

Background: Metabolic enzymes have been hypothesized to assemble into complex to respond to cellular metabolism changes.
Results: De novo purine biosynthesis increases in purinosome-containing cells.
Conclusion: Purine metabolism is adjusted by purinosome assembly.
Significance: This study indicates that purinosome is a functional multienzyme complex.
Keywords: Mass Spectrometry (MS), Metabolomics, Nucleoside/Nucleotide Biosynthesis, Protein Complex, Purine, Metabolic Flux, Purinosome
Abstract
Enzymes in the de novo purine biosynthesis pathway are recruited to form a dynamic metabolic complex referred to as the purinosome. Previous studies have demonstrated that purinosome assembly responds to purine levels in culture medium. Purine-depleted medium or 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) treatment stimulates the purinosome assembly in HeLa cells. Here, several metabolomic technologies were applied to quantify the static cellular levels of purine nucleotides and measure the de novo biosynthesis rate of IMP, AMP, and GMP. Direct comparison of purinosome-rich cells (cultured in purine-depleted medium) and normal cells showed a 3-fold increase in IMP concentration in purinosome-rich cells and similar levels of AMP, GMP, and ratios of AMP/GMP and ATP/ADP for both. In addition, a higher level of IMP was also observed in HeLa cells treated with DMAT. Furthermore, increases in the de novo IMP/AMP/GMP biosynthetic flux rate under purine-depleted condition were observed. The synthetic enzymes, adenylosuccinate synthase (ADSS) and inosine monophosphate dehydrogenase (IMPDH), downstream of IMP were also shown to be part of the purinosome. Collectively, these results provide further evidence that purinosome assembly is directly related to activated de novo purine biosynthesis, consistent with the functionality of the purinosome.
Introduction
Purine nucleotides have very important and diverse functions inside the cell. They are building blocks of DNA and RNA, energy carriers, and cell signaling molecules, and the play central roles in metabolism. Purine nucleotides are biosynthesized by the de novo pathway and recycled from their free bases by the salvage pathway. IMP serves as a purine nucleotide precursor, which can be synthesized from 5-phospho-α-d-ribose 1-diphosphate (PRPP)3 by the de novo purine biosynthesis pathway or made from hypoxanthine by the salvage pathway. The de novo pathway contains 10 highly conserved chemical steps that require the coordination of six enzymes in humans. It has long been hypothesized that these six enzymes form a functional multienzyme complex in the cell similar to what has been observed previously with other sequential metabolic enzymes (1–3). Many advantages for metabolic multienzyme complexes have been proposed in the literature. The first is kinetic in that these complexes increase metabolic flux by increasing the local concentration of a substrate/intermediate or reducing the diffusion process of intermediate substrates. In addition to the kinetic advantage, colocalization of metabolic proteins may protect or stabilize chemically unstable intermediates and separate the intermediates from competing chemical and enzymatic reactions (4–7). The idea of increasing catalytic efficiency has been verified by in vitro experiments, such as coimmobilized glycolysis multienzymes, a three-enzyme system for oxaloacetate production, and a cAMP degradation multienzyme sequence (8–10).
Our studies based on live-cell fluorescence microscopy have shown the colocalization of these six enzymes into clusters, referred to as purinosomes, under purine-depleted (P−) conditions. A hypothesis was proposed that purinosomes form to satisfy the cellular demand for purines through increasing de novo purine biosynthesis (11). Additional studies surrounding purinosome composition, characterization, and regulation have been conducted (12–16). However, alteration of intracellular purine nucleotide pools in purinosome-containing cells, specifically whether de novo synthesis flux of the individual purines, such as IMP, AMP, and GMP, is increased, has never been reported.
Purine nucleotide profiling is an analytical challenge because of their high polarity, instability, and chemical similarities under traditional reverse phase methodologies (17). Here we applied hydrophilic interaction liquid chromatography-based ultra high pressure liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI-MS/MS) to specifically quantify each individual cellular purine nucleotide in purinosome-rich HeLa cells (cultured in P− medium) and in normal HeLa cells (cultured in non-dialyzed FBS-based purine-rich (P+) medium), including IMP, the product of the de novo pathway, and two direct products of subsequent branch pathways, AMP and GMP, as well as their corresponding phosphorylated species: ADP, ATP, GDP, and GTP. The observed differences in nucleotide cellular concentrations between purinosome-rich cells and normal cells were further confirmed by 1H NMR global metabolite scanning. Previous studies have measured the metabolic rate of the de novo pathway in CHO fibroblast cell lines in purine-free medium by the incorporation of [14C]glycine in all cellular purines (18, 19). Using the same method, our laboratory reported that the de novo biosynthesis rate of total purines is about 42% greater in purine-depleted medium when compared with purine-rich medium (13). However, the pool of purines inside a cell contains many different species, such as nucleotides, nucleosides, and free bases. Purine nucleotides are products of multiple biological processes: de novo biosynthesis, purine nucleotide cycle and salvage pathway recycling, degradation, and urinary excretion. To determine the metabolic rate of the de novo pathway more accurately, the incorporation of [15N]glycine into IMP, AMP, and GMP, respectively, through the de novo pathway was measured by super resolution mass spectrometry with an LC-Orbitrap in this study.
EXPERIMENTAL PROCEDURES
Cell Culture and Harvest
Cell culture followed the same protocol published before (11). Briefly, cells were cultured in a humidified atmosphere 5% CO2 incubator at 37 °C. For cells cultured in purine-depleted medium, all cells were regularly maintained in RPMI 1640 (Mediatech, Inc.) supplemented with 10% dialyzed FBS (Atlanta Biologicals) for three passages (about 1 week) before harvesting. FBS was dialyzed against 0.9% NaCl at 4 °C for 2 days using a 25,000 molecular weight cut off dialysis membrane to remove purines. Cells in the [15N]glycine incorporation experiment were maintained in minimum essential medium Eagle (Mediatech, Inc.) supplemented with 10% FBS or 10% dialyzed FBS, respectively. Minimum essential medium Eagle does not contain glycine. Cells were grown in 100 × 20-mm tissue culture dishes (Falcon). The cell number per culture for both P+ and P− media is ∼2 × 106. Cells were harvested by 0.25% trypsin/2.21 mm EDTA solution (Mediatech, Inc.) and washed by PBS three times. Any metabolic activity of HeLa cells was quenched by adding 300 μl of cold methanol (preincubated at −80 °C) into the cell pellet. Cells could be kept in the −80 °C freezer before the next step.
Immunofluorescence Imaging of Fixed Cells
A total of 2 × 105 HeLa cells grown in purine-rich and purine-depleted media were plated on a 35-mm2 glass bottom plate (MatTek) and incubated at 37 °C (5% CO2) for 16 h. Adherent cells were fixed with freshly prepared 4% (v/v) formaldehyde (Electron Microscopy Sciences) prepared in 1× Dulbecco's PBS (Corning) for 10 min in the dark at room temperature and permeabilized with 0.2% Triton X-100 prepared in 1× Dulbecco's PBS for 10 min at room temperature. Cells were blocked with 5% normal goat serum (Jackson ImmunoResearch Laboratories) for 30 min at room temperature. Primary antibodies for phosphoribosyl pyrophosphate amidotransferase (PPAT) (LifeSpan BioSciences, catalogue number LS-C173822) and formylglycinamidine ribonucleotide synthase (FGAMS) (Bethyl Laboratories; catalogue number A304-218A) were directly labeled using Lightning-Link antibody conjugation kits (Novus Biologics) with Atto488 and Cy3, respectively. Cells were stained with a 1:100 dilution of Atto488-labeled PPAT antibody and a 1:250 dilution of Cy3-labeled FGAMS antibody for 2 h at room temperature in the dark. All antibody dilutions were performed in 1× Dulbecco's PBS. Cells were washed six times with 1 ml of 1× Dulbecco's PBS and placed into Hanks' balanced salt solution prior to imaging. Imaging was performed with a 60× Nikon Apo TIRF oil immersion objective (Nikon) using a S484/15x excitation filter and a S517/30m emission filter for Atto488 detection and a S555/25x excitation filter and a S605/40m emission filter for Cy3 detection (Chroma Technology).
Metabolite Extraction for LC-MS
The metabolites were extracted according to a procedure slightly modified from previous references (20, 21). Basically, collected HeLa cells were added with 500 μl of 80% methanol (20% water) solvent and lysed by a freeze-thaw cycle repeated three times. Then the mixture was centrifuged at 5700 × g, 4 °C for 15 min, and the supernatant was collected. For an additional extraction, 400 μl of 80% methanol (20% water) solvent were added to the cell pellet followed by centrifugation at 5700 × g, 4 °C for 5 min. This extraction was repeated twice. All the supernatants were combined and centrifuged at 7000 × g for at least 20 min to remove the insoluble material.
Ultra High Pressure Liquid Chromatography Coupled with Electrospray Ionization Tandem Mass Spectrometry (UPLC-ESI-MS/MS)
Chromatographic separation of nucleotides from HeLa cell lysates was achieved with hydrophilic interaction liquid chromatography using an ACQUITY BEH amide (2.1 × 100 mm, 1.7-μm particle size) column (Waters Corp, Milford, MA) thermostated at 40 °C. A mobile phase gradient composed of 10 (component A) and 90% (component B) aqueous acetonitrile containing 10 mm ammonium acetate (pH = 9.0) was delivered at a flow rate of 200 μl/min using an ACQUITY UPLC system (Waters Corp). Metabolites were eluted by increasing the percentage of component B in the solvent system from 1 to 40% (v/v) over 12 min. Mass spectrometry-based targeted profiling of nucleotides was performed on a Waters TQ-S triple quadrupole system operating in positive ionization mode with capillary and cone voltages set at 3200 and 30 V, respectively. Source and desolvation temperatures were maintained at 150 and 250 °C, respectively. Nitrogen was used as both cone gas (150 liters/h) and desolvation gas (800 liters/h), while argon (0.15 ml/min) was used as the collision gas.
1H NMR Sample Preparation
All of the frozen cell samples (with wet weight of 80 mg) were homogenized in 600 μl of ice-cold methanol/H2O (2:1 v/v) extraction buffer followed by three freeze-thaw cycles with rapid and vigorous manual mixing after each cycle. The samples were extracted using the Precellys tissue homogenizer (Bertin Technologies, Rockville, MD) followed by sonification on wet ice for 10 min. The supernatant was collected by centrifugation for 10 min at 18,100 × g and 4 °C. The remaining solid residues were further extracted twice using the same procedure. The combined supernatants from the three extractions were dried after removing methanol in vacuo. Each of the aqueous cell extracts was respectively reconstituted in 600 μl of phosphate buffer (0.1 m K2HPO4/NaH2PO4, pH 7.4) containing 50% D2O and 0.005% 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionic acid as chemical shift reference (δ 0.00 ppm). After vortexing and centrifugation (18,100 × g, 10 min, 4 °C), a total of 550 μl of the supernatants was pipetted into 5-mm NMR tubes for NMR analysis.
1H NMR Spectroscopy
1H NMR spectra of cell extracts were recorded at 298 K on a Bruker Avance III 600-MHz spectrometer, operating at 600.13 MHz for 1H, equipped with a Bruker inverse detection cryogenic probe (Bruker, Biospin, Germany). For all the samples, a standard water-suppressed one-dimensional NMR spectrum was recorded using the first increment of the NOESY pulse sequence (recycle delay-90°-t1-90°-tm-90°-acquisition) with a recycle delay of 2 s, t1 of 3 μs, and a mixing time, tm, of 100 ms. Typically, the 90° pulse length was adjusted to ∼10 μs and 64 transients were collected into 32,000 data points for each spectrum with a spectral width of 20 ppm. For the purpose of NMR signal assignments, a range of two-dimensional NMR spectra including 1H-1H COSY and total correlation spectroscopy (TOCSY), 1H-13C heteronuclear single quantum correlation (HSQC), and HMBC were acquired and processed for selective samples.
1H NMR Data Processing and Multivariate Data Analysis
All the 1H NMR spectra were manually corrected for phase and baseline distortions using TOPSPIN (version 3.0, Bruker Biospin) and referenced to the 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionic acid signal (δ 0.0). The spectral region δ 0.5–9.5 was automatically integrated into regions with equal widths of 0.004 ppm (2.4 Hz) using the AMIX software package (V3.8, Bruker Biospin). The region δ 4.7–5.2 was removed to avoid the effects of imperfect water suppression. Each integral region was normalized to the total sum of all integrals for each spectrum to compensate for the overall concentration differences prior to statistical data analysis.
The multivariate data analyses were carried out using the SIMCAP+ software package (version 11.0, Umetrics). Principal component analysis was performed for each treatment to generate an overview of the data distribution (e.g. clustering and outliers). A supervised multivariate data analytical tool, the orthogonal projection to latent structures discriminant analysis (OPLS-DA), was subsequently applied to the analysis of 1H NMR spectral data scaled to unit variance. The OPLS-DA models were validated using a 7-fold cross validation method, and the quality of the model was described by the parameters R2X, representing the total explained variations, and Q2, indicating the model predictability related to its statistical validity. The validity of all models was additionally ensured with coefficient of variation analysis of variance approach (p < 0.05) and permutation tests (200 permutations). The model was interpreted by back-scale transformed loadings with incorporated color-coded correlation coefficients of the metabolites responsible for the differentiation. The color plot was obtained with version 7.1 of the MATLAB (The MathWorks Inc., Natick, MA) environment using an in-house developed script. The color of correlation coefficient indicates the importance of the metabolite contributing to the differentiation between classes, with a “hot” color (e.g. red) indicating a higher importance than a “cold” color (e.g. blue). In this study, a correlation coefficient of |r| > 0.602 (i.e. r > 0.602 or r <−0.602) was used as the threshold value for the statistical significance based on the discrimination significance at the level of p < 0.05.
HPLC-MS
Samples were analyzed by HPLC-MS using a modified version of an ion pairing reversed phase negative ion electrospray ionization method (22). Samples were separated on a Supelco Titan C18 column (100 × 2.1 mm, 1.9-μm particle size) (Bellefonte, PA) using a water-methanol gradient with tributylamine added to the aqueous mobile phase. The LC-MS platform consisted of a Dionex Ultimate 3000 quaternary HPLC pump, a Dionex 3000 column compartment, a Dionex 3000 autosampler, and an Exactive plus orbitrap mass spectrometer controlled by Xcalibur 2.2 software (all from Thermo Fisher Scientific). The HPLC column was maintained at 30 °C and a flow rate of 200 μl/min. Solvent A was 3% aqueous methanol with 10 mm tributylamine and 15 mm acetic acid; solvent B was methanol. The gradient was 0 min, 0% B; 5 min, 20% B; 7.5 min, 20% B; 13 min, 55% B; 15.5 min, 95% B, 18.5 min, 95% B; 19 min, 0% B; 25 min 0% B. The orbitrap was operated in negative ion mode at maximum resolution (140,000) and scanned from m/z 85 to m/z 1000. The integrated areas of the reconstructed ion chromatograms for the monoisotopic and 15N isotopologues for the [M-H]− molecular ion were measured to calculate the 15N incorporation.
Cloning of Plasmids
Adenylosuccinate synthase (ADSS) (Clone ID HsCD00295729) and inosine monophosphate dehydrogenase 2/IMPDH2 (Clone ID Hscd00042407) template cDNA were obtained from the Arizona State University Biodesign Institute plasmid repository (DNASU). Plasmids containing target gene were amplified by PCR using Pfu DNA polymerase (Stratagene). Restriction sites were used in primers: NheI and EcoRI for ADSS and NheI and SacI for IMPDH2. The PCR products were ligated into the modified pmEGFP-N1 vector as described before (11). Plasmids were transformed into XL1-Blue competent cells for propagation and isolated by the QIAprep spin miniprep kit (Qiagen).
Transfection and Live-cell Fluorescence Imaging
HeLa cells were maintained in purine-rich or purine-depleted media as described previously (11). Cells were transiently transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Prior to imaging, all samples were washed three times for 5 min with balanced salt solution (20 mm HEPES (pH 7.4), 135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, and 5.6 mm glucose). Cells were imaged at ambient temperature (∼25 °C) on a Nikon TE-2000E inverted fluorescence microscope with a 60× Nikon Apo TIRF oil immersion objective using an S484/15x excitation filter and S517/30m emission filter for GFP and an S555/25x excitation filter and an S605/40m emission filter for OFP. The degree of colocalization of two enzymes in each merge image was determined by Pearson's linear correlation coefficient by ImageJ Colocalization Indices plugins. All images were created using the ImageJ program and were in some cases cropped, inverted, or shown in color for clarity, but were otherwise unmodified.
Western Blot
Cells cultured under purine-rich and purine-depleted media were lysed by M-PER mammalian protein extraction reagent (Thermo Scientific). Primary antibodies for six pathway enzymes were purchased from either Abcam (Cambridge, MA) or LifeSpan Bioscience (Seattle, WA). Quantification of protein expression levels from Western blots was carried out using a Bio-Rad ChemiDoc XRS+system (Bio-Rad). For each enzyme, three independent blots containing samples from P+ medium and P− medium were repeated. Student's t test (n = 3) was done to determine the statistical significance between two culture conditions.
RESULTS
Purinosome-rich HeLa Cells Have a Higher Level of IMP Measured by Targeted UPLC-ESI-MS/MS
To elucidate the effect of purinosome formation on the cellular purine nucleotide levels, a multiple reaction monitoring mass spectrometry-based method capable of measuring 10 well known cellular purine nucleotides simultaneously was developed. Changes in the purine nucleotide pools between cells cultured under the purine-depleted condition and normal cells were quantified (Table 1). Purine depletion was achieved through extensive dialysis of FBS and culturing cells in the growth medium with dialyzed FBS for 168 h (1 week). Purinosome formation by the endogenous enzymes under a purine-depleted condition was further established by the colocalization of two pathway enzymes, PPAT and FGAMS, in the immunostaining image (Fig. 1a). As shown in Table 1, ATP is the most abundant compound in both growth conditions (2.33 nmol/million cells in the purine-rich condition and 2.74 nmol/million cells in the purine-depleted condition) followed by ADP, AMP, GTP, GDP, and GMP. These results are consistent with published normal cellular purine nucleotide levels (23, 24). The ITP level is influenced by the ITP futile cycle and the activity of ATP deaminase (25, 26) and is higher than GTP and nearly half of that of ATP. Unfortunately, additional data on ITP cellular levels were not found. The precursors to ATP and GTP, IMP, XMP, and AMPS are in lower abundance relative to ATP and GTP. By comparison, the levels of all metabolites analyzed in purinosome-rich cells were relatively higher than the normally cultured cells. To validate the purine concentration changes between the two conditions, statistical analysis of the 12 cell samples tested was performed. The data showed the IMP concentration to be 2.8-fold higher under purine-depleted conditions than that of purine-rich conditions (p < 0.001). AMP and GMP are slightly higher (p > 0.05), as are ADP and GDP levels. In addition, the average ratio of ATP to ADP remains constant for the two groups (1.96 in purine-rich condition, 1.98 in purine-depleted condition), and the average ratio of AMP to GMP has only an 8% difference (8.50 in purine-rich condition, 7.82 in purine-depleted condition) (Fig. 1b), suggesting that the cellular energy status as determined by the ATP/ADP ratio and the intracellular balance of adenine and guanine nucleotides does not change for cells under the purine-depleted conditions.
TABLE 1.
Concentration of purine nucleotides in HeLa cells under purine-rich (P+) and purine-depleted (P−) conditions, determined by UPLC-ESI-MS/MS
Data are shown as mean ± S.D. Asterisks indicate the statistical difference between two culture conditions: ***, p < 0.001 (Student's t test) n = 12.
| P+ | P− | Ratio of P−/P+ | |
|---|---|---|---|
| nmol/million cells | nmol/million cells | ||
| AMP | 0.17 ± 0.03 | 0.23 ± 0.07 | 1.4 |
| ADP | 1.19 ± 0.19 | 1.40 ± 0.43 | 1.2 |
| ATP | 2.33 ± 0.44 | 2.74 ± 0.87 | 1.2 |
| GMP | 0.02 ± 0.01 | 0.03 ± 0.01 | 1.5 |
| GDP | 0.28 ± 0.03 | 0.35 ± 0.05 | 1.3 |
| GTP | 0.72 ± 0.11 | 0.88 ± 0.16 | 1.2 |
| IMP | 0.02 ± 0.01 | 0.06 ± 0.03 | 3.0*** |
| ITP | 1.05 ± 0.11 | 1.25 ± 0.22 | 1.2 |
| XMP | 0.05 ± 0.01 | 0.08 ± 0.01 | 1.6 |
| AMPS | 0.11 ± 0.01 | 0.14 ± 0.01 | 1.3 |
FIGURE 1.

a, immunofluorescence imaging of endogenous PPAT and FGAMS colocalization in fixed HeLa cells. HeLa cells cultured in purine-depleted growth media were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100 prior to being stained with Atto488-labeled PPAT and Cy3-labeled FGAMS antibodies. The degree of colocalization of PPAT and FGAMS was determined by Pearson's linear correlation coefficient: 0.93. Scale bar: 10 μm. b, the ratios of cellular concentrations of ATP/ADP and AMP/GMP remain the same between the two culture conditions. c, the IMP level of HeLa cells treated with DMAT is about 1.5 times higher than normal HeLa cells. *, p < 0.01 (Student's t test) n = 10. Data are presented as the mean ± S.D.
The formation of purinosomes can also be induced by other factors beyond purine depletion. For example, a previous study showed that treatment by DMAT, a casein kinase-2 (CK2) inhibitor, in HeLa cells induces purinosome formation (12, 14). To test whether DMAT treatment also enhances the cellular level of IMP, HeLa cells were cultured in purine-rich medium in the presence and absence of DMAT (10 μm) for 2 h followed by application of the same MS measurement to these cells. As shown in Fig. 1c, a 1.5-fold higher IMP response was observed in HeLa cells treated with DMAT when compared with HeLa cells in the absence of DMAT (p < 0.01, n = 10).
Higher Levels of IMP and AMP in Purinosome-rich HeLa Cells Are Confirmed by 1H NMR Metabolite Scanning
To validate the results obtained from the UPLC-ESI-MS/MS measurements, 1H NMR spectroscopy combined with multivariate data analysis was used. A total of 48 metabolites were identified; the metabolic changes in HeLa cells cultured under purine-rich and purine-depleted conditions were analyzed (n = 10) by the orthogonal projection to latent structures discriminant analysis (OPLS-DA). The OPLS-DA scores plot showed clear distinctions between the metabolites in cells in the purine-rich group and cells in the purine-depleted group (data not shown). Differences of intracellular purine levels between the two conditions were shown in Fig. 2. When compared with cells cultured in the purine-rich medium, HeLa cells under purine starvation showed elevated levels of IMP and AMP. The downstream catabolic intermediates, adenosine, guanosine, and inosine, were also higher in the purine-depleted condition. In addition, a lower level of hypoxanthine was observed inside cells under purine-depleted conditions.
FIGURE 2.
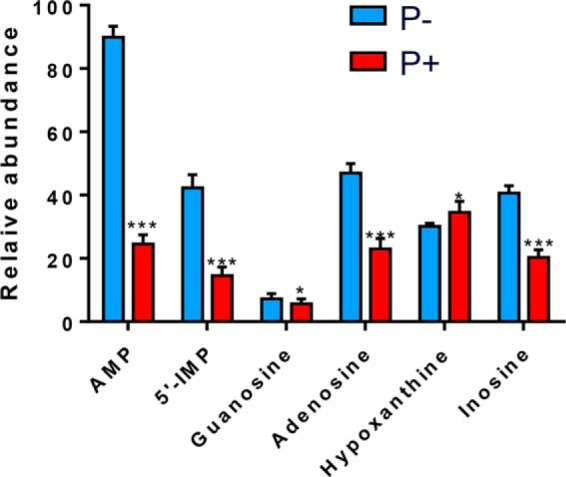
Relative abundance of intercellular purines in 1H NMR metabolite scanning (AMP, 5′-IMP, guanosine, adenosine, hypoxanthine, and inosine) in HeLa cells cultured in purine-depleted medium (shown in blue) and purine-rich medium (shown in red). Data are presented as the mean ± S.D. *, p < 0.05, **, p < 0.01, ***, p < 0.001 (2-tailed Student's t test), n = 10.
The Initial Rate of [15N]Glycine Incorporation into IMP Is Higher in Purinosome-rich HeLa Cells
IMP is the product of de novo purine biosynthesis and the precursor for AMP and GMP biosynthesis. Higher cellular IMP levels were observed in purinosome-rich cells in our test. There are many mechanisms to generate IMP inside the cell (Fig. 3). To find out whether de novo biosynthesis contributes to the increased IMP level, a stable isotope labeling assay was applied. HeLa cells were pulsed with [15N]glycine, a molecule incorporated into the purine base ring by the enzyme glycinamide ribonucleotide synthetase (GART) in step 2 of de novo purine biosynthesis. The incorporation of a heavy nitrogen into IMP molecules was measured over time and calculated using the amount of [15N]IMP divided by the amount of monoisotopic molecules (15N-12C and 14N-12C), and the flux was estimated by calculating the initial incorporation rate of 15N into IMP. To begin, a series of starvation experiments was performed to investigate how different starvation times affect glycine incorporation. In these experiments, cells were maintained in purine-depleted medium for 24, 72, and 168 h prior to incubation with [15N]glycine. The result showed more 15N incorporation into IMP at longer starvation times. Therefore, the incorporation rate of IMP was measured for cells grown under normal condition (purine-rich medium) and cells having been starved for 168 h in purine-depleted medium. Fig. 4a shows 15N incorporation as a function of time. Incorporation levels for the two conditions were the same after about 1.5 h. To calculate the rate, the incorporation within 1 h was fitted to a linear function. The initial incorporation rate in purine-depleted medium is 12.5% per hour when compared with 8.5% per hour for cells under normal culturing condition, which amounts to about a 50% increase in the rate for purine-depleted cells.
FIGURE 3.
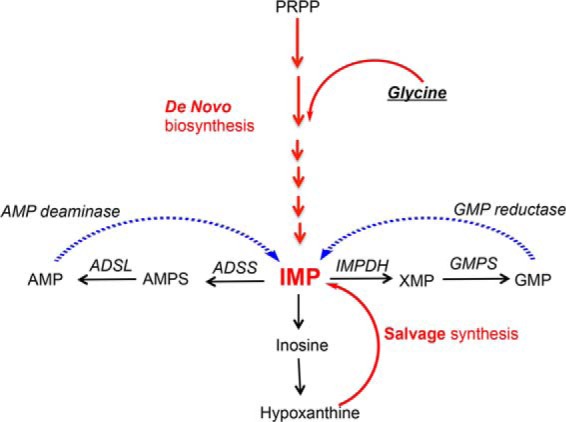
Simplified IMP biosynthesis and catabolic pathways in humans. IMP can be synthesized from PRPP by the de novo purine biosynthesis pathway or made from hypoxanthine by the salvage pathway. IMP can be further transformed into AMP and GMP or recycled back by AMP deaminase and GMP reductase. ADSL, adenylosuccinate lyase; GMPS, GMP synthase.
FIGURE 4.

Incorporation of [15N]glycine into purines by the de novo biosynthesis pathway. a, the incorporation of [15N]glycine into IMP. The incorporation was plotted using percentage of 15N incorporated purines as a function of time of incubation. HeLa cells cultured in P− media (in blue) showed a faster incorporation rate than cells in P+ media (in red) within 1.5 h. The initial incorporation rate for IMP, AMP, and GMP was calculated according to the slope of the linear relationship of the incorporation plot within 1 h. The initial rate of cells in purine-depleted condition was increased by ∼47% for IMP biosynthesis. b, the incorporation of [15N]glycine into AMP. c, the incorporation of [15N]glycine into GMP. Data are presented as the mean ± S.D.
Downstream de Novo Biosynthesis Enzymes, ADSS and IMPDH, Co-clustering with FGAMS inside the Purinosome
Similarly to IMP, increased 15N incorporation was observed for AMP and GMP in purine-depleted cells (Fig. 4, b and c). AMP and GMP are synthesized through two independent pathways downstream of IMP. IMP dehydrogenase (IMPDH) catalyzes the rate-limiting step in the GMP synthesis and ADSS in AMP synthesis (Fig. 3). As shown in the glycine incorporation experiment for cells cultured under purine depletion in the first hour, the AMP synthesis rate is ∼70% greater; the GMP synthesis rate is about 20% higher. The initial increase in de novo synthesis of AMP and GMP to meet cellular demands for purines under purine-depleted conditions suggested that enzymes downstream of IMP dehydrogenase might be involved in purinosome assembly. To verify the involvement of the downstream pathway in purinosome formation, fluorescence imaging studies were performed to investigate colocalization of IMPDH and ADSS with the purinosome marker FGAMS in transiently transfected HeLa cells. Both IMPDH and ADSS co-clustered with FGAMS in HeLa cells in purine-depleted medium as shown in the images (Fig. 5, a–f). In addition, IMPDH formed a filament structure under starvation conditions, which has been previously reported by other groups (27, 28). We found that purinosome bodies and filament structures are different phenomena, and they are not colocalized with each other inside the cytoplasm (Fig. 5, g and h).
FIGURE 5.

Colocalization of transiently transfected ADSS-GFP or IMPDH-GFP with FGAMS-OFP in HeLa cells grown in purine-depleted media and formation of IMPDH filaments (rods) in HeLa cells. a–f, ADSS-GFP (b) and IMPDH-GFP (e) were co-transfected with FGAMS-OFP (a and d); c and f are merged images. The degree of colocalization was determined by the Pearson's linear correlation coefficient: 0.88 (ADSS and FGAMS) and 0.82 (IMPDH and FGAMS). g, IMPDH-GFP transiently transfected HeLa cells showing the filament or rod structures, as indicated by white arrows. h, the filament macrostructure formed in the right cell (white arrows) is different from the purinosome spots (red arrows) in the left cell. All images were taken on the Nikon TE-2000E inverted fluorescence microscope. Scale bar, 10 μm.
The Expression Levels of Core Enzyme Members Do Not Change When Cultured in the Purine-depleted Medium
The MS and NMR metabolomic studies indicate activation of the de novo purine biosynthesis pathway in purinosome-rich HeLa cells. To find out whether pathway enzyme expression levels are up-regulated to meet the requirement for the activation of the pathway, we used Western blots to compare the endogenous expression of each enzyme in the pathway between the two conditions studied (Fig. 6). No significant difference in expression levels for five out of six enzymes was observed, except for the enzyme that catalyzes the last two steps in the pathway: aminoimidazole carboxamide ribonucleotide transformylase-IMP cyclohydrolase (ATIC). According to the statistical analysis of different Western blots, ATIC expression level in cells under purine-depleted medium is 1.7-fold higher when compared with purine-rich condition (p < 0.05); all others are statistically the same.
FIGURE 6.

Western blot analysis of enzymes in the purine de novo biosynthesis pathway in HeLa cells. Quantification of protein expression levels from three independent blots was determined by calculating the optical density of the target blot. There was no difference in levels for five out of six enzymes between cells cultured in P+ and P− media. ATIC in cells under P− condition has higher expression according to the Student's t test between P+ and P− conditions (p < 0.05). GAPDH or β-actin was used as the loading control. PAICS, carboxyaminoimidazole ribonucleotide synthase-succinyl aminoimidazole carboxamide ribonucleotide synthetase; TriGART, trifunctional GARS-GAR transformylase-aminoimidazole ribonucleotide synthetase; ASL, adenylosuccinate lyase.
DISCUSSION
In this study, we have shown an ∼3-fold difference in the static cellular IMP levels between purinosome-rich cells cultured in purine-depleted medium and normal HeLa cells. In addition, we treated cells with DMAT, a CK2 inhibitor that stimulates purinosome formation. By comparing cells treated with DMAT with normal cells, we again observed a higher level of IMP in purinosome-rich cells, comparable with cells cultured in purine-depleted medium. Furthermore, we have provided evidence that the rate of de novo biosynthesis of IMP is increased 50% when cells are cultured under purine-depleted condition. In addition, the live-cell fluorescence images showed that two rate-limiting enzymes, ADSS and IMPDH, which are members of downstream pathways leading to AMP/GMP, are also found in purinosome clusters. Collectively, these results support our assumption that purine metabolism is adjusted in purinosome-containing cells.
Intracellular IMP Level
Removing the purines from a culturing medium might be expected to result in a purine deficiency inside cells. However, the UPLC-ESI-MS/MS- and NMR-based metabolomics analyses of HeLa cells showed a significantly higher level of IMP in cells under purine starvation. In these experiments, HeLa cells were maintained in purine-depleted and purine-rich media for at least 1 week (three passages). Our result showed that the static cellular IMP level of cells cultured in purine-depleted medium is higher. IMP metabolism is complicated, and many pathways contribute to the final net concentration of IMP inside cells (Fig. 3). First of all, IMP is the precursor for the further biosynthesis of AMP and GMP and their ultimate conversion to ATP/GTP. Meanwhile, the GMP reductase returns GMP to IMP for conversion to AMP, to maintain the intracellular balance of adenine and guanine nucleotides. Additionally, IMP is regenerated from AMP in the purine nucleotide cycle by AMP deaminase, which is also a part of maintaining intracellular balance. As we have shown in Fig. 1, the ratio of AMP to GMP and ATP/ADP (as the cellular energy marker) is comparable in cells grown in either purine-rich or purine-depleted conditions. Therefore, neither the GMP reduction pathway nor the AMP deamination pathway appears to contribute to the higher level of IMP. Furthermore, a lower level of hypoxanthine in cells under purine-depleted conditions was observed in the 1H NMR study. It is possible that membrane-permeable purines leak out from the cells in the absence of extracellular purines. However, higher levels of adenosine, inosine, and guanosine in purine-depleted conditions were observed in this study. Therefore, it is most likely that hypoxanthine has been decreased by the salvage pathway in cells under purine-depleted conditions. In the absence of adequate purines (intra- and extracellular), the cellular salvage pathway through IMP to AMP/GMP is inadequate because hypoxanthine cannot be replenished effectively under purine-depleted condition. Consequently, the HeLa cells then have to rely heavily on the de novo biosynthesis pathway.
To find out the relative contribution of the de novo biosynthesis pathway to IMP levels in purinosome-containing cells, the de novo purine synthesis rate was measured by the incorporation of [15N]glycine into IMP in both non-dialyzed and purine-depleted conditions. In these experiments, an excess amount of heavy labeled glycine when compared with endogenous glycine was used to mitigate the influence of endogenous glycine levels on the initial incorporation rate. For the purine-depleted condition, HeLa cells were starved in the dialyzed FBS medium for 168 h before adding [15N]glycine into the medium. The initial incorporation rate of glycine into IMP synthesis is 50% higher in cells under purine-depleted conditions than purine-rich conditions. These results along with the higher IMP levels indicate that de novo purine biosynthesis is turned on in response to the purine starvation.
Purinosomes Form to Increase Purine de Novo Biosynthesis
It is uncertain as to how this pathway is activated. Western blots showed that the expression levels of five out of six pathway enzymes in cells cultured in purine-depleted medium are not differentially expressed with the exception of ATIC, which has been reported to also serve as a rate-limiting enzyme for purine biosynthesis to convert accumulated levels of aminoimidazole carboxamide ribonucleotide (AICAR) to IMP (29, 30). ATIC also is at the branch point of the whole pathway; a higher level of ATIC might direct the flux toward AMP and GMP. In addition, cells treated with DMAT to stimulate purinosome formation are shown to have higher cellular IMP levels than normal cells as well. Collectively, these results suggest that the increased production of cellular IMP can be correlated with purinosome formation. This conclusion is consistent with our hypothesis that purinosomes form to synthesize purines more effectively in response to purine starvation when cells are cultured in the purine-depleted medium. Meanwhile, events downstream of the IMP biosynthesis pathway were reflected in the [15N]glycine incorporation. According to previous studies, the rate-limiting steps for synthesis of GMP and AMP are catalyzed by IMPDH and ADSS, respectively. IMPDH has a Vmax = 5.58 μmol/min/mg, Km (IMP) = 36 μm (31), and ADSS has a Vmax = 0.2 μmole/min/mg, Km (IMP) = 37 μm (32, 33), so the partitioning to GMP relative to AMP should be ∼25 times faster. The AMP and GMP incorporation showed a similar trend; the ratio of 15N/14N of AMP did not reach equilibrium until 4 h after incorporation, whereas GMP reached equilibrium within 90 min (data not shown). These observations verified the accuracy of the method used in this study. Direct comparison of [15N]glycine incorporation rates between the two conditions studied showed increases in the production of AMP and GMP under purine-depleted conditions and in turn raised the possibility that enzymes involved in AMP and GMP de novo biosynthesis are also inside the purinosome. Indeed live-cell fluorescence microscopy experiments showed the colocalization of IMPDH-GFP and ADSS-GFP with FGAMS-OFP, confirming their involvement in the purinosome. Collectively, our results indicate that the purinosome is a functional complex formed to increase the de novo metabolic flux to complement the salvage pathway when purine demand is high, and moreover, contains all the enzymes to catalyze the 12 steps conversion of PRPP to AMP/GMP.
The purinosome appears to be an example of “enzyme clustering” to accelerate the processing of intermediates through metabolic channeling (34). The efficiency of such a system depends on the steady-state densities of the enzymes in the cluster, which in turn depends on the number and physical dimensions of the enzyme, their catalytic constants, and the diffusion coefficients of the proteins and substrates. In model calculations, simple clusters involving two enzymes (ours involves six) can easily achieve the observed 1.5-fold rate acceleration. The question is why do the clusters form? Two possibilities remain to be explored. The first is reversible posttranslational modification of one or more member of these enzymes; the second is substrate concentration-dependent increase in diffusion of the enzymes in the presence of their respective substrates to drive complex formation (Brownian ratchet) (35–37). Both are being studied.
Acknowledgments
We thank Dr. Bruce Stanley and Anne Stanley at Penn State Hershey College of Medicine core research facility for assistance with the initial measurement of purine metabolites.
This work was supported, in whole or in part, by National Institutes of Health Grants GM024129 (to S. J. B.) and ES022186 (to A. D. P.).
This article was selected as a Paper of the Week.
- PRPP
- 5-phospho-α-d-ribose 1-diphosphate
- P−
- purine-depleted
- P+
- purine-rich
- UPLC-ESI-MS/MS
- ultra high pressure liquid chromatography coupled with electrospray ionization tandem mass spectrometry
- PPAT
- phosphoribosyl pyrophosphate amidotransferase
- FGAMS
- formylglycinamidine ribonucleotide synthase
- DMAT
- 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole
- OPLS-DA
- the orthogonal projection to latent structures discriminant analysis
- IMPDH
- inosine monophosphate dehydrogenase
- ADSS
- adenylosuccinate synthase
- OFP
- orange fluorescence protein
- ATIC
- aminoimidazole carboxamide ribonucleotide transformylase-IMP cyclohydrolase.
REFERENCES
- 1. Rowe P. B., McCairns E., Madsen G., Sauer D., Elliott H. (1978) De novo purine synthesis in avian liver: co-purification of the enzymes and properties of the pathway. J. Biol. Chem. 253, 7711–7721 [PubMed] [Google Scholar]
- 2. Rowe P. B., Wyngaarden J. B. (1968) Glutamine phosphoribosylpyrophosphate amidotransferase: purification, substructure, amino acid composition, and absorption spectra. J. Biol. Chem. 243, 6373–6383 [PubMed] [Google Scholar]
- 3. Smith G. K., Mueller W. T., Wasserman G. F., Taylor W. D., Benkovic S. J. (1980) Characterization of the enzyme complex involving the folate-requiring enzymes of de novo purine biosynthesis. Biochemistry 19, 4313–4321 [DOI] [PubMed] [Google Scholar]
- 4. Wombacher H. (1983) Molecular compartmentation by enzyme cluster formation: a view over current investigations. Mol. Cell. Biochem. 56, 155–164 [DOI] [PubMed] [Google Scholar]
- 5. Srere P. A. (1987) Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56, 89–124 [DOI] [PubMed] [Google Scholar]
- 6. Ovádi J. (1991) Physiological significance of metabolic channelling. J. Theor. Biol. 152, 1–22 [PubMed] [Google Scholar]
- 7. Mathews C. K. (1993) The cell: bag of enzymes or network of channels? J. Bacteriol. 175, 6377–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Srere P. A., Mattiasson B., Mosbach K. (1973) An immobilized three-enzyme system: a model for microenvironmental compartmentation in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 70, 2534–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Luca M., Kricka L. J. (1983) Coimmobilized multienzymes: an in vitro model for cellular processes. Arch. Biochem. Biophys. 226, 285–291 [DOI] [PubMed] [Google Scholar]
- 10. Wombacher H. (1980) Evidence for a membrane-bound multienzyme sequence degrading cyclic adenosine 3′:5′-monophosphate. Arch. Biochem. Biophys. 201, 8–19 [DOI] [PubMed] [Google Scholar]
- 11. An S., Kumar R., Sheets E. D., Benkovic S. J. (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103–106 [DOI] [PubMed] [Google Scholar]
- 12. An S., Kyoung M., Allen J. J., Shokat K. M., Benkovic S. J. (2010) Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. J. Biol. Chem. 285, 11093–11099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. An S., Deng Y., Tomsho J. W., Kyoung M., Benkovic S. J. (2010) Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc. Natl. Acad. Sci. U.S.A. 107, 12872–12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verrier F., An S., Ferrie A. M., Sun H., Kyoung M., Deng H., Fang Y., Benkovic S. J. (2011) GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat. Chem. Biol. 7, 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deng Y., Gam J., French J. B., Zhao H., An S., Benkovic S. J. (2012) Mapping protein-protein proximity in the purinosome. J. Biol. Chem. 287, 36201–36207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. French J. B., Zhao H., An S., Niessen S., Deng Y., Cravatt B. F., Benkovic S. J. (2013) Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. U.S.A. 110, 2528–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cordell R. L., Hill S. J., Ortori C. A., Barrett D. A. (2008) Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 871, 115–124 [DOI] [PubMed] [Google Scholar]
- 18. Yamaoka T., Kondo M., Honda S., Iwahana H., Moritani M., Ii S., Yoshimoto K., Itakura M. (1997) Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 272, 17719–17725 [DOI] [PubMed] [Google Scholar]
- 19. Yamaoka T., Yano M., Kondo M., Sasaki H., Hino S., Katashima R., Moritani M., Itakura M. (2001) Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J. Biol. Chem. 276, 21285–21291 [DOI] [PubMed] [Google Scholar]
- 20. Danielsson A. P., Moritz T., Mulder H., Spégel P. (2010) Development and optimization of a metabolomic method for analysis of adherent cell cultures. Anal. Biochem. 404, 30–39 [DOI] [PubMed] [Google Scholar]
- 21. Dettmer K., Nürnberger N., Kaspar H., Gruber M. A., Almstetter M. F., Oefner P. J. (2011) Metabolite extraction from adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 399, 1127–1139 [DOI] [PubMed] [Google Scholar]
- 22. Lu W., Clasquin M. F., Melamud E., Amador-Noguez D., Caudy A. A., Rabinowitz J. D. (2010) Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 24. Pabst M., Grass J., Fischl R., Léonard R., Jin C., Hinterkörner G., Borth N., Altmann F. (2010) Nucleotide and nucleotide sugar analysis by liquid chromatography-electrospray ionization-mass spectrometry on surface-conditioned porous graphitic carbon. Anal. Chem. 82, 9782–9788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zamzami M. A., Duley J. A., Price G. R., Venter D. J., Yarham J. W., Taylor R. W., Catley L. P., Florin T. H., Marinaki A. M., Bowling F. (2013) Inosine triphosphate pyrophosphohydrolase (ITPA) polymorphic sequence variants in adult hematological malignancy patients and possible association with mitochondrial DNA defects. J. Hematol. Oncol. 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whittam R. (1960) The high permeability of human red cells to adenine and hypoxanthine and their ribosides. J. Physiol. 154, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gunter J. H., Thomas E. C., Lengefeld N., Kruger S. J., Worton L., Gardiner E. M., Jones A., Barnett N. L., Whitehead J. P. (2008) Characterisation of inosine monophosphate dehydrogenase expression during retinal development: differences between variants and isoforms. Int. J. Biochem. Cell Biol. 40, 1716–1728 [DOI] [PubMed] [Google Scholar]
- 28. Thomas E. C., Gunter J. H., Webster J. A., Schieber N. L., Oorschot V., Parton R. G., Whitehead J. P. (2012) Different characteristics and nucleotide binding properties of inosine monophosphate dehydrogenase (IMPDH) isoforms. PLoS One 7, e51096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sidi Y., Mitchell B. S. (1985) Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J. Clin. Invest. 76, 2416–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang W., Fridman A., Blackledge W., Connelly S., Wilson I. A., Pilz R. B., Boss G. R. (2009) The phosphatidylinositol 3-kinase/Akt cassette regulates purine nucleotide synthesis. J. Biol. Chem. 284, 3521–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Labesse G., Alexandre T., Vaupré L., Salard-Arnaud I., Him J. L., Raynal B., Bron P., Munier-Lehmann H. (2013) MgATP regulates allostery and fiber formation in IMPDHs. Structure 21, 975–985 [DOI] [PubMed] [Google Scholar]
- 32. Sun H., Li N., Wang X., Chen T., Shi L., Zhang L., Wang J., Wan T., Cao X. (2005) Molecular cloning and characterization of a novel muscle adenylosuccinate synthetase, AdSSL1, from human bone marrow stromal cells. Mol. Cell. Biochem. 269, 85–94 [DOI] [PubMed] [Google Scholar]
- 33. Van der Weyden M. B., Kelly W. N. (1974) Human adenylosuccinate synthetase. Partial purification, kinetic and regulatory properties of the enzyme from placenta. J. Biol. Chem. 249, 7282–7289 [PubMed] [Google Scholar]
- 34. Castellana M., Wilson M. Z., Xu Y., Joshi P., Cristea I. M., Rabinowitz J. D., Gitai Z., Wingreen N. S. (2014) Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotechnol. 32, 1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muddana H. S., Sengupta S., Mallouk T. E., Sen A., Butler P. J. (2010) Substrate catalysis enhances single-enzyme diffusion. J. Am. Chem. Soc. 132, 2110–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sengupta S., Dey K. K., Muddana H. S., Tabouillot T., Ibele M. E., Butler P. J., Sen A. (2013) Enzyme molecules as nanomotors. J. Am. Chem. Soc. 135, 1406–1414 [DOI] [PubMed] [Google Scholar]
- 37. Sengupta S., Spiering M. M., Dey K. K., Duan W., Patra D., Butler P. J., Astumian R. D., Benkovic S. J., Sen A. (2014) DNA polymerase as a molecular motor and pump. ACS Nano 8, 2410–2418 [DOI] [PubMed] [Google Scholar]