two groups. A Mann-Whitney U test was used to analyze bleb area because those data did not have a normal distribution. For comparisons of multiple groups, a one-way analysis of variance (ANOVA) was performed with a Dunnett’s multiple-comparison test for post hoc analysis to compare each group with the corresponding control group. P values of <0.05 were considered significant.
SUPPLEMENTAL MATERIAL
Rhodamine-phalloidin staining of human corneal epithelial cells following a 3-h incubation in DMSO (control, left panel) or 5 µM latrunculin (right panel). Download
{kind=link}
Representative images of blebbing human corneal epithelial cells following infection with P. aeruginosa and in the presence of r-blebbistatin or s-blebbistatin (40 µM). The assay was performed as described in the Fig. 1c legend. Download
{kind=link}
Blebbing frequency and P. aeruginosa occupation of blebs induced by treatment with blebbistatin (40 µM). The assay was performed as described in the Fig. 1c legend. Download
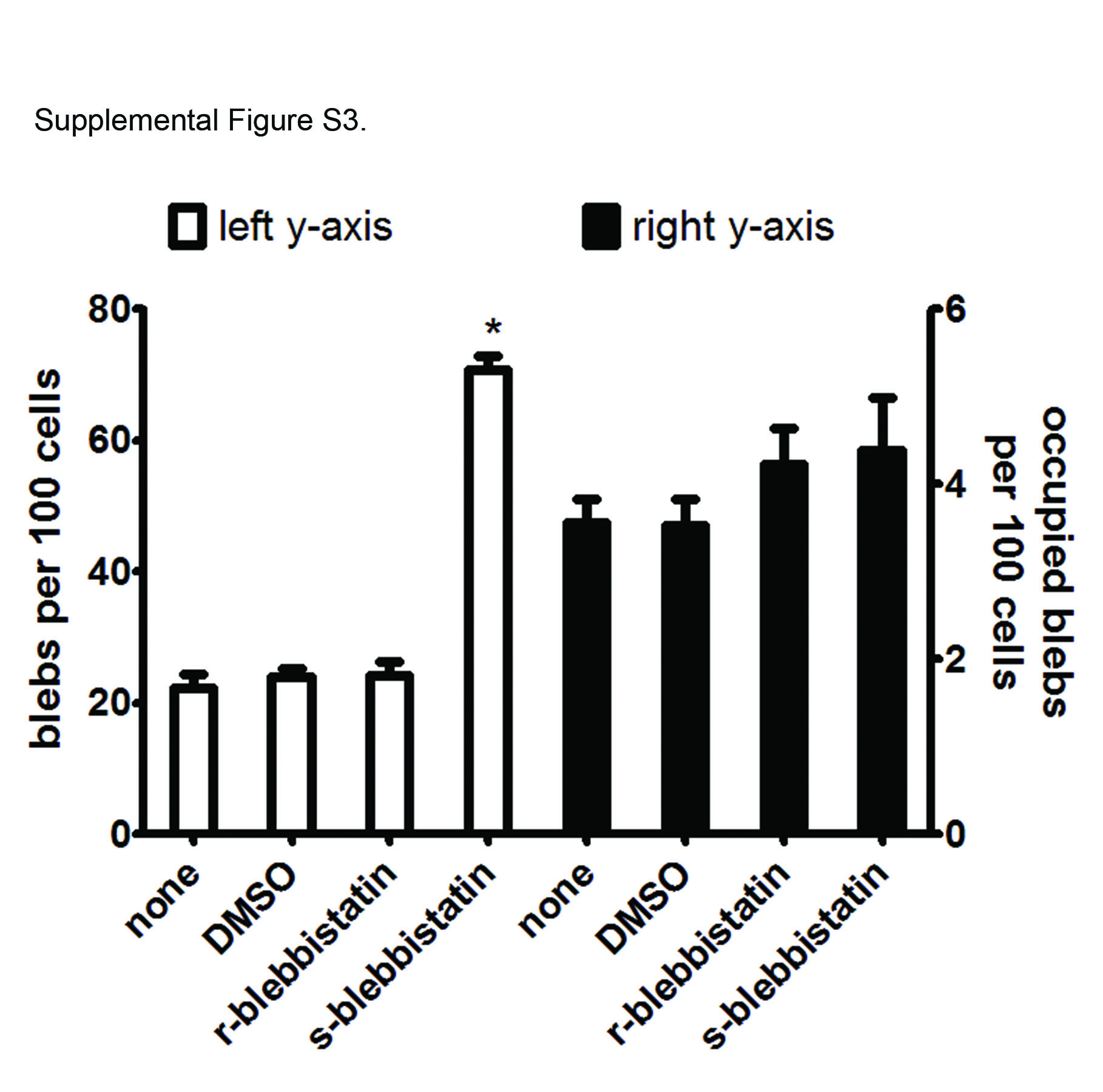{kind=link}
P. aeruginosa-induced blebbing was prevented by over-expression of GFP-CFTR in CF cells. CF cells were transfected with pGFP-CFTR (green) and infected (MOI = 100) with PAO1-mCherry (red). Z stacks were imaged using confocal microscopy (maximum projections of the green channel are shown overlaying a single z slice in the red and phase channels). Cells that did not express GFP-CFTR (top row) were induced to form blebs containing bacteria, while cells in the same dish that expressed GFP-CFTR (middle row) failed to form blebs. The assay was repeated in three independent experiments, with a composite of 196 untransfected cells analyzed (29% ± 3% blebbing) and 23 transfected cells analyzed (0% ± 0% blebbing). Data are presented as means ± standard deviations. pEGFP was used as a transfection control for the effects of GFP over-expression and toxicity on P. aeruginosa-induced bleb formation. Blebbing was induced when cells over-expressing GFP were infected with P. aeruginosa (bottom row). Images of EGFP-expressing cells are only of cells expressing a higher GFP signal intensity visualized using microscope settings identical to those used for cells expressing GFP-CFTR. Download
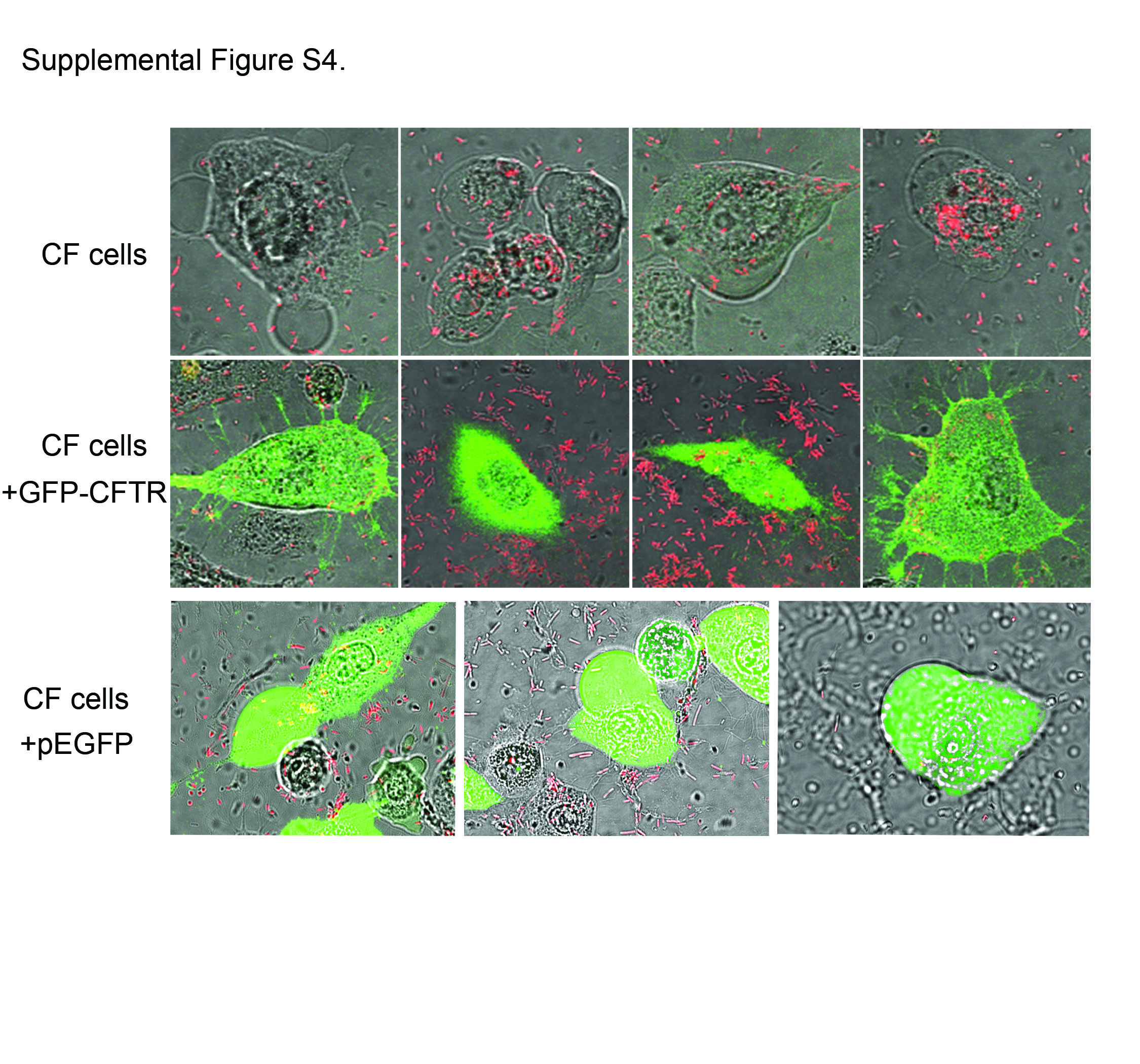{kind=link}
Survival of intracellular bacteria in the presence of 600 mOsM mannitol added to the media following bacterial internalization. (a) Effect of hyperosmotic media added at 3 h postinoculation on the number of intracellular bacteria at 6 h in human corneal epithelial cells. (b) Effect of hyperosmotic media on the number of intracellular bacteria in RAW 246.7 macrophage cells determined using a modified survival assay (see Materials and Methods). Data represent the results of representative assays. Data are presented as means ± standard deviations (P < 0.05, Students t test). Download
{kind=link}
Rhodamine-phalloidin staining of CF bronchial epithelial cells at various time points during infection with P. aeruginosa (MOI = 100). Gentamicin was added at 3 h post-inoculation. Identical imaging settings were used for all images. Download
{kind=link}
P. aeruginosa-induced bleb initiation in iso-osmolar media following suppression of blebbing in 600 mOsM mannitol. CF cells were infected with PAO1 for 3 h and then treated with gentamicin and 600 mOsM mannitol for an additional 3 h prior to imaging. Shortly before imaging began, hypo-osmolar media were added to the chamber and blebbing was modulated using a range of 600 mOsM media to 10% hypo-osmolar media. Live images were captured at 30 fps and sped up to 120 fps. Movie titles were added using iMovie software. Download
P. aeruginosa enters bleb following artificial bleb initiation in hypo-osmolar media. CF cells were infected with PAO1 for 3 h and then treated with gentamicin and 600 mOsM mannitol for an additional 3 h prior to imaging. Blebs were artificially induced at 6 h by adding hypo-osmolar media, and imaging began shortly thereafter. Live images were captured at 30 fps and sped up to 240 fps. Movie titles were added using iMovie software. Download
ACKNOWLEDGMENTS
We thank Dara Frank (MCW) for providing the T3SS mutant of PAO1. We thank Peter Haggie (UCSF) for generously providing the GFP-CFTR plasmid. We thank Aimee Wessel (University of Texas at Austin) for the pUCP18-mCherry plasmid.
This work was funded by the National Institutes of Health (AI079192 to S.M.J.F.) and a T32 training grant (EY007043 to the UC Berkeley Vision Sciences program to A.L.J.).
A.L.J., D.T., D.J.E., and S.M.J.F. designed the experiments. A.L.J., D.T., O.O.O., S.A.W., S.W.C., E.R.W., and K.Q. performed the experiments. A.L.J., D.T., D.J.E., and S.M.J.F. analyzed the experiments. A.L.J., D.J.E., and S.M.J.F. wrote the manuscript.
S.M.J.F. is a paid consultant with Allergan. That activity is unrelated to the work presented in the manuscript.
Footnotes
Citation Jolly AL, Takawira D, Oke OO, Whiteside SA, Chang SW, Wen ER, Quach K, Evans DJ, Fleiszig SMJ. 2015. Pseudomonas aeruginosa-induced bleb-niche formation in epithelial cells is independent of actinomyosin contraction and enhanced by loss of cystic fibrosis transmembrane-conductance regulator osmoregulatory function. mBio 6(2):e02533-14. doi:10.1128/mBio.02533-14.
REFERENCES
- 1.Ratjen F, Döring G. 2003. Cystic fibrosis. Lancet 361:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 2.Treggiari MM, Rosenfeld M, Retsch-Bogart G, Gibson R, Ramsey B. 2007. Approach to eradication of initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 42:751–756. doi: 10.1002/ppul.20665. [DOI] [PubMed] [Google Scholar]
- 3.Lovewell RR, Patankar YR, Berwin B. 2014. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 306:L591–L603. doi: 10.1152/ajplung.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campodónico VL, Gadjeva M, Paradis-Bleau C, Uluer A, Pier GB. 2008. Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol Med 14:120–133. doi: 10.1016/j.molmed.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleiszig SM, Wiener-Kronish JP, Miyazaki H, Vallas V, Mostov KE, Kanada D, Sawa T, Yen TS, Frank DW. 1997. Pseudomonas aeruginosa-mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzyme S. Infect Immun 65:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angus AA, Evans DJ, Barbieri JT, Fleiszig SM. 2010. The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun 78:4500–4510. doi: 10.1128/IAI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tam C, LeDue J, Mun JJ, Herzmark P, Robey EA, Evans DJ, Fleiszig SM. 2011. 3D quantitative imaging of unprocessed live tissue reveals epithelial defense against bacterial adhesion and subsequent traversal requires MyD88. PLoS One 6:e24008. doi: 10.1371/journal.pone.0024008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleiszig SM, Zaidi TS, Fletcher EL, Preston MJ, Pier GB. 1994. Pseudomonas aeruginosa invades corneal epithelial cells during experimental infection. Infect Immun 62:3485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pier GB, Grout M, Zaidi TS. 1997. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci U S A 94:12088–12093. doi: 10.1073/pnas.94.22.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaidi TS, Lyczak J, Preston M, Pier GB. 1999. Cystic fibrosis transmembrane conductance regulator-mediated corneal epithelial cell ingestion of Pseudomonas aeruginosa is a key component in the pathogenesis of experimental murine keratitis. Infect Immun 67:1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaidi T, Bajmoczi M, Zaidi T, Golan DE, Pier GB. 2008. Disruption of CFTR-dependent lipid rafts reduces bacterial levels and corneal disease in a murine model of Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci 49:1000–1009. doi: 10.1167/iovs.07-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SM. 2008. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun 76:1992–2001. doi: 10.1128/IAI.01221-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heimer SR, Evans DJ, Stern ME, Barbieri JT, Yahr T, Fleiszig SM. 2013. Pseudomonas aeruginosa utilizes the type III secreted toxin ExoS to avoid acidified compartments within epithelial cells. PLoS One 8:e73111. doi: 10.1371/journal.pone.0073111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charras GT. 2008. A short history of blebbing. J Microsc 231:466–478. doi: 10.1111/j.1365-2818.2008.02059.x. [DOI] [PubMed] [Google Scholar]
- 16.Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. 2008. Life and times of a cellular bleb. Biophys J 94:1836–1853. doi: 10.1529/biophysj.107.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goudarzi M, Banisch TU, Mobin MB, Maghelli N, Tarbashevich K, Strate I, van den Berg J, Blaser H, Bandemer S, Paluch E, Bakkers J, Tolić-Nørrelykke IM, Raz E. 2012. Identification and regulation of a molecular module for bleb-based cell motility. Dev Cell 23:210–218. doi: 10.1016/j.devcel.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Brugués J, Maugis B, Casademunt J, Nassoy P, Amblard F, Sens P. 2010. Dynamical organization of the cytoskeletal cortex probed by micropipette aspiration. Proc Natl Acad Sci U S A 107:15415–15420. doi: 10.1073/pnas.0913669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tinevez J-Y, Schulze U, Salbreux G, Roensch J, Joanny J-F, Paluch E. 2009. Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A 106:18581–18586. doi: 10.1073/pnas.0903353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paluch EK, Raz E. 2013. The role and regulation of blebs in cell migration. Curr Opin Cell Biol 25:582–590. doi: 10.1016/j.ceb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bray D, White JG. 1988. Cortical flow in animal cells. Science 239:883–888. doi: 10.1126/science.3277283. [DOI] [PubMed] [Google Scholar]
- 22.Barros LF, Kanaseki T, Sabirov R, Morishima S, Castro J, Bittner CX, Maeno E, Ando-Akatsuka Y, Okada Y. 2003. Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ 10:687–697. doi: 10.1038/sj.cdd.4401236. [DOI] [PubMed] [Google Scholar]
- 23.Alaoui-El-Azher M, Jia J, Lian W, Jin S. 2006. ExoS of Pseudomonas aeruginosa induces apoptosis through a Fas receptor/caspase 8-independent pathway in HeLa cells. Cell Microbiol 8:326–338. doi: 10.1111/j.1462-5822.2005.00624.x. [DOI] [PubMed] [Google Scholar]
- 24.Jia J, Wang Y, Zhou L, Jin S. 2006. Expression of Pseudomonas aeruginosa toxin ExoS effectively induces apoptosis in host cells. Infect Immun 74:6557–6570. doi: 10.1128/IAI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman MR, Jia J, Zeng L, Ha U, Chow M, Jin S. 2000. Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS. Microbiology 146:2531–2541. [DOI] [PubMed] [Google Scholar]
- 26.Krejbich-Trotot P, Denizot M, Hoarau J-J, Jaffar-Bandjee M-C, Das T, Gasque P. 2011. Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. FASEB J 25:314–325. doi: 10.1096/fj.10-164178. [DOI] [PubMed] [Google Scholar]
- 27.Yanase Y, Carvou N, Frohman MA, Cockcroft S. 2010. Reversible bleb formation in mast cells stimulated with antigen is Ca2+/calmodulin-dependent and bleb size is regulated by ARF6. Biochem J 425:179–193. doi: 10.1042/BJ20091122. [DOI] [PubMed] [Google Scholar]
- 28.Gutjahr MC, Rossy J, Niggli V. 2005. Role of Rho, Rac, and Rho-kinase in phosphorylation of myosin light chain, development of polarity, and spontaneous migration of Walker 256 carcinosarcoma cells. Exp Cell Res 308:422–438. doi: 10.1016/j.yexcr.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Barros LF, Hermosilla T, Castro J. 2001. Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol A Mol Integr Physiol 130:401–409. doi: 10.1016/S1095-6433(01)00438-X. [DOI] [PubMed] [Google Scholar]
- 30.Braunstein GM, Zsembery A, Tucker TA, Schwiebert EM. 2004. Purinergic signaling underlies CFTR control of human airway epithelial cell volume. J Cyst Fibros 3:99–117. doi: 10.1016/j.jcf.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Seidler U, Bachmann O, Jacob P, Christiani S, Blumenstein I, Rossmann H. 2001. Na+/HCO3- cotransport in normal and cystic fibrosis intestine. JOP 2(4 Suppl):247–256. http://www.joplink.net/prev/200107/18.html. [PubMed] [Google Scholar]
- 32.Valverde MA, O’Brien JA, Sepúlveda FV, Ratcliff RA, Evans MJ, Colledge WH. 1995. Impaired cell volume regulation in intestinal crypt epithelia of cystic fibrosis mice. Proc Natl Acad Sci U S A 92:9038–9041. doi: 10.1073/pnas.92.20.9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vázquez E, Nobles M, Valverde MA. 2001. Defective regulatory volume decrease in human cystic fibrosis tracheal cells because of altered regulation of intermediate conductance Ca2+-dependent potassium channels. Proc Natl Acad Sci U S A 98:5329–5334. doi: 10.1073/pnas.091096498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moyer BD, Loffing J, Schwiebert EM, Loffing-Cueni D, Halpin PA, Karlson KH, Ismailov II, Guggino WB, Langford GM, Stanton BA. 1998. Membrane trafficking of the cystic fibrosis gene product, cystic fibrosis transmembrane conductance regulator, tagged with green fluorescent protein in Madin-Darby canine kidney cells. J Biol Chem 273:21759–21768. doi: 10.1074/jbc.273.34.21759. [DOI] [PubMed] [Google Scholar]
- 35.Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. 2001. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miao EA, Rajan JV, Aderem A. 2011. Caspase-1-induced pyroptotic cell death. Immunol Rev 243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wall DM, McCormick BA. 2014. Bacterial secreted effectors and caspase-3 interactions. Cell Microbiol 16:1746–1756. doi: 10.1111/cmi.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chambers LA, Rollins BM, Tarran R. 2007. Liquid movement across the surface epithelium of large airways. Respir Physiol Neurobiol 159:256–270. doi: 10.1016/j.resp.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wunderlich P, Kemmer C, Fischer R, Sprössig C, Leupold W, Burkhardt J, Gottschalk B. 1986. Bronchial secretions and bronchial mucosa in children with cystic fibrosis: comparison of bronchoscopic, biochemical, bacteriological, microscopic and ultrastructural findings. Acta Paediatr Hung 27:123–131. [PubMed] [Google Scholar]
- 40.Bajmoczi M, Gadjeva M, Alper SL, Pier GB, Golan DE. 2009. Cystic fibrosis transmembrane conductance regulator and caveolin-1 regulate epithelial cell internalization of Pseudomonas aeruginosa. Am J Physiol Cell Physiol 297:C263–C277. doi: 10.1152/ajpcell.00527.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacEachran DP, Ye S, Bomberger JM, Hogan DA, Swiatecka-Urban A, Stanton BA, O’Toole GA. 2007. The Pseudomonas aeruginosa secreted protein PA2934 decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect Immun 75:3902–3912. doi: 10.1128/IAI.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pier GB, Grout M, Zaidi TS, Goldberg JB. 1996. How mutant CFTR may contribute to Pseudomonas aeruginosa infection in cystic fibrosis. Am J Respir Crit Care Med 154:S175–S182. doi: 10.1164/ajrccm/154.4_Pt_2.S175. [DOI] [PubMed] [Google Scholar]
- 43.Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, Gallan AJ, Shevchenko PD, Bindokas VP, Nelson DJ. 2009. Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem 284:35926–35938. doi: 10.1074/jbc.M109.057372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. 2006. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 45.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB, Zabner J, Welsh MJ. 2010. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggiò MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, Ascenzioni F. 2011. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 6:e19970. doi: 10.1371/journal.pone.0019970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bessich JL, Nymon AB, Moulton LA, Dorman D, Ashare A. 2013. Low levels of insulin-like growth factor-1 contribute to alveolar macrophage dysfunction in cystic fibrosis. J Immunol 191:378–385. doi: 10.4049/jimmunol.1300221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, al-Awqati Q. 1991. Defective acidification of intracellular organelles in cystic fibrosis. Nature 352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 49.Poschet JF, Skidmore J, Boucher JC, Firoved AM, Van Dyke RW, Deretic V. 2002. Hyperacidification of cellubrevin endocytic compartments and defective endosomal recycling in cystic fibrosis respiratory epithelial cells. J Biol Chem 277:13959–13965. doi: 10.1074/jbc.M105441200. [DOI] [PubMed] [Google Scholar]
- 50.Teichgräber V, Ulrich M, Endlich N, Riethmüller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kürthy G, Schmid KW, Weller M, Tümmler B, Lang F, Grassme H, Döring G, Gulbins E. 2008. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Li X, Grassmé H, Döring G, Gulbins E. 2010. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J Immunol 184:5104–5111. doi: 10.4049/jimmunol.0902851. [DOI] [PubMed] [Google Scholar]
- 52.Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, Radzioch D, Lukacs GL. 2009. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell 20:3125–3141. doi: 10.1091/mbc.E09-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haggie PM, Verkman AS. 2009. Unimpaired lysosomal acidification in respiratory epithelial cells in cystic fibrosis. J Biol Chem 284:7681–7686. doi: 10.1074/jbc.M809161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, Grinstein S. 2010. A cation counterflux supports lysosomal acidification. J Cell Biol 189:1171–1186. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fahy JV, Dickey BF. 2010. Airway mucus function and dysfunction. N Engl J Med 363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chawla R. 2008. Epidemiology, etiology, and diagnosis of hospital-acquired pneumonia and ventilator-associated pneumonia in Asian countries. Am J Infect Control 36:S93–S100. doi: 10.1016/j.ajic.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 57.Engel JN. 2003. Severe infections caused by Pseudomonas aeruginosa. Kluwer Academic/Plenum Press, New York, NY. [Google Scholar]
- 58.Nair GB, Niederman MS. 2013. Nosocomial pneumonia: lessons learned. Crit Care Clin 29:521–546. doi: 10.1016/j.ccc.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Cheng KH, Leung SL, Hoekman HW, Beekhuis WH, Mulder PG, Geerards AJ, Kijlstra A. 1999. Incidence of contact-lens-associated microbial keratitis and its related morbidity. Lancet 354:181–185. doi: 10.1016/S0140-6736(98)09385-4. [DOI] [PubMed] [Google Scholar]
- 60.Molina DN, Colón M, Bermúdez RH, Ramírez-Ronda CH. 1991. Unusual presentation of Pseudomonas aeruginosa infections: a review. Bol Asoc Med P R 83:160–163. [PubMed] [Google Scholar]
- 61.Hogardt M, Heesemann J. 2010. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol 300:557–562. doi: 10.1016/j.ijmm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohan RR, Mohan RR, Kim W-J, Wilson SE. 2000. Modulation of TNF-alpha-induced apoptosis in corneal fibroblasts by transcription factor NF-kappaB. Invest Ophthalmol Vis Sci 41:1327–1336. [PubMed] [Google Scholar]
- 64.Cozzi A, Levi S, Corsi B, Santambrogio P, Campanella A, Gerardi G, Arosio P. 2003. Role of iron and ferritin in TNFalpha-induced apoptosis in HeLa cells. FEBS Lett 537:187–192. doi: 10.1016/S0014-5793(03)00114-5. [DOI] [PubMed] [Google Scholar]
- 65.Melis N, Tauc M, Cougnon M, Bendahhou S, Giuliano S, Rubera I, Duranton C. 2014. Revisiting CFTR inhibition: a comparative study of CFTRinh-172 and GlyH-101 inhibitors. Br J Pharmacol 171:3716–3727. doi: 10.1111/bph.12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rhodamine-phalloidin staining of human corneal epithelial cells following a 3-h incubation in DMSO (control, left panel) or 5 µM latrunculin (right panel). Download
Representative images of blebbing human corneal epithelial cells following infection with P. aeruginosa and in the presence of r-blebbistatin or s-blebbistatin (40 µM). The assay was performed as described in the Fig. 1c legend. Download
Blebbing frequency and P. aeruginosa occupation of blebs induced by treatment with blebbistatin (40 µM). The assay was performed as described in the Fig. 1c legend. Download
P. aeruginosa-induced blebbing was prevented by over-expression of GFP-CFTR in CF cells. CF cells were transfected with pGFP-CFTR (green) and infected (MOI = 100) with PAO1-mCherry (red). Z stacks were imaged using confocal microscopy (maximum projections of the green channel are shown overlaying a single z slice in the red and phase channels). Cells that did not express GFP-CFTR (top row) were induced to form blebs containing bacteria, while cells in the same dish that expressed GFP-CFTR (middle row) failed to form blebs. The assay was repeated in three independent experiments, with a composite of 196 untransfected cells analyzed (29% ± 3% blebbing) and 23 transfected cells analyzed (0% ± 0% blebbing). Data are presented as means ± standard deviations. pEGFP was used as a transfection control for the effects of GFP over-expression and toxicity on P. aeruginosa-induced bleb formation. Blebbing was induced when cells over-expressing GFP were infected with P. aeruginosa (bottom row). Images of EGFP-expressing cells are only of cells expressing a higher GFP signal intensity visualized using microscope settings identical to those used for cells expressing GFP-CFTR. Download
Survival of intracellular bacteria in the presence of 600 mOsM mannitol added to the media following bacterial internalization. (a) Effect of hyperosmotic media added at 3 h postinoculation on the number of intracellular bacteria at 6 h in human corneal epithelial cells. (b) Effect of hyperosmotic media on the number of intracellular bacteria in RAW 246.7 macrophage cells determined using a modified survival assay (see Materials and Methods). Data represent the results of representative assays. Data are presented as means ± standard deviations (P < 0.05, Students t test). Download
Rhodamine-phalloidin staining of CF bronchial epithelial cells at various time points during infection with P. aeruginosa (MOI = 100). Gentamicin was added at 3 h post-inoculation. Identical imaging settings were used for all images. Download
P. aeruginosa-induced bleb initiation in iso-osmolar media following suppression of blebbing in 600 mOsM mannitol. CF cells were infected with PAO1 for 3 h and then treated with gentamicin and 600 mOsM mannitol for an additional 3 h prior to imaging. Shortly before imaging began, hypo-osmolar media were added to the chamber and blebbing was modulated using a range of 600 mOsM media to 10% hypo-osmolar media. Live images were captured at 30 fps and sped up to 120 fps. Movie titles were added using iMovie software. Download
P. aeruginosa enters bleb following artificial bleb initiation in hypo-osmolar media. CF cells were infected with PAO1 for 3 h and then treated with gentamicin and 600 mOsM mannitol for an additional 3 h prior to imaging. Blebs were artificially induced at 6 h by adding hypo-osmolar media, and imaging began shortly thereafter. Live images were captured at 30 fps and sped up to 240 fps. Movie titles were added using iMovie software. Download