

ABSTRACT
Many RNA viruses are detected by retinoic acid-inducible gene i (RIG-I), a cytoplasmic sensor that triggers an antiviral response upon binding non-self-RNA that contains a stretch of double-stranded RNA (dsRNA) bearing a base-paired 5′ ppp nucleotide. To gain insight into how RIG-I discriminates between self-RNA and non-self-RNA, we used duplexes whose complementary bottom strand contained both ribo- and deoxynucleotides. These duplexes were examined for their binding to RIG-I and their relative abilities to stimulate ATPase activity, to induce RIG-I dimerization on the duplex, and to induce beta interferon (IFN-β) expression. We show that the chemical nature of the bottom strand is not critical for RIG-I binding. However, two key ribonucleotides, at positions 2 and 5 on the bottom strand, are minimally required for the RIG-I ATPase activity, which is necessary but not sufficient for IFN-β stimulation. We find that duplexes with shorter stretches of dsRNA, as model self-RNAs, bind less stably to RIG-I but nevertheless have an enhanced ability to stimulate the ATPase. Moreover, ATPase activity promotes RIG-I recycling on RIG-I/dsRNA complexes. Since pseudo-self-RNAs bind to RIG-I less stably, they are preferentially recycled by ATP hydrolysis that weakens the helicase domain binding of dsRNA. Our results suggest that one function of the ATPase is to restrict RIG-I signaling to its interaction with non-self-RNA. A model of how this discrimination occurs as a function of dsRNA length is presented.
IMPORTANCE
The innate immune response to pathogens is based on the discrimination between self-RNA and non-self-RNA. The main determinants of this detection for RNA viruses are specific pathogen-associated molecular patterns (PAMPs) of RNA, which are detected by dedicated cytoplasmic pattern recognition receptors (PRRs). RIG-I is a PRR that specifically detects short viral dsRNAs amid a sea of cellular RNAs. Here we study the determinants of this discrimination and how RIG-I ATPase activity, the only enzymatic activity of this sensor, contributes to its activation in a manner restricted to its interaction with non-self-RNAs. We also show how the innate immune response evolves during infection via IFN expression, from a state in which discrimination of self-RNA from non-self-RNA is most important to one in which this discrimination is sacrificed for the effectiveness of the antiviral response.
INTRODUCTION
One important feature of the innate immune response is its ability to detect foreign (non-self) nucleic acids. Various cellular receptors, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), and retinoic acid-inducible gene i (RIG-I)-like receptors (RLRs), are involved in this detection.
The cytoplasmic RLR family, which includes retinoic acid-inducible gene i (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and Laboratory of Genetics and Physiology 2 (LGP2), is involved in the detection of specific RNAs in the cytoplasm of host cells (1). These sensors belong to a family of duplex RNA-activated ATPases (2), sharing a C-terminal domain (CTD) that is involved in RNA recognition (3, 4) and a central DExD/H-box helicase (Hel) domain responsible for the ATPase activity (5). Additionally, RIG-I and MDA5 have at their N terminus two caspase activation and recruitment domains (CARDs) that are involved in the transmission of the activation signal downstream, which leads to the production of type I interferon (IFN), cytokines, and chemokines (6).
RIG-I specifically responds to at least two kinds of RNAs: short double-stranded RNAs (dsRNAs) and long dsRNAs (7–9). For short dsRNAs, RIG-I activation relies on the presence of a base-paired 5′ ppp nucleotide. The minimal length of the dsRNAs required for RIG-I activation (10 or 19 bp) is still a subject of debate (10, 11), but both lengths are compatible with the intramolecular panhandles formed by the genomes and antigenomes of negative-strand RNA viruses, such as orthomyxoviruses, arenaviruses, and bunyaviruses (12). With longer dsRNAs, the presence of a 5′ ppp is not required, as poly(I·C) and other dsRNAs lacking a 5′ ppp can activate RIG-I (13–15). On the basis of the RIG-I structure (16–18), a model of RIG-I activation has been proposed in which both strands of the dsRNA specifically interact with different domains of the protein (19). Residues involved in these contacts have been determined based on structural data and remain to be functionally validated. In the absence of RNA, RIG-I is found in an autorepressed state. Upon 5′ ppp-dsRNA binding to the CTD, a conformational change takes place, allowing the helicase (Hel) domains to close and form an ATP binding pocket. This in turn leads to the exposure of the CARDs, which are now available for downstream signaling. The precise role of ATP binding and hydrolysis in this process, however, remains unclear.
A better understanding of the RNA features required for its binding to, and activation of, RIG-I would shed light on the molecular signatures of a viral infection and on the capacity of RIG-I to discriminate between self-RNA and non-self-RNA and would refine the conception of the mechanism of RIG-I activation. Viruses have developed strategies to prevent RIG-I activation. One of these strategies is to minimize their visibility, i.e., to limit their detection by generating viral RNAs that mimic cellular RNAs. The result is never perfect, and cells have developed mechanisms that exploit the remaining differences. One aim of this study was to characterize the differences that are important in detection of viral pathogen-associated molecular patterns (PAMPs) by RIG-I. The work focuses on the role of the bottom strand of short RNA duplexes in RIG-I activation. We confirm the importance of the 5′ ppp structure for RIG-I binding and demonstrate that the nature of the bottom strand (ribo- or deoxyribonucleotides) is not critical for binding. In contrast, the nature of this bottom strand is essential for the ATPase activity. Two key ribonucleotides, at positions 2 and 5, are minimally required for ATPase activity, which is further modulated by nucleotides between positions 6 and 13. Although dispensable for RNA binding, this ATPase activity is found to be required for RIG-I/dsRNA recycling, and this recycling occurs preferentially on short dsRNAs that are more similar to cellular RNAs. In cells, IFN-β induction requires a minimum RNA duplex of 13 bp. However, when cells are first primed with IFN-β, the minimum duplex length is reduced to 8 to 10 bp: this shift in pattern recognition highlights the profound changes induced by IFN in response to viral infection.
RESULTS
A base-paired 5′ ppp dsRNA structure is required for RIG-I binding, irrespective of the bottom-strand composition.
The first prerequisite for RIG-I activation is RNA binding. To explore the contribution of the dsRNA bottom strand in this step, we performed RNA pulldown assays using different dsRNAs. The top (5′ ppp) strand was made in vitro using a DNA template devoid of A residues in a T7 RNA polymerase reaction mixture lacking UTP. This prevents formation of contaminating dsRNA, as T7 RNA polymerase can also use the newly formed RNA as a template (20, 21) (see Materials and Methods). The 5′ ppp single-stranded RNA (ssRNA) was then annealed with various synthetic (5′ OH) bottom strands (Table 1). (7, 9–11). This 5′ ppp nucleotide must be base paired, as a 5′ overhang strongly decreased binding whereas a 3′ overhang was tolerated (Fig. 1B). Consistent with this, an RNA hybrid with a mismatch at position 1 (30r mis.1) bound much less effectively to RIG-I (Fig. 1E). The nature of the bottom strand appears not to be important, since hybrids containing a full deoxyribonucleotide bottom strand (20d; Fig. 1D) and a chimeric bottom strand, composed of both ribo- and deoxyribonucleotides (5r-15d to 60r; Fig. 1C), all bound similarly to RIG-I. In agreement with previous results (4, 22, 23), we observed that the CTD was the main determinant for binding to our hybrid duplexes. Consistently, removal of the CARDs (ΔN; see Fig. S1B in the supplemental material) did not affect RNA binding. Moreover, both RIG-I ΔCTD (ΔC; see Fig. S1A) and RIG-I ΔCARD ΔCTD (ΔNΔC; see Fig. S1B) did not bind RNA above the background levels (although the high background of the ΔNΔC mutant may have biased the result). Taken together, these results argue for end recognition by the RIG-I CTD of duplexes with a base-paired 5′ ppp nucleotide, regardless of the nature of the bottom strand. Given that RIG-I helicase mutants lacking ATPase activity (K270A and D372A; see Fig. S2C) still bind RNA (see Fig. S1C and D), the results indicate that ATPase activity is not required for binding.
TABLE 1 .
Graphical representation of the principal RNA hybrids used in this work and their respective activities in the different tests used in this studya
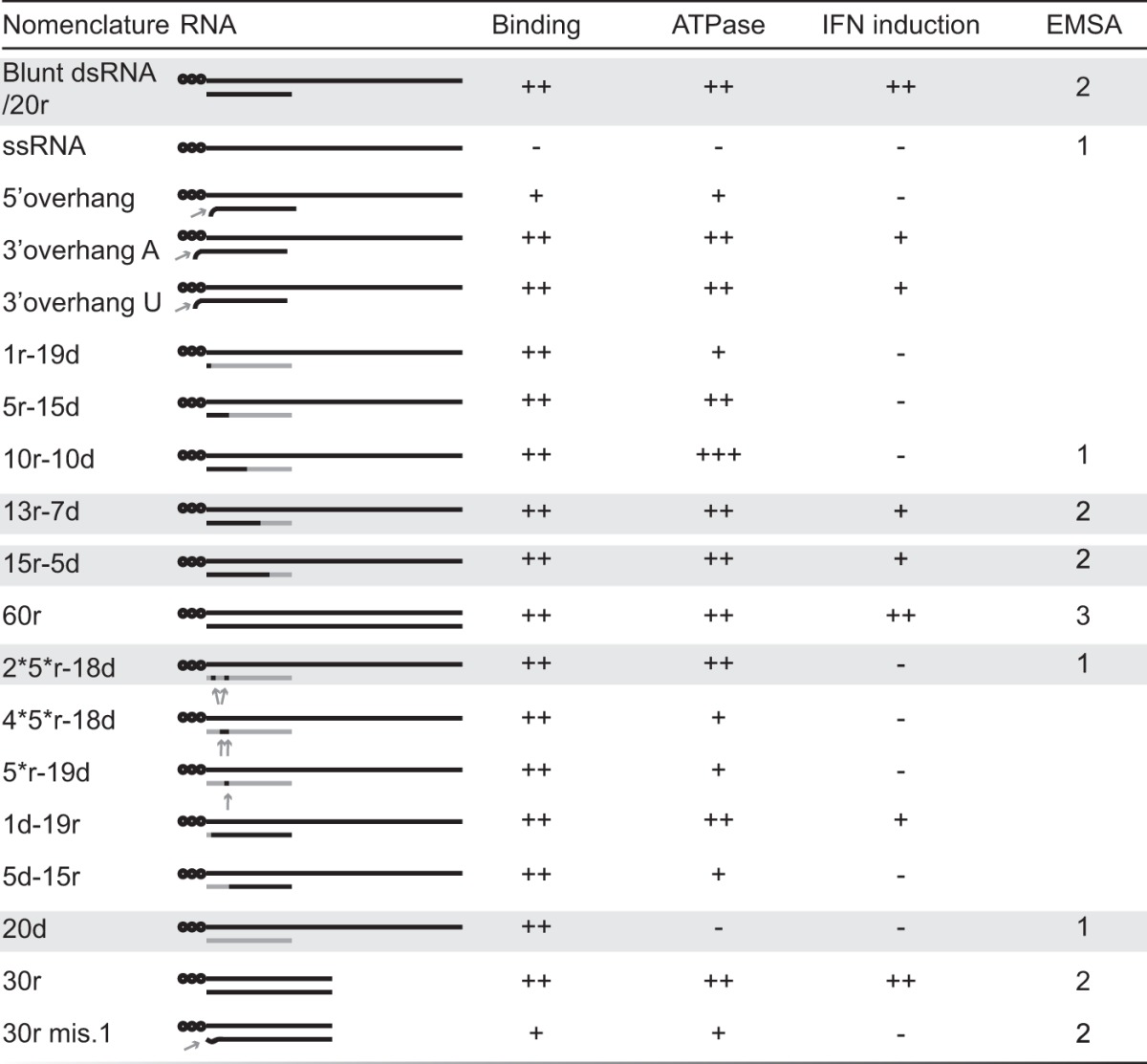
The principal RNA hybrids used in this work are shown at the left side of the table, and their respective activities in the different tests used in this study are shown on the right. Numbers in the EMSA column represent the maximum numbers of RIG-I molecules found bound to the corresponding RNA. Black stands for ribonucleotides, and gray stands for deoxyribonucleotides. Unless specified by 5′ OH, all the hybrids carry a 5′ ppp (represented by black dots). RNA hybrids are named according to their length and composition in ribo- and deoxyribonucleotide of the bottom strand. RNAs highlighted in gray represent the key molecules supporting our conclusions.
FIG 1 .
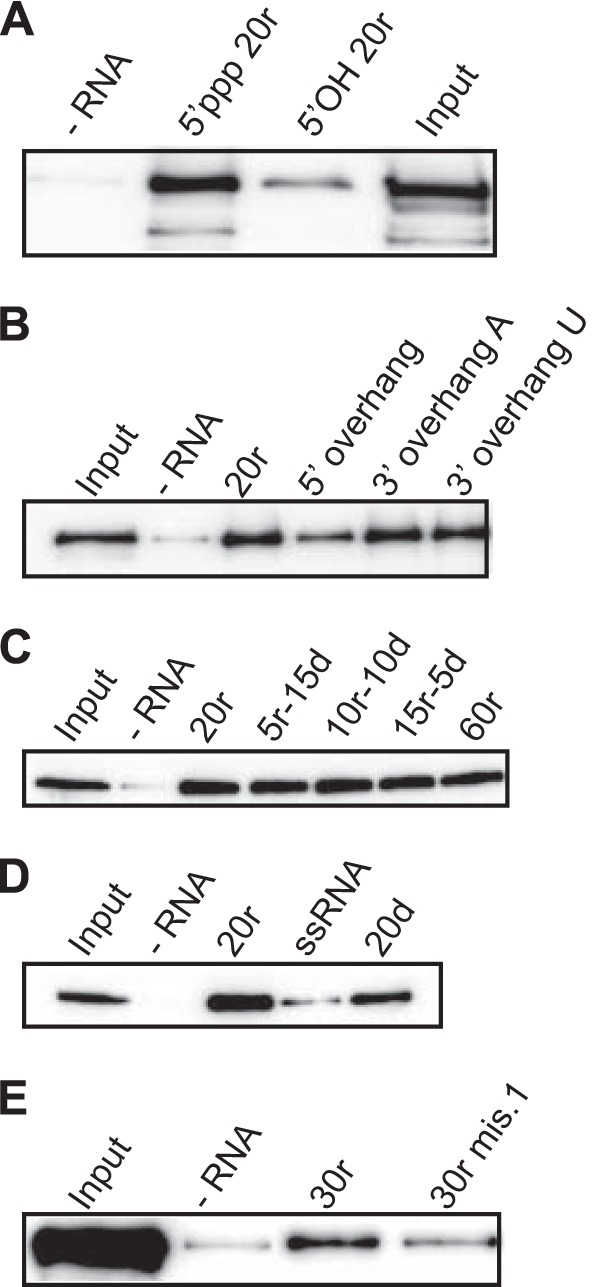
RIG-I binds base-paired 5′ ppp-double-strand molecules independently of the nature of the bottom strand. (A to D) RNA pulldown assays were performed using 13 pmol of various biotinylated double-stranded molecules (as indicated) and purified his-RIG-I (60 nM) in the absence of ATP and MgCl2. (A) Importance of the 5′ ppp. (B and E) Importance of a base-paired 5′ end. (C and D) Nature of the bottom strand. The amount of RIG-I pulled down was analyzed by Western blotting using an anti-RIG-I antibody (see also Fig. S1 in the supplemental material).
Key residues on the bottom strand are involved in RIG-I ATPase activity.
ATPase is required for RIG-I activation (5; reviewed in reference 2). However, its role in RIG-I activation is still unclear, and the precise residues on the bottom strand involved in ATPase activity are unknown. In order to identify the minimal requirements for RIG-I ATPase activity, we tested our hybrids in an ATPase assay. In line with its requirement for RNA binding, the CTD is necessary for the ATPase activity (24 and data not shown). Additionally, and in agreement with results of pulldown experiments, a base-paired end is also required, as molecules with a 5′ overhang or a mismatch at position 1 or ssRNA exhibited decreased ATPase activity (Fig. 2B and G). Surprisingly, in marked contrast to pulldown experiments that measure stable binding, the 5′ ppp was not essential for ATPase activity in vitro, since a 5′ OH duplex was as efficient (Fig. 2A; see also Fig. S2A in the supplemental material). In this case, since the 5′ ppp is necessary for the stability of the RNA/RIG-I complex in pulldown assays and subsequent IFN-β stimulation, stable RNA binding appears not to be required for in vitro ATPase activity (see Discussion). These results nevertheless emphasize the role of the CTD in interacting with the blunt end of the dsRNA (independently of the 5′ ppp) as the initial step for ATPase activity.
FIG 2 .

Ribonucleotides on the bottom strand are required for RIG-I ATPase activity. (A to G) Purified his-RIG-I (200 nM) was incubated with [γ-32P]ATP in the presence of increasing amounts (4 to 250 nM) of various RNA or RNA/DNA hybrids as indicated (only the data from the 250 nM concentration are shown here; the complete range is shown in Fig. S2 in the supplemental material). (A) Importance of the 5′ ppp. (B and G) Importance of a base-paired 5′ end. (C to F) Nature of the bottom strand. Reactions were revealed and quantified by phosphorimaging. ATPase data are presented as relative (Rel.) ATPase activities normalized to the ATPase activity of RIG-I in the absence of RNA. Data are represented as means ± standard errors of the means (SEM) (n = 4) of the 250 nM RNA concentration. Significance: NS, P > 0.05; *, 0.01 ≤ P < 0.05; **, 0.001 ≤ P < 0.01. See also Fig. S2.
In contrast to the binding requirements, key ribonucleotides on the bottom strand are needed for ATPase activity. A hybrid with a bottom strand made entirely of deoxyribonucleotides was a very poor inducer of the ATPase despite normal binding to RIG-I (20d, Fig. 2C; see also Fig. S2A in the supplemental material). The effective binding of this duplex to RIG-I was confirmed by its ability to interfere with the ATPase activity induced by a bona fide dsRNA (see Fig. S2B). Increasing the number of ribonucleotides on the bottom strand from its 3′ end progressively increased ATPase activity until +5 ribonucleotides (5r-15d) was reached, at which point it was equivalent to that triggered by pure dsRNA (Fig. 2C; see also Fig. S2A). When these results are combined with those obtained for hybrids in which an RNA bottom strand contained increasing numbers of deoxyribonucleotides from the 3′ end (Fig. 2E; see also Fig. S2A), it appears that two residues, at positions 2 and 5, represent key residues for the recovery of normal ATPase activity. By testing more hybrids, we confirmed that these two ribonucleotides, in a background of deoxyribonucleotide bottom strand, are necessary and sufficient to restore ATPase activity (2*5*r-18d, Fig. 2D; see also Fig. S2A). According to structural data (16, 25), this corresponds to the binding of HEL1 and HEL2 to ribonucleotides 2 and 5 of the bottom strand, respectively. As both RNA:RNA and RNA:DNA hybrids are A-type helices, this suggests that the helicase domains need to interact with these 2′ hydroxyls for optimal ATPase activity.
Surprisingly, a further increase in the number of ribonucleotides on the bottom strand from 6 to 10, a length that corresponds to coverage by one RIG-I molecule (16, 18, 25, 26), resulted in ATPase activity that was significantly enhanced relative to that promoted by pure dsRNA (Fig. 2F; see also Fig. S2A in the supplemental material). According to structural data (16, 25), these additional ribonucleotides probably make contact with the HEL2i domain. Remarkably, further lengthening of the bottom strand beyond 10 nucleotides led to a return to the ATPase levels promoted by pure dsRNA (Fig. 2F; see also Fig. S2A and Discussion).
Further components on the bottom strand are necessary for full RIG-I activation.
The ultimate aim of the RIG-I pathway is the activation of downstream genes, such as IFN-encoding genes. To investigate the involvement of the bottom strand in this process, we tested our hybrids in A549 cells using reporter gene assays (Fig. 3A). As ATPase is required for RIG-I activation, the 20d hybrid that is unable to induce ATPase did not activate IFN-β (Fig. 3B and D; see also Fig. S3A in the supplemental material). In addition, molecules with a low ATPase level, such as 2d-18r to 5d-15r, induced IFN-β to a lower extent than the reference RNA (Fig. 3D). However, there was no strict correlation between the ability of the hybrids to induce ATPase and RIG-I activation. In particular, the 2*5*r-18d hybrid, which represents the minimal requirements for normal ATPase, did not induce IFN-β either in our reporter gene system (Fig. 3B) or in A549 cells expressing green fluorescent protein (GFP) under the control of the IFN-β promoter (see also Fig. S3A). The absence of IFN-β induction with this molecule was not due to its potential instability in vivo, since cotransfection with a bona fide RNA PAMP, the 2*5*r-18d hybrid, interfered with RIG-I activation and induced decreased IFN-β induction, as shown previously for the 5′ overhang duplexes (Fig. 3E) (37). In this experiment, increasing the amount of transfected 20r RNA increased IFN-β stimulation. This was in marked contrast to cotransfection with 5′ overhang or 2*5*r-18d molecules, which led to decreased IFN-β stimulation (Fig. 3E). This ruled out possible interference at the level of transfection.
FIG 3 .
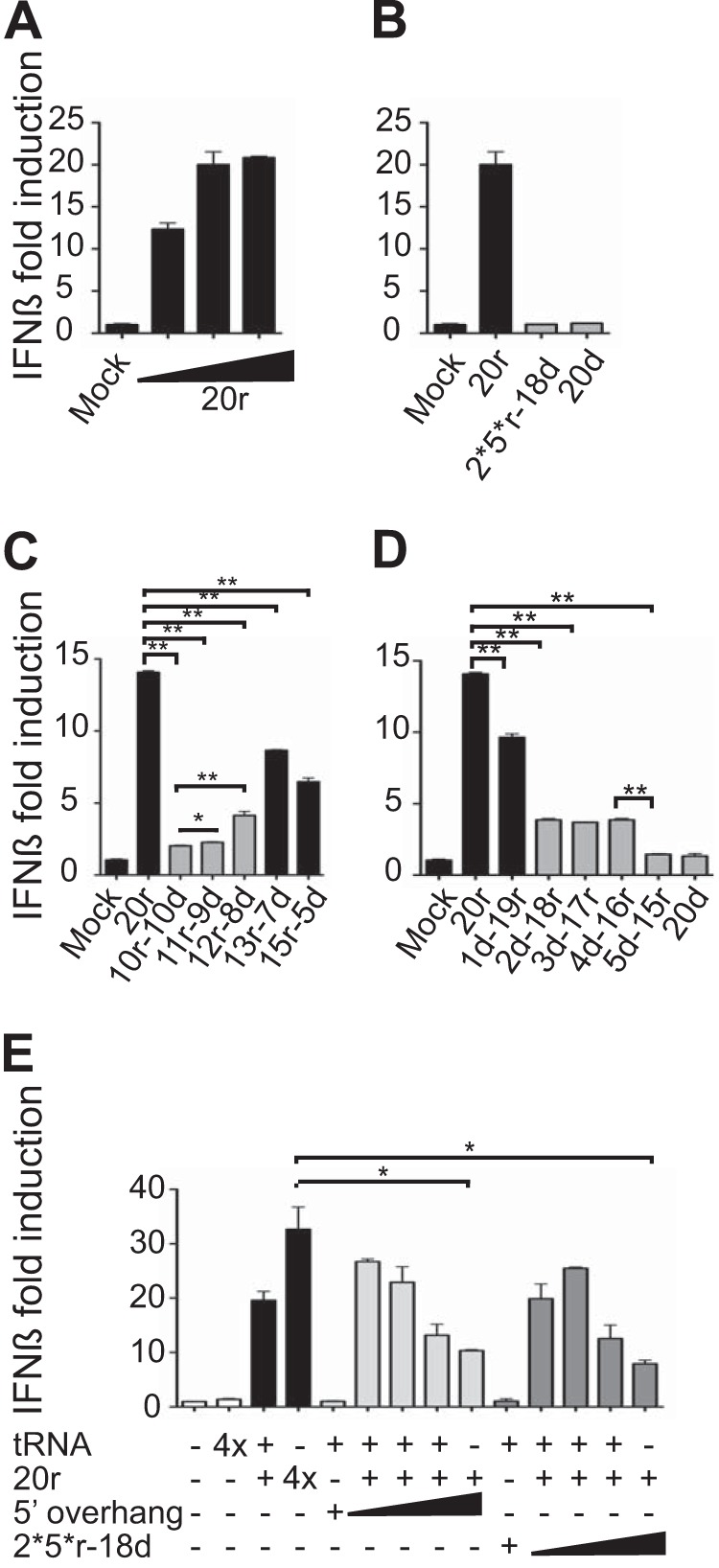
IFN-β promoter activation requires a minimal length of RNA duplex. A549 cells were transfected with an IFN-β-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase prior to transfection with various RNAs or RNA/DNA hybrids as indicated. Luciferase activity was determined 20 h post-RNA transfection and normalized to Renilla luciferase activity and is reported as the fold increase compared to the level seen with the mock control without RNA. (A) IFN-β activation stimulated by increasing amounts of our reference RNA (20r; 5′ ppp-blunt-ended dsRNA 20 nt in length; 200, 400, and 800 ng). (B to D) IFN-β activation stimulated by 400 ng of various RNA/DNA hybrids as indicated. (E) Interference experiment. A549 cells were transfected with 200 ng (1×) of our reference RNA (20r) and increasing amounts of various hybrids as indicated (0.5 and 1; 2× and 4×). The total amount of RNA transfected was kept constant, with tRNA tested as inactive with the highest concentration (4× = 800 ng). Data are represented as means ± SD (n = 2). Significance: NS, P > 0.05; *, 0.01 ≤ P < 0.05; **, 0.001 ≤ P < 0.01. See also Fig. S3 in the supplemental material.
If some hybrid duplexes can bind RIG-I and induce normal ATPase activity and yet do not activate IFN-β, what is the possible role of the ATPase under these conditions? One hypothesis could be that, if RIG-I binds a non-bona fide RNA PAMP (such as self-RNA), the ATPase activity functions in RIG-I recycling. In order to test this hypothesis, a simple competition experiment was performed. RNA pulldown assays were carried out in which His-tagged RIG-I was first bound to biotinylated RNA and, in a second step, competed with an excess of the same non-biotinylated RNA in the presence of ATP with or without MgCl2 (Fig. 4A) or in the presence and absence of both ATP and MgCl2 (Fig. 4B). Mg2+, which coordinates the β and γ phosphates of ATP in ATPases, is an essential cofactor for activity (28) (Fig. S4A in the supplemental material shows that there is no ATPase activity in the absence of MgCl2). As shown in Fig. 4A, addition of MgCl2 and, presumably, the resulting hydrolysis of ATP strongly increased RIG-I exchange on both the 10r-10d and 20r RNA duplexes. ATP and MgCl2 by themselves had no effect on RIG-I binding in the absence of competitor as seen in Fig. 4B (lane 4 versus lane 3). RIG-I recycling is observed only when the RNA competitor is added (Fig. 4B).
FIG 4 .
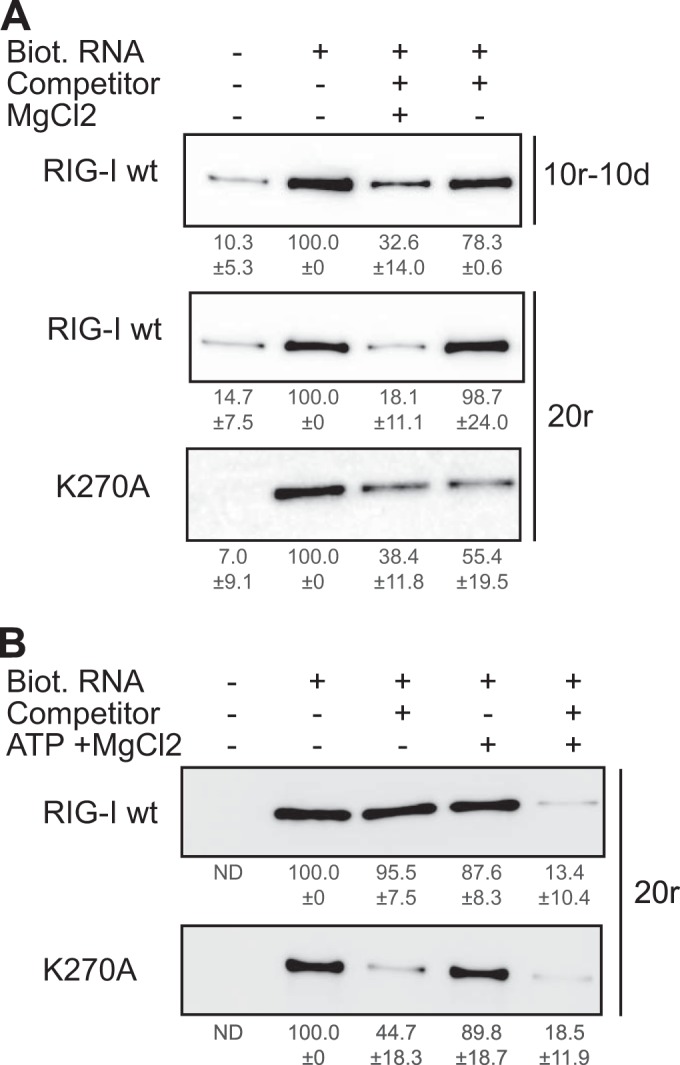
Rig-I ATPase activity promotes RNA recycling. (A) RNA pulldown assays were performed using 13 pmol of biotinylated (Biot.) 10r-10d and 20r hybrids (as indicated) and purified his-RIG-I (60 nM) in the presence of ATP and absence of MgCl2. Competition experiments were performed by adding 52 pmol of the same non-biotinylated RNA molecule (competitor) to the reaction mix in the presence or absence of MgCl2 (as indicated). (B) RNA pulldown assays were performed using 13 pmol of biotinylated 20r hybrids and purified his-RIG-I (60 nM) in the absence of ATP and MgCl2. Competition experiments were performed by adding 52 pmol of the same non-biotinylated RNA molecule (competitor) to the reaction mix in the presence or absence of MgCl2 (as indicated). The amounts of RIG-I remaining on the beads were determined by Western blotting using an anti-RIG-I antibody and quantified. RIG-I quantifications are indicated as the average percentage ± SD. (A) n = 3 for 10r-10d/RIG-I wt; n = 6 for 20r/RIG-I wt and 20r/K270A. (B) n = 5 for 20r/RIG-I wt; n = 3 for 20r/K270A.
As a control, the same experiments were carried out with the K270A RIG-I mutant that cannot hydrolyze ATP (see Fig. S2C in the supplemental material). Although the lysine of the Walker A motif, in combination with the main chain NH groups, is considered essential for ATP binding (29), and this lysine directly contacts the γ-PO4 of ATP (27), Peisley et al. have recently reported that K270A RIG-I nevertheless binds ATP in vitro (30) but presumably not with the same stability as the wild-type (wt) protein. In this case, RIG-I exchange was found to occur readily, and equally in the presence and absence of MgCl2 and ATP (Fig. 4), presumably due to less stable binding. The notion that one function of the ATPase is to promote RIG-I recycling on the RNA is also consistent with the finding that even hybrids with enhanced ATPase activity remain inactive under basal conditions (Fig. 5A [minus IFN-β] and data not shown).
FIG 5 .
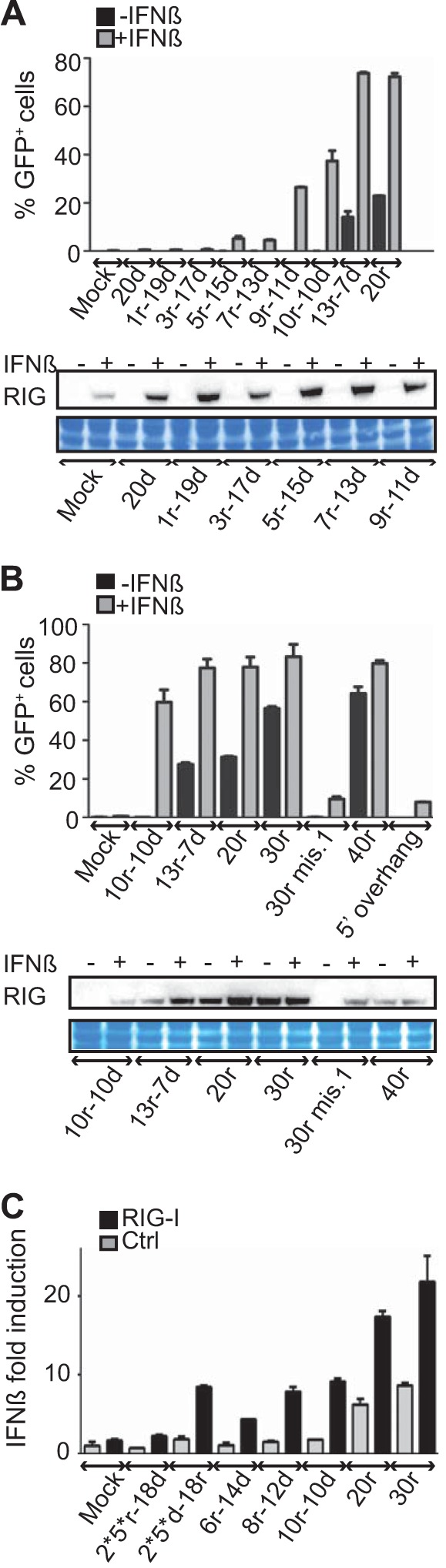
IFN-β priming and ectopic expression of RIG-I change the pattern and the sensitivity of the IFN-β stimulation. (A and B) A549/pr(IFN-β). GFP reporter cells were treated or not with IFN-β for 6 h prior to transfection with 500 ng of various RNA/DNA hybrids (as indicated). Data are plotted as the percentage of GFP-positive cells measured by flow cytometry and are represented as means ± SD (n = 2). (Bottom panels) Intracellular level of RIG-I analyzed by Western blotting. As a loading control, the membrane was stained with Coomassie blue after immunoblotting. (C) A549 cells were transfected with an IFN-β-driven firefly luciferase reporter and a plasmid constitutively expressing Renilla luciferase. Additionally, cells were also transfected with a plasmid expressing RIG-I (RIG-I) or an empty vector (Ctrl.). After 24 h, the cells were transfected with 400 ng of various RNA or RNA/DNA hybrids as indicated. Luciferase activity was determined 20 h post-RNA transfection and normalized to Renilla luciferase activity and is reported as the fold increase compared to the level seen with the mock control without RNA. Data are represented as means ± SD (n = 2). The low transfection efficacy of A549 cells does not permit us to observe a significant increase in RIG-I expression by Western blot analysis (data not shown).
The effect of IFN priming.
Interestingly, the lack of IFN-β stimulation by many of our hybrids can be reversed if cells are treated with IFN-β (primed) before RNA transfection (Fig. 5A and B). The 9r-11d and 10r-10d hybrids, inactive under unprimed conditions, become partially active after IFN priming (Fig. 5A). Likewise, the 13r-7d and 20r duplexes also showed enhanced stimulation in cells primed with IFN-β (Fig. 5A and B). In contrast, duplexes with a mismatch at position 1 (30r mis.1), as in the Ebola virus genome panhandle (Zaire Ebola virus, complete genome, gi |10313991|ref|NC_002549.1|), or with a 5′ overhang, as seen in the panhandle of some arenaviruses (31), remained inactive even after IFN priming (Fig. 5B). Thus, the genome configurations detailed above may represent an efficient viral strategy to escape RIG-I activation.
One consequence of IFN-β priming is the increase of expression of interferon-stimulated genes (ISG), including that of RIG-I itself (Fig. 5A and B, bottom panel). Similar increases in RIG-I expression were observed under physiological conditions when cells were transfected with stimulatory RNAs (Fig. 5B, bottom panel). To examine whether this increased RIG-I expression was responsible for changing the minimum dsRNA length required for effective IFN-β stimulation (Fig. 5A), A549 cells were transfected with plasmids expressing RIG-I prior to transfection with various duplexes. The results (Fig. 5C) show that both the 8r-12d and 10r-10d duplexes, inactive under unprimed conditions, induced IFN-β when RIG-I was overexpressed ectopically. Also, and as seen with IFN-β priming, RIG-I overexpression showed enhanced stimulation with the 20r and 30r duplexes (Fig. 5C). The 2*5*r-18d duplex, which represents the minimal requirement for normal RIG-I ATPase activity, nevertheless remained largely inactive under the overexpressed RIG-I conditions (Fig. 5C). Interestingly, its mirror molecule, 2*5*d-18r (with deoxynucleotides at positions 2 and 5 in an otherwise ribonucleotide-based backbone), exhibited decreased ATPase activity (Fig. 2H) and was inactive in inducing IFN-β under basal conditions (Fig. 5C), highlighting once more the role of the ATPase in RIG-I activation.
The minimal length of dsRNA that activates RIG-I in unprimed cells is consistent with the formation of a 2-RIG-I/dsRNA complex.
According to the structural data, the dsRNA region of 10r-10d is sufficient to fully interact with one RIG-I molecule. However, this hybrid does not trigger IFN-β under unprimed conditions. One hypothesis to explain the minimal length beyond 10r (as dsRNA) required to activate RIG-I is that a 2-RIG-I/dsRNA complex is required (32). We therefore performed electrophoretic mobility shift assays (EMSA) using our hybrid molecules. First, in contrast to the results seen with RNA pulldown, RIG-I binds to ssRNA in EMSA (Fig. 6D and data not shown). Only a 1-RIG-I/RNA complex is observed, and this likely reflects the capacity of the CTD to bind the 5′ ppp group. The fact that this binding is seen only in EMSA could be due to the “caging effect,” in which the tight environment of the acrylamide gel retains the link between RNA and protein even if this link is weak (33). The 20r duplex, as well as the 2*5*r-18r duplexes, was found to bind two RIG-Is (Fig. 6A, B, and E), in contrast to the 20d, 2*5*r-18d, and 10r-10d hybrids, where the 1-RIG-I/RNA complex was mainly observed (Fig. 6A, B, C and E). As the total length of these hybrids is a constant 20 bp, RIG-I does not bind on RNA/DNA hybrids other than at the 5′ ppp end. No difference was observed in the presence or absence of the ATP and MgCl2 needed for ATPase activity (Fig. 6A; see also Fig. S4A in the supplemental material). Moreover, two RIG-I ATPase mutants (K270A and D372A) also formed 2-RIG-I/RNA complexes with the same efficiency as the wt (see Fig. S4E). Thus, in contrast to previously published data (34), this complex formation is independent of ATPase activity, suggesting that RIG-I translocation is not involved in the formation of such duplexes (35). As expected, the CARDs were dispensable (ΔN; see Fig. S4C) but the CTD was absolutely required for binding, as complex formation was not observed with ΔCTD RIG-I (ΔC, ΔNΔC; see Fig. S4C) even in this very sensitive system. Interestingly, the CTD was also required for the binding of the second RIG-I, as no additional band corresponding to a heteroduplex RIG-I:RIG-I-ΔC/RNA was observed under conditions where a monomeric RIG-I/RNA complex was first made (see Fig. S4D). Supershift experiments confirmed that this complex was specifically formed by RIG-I (see Fig. S4B).
FIG 6 .
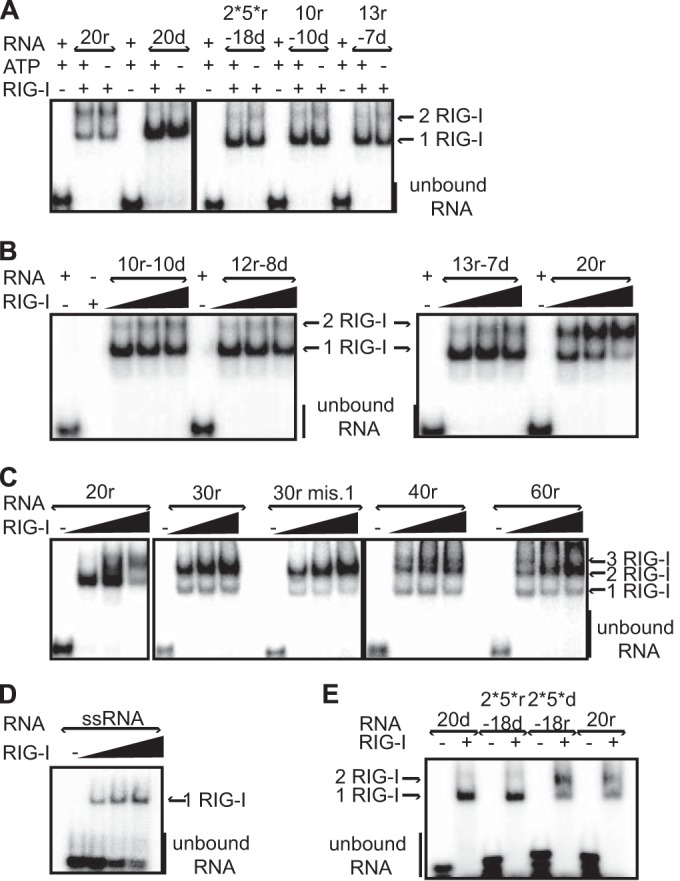
RIG-I oligomerization on RNA hybrids is RNA length dependent and ATPase independent. EMSA analysis was performed by incubating various radiolabeled RNA hybrids (250 nM) with purified his-RIG-I (2.5 μM (A and E); with increasing amount of RIG-I (4, 6, or 8 μM) in the presence of ATP and MgCl2 (B to D)). (A) RIG-I binding and oligomerization are independent of the ATPase activity. (B) dsRNA length dependence for RIG-I oligomerization. (C) The longer the dsRNA hybrid, the more easily 2-RIG-I/RNA or 3-RIG-I/RNA complexes are formed. (D) RIG-I binds ssRNA. (E) Importance of ribonucleotide content. The 2*5*d-18r molecule forms a RIG-I dimer, in contrast to its mirror molecule, 2*5*r-18d. Reactions were analyzed on native gels and revealed by phosphorimaging. See also Fig. S4 in the supplemental material.
Increasing the length of the hybrid from 20 to 30 ribonucleotides only increased the efficiency of 2-RIG-I complex formation, without leading to binding of an additional RIG-I (Fig. 6C). Upon further lengthening of the duplex to 40r and 60r, an additional complex, probably a 3-RIG-I/RNA complex, could now be observed (Fig. 6C). Introducing a mismatch at the first position of a 30r duplex (30r mis.1, which alters the blunt-end nature of the ds structure) did not affect 2-RIG-I/RNA complex formation (Fig. 6C). However, as in the presence of a 5′ overhang, this duplex had slightly less ATPase activity (Fig. 2G) and virtually no ability to induce IFN-β in vivo (Fig. 5B). In EMSA, increasing the amount of RNA above that required for 2-RIG-I/RNA complex formation resulted in a decrease of the level of the 2-RIG-I/RNA complex in favor of the 1-RIG-I/RNA complex, probably due to an excess of the free 5′ ppp ends that are preferentially bound (see the right panel in Fig. S4F in the supplemental material). These results are consistent with the 2-RIG-I/RNA complexes effectively formed by the binding of 2 RIG-I molecules and were not due to a conformational change of a 1-RIG-I/RNA complex that altered its electrophoretic mobility.
Direct comparison of various hybrids for their abilities to form 2-RIG-I/RNA complexes in the presence of increasing amounts of RIG-I shows that the longer the ribonucleotide hybrid, the more efficient the 2-RIG-I/RNA complex formation (i.e., this complex was formed at lower RIG-I concentrations; Fig. 6B and C). This may reflect the higher stability of such complexes. The length of these hybrids also correlates with their ability to induce IFN-β in our unprimed cells (Fig. 3C; see also Fig. S3B in the supplemental material). Taken together, these results indicate that for short dsRNAs, a minimum length of ribonucleotide duplex is required for 2-RIG-I/RNA complex formation and IFN-β activation. This complex formation requires the CTD but not the CARDs (see Fig. S4C).
DISCUSSION
Taken together, our results and the available structural data allow us to refine a model for the contacts between the functional domains within RIG-I and a short 5′ ppp-dsRNA (Fig. 7A). In this model, RIG-I activation depends on the length of the dsRNA (Fig. 7B). If a tetrameric RIG-I/RNA complex is the minimal structural requirement for activation (4), such a complex could be assembled as a single tetramer, 2 dimers, or 4 monomers, depending on the length of the dsRNA. The probability of forming such tetrameric complexes depends on the stability of the RIG-I/RNA complex, the rate of RIG-I recycling, and the RIG-I concentration.
FIG 7 .

RIG-I binding and activation. (A) Schematic representation of the contacts of various RIG-I motifs to 5′ ppp-dsRNA (adapted from reference 19). Domains on the 5′ ppp-dsRNA involved in binding to RIG-I, ATPase activity of RIG-I, and stability of the RIG-I/RNA complex are indicated. (B) Model of RIG-I activation depending on the length of the dsRNA. If a tetrameric RIG-I/RNA complex is the minimal structure required for RIG-I activation (42), 3 different scenarios (1 tetramer, 2 dimers, or 4 monomers) that are dependent on the length of the dsRNA can be envisaged. The probability of forming such tetrameric complexes depends on the stability of the RIG-I/RNA complex, including the rate of recycling of RIG-I in this complex. A direct consequence of this is that it also depends on the concentration of RIG-I within the cytoplasm.
RIG-I activation leading to IFN-β stimulation requires at least 3 steps: RNA recognition, ATPase activity, and exposure of the CARDs for downstream signaling. RIG-I oligomerization and binding to K63 polyubiquitin (poly-Ub) chains (mediated by TRIM25) are also part of this pathway (36). Given the risk of an unintended autoimmune reaction, the entire activation mechanism must be tightly controlled.
In our RNA pulldown assays, short dsRNAs, such as those present in viral RNA panhandle structures, are found to bind RIG-I because of their intrinsic double-stranded nature, independently of bottom strand composition (i.e., deoxy- or ribonucleotide; Fig. 1C and D). A base-paired 5′ ppp nucleotide is also required for RIG-I recognition, since a 5′ overhang or a mismatch at position 1 decreases RIG-I binding (Fig. 1A and B and data not shown). The absence of binding in EMSA with the helicase domain alone (with or without the CARDs) highlights the importance of the CTD for RNA binding (Fig. 7A; see also Fig. S4C in the supplemental material). Notably, this binding is also independent of ATP hydrolysis (see Fig. S1D), in contrast to the dense loading of RIG-I onto longer dsRNAs to form RIG-I:dsRNA filaments (30, 34).
Based on crystal structures of RIG-I with or without dsRNA and a non-hydrolyzable form of ATP, the transition from autorepressed to activated RIG-I involves closure of its helicase domains upon binding dsRNA and ATP, which simultaneously disrupts the CARD:helicase interaction, as the CARDs and dsRNA bind in part to the same helicase surface. The closure of the helicase domains, however, creates a pocket not only for ATP binding but also for its hydrolysis. The function of this, the only known catalytic activity of RIG-I, is poorly understood. Although ATPase is essential for RIG-I signaling, there is nevertheless no strict correlation between the ability of various duplexes to stimulate the ATPase and their ability to induce IFN. Some duplexes, e.g., 10r-10d, stimulate the ATPase even more potently than pure dsRNA (20r) and yet are unable to induce IFN. Moreover, there is sometimes a seemingly contradictory negative relationship between stable RNA binding and ATPase induction; e.g., duplex mismatches decrease RIG-I activation but increase ATPase levels, again to levels above that of pure dsRNA (37). A possible explanation of these unexpected findings is that, as the ATPase active site is formed by the closure of 2 helicase domains in coordination with dsRNA binding, if a rate-limiting step in ATP hydrolysis is removal of ADP and Pi from the active site, duplex instability due to mismatches or deoxyribonucleotides can also increase ATPase levels by facilitating the discharge of ADP and Pi from the active site. ATP turnover is thus enhanced, increasing ATPase activity. Thus, in some cases, weaker duplex binding can apparently be more than compensated for by increased ATP turnover, leading to enhanced ATPase activity. In line with this, we show that ATPase increased RIG-I recycling (Fig. 4).
As ATPase is also required for RIG-I activation, this may appear incompatible with the ATPase recycling activity. One way to reconcile these two roles of the ATPase (essential for activation and for RNA recycling) is to postulate that there is a competition between recycling and RIG-I oligomerization. This competition may be particularly important for short dsRNAs that are similar to self-RNAs. RIG-I complex formation depends on the length of the duplex (in agreement with references 4 and 34) and RIG-I abundance. Very short (8- to 12-bp) dsRNAs require higher concentrations of RIG-I for complex formation and activation, presumably because recycling outweighs complex formation. When the dsRNA is even shorter, i.e., indistinguishable from self-RNAs, recycling always prevails and RIG-I is never activated. ATPase activity is presumably responsible for the same RIG-I conformational changes needed for both complex formation and recycling. If a RIG-I tetrameric complex is not formed (RNA too short, RIG-I concentration too low), ATPase leads to recycling. If a RIG-I oligomeric complex is formed (longer RNA, higher concentration of RIG-I), RIG-I is not recycled, presumably because the complex is stable. In this way, the presence of ATPase leads to the formation of stable, active complexes. However, none of the circumstances outlined above exclude the possibility that ATP hydrolysis is required for other functions in IFN induction.
Given the abundance of cellular RNAs and the serious risk that inappropriate induction of IFN can lead to an autoimmune response, RIG-I signaling must be very tightly controlled, occurring only when confronted with non-self-RNA. Should self-RNA be bound, however infrequently, there is likely to be a mechanism to free this RIG-I from its inappropriate ligand, and the finding that ATP hydrolysis promotes RIG-I exchange on preformed RIG-I:dsRNA complexes (Fig. 4) is likely to be germane in this context. This exchange is possible presumably because the RNA is less tightly bound to the helicase in the absence of the ATP that helps to maintain its closure. One function of the RIG-I ATPase would then be to dissociate RIG-I from inappropriate ligands, as well as from those with a dsRNA less than 13 bp in length in unprimed cells (see below). The fact that duplexes with reduced stability (a possible indicator of self-RNAs) more strongly stimulate the ATPase is in line with this exchange function of RIG-I.
The minimum length of dsRNA required for IFN-β stimulation is still a subject of debate, with estimates ranging from 10 bp (11) to 19 bp (10). Recently, two groups reported that dsRNAs as short as 10 bp, a length that can accommodate only one RIG-I, can activate IFN-β (38, 39). These experimental differences can have several explanations. The structures of the RNAs used in the latter studies were different, as the Goulet and Kohlway groups used RNAs with stem-loop structures, which might affect stability of the RIG-I:RNA complex, especially within cells. Our experiments suggest that variations in cellular RIG-I concentrations of cultured cells could also explain these divergent results. Even 1-RIG-I/10r-10d duplexes can oligomerize in trans, to form the essential tetrameric RIG-I/poly-Ub chain complex (Fig. 7B, left panel), but this would require higher RIG-I concentrations than those required for 2-RIG-I/20r duplexes or longer duplexes (as found in Fig. 5), especially as RIG-I on 10r-10d duplexes recycles more rapidly than RIG-I on 20r duplexes (Fig. 7B, middle panel). The notion that the minimum length of dsRNA required for IFN induction can be modified by cellular RIG-I concentrations is suggested by the finding that when our cells were primed, i.e., pretreated with IFN-β before RNA transfection, short duplex molecules such as 9r-11d, 10r-10d, 11r-9d, and 12r-8d, which are inactive in unprimed cells, became active (Fig. 5A and B and data not shown). One consequence of this priming is the increased expression of RIG-I, as RIG-I itself is an ISG (Fig. 5). A similar result was obtained with RIG-I overexpression, which apparently also brings about a qualitative shift in the nature of the RNA recognized as a PAMP (Fig. 5C). When RIG-I concentrations increase, formation of active tetrameric RIG-I complexes is favored, mainly for very short dsRNAs (between 8 and 12 bp in length) that carry only one RIG-I and are presumably more prone to RIG-I recycling (Fig. 7B).
This scheme is consistent with the notion that uninfected cells need to be highly selective in recognizing RNAs as PAMPs, so as to discriminate between self-RNA and non-self-RNAs. Once RIG-I is activated upon viral infection and IFN produced, the increased levels of PRRs render cells less discriminatory toward PAMPs but more efficient in mounting an antiviral response.
MATERIALS AND METHODS
Plasmids.
pβ-IFN-fl-lucter contains the firefly luciferase gene driven by the human IFN-β promoter as described previously (40). pTK-rl-lucter contains the Renilla luciferase gene (Promega) driven by the herpes simplex virus TK promoter. pEBS-tom encodes a red fluorescent protein. pET28-His10Smt3 containing a wt or mutant human RIG-I gene was engineered as described previously (24).
In vitro RNA synthesis.
The template for T7 RNA polymerase synthesis of 5′ ppp-ssRNA (top strand) was prepared by annealing oligonucleotides as follows: 5′-TAATACGACTCACTATAgcgcaccggggaaccaaggcgaacacggacacgcaacaaacgagaccgacaacagacagga, representing the T7 polymerase promoter (bold) and the −1 to +59 junin virus 5′ RNA sequence (lowercase) in which U residues at the underlined positions were replaced by the indicated nucleotides to prevent synthesis of double-stranded RNA in a T7 polymerase reaction mixture lacking U residues, and its complementary oligonucleotide, and 5′-tcctgtctgttgtcggtctcgtttgttgcgtgtccgtgttcgccttggttccccggtgcgcTATAGTGAGTCGTATTA.
After annealing, dsDNAs were purified with a QIAquick PCR purification kit (Qiagen) and 12 pmol was used for the in vitro transcription performed with T7 MEGAshortscript (Ambion) according to the manufacturer’s instructions (in the absence of UTP). Biotinylated ssRNA was obtained by T7 polymerase transcription in the presence of a 3:1 molar ratio of CTP and biotin-11-CTP (Roche 04739205001). Radiolabeled ssRNA was obtained by T7 polymerase transcription in the presence of a 50:1 molar ratio of CTP and [α-32P]CTP (Hartmann Analytic). Fluorescent ssRNA was obtained by performing in vitro transcription in the presence of a 2:3 molar ratio of CTP and cyanine 5-CTP (PerkinElmer NEL581001EA).
To obtain 5′ OH-ssRNA, in vitro transcripts were treated with alkaline phosphatase (Roche 11097075001) according to the manufacturer’s instructions. Total T7 transcripts were digested with turbo DNase for 15 min at 37°C and purified on NucAway Spin columns (Ambion) to remove unincorporated nucleotides and DNA fragments.
Double-stranded RNA preparation.
5′ ppp-ssRNA or 5′ OH-ssRNA (synthetic or obtained by alkaline phosphatase treatment) was mixed with the indicated synthetic complementary 5′ OH oligoribonucleotides in a 1 to 2 M ratio in a final volume of 100 µl (300 mM NaCl, 50 mM Tris [pH 7.5], 1 mM EDTA), heated 1 min at 95°C, and progressively cooled to room temperature (RT).
Transfection and measurement of IFN-β promoter activity.
A549 cells (human alveolar adenocarcinoma cell line) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Pen/Strep). A total of 100,000 cells were plated into 6-well plates and were transfected 24 h later with 1.5 µg of pβ-IFN-fl-lucter, 0.5 µg of pTK-rl-lucter, and 0.5 µg of pEBS-tom (used as a transfection control), using Gene Juice transfection reagent (Novagen). Twenty-four hours later, cells were transfected (or not) with 400 ng (or as otherwise indicated) of 5′ ppp-dsRNA using TransMessenger transfection reagent (Qiagen) according to the manufacturer’s instructions. Twenty hours later, cells were harvested and cell lysates were used to measure firefly and Renilla luciferase activity (dual-luciferase reporter assay system, Glomax 20/20 luminometer; Promega).
Recombinant RIG-I expression.
The pET28-His10Smt3-RIG-I wt or mutant plasmids were transformed into Escherichia coli BL21 cells. Cultures (500 ml) derived from single transformants were grown at 37°C in LB medium containing 50 μg/ml kanamycin to an OD600 of 0.6. The cultures were adjusted to 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and 2% ethanol and further incubated for 20 h at 17°C. Cells were harvested by centrifugation, and recombinant RIG-I protein was purified from bacteria as previously described (24). Protein concentrations were determined using the Bio-Rad dye-binding method with bovine serum albumin (BSA) as the standard.
RNA pulldown assay.
Strepavidin agarose beads (Invitrogen SA100-04) were preequilibrated with blocking buffer (base buffer [20 mM HEPES {pH 7.9}, 15% glycerol, 0.05% Nonidet P-40, 50 mM NaCl, 0.2 mg/ml tRNA; Roche Applied Science, 10109495001], and 2 mM dithiothreitol [DTT]) plus 100 mM NaCl, 0.1 mg/ml glycogen, and 5 mg/ml BSA for 2 h at 4°C. For each assay, 13 pmol of biotinylated RNA was incubated with the beads in binding buffer (base buffer plus 2 mM DTT, 1% protease inhibitor mixture [Sigma, P8340], and 100 U/ml RNasin [Promega N2515]) for 2 h at 4°C. After two washes with washing buffer (base buffer plus 2 mM DTT and 0.05 mg/ml tRNA), the beads were incubated with 1.25 µg (12 pmol) of purified recombinant RIG-I in binding buffer with or without 2 mM MgCl2 and 0.5 mM ATP as indicated. After 15 min at 37°C, the beads were washed three times with washing buffer followed by elution in SDS protein sample buffer. The reactions were analyzed by Western blotting probing the membrane with anti-RIG-I antibody.
Electrophoretic mobility shift assay.
Increasing amounts of purified recombinant RIG-I were incubated with 250 nM radiolabeled RNA in a final volume of 20 µl (20 mM HEPES [pH 7.5], 75 mM NaCl, 2.5 mM MgCl2, 2 mM DTT, 1 mM ATP) for 30 min at RT. After addition of 5× native loading buffer (300 mM Tris [pH 6.8], 50% glycerol, 0.05% bromophenol blue), reactions were analyzed on Tris-borate-EDTA (TBE) acrylamide 4% to 12% gradient nondenaturing gel. The radioactivity was revealed by phosphorimaging (Typhoon; GE Healthcare Life Sciences).
Measurement of RIG-I ATPase activity.
Increasing amounts (4 to 250 nM) of various RNA molecules were incubated with 200 nM purified recombinant RIG-I and [γ-32P]ATP (Hartmann Analytic) in a final volume of 15 µl (50 mM Tris acetate [pH 6], 5 mM DTT, 1.5 mM MgCl2) for 15 min at 37°C. Reactions were then stopped with 1 mM formic acid, and 2.5 µl of each reaction mixture was spotted onto TLC PEI cellulose F plates (Merck 1.05579.0001) and applied to a migration buffer (0.5 M LiCl, 1 N formic acid) to separate released 32PO4 and non-hydrolyzed ATP. 32PO4 release was measured in a phosphorimager (Typhoon; GE Healthcare Life Sciences) and quantified with ImageQuantTL software (GE Healthcare Life Sciences).
Transfection and measurement of IFN-β promoter activity.
A549/pr(IFN-β) GFP reporter cells (41) were transfected as described above. Twenty hours later, GFP expression was monitored and pictures were acquired using an Evos FL epifluorescence microscope. Twenty-four hours posttransfection, cells were harvested and the percentage of GFP-positive cells was determined by flow cytometry on 20,000 cells (i.e., 10% of the harvested cells) using a BD Accuri C6 Cytometer. Data were analyzed using CFlow Plus software (Accuri, version 1.0.264.15).
Antibodies.
The following primary antibodies were used: anti-RIG-I (Alexis 210-932C100) (1:1,000) and anti-His (H1029; Sigma) (1:2,000). Immunoblot analyses were developed with the following secondary antibody: anti-mouse IgG horseradish peroxidase-conjugated whole antibody (Bio-Rad) (1:3,000).
Statistical analysis.
Unpaired t tests were performed using GraphPad Prism version 6.00 (GraphPad Software).
Quantifications.
Western blot quantifications were performed using ImageJ version 1.44p (W. S. Rasband; ImageJ, U. S. National Institutes of Health, Bethesda, MD, http://imagej.nih.gov/ij/, 1997 to 2014).
SUPPLEMENTAL MATERIAL
RIG-I binds RNA through its CTD and independently of its ATPase activity. (A to D) RNA pulldown assays were performed using 13 pmol of biotinylated reference RNA (20r) and wt RIG-I and various his-RIG-I mutants (60 nM). (A and B) RIG-I binds to 5′ ppp-dsRNA through its CTD. Reactions were performed with wt, ΔCTD (ΔC), ΔCARD (ΔN), or ΔCARD ΔCTD (ΔNΔC) RIG-I in the absence of ATP and MgCl2. (C and D) Rig-I ATPase activity is not required for RNA binding. Reactions were performed with wt RIG-I or an ATPase mutant (K270A or D372A) in the absence (C) or presence (D) of ATP and MgCl2. Reactions were analyzed by Western blotting using an anti-histidine antibody. Download
Control ATPase reactions. (A) Complete ATPase reactions. Purified his-RIG-I (200 nM) was incubated with [γ-32P]ATP in the presence of increasing amounts (4 to 250 nM) of different RNAs or RNA/DNA hybrids as indicated (only the 250 nM concentration data are shown in Fig. 2). (B) The 20d hybrid interferes with the RIG-I ATPase induced by 20r. Increasing amounts (60, 180, 360, and 540 nM) of the 20d hybrid were added to the ATPase reaction performed with a 60 nM concentration of our reference RNA (5′ ppp-blunt dsRNA; 20r). The 20d molecule is inactive per se. As a control, increasing amounts of 20r were added to itself and did not interfere with RIG-I ATPase activity. Data are represented as means ± SEM (n = 3). Significance: NS, P > 0.05; **, 0.001 ≤ P < 0.01. (C) ATPase activity of RIG-I mutants. wt RIG-I or mutant RIG-I (K270A or D372A) (200 nM) was tested using different RNA ligands (250 nM). Reactions were revealed and quantified by phosphorimaging. ATPase data are presented as relative ATPase activities normalized to the ATPase activity of RIG-I in the absence of RNA. Download
The IFN-β promoter activation is strictly dependent on the length of the dsRNA hybrid. (A and B) A549/pr(IFN-β). GFP reporter cells were transfected with 800 ng of different RNA or RNA/DNA hybrids as indicated. At 24 h posttransfection, cells were harvested and subjected to flow cytometry analysis to determine GFP expression. (A) The presence of ATPase is not sufficient to induce IFN-β activation. (B) A sequence of 13 ribonucleotides represents the minimum dsRNA length for IFN-β activation under normal (i.e., unprimed) conditions. The top panel shows white-light (WL) and fluorescence (GFP) microscopy pictures. The bottom panel shows flow cytometry data. Data are represented as means ± SD (n = 2). Download
RIG-I oligomerizes through its CTD and independently of its ATPase activity. (A) Rig-I ATPase activity requires MgCl2. Purified RIG-I (200 nM) was incubated with [γ-32P]ATP in the presence of 5′ ppp-blunt-ended dsRNA (4 to 250 nM), with or without MgCl2. Reactions were revealed and quantified by phosphorimaging. (B) The observed RNA shifts in EMSA were specific to RIG-I. A constant amount (250 nM) of radiolabeled RNA was incubated with his-RIG-I (2.5 µM) or his-RIG-I and anti-His antibody in the presence of ATP and MgCl2. (C to F) Oligomerization of various variants of RIG-I. (C) Radiolabeled RNA (250 nM) was incubated with RIG-I ΔCARD (ΔN), ΔCTD (ΔC), and ΔCARDΔCTD (ΔNΔC) at 2.5 µM, in the presence or absence of ATP and MgCl2. (D) Radiolabeled RNA (125 nM) was sequentially incubated with RIG-I wt (1.25 µM) and with 2.5 µM RIG-I wt or ΔCTD (ΔC), in the absence of ATP and MgCl2. (E) Radiolabeled RNA (250 nM) was incubated with wt RIG-I or an ATPase mutant (K270A or D372A) at 1, 2, or 3 µM in the presence of ATP and MgCl2. (F) (Left panel) RIG-I oligomerization depends on the protein/RNA ratio. (Right panel) A constant amount (250 nM) of radiolabeled RNA was incubated with increasing amounts (0.4, 0.8, 1.6, and 3.2 µM) of RIG-I. A constant amount (2.5 µM) of RIG-I was incubated with increasing amounts (250 nM, 500 nM, 1 µM, and 2 µM) of radiolabeled RNA. Reactions were analyzed on native gel and revealed by phosphorimaging. Download
ACKNOWLEDGMENTS
This work was supported by the Swiss National Science Foundation, grant 31003A_135467.
Thanks to Steve Goodbourn and Richard Randall for providing the GFP-IFN β-reporter A549 cells. We thank Stéphane Hausmann and Jean-Baptiste Marq for excellent technical advices and assistance. We also thank Daniel Kolakofsky, Laurent Roux, and Joseph Curran for their precious help discussing the project and critically reading the manuscript.
Footnotes
Citation Anchisi S, Guerra J, Garcin D. 2015. RIG-I ATPase activity and discrimination of self-RNA versus non-self-RNA. mBio 6(2):e02349-14. doi:10.1128/mBio.02349-14.
REFERENCES
- 1.Ranjan P, Bowzard JB, Schwerzmann JW, Jeisy-Scott V, Fujita T, Sambhara S. 2009. Cytoplasmic nucleic acid sensors in antiviral immunity. Trends Mol Med 15:359–368. doi: 10.1016/j.molmed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Luo D, Kohlway A, Pyle AM. 2013. Duplex RNA activated ATPases (DRAs): platforms for RNA sensing, signaling and processing. RNA Biol 10:111–120. doi: 10.4161/rna.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. 2008. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell 29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 4.Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M Jr, Inagaki F, Fujita T. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell 29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 6.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 7.Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 8.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 9.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 10.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, Endres S, Rothenfusser S. 2009. 5′-Triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci USA 106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruigrok RW, Crépin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis e Sousa C. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang SY, Sun HY, Lee KH, Oh BH, Cha YJ, Kim BH, Yoo JY. 2012. 5′-Triphosphate-RNA-independent activation of RIG-I via RNA aptamer with enhanced antiviral activity. Nucleic Acids Res 40:2724–2733. doi: 10.1093/nar/gkr1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. 2011. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 17.Leung DW, Amarasinghe GK. 2012. Structural insights into RNA recognition and activation of RIG-I-like receptors. Curr Opin Struct Biol 22:297–303. doi: 10.1016/j.sbi.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. 2011. Structural insights into RNA recognition by RIG-I. Cell 147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolakofsky D, Kowalinski E, Cusack S. 2012. A structure-based model of RIG-I activation. RNA 18:2118–2127. doi: 10.1261/rna.035949.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konarska MM, Sharp PA. 1989. Replication of RNA by the DNA-dependent RNA polymerase of phage T7. Cell 57:423–431. doi: 10.1016/0092-8674(89)90917-3. [DOI] [PubMed] [Google Scholar]
- 21.Marq JB, Hausmann S, Luban J, Kolakofsky D, Garcin D. 2009. The double-stranded RNA binding domain of the vaccinia virus E3L protein inhibits both RNA- and DNA-induced activation of interferon beta. J Biol Chem 284:25471–25478. doi: 10.1074/jbc.M109.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P. 2010. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18:1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, Hartmann G, Patel DJ. 2010. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat Struct Mol Biol 17:781–787. doi: 10.1038/nsmb.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hausmann S, Marq JB, Tapparel C, Kolakofsky D, Garcin D. 2008. RIG-I and dsRNA-induced IFNbeta activation. PLoS One 3:e3965. doi: 10.1371/journal.pone.0003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M Jr, Patel SS, Marcotrigiano J. 2011. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Civril F, Bennett M, Moldt M, Deimling T, Witte G, Schiesser S, Carell T, Hopfner KP. 2011. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep 12:1127–1134. doi: 10.1038/embor.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Story RM, Weber IT, Steitz TA. 1992. The structure of the E. coli recA protein monomer and polymer. Nature 355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 28.Berg JM, Tymoczko JL, Stryer L. 2002. Biochemistry, 5th ed. W. H. Freeman, New York, NY. [Google Scholar]
- 29.Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 30.Peisley A, Wu B, Yao H, Walz T, Hur S. 2013. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell 51:573–583. doi: 10.1016/j.molcel.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Garcin D, Kolakofsky D. 1992. Tacaribe arenavirus RNA synthesis in vitro is primer dependent and suggests an unusual model for the initiation of genome replication. J Virol 66:1370–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckham SA, Brouwer J, Roth A, Wang D, Sadler AJ, John M, Jahn-Hofmann K, Williams BR, Wilce JA, Wilce MC. 2013. Conformational rearrangements of RIG-I receptor on formation of a multiprotein:dsRNA assembly. Nucleic Acids Res 41:3436–3445. doi: 10.1093/nar/gks1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fried MG, Liu G. 1994. Molecular sequestration stabilizes CAP-DNA complexes during polyacrylamide gel electrophoresis. Nucleic Acids Res 22:5054–5059. doi: 10.1093/nar/22.23.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel JR, Jain A, Chou YY, Baum A, Ha T, García-Sastre A. 2013. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 14:780–787. doi: 10.1038/embor.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. 2009. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science 323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 37.Marq JB, Hausmann S, Veillard N, Kolakofsky D, Garcin D. 2011. Short double-stranded RNAs with an overhanging 5′ ppp-nucleotide, as found in arenavirus genomes, act as RIG-I decoys. J Biol Chem 286:6108–6116. doi: 10.1074/jbc.M110.186262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goulet ML, Olagnier D, Xu Z, Paz S, Belgnaoui SM, Lafferty EI, Janelle V, Arguello M, Paquet M, Ghneim K, Richards S, Smith A, Wilkinson P, Cameron M, Kalinke U, Qureshi S, Lamarre A, Haddad EK, Sekaly RP, Peri S, Balachandran S, Lin R, Hiscott J. 2013. Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog 9:e1003298. doi: 10.1371/journal.ppat.1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohlway A, Luo D, Rawling DC, Ding SC, Pyle AM. 2013. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep 14:772–779. doi: 10.1038/embor.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King P, Goodbourn S. 1994. The beta-interferon promoter responds to priming through multiple independent regulatory elements. J Biol Chem 269:30609–30615. [PubMed] [Google Scholar]
- 41.Chen S, Short JA, Young DF, Killip MJ, Schneider M, Goodbourn S, Randall RE. 2010. Heterocellular induction of interferon by negative-sense RNA viruses. Virology 407:247–255. doi: 10.1016/j.virol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. 2012. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RIG-I binds RNA through its CTD and independently of its ATPase activity. (A to D) RNA pulldown assays were performed using 13 pmol of biotinylated reference RNA (20r) and wt RIG-I and various his-RIG-I mutants (60 nM). (A and B) RIG-I binds to 5′ ppp-dsRNA through its CTD. Reactions were performed with wt, ΔCTD (ΔC), ΔCARD (ΔN), or ΔCARD ΔCTD (ΔNΔC) RIG-I in the absence of ATP and MgCl2. (C and D) Rig-I ATPase activity is not required for RNA binding. Reactions were performed with wt RIG-I or an ATPase mutant (K270A or D372A) in the absence (C) or presence (D) of ATP and MgCl2. Reactions were analyzed by Western blotting using an anti-histidine antibody. Download
Control ATPase reactions. (A) Complete ATPase reactions. Purified his-RIG-I (200 nM) was incubated with [γ-32P]ATP in the presence of increasing amounts (4 to 250 nM) of different RNAs or RNA/DNA hybrids as indicated (only the 250 nM concentration data are shown in Fig. 2). (B) The 20d hybrid interferes with the RIG-I ATPase induced by 20r. Increasing amounts (60, 180, 360, and 540 nM) of the 20d hybrid were added to the ATPase reaction performed with a 60 nM concentration of our reference RNA (5′ ppp-blunt dsRNA; 20r). The 20d molecule is inactive per se. As a control, increasing amounts of 20r were added to itself and did not interfere with RIG-I ATPase activity. Data are represented as means ± SEM (n = 3). Significance: NS, P > 0.05; **, 0.001 ≤ P < 0.01. (C) ATPase activity of RIG-I mutants. wt RIG-I or mutant RIG-I (K270A or D372A) (200 nM) was tested using different RNA ligands (250 nM). Reactions were revealed and quantified by phosphorimaging. ATPase data are presented as relative ATPase activities normalized to the ATPase activity of RIG-I in the absence of RNA. Download
The IFN-β promoter activation is strictly dependent on the length of the dsRNA hybrid. (A and B) A549/pr(IFN-β). GFP reporter cells were transfected with 800 ng of different RNA or RNA/DNA hybrids as indicated. At 24 h posttransfection, cells were harvested and subjected to flow cytometry analysis to determine GFP expression. (A) The presence of ATPase is not sufficient to induce IFN-β activation. (B) A sequence of 13 ribonucleotides represents the minimum dsRNA length for IFN-β activation under normal (i.e., unprimed) conditions. The top panel shows white-light (WL) and fluorescence (GFP) microscopy pictures. The bottom panel shows flow cytometry data. Data are represented as means ± SD (n = 2). Download
RIG-I oligomerizes through its CTD and independently of its ATPase activity. (A) Rig-I ATPase activity requires MgCl2. Purified RIG-I (200 nM) was incubated with [γ-32P]ATP in the presence of 5′ ppp-blunt-ended dsRNA (4 to 250 nM), with or without MgCl2. Reactions were revealed and quantified by phosphorimaging. (B) The observed RNA shifts in EMSA were specific to RIG-I. A constant amount (250 nM) of radiolabeled RNA was incubated with his-RIG-I (2.5 µM) or his-RIG-I and anti-His antibody in the presence of ATP and MgCl2. (C to F) Oligomerization of various variants of RIG-I. (C) Radiolabeled RNA (250 nM) was incubated with RIG-I ΔCARD (ΔN), ΔCTD (ΔC), and ΔCARDΔCTD (ΔNΔC) at 2.5 µM, in the presence or absence of ATP and MgCl2. (D) Radiolabeled RNA (125 nM) was sequentially incubated with RIG-I wt (1.25 µM) and with 2.5 µM RIG-I wt or ΔCTD (ΔC), in the absence of ATP and MgCl2. (E) Radiolabeled RNA (250 nM) was incubated with wt RIG-I or an ATPase mutant (K270A or D372A) at 1, 2, or 3 µM in the presence of ATP and MgCl2. (F) (Left panel) RIG-I oligomerization depends on the protein/RNA ratio. (Right panel) A constant amount (250 nM) of radiolabeled RNA was incubated with increasing amounts (0.4, 0.8, 1.6, and 3.2 µM) of RIG-I. A constant amount (2.5 µM) of RIG-I was incubated with increasing amounts (250 nM, 500 nM, 1 µM, and 2 µM) of radiolabeled RNA. Reactions were analyzed on native gel and revealed by phosphorimaging. Download