

Background: PDE3A is part of a SERCA2 signaling complex in cardiac myocytes.
Results: PDE3, not PDE4, regulates the activation of SERCA2 by PKA in human myocardium; phosphorylation of PDE3A1 at Ser-292/Ser-293 promotes its integration into the SERCA2 signaling complex.
Conclusion: PDE3A1 regulates cAMP-mediated control of SERCA2 through its phosphorylation-dependent interaction with SERCA2.
Significance: Targeted disruption of the PDE3A1-SERCA2 interaction may provide a new therapeutic approach for heart failure.
Keywords: A-kinase Anchoring Protein (AKAP), Cyclic AMP (cAMP), Immunohistochemistry, Protein Kinase A (PKA), Subcellular Fractionation, Cyclic Nucleotide Phosphodiesterase, PDE3A, Phospholamban, SERCA2
Abstract
Cyclic nucleotide phosphodiesterase 3A (PDE3) regulates cAMP-mediated signaling in the heart, and PDE3 inhibitors augment contractility in patients with heart failure. Studies in mice showed that PDE3A, not PDE3B, is the subfamily responsible for these inotropic effects and that murine PDE3A1 associates with sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2), phospholamban (PLB), and AKAP18 in a multiprotein signalosome in human sarcoplasmic reticulum (SR). Immunohistochemical staining demonstrated that PDE3A co-localizes in Z-bands of human cardiac myocytes with desmin, SERCA2, PLB, and AKAP18. In human SR fractions, cAMP increased PLB phosphorylation and SERCA2 activity; this was potentiated by PDE3 inhibition but not by PDE4 inhibition. During gel filtration chromatography of solubilized SR membranes, PDE3 activity was recovered in distinct high molecular weight (HMW) and low molecular weight (LMW) peaks. HMW peaks contained PDE3A1 and PDE3A2, whereas LMW peaks contained PDE3A1, PDE3A2, and PDE3A3. Western blotting showed that endogenous HMW PDE3A1 was the principal PKA-phosphorylated isoform. Phosphorylation of endogenous PDE3A by rPKAc increased cAMP-hydrolytic activity, correlated with shift of PDE3A from LMW to HMW peaks, and increased co-immunoprecipitation of SERCA2, cav3, PKA regulatory subunit (PKARII), PP2A, and AKAP18 with PDE3A. In experiments with recombinant proteins, phosphorylation of recombinant human PDE3A isoforms by recombinant PKA catalytic subunit increased co-immunoprecipitation with rSERCA2 and rat rAKAP18 (recombinant AKAP18). Deletion of the recombinant human PDE3A1/PDE3A2 N terminus blocked interactions with recombinant SERCA2. Serine-to-alanine substitutions identified Ser-292/Ser-293, a site unique to human PDE3A1, as the principal site regulating its interaction with SERCA2. These results indicate that phosphorylation of human PDE3A1 at a PKA site in its unique N-terminal extension promotes its incorporation into SERCA2/AKAP18 signalosomes, where it regulates a discrete cAMP pool that controls contractility by modulating phosphorylation-dependent protein-protein interactions, PLB phosphorylation, and SERCA2 activity.
Introduction
Enzymes in the cyclic nucleotide phosphodiesterase 3 (PDE3)2 family of cyclic nucleotide phosphodiesterases have an important role in regulating cAMP-mediated signaling in cardiac myocytes. Inhibitors of these enzymes are used to increase the force of contraction in patients with heart failure. The observation that inotropic responses to PDE3 inhibition are preserved in PDE3B-KO mice but not in PDE3A-KO mice indicates that PDE3A isoforms are specifically involved in the regulation of contractility (1, 2). Furthermore, in mouse myocardium, the PDE3A1 isoform was found to be a component of a SERCA2-PLB-AKAP18 multiprotein complex or “signalosome” that regulates the transport of cytoplasmic Ca2+ into the sarcoplasmic reticulum (2–3). By potentiating cAMP-dependent phosphorylation of phospholamban in this signalosome, PDE3 inhibition stimulates Ca2+ uptake and increases the amplitude of intracellular Ca2+ cycling. In renal collecting duct principal cells, incorporation of PDE4D into a signalosome containing AKAP18δ and aquaporin-2 (AQP2) plays an important role in cAMP/PKA-mediated insertion of the AQP2 water channel into the cell membrane and regulation of vasopressin-induced water reabsorption (4).
Many of the interactions of PDE3 isoforms with other proteins have been shown to be dependent upon their phosphorylation. This has been well established in the case of PDE3B (5–12). Some of the protein-protein interactions of PDE3A isoforms have also been shown to be phosphorylation-dependent, including those with 14-3-3 (13–15). More recently, it has become clear that different isoforms of PDE3A, whose amino acid sequences are identical except for varying lengths of N-terminal sequence, differ significantly with respect to these interactions (15). PDE3A1 is found exclusively in intracellular membranes of cardiac myocytes and is preferentially phosphorylated at the 14-3-3-binding site Ser-312 in response to PKA activation, whereas PDE3A2, which lacks the N-terminal 154 amino acid sequence of PDE3A1, is preferentially phosphorylated at the alternative 14-3-3-binding site Ser-428 in response to PKC activation (15). PKA activation and PKC activation were found to promote the association of PDE3A1 and PDE3A2 with distinct protein interactomes (15).
In the experiments described below, we identified components of the SERCA2-PLB-AKAP18 signalosome in human myocardium. We showed that PDE3, and not PDE4 (that has also been shown to interact with PLB in human cardiac myocytes (16)), regulates the phosphorylation of PLB and the stimulation of SERCA2 activity and Ca2+ transport by cAMP in human SR fractions. The integration of PDE3A1 into the SERCA2-regulatory signalosome involves direct interactions with both AKAP18 and SERCA2 and is phosphorylation-dependent. Interaction of PDE3A1 with SERCA2 is dependent upon the phosphorylation of Ser-292/Ser-293, a multikinase site within the unique N-terminal extension of PDE3A1.
MATERIALS AND METHODS
Use of human heart samples in these studies was approved by review committees at NHLBI, NIH, and the University of Utah.
Materials
[γ-32P]ATP (3000 Ci/mmol) was obtained from MP Biomedicals (Solon, OH); rPKAc (catalog #14-440) and rSERCA2 (catalog #H00000488-P01) were obtained from Calbiochem and Abnova, respectively. Anti-PLB (ab2865), -p-PLB (ab15000), -AKAP-LBC (ab56917), and -AKAP18 (ab30987) antibodies were from Abcam (Cambridge, MA). Antibodies to PP2A (610556), PKA regulatory subunit (PKARII; 610626), and PKAc (610981) were from BD Biosciences. Anti-PP1 antibody (sc7482) and anti-caveolin-3 antibody (sc5310) were from Santa Cruz Biotechnology; anti-SERCA2 antibody (MA3-910) was from Affinity Bioreagents. Anti-phospho-PKA substrate antibody (9621) was from Cell Signaling; anti-desmin antibody (M0760) was from Dako. Protein G magnetic beads (88848) were from Thermo Fisher Scientific (Rockford, IL); mouse anti-FLAG® M2 magnetic beads (M8823) and mouse anti-FLAG M2-peroxidase HRP antibody (A8592) were from Sigma; mouse anti-His mAb magnetic beads (L00275) were from Genscript. Anti-myomesin antibody (mMac myomesin B4) was obtained from Developmental Studies Hybridoma Bank (University of Iowa). Rabbit polyclonal antibody to human PDE3A (accession number AAA35912) was generated against peptide corresponding to amino acids 1127–1141 (GKPRGEEIPTQKPDQ) CT domain (C-terminal domain) and was affinity-purified as described below. For immunohistochemical studies, the secondary antibodies, Alexa Fluor 488 or Alexa Fluor 594, were from Molecular Probes. For SDS-PAGE and Western blots, HRP-labeled secondary antibody and SuperSignal® Westpico and Westfemto chemiluminescent reagents were from Thermo Fisher Scientific. Signals were detected with Image reader LAS3000 (GE Healthcare). Other materials were obtained as indicated.
Methods
Affinity Purification of Anti-human PDE3A-CT Antibody
Approximately 30 mg of PDE3A-CT peptide (GKPRGEEIPTQKPDQ) was coupled to 3 ml of Affi-Gel 10 (Bio-Rad) beads that were used to purify anti-PDE3A-CT rabbit polyclonal antibody. Immunized serum was diluted (10 times) with 10 mm Tris buffer, pH 7.5. Affi-Gel 10-PDE3A-CT-peptide beads and the diluted serum were rotated end-over-end for 16 h at 4 °C. After centrifugation, the immunobeads were washed (3 times, 20 bed volumes) with 10 mm Tris, pH 7.5. Anti-PDE3A antibody was eluted from the beads by adding 5 ml of elution buffer (100 mm glycine buffer, pH 2.5) and by rotating the mixture for 3 min. The eluted antibody was neutralized immediately with 1 m Tris, pH 7.5, and dialyzed at 4 °C against 1 liter of PBS overnight. Dialyzed antibody was reconstituted with 50% glycerol and kept at −80 °C. Affinity-purified anti-PDE3A-CT antibodies were used for all studies in this report.
Immunohistochemistry
Frozen heart blocks were prepared from snap-frozen normal human heart samples obtained from Capital Biosciences, Inc, and sections were made at a thickness of 10 μm using a microtome at −25 °C. Paraformaldehyde-fixed cryostat heart sections were washed in PBS (3 × 5 min) and blocked and permeabilized (6 h, 4 °C) in blocking buffer (PBS containing 10% donkey serum and 0.05% Triton X-100). Slides were incubated in blocking buffer with primary antibody overnight and washed with PBS (3 × 5 min) before incubating in blocking buffer for 2 h with the secondary antibodies, Alexa Fluor 488 or Alexa Fluor 594. Anti-PDE3A-CT antibody, generated against human PDE3A aa 1127–1141 (GKPRGEEIPTQKPDQ), was used in these studies. As controls, slides were incubated with nonimmune IgG or with primary anti-PDE3A-CT antibodies incubated with blocking (immunizing) peptide before staining with secondary antibody. For peptide blocking/competition experiments, ∼20 μg of anti-PDE3A-CT antibody was combined with 100 μg of blocking peptide in a small volume (500 μl) of PBS and incubated (2 h at room temperature or overnight at 4 °C). After blocking with the immunizing peptides, antibody/peptide mixtures were diluted into blocking buffer and used for staining of samples. Slides were viewed with a Zeiss LSM510 laser scanning confocal microscope.
Preparation of Subcellular Fractions of Human Myocardium
Human myocardium was obtained from the left ventricular free wall of explanted hearts from patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation (University of Utah). Normal human heart samples were also obtained from Capital Biosciences, Inc. (Rockville, MD). Heart tissues were quickly washed in ice-cold PBS, chopped with scissors, and homogenized (4 ml/g of tissue) at 4 °C in buffer A (0.29 m sucrose, 10 mm MOPS, 2 mm EGTA, Roche Applied Science protease inhibitor mixture, pH 7.0) with a rotor-stator homogenizer (Omni International, Marietta, GA) at 30,000 rpm (60–80 s on ice) followed by homogenization (on ice, 20 strokes in a glass Dounce homogenizer). Homogenates were centrifuged (11,000 × g, 15 min) in a Beckman JA-20 rotor. Supernatants were further centrifuged (150,000 × g, 1 h) in a Beckman 55.2 Ti rotor, yielding cytosolic fractions and pellets. Pellets were resuspended by hand homogenization (glass-glass) in two volumes (relative to starting material) of buffer A without EGTA. After recentrifugation (150,000 × g for 1 h), pellets, i.e. “myocardial membrane fractions,” were suspended in buffer A (without EGTA) using a Dounce homogenizer and stored at −80 °C. Each preparation was made from combined tissues from at least three different explanted hearts. For some experiments myocardial membrane fractions were suspended in buffer B (50 mm HEPES, 50 mm sucrose, 1 mm EDTA, 10 mm pyrophosphate, 5 mm NaF, 100 mm NaCl, 5 mm MgCl2, 0.1 μm okadaic acid, Roche Applied Science protease inhibitor mixture, pH 7.5). Myocardial membranes were then solubilized by homogenization (using a Dounce homogenizer, 20 strokes) and incubation/rotation of homogenates with Nonidet P40 (v/v, 1% final) (Thermo Fisher Scientific) for 1 h at 4 °C. Solubilized membrane proteins (supernatants) were obtained by centrifugation (24,000 rev/min (98,500 × g)) for 30 min at 4 °C using a Beckman SW41 Ti rotor.
For isolation of SR vesicles, 11,000 × g supernatants were centrifuged (45 min, 43,666 × g) in a Beckman SW41 Ti rotor, as described (17). The resultant pellets were resuspended in buffer containing 0.6 m KCl and 20 mm Tris, pH 6.8, and centrifuged (45 min, 43,666 × g). The final washed pellets containing SR fractions were suspended in buffer A and stored at −80 °C.
cAMP PDE Assay
PDE3 activity (that portion of total PDE activity inhibited by 1.0 μm cilostamide, a selective PDE3 inhibitor), was measured by modification of our published method (18) using 0.1 μm [3H]cAMP (35000 cpm) as substrate. PDE activity is expressed as pmol of cAMP hydrolyzed/min/mg.
Immunoprecipitation and Immunoblotting
In experiments with solubilized myocardial membrane fractions and Superose 6 column fractions (Figs. 3 and 5), fractions were cleared by incubation for 1 h at 4 °C with 5 μg of rabbit non-immune IgG and then with 50 μl of Protein G magnetic beads for 30 min before placing the tubes into a magnetic stand to collect the beads against the side of the tube. Cleared fractions were transferred to new tubes and incubated overnight at 4 °C with specified antibodies or with non-immune IgG followed by incubation with fresh Protein G magnetic beads for 1 h before placing the tubes into a magnetic stand to collect the beads. In experiments with recombinant proteins (Figs. 6 and 7), fractions (reaction mixtures) containing FLAG-tagged human PDE3A variants (Fig. 6) or His-tagged AKAP18δ (Fig. 7) were cleared as described above but incubated with 5 μg of mouse non-immune IgG and then with 50 μl Protein G Magnetic beads before placing the tubes in a magnetic stand to separate the beads from the cleared fractions for immunoprecipitation. The cleared fractions were transferred to new tubes and incubated with mouse anti-FLAG M2 magnetic beads or mouse anti-His mAb magnetic beads before placing the tubes into a magnetic stand to collect the beads.
FIGURE 3.

Analysis of PDE3A isoforms by S6 gel filtration chromatography of solubilized human myocardial membrane or cytosolic fractions. Upper panels: A and B, solubilized myocardial (myc) membranes (A) and cytosolic (cyt) fractions (Fr; B) were prepared (3 mg of protein, 1 ml) and subjected to chromatography on Superose 6 columns. Portions (10 μl) of fractions (0.5 ml) were assayed for PDE3 activity (▴) (pmol of cAMP hydrolyzed/min/0.5 ml) and protein content (milliabsorption units (mAU)) 280 nm) (●). Molecular mass standard peaks are indicated: 1, thyroglobulin (670 kDa); 2, γ-globulin (158 kDa); 3, ovalbumin (44 kDa); 4, myoglobin (17 kDa); 5, vitamin B12 (1.35 kDa). IB, immunoblot. Lower panels: A and B, portions (20 μl) of the indicated fractions were subjected to SDS/PAGE and immunoblotted with anti-PDE3A-CT antibodies. One representative experiment is shown (n = 3). C, as described under “Methods,” portions of pooled HMW (H) and LMW (L) fractions (Fig. 3, A and B) from cytosol (C) and solubilized membrane (M) fractions were cleared by incubation with 5 μg non-immune rabbit IgG and 50 μl of Protein G magnetic beads. The beads were removed by placing the tubes in a magnetic stand, and the cleared fractions were then incubated with 10 μg of non-immune IgG (IgG) or 10 μg of anti-PDE3A-CT (PDE3A) as indicated. Fractions were subjected to immunoprecipitation (IP) with 50 μl Protein G magnetic beads, which were separated from the HMW and LMW fractions as described above. The beads were washed, and proteins were eluted from the beads by boiling with Laemmli SDS sample buffer. Portions (15–20 μl) of the eluted proteins were subjected to SDS/PAGE and immunoblotted with anti-phospho-PKA-substrate (upper panel) and anti-PDE3A-CT (lower panel) antibodies. PDE3A and pPDE3A were detected in immunoprecipitates from cleared HMW and LMW fractions incubated with anti-PDE3A-CT but not non-immune IgG. Bar graph, right panel, pPDE3A1/PDE3A1 (p3A1/3A1) and pPDE3A2/PDE3A2 (p3A2/3A2) ratios in HMW and LMW peaks indicated that endogenous PDE3A1 (HMW peak) was the most highly PKA-phosphorylated isoform and demonstrated an ≈8-fold increase in phosphorylation of PDE3A1 and ∼5-fold increase in phosphorylation of PDE3A2 in HMW peaks compared with LMW peaks (*, p < 0.01). Results are representative of three individual experiments. D, as described under “Methods,” PDE3A was immunoprecipitated by incubation of cleared solubilized myocardial membranes with anti-PDE3A-CT and Protein G magnetic beads. The beads containing immunoprecipitated PDE3A were separated from the solubilized membranes, washed, and incubated as indicated without (Ctrl) or with 250 units of rPKAc or 250 units rPKAc plus 10 μm PKI (PKAc inhibitor peptide) in phosphorylation buffer containing 200 μm ATP and 5 mm MgCl2 supplemented with (upper panel) or without (lower panel) [γ-32P]ATP. Upper panel, after incubation in phosphorylation buffer containing [γ-32P]ATP, the beads containing immunoprecipitated PDE3A were separated from reaction mixtures and washed, and 32P-labeled proteins were eluted from the beads by boiling with Laemmli SDS sample buffer. Portions (15–20 μl) of the eluted proteins were subjected to SDS/PAGE. γ-32P phosphorylation of PDE3A was detected by scanning the wet gels by phosphorimaging (GE Healthcare). Phosphorylation was blocked by PKI. Middle panel, proteins on wet gels were then electrophoretically transferred to nitrocellulose membranes, and PDE3A was identified by Western blotting using anti-PDE3A-CT antibody. Lower panel, PDE3 activity was assayed in PDE3A immunoprecipitates incubated with or without rPKAc and PKI in the absence of [γ-32P]ATP. Results are expressed as pmol of cAMP hydrolyzed/min. Shown are representative data from three independent experiments. rPKAc significantly increased PDE3A activity (p < 0.01); activation was blocked by PKI (PKAc inhibitor peptide).
FIGURE 5.
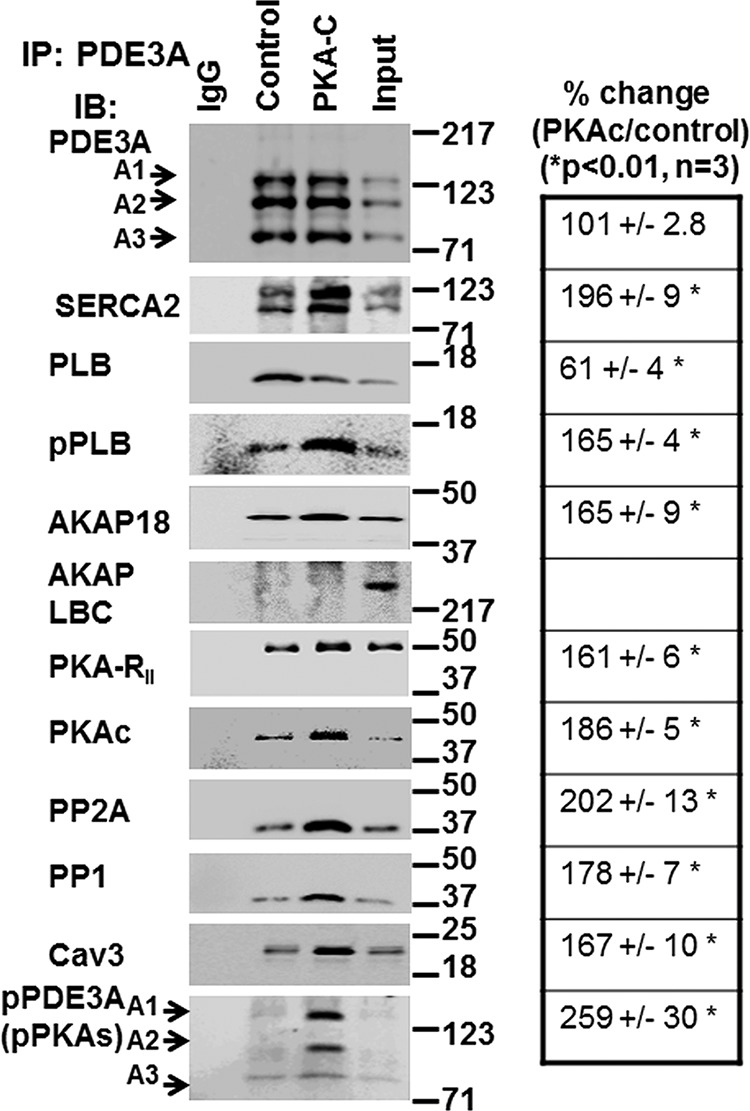
rPKAc promotes interactions of PDE3A with components of the SERCA2 regulatory signalosome. Solubilized myocardial membranes were prepared (3 mg of protein, 1 ml) and subjected to chromatography on Superose 6 columns as in Fig. 3A. Membrane LMW fractions were pooled from two different experiments (Fig. 3A) and concentrated via Centriprep YM-3. Pooled, concentrated fractions were split into three fractions and incubated in phosphorylation buffer with 200 μm ATP and 5 mm MgCl2 for 1 h at 30 °C in the absence (IgG, Control) or presence (PKA-C) of rPKAc. At the completion of these reactions, fractions were cleared with non-immune IgG and Protein G magnetic beads as described above and then incubated (overnight, 4 °C) with non-immune IgG (IgG) or anti-PDE3A-CT antibody (control, PKA-C) before incubation and immunoprecipitation (IP) with Protein G magnetic beads (1 h, 4 °C). Proteins associated with the Protein G magnetic beads were eluted by boiling in 200 μl of Laemmli SDS sample buffer. Samples (15 μl) were subjected to SDS/PAGE and immunoblotted (IB) with specific antibodies as shown. Input membrane proteins (10 μg) were also loaded on the gels as positive controls. Representative results from three independent experiments are shown. Similar amounts of PDE3A were immunoprecipitated in the control fractions and in fractions incubated with rPKAc. Band intensities of immunoprecipitated PDE3A and its interacting signaling molecules were analyzed using an LAS3000 analyzer and presented as binding percentage ratios of signaling molecules (rPKAc/control). For PDE3A, band intensities of pPDEA1/pPDE3A2/pPDE3A3 in PKAc/control percentage ratios were calculated. *, p < 0.01 versus control (n = 3 independent experiments) Cav3, caveolin 3.
FIGURE 6.

rPKAc phosphorylates rhPDE3A and increases its interactions with rSERCA2. A, schemes representing protein domains and phosphorylation sites in FLAG-tagged-PDE3A isoforms (NHR1, transmembrane domain (obligatory membrane insertion domain); NHR2, membrane association domain; CCR, conserved C-terminal catalytic region; P1–4, predicted PKA phosphorylation sites; rhPDE3A and truncated mutants (PDE3A2, PDE3A-Δ510, PDE3A-RD (regulatory domain: aa 146–484)) and protein domains in rSERCA2 (1042 amino acids, including 3–77 cation transporter N termini; 93–341, E1-E2 ATPase; 345–724, haloacid dehydrogenase like hydrolase; 819–991, cation transporter ATPase C terminus). B, putative PKA phosphorylation sites in PDE3A designated with asterisks. C and D, purified rSERCA2 (150 ng) (Abnova) and 50 units of FLAG-tagged rhPDE3A1 or FLAG-tagged truncated mutants ((rhPDE3A2, rhPDE3A-Δ510, and rhPDE3A-RD) (C) or FLAG-tagged mutants lacking the PKA putative phosphorylation sites of rhPDE3A1 (S292A/S293A (P1), S312A (P2), S428A (P3), S438A (P4), S292A/S293A/S312A/S438A (P5)) (D) were incubated for 30 min at 30 °C in phosphorylation buffer containing 5 mm MgCl2 and 200 μm ATP in the presence or absence of rPKAc. As described under “Methods,” proteins were immunoprecipitated (IP) with anti-FLAG M2 magnetic beads. Immunoprecipitated proteins bound to the anti-FLAG-M2 magnetic beads were eluted by boiling in Laemmli SDS buffer (200 μl), and samples (15–20 μl) of eluted proteins as well as of reaction mixtures (lowest panel) were subjected to SDS-PAGE and Western immunoblotting (IB) as indicated with anti-SERCA2, anti-PKA substrate, and anti-FLAG-HRP primary antibodies and anti-mouse-HRP or anti-rabbit-HRP secondary antibodies as needed. Lowest panels, input control (SERCA2). Shown are representative blots from three independent experiments.
FIGURE 7.

rPKAc-induced phosphorylation of rhPDE3A increases its interaction with rat rAKAP18δ. A, schemes representing hPDE3A1 protein domains (NHR1, trans-membrane domain (obligatory membrane insertion domain); NHR2, membrane association domain; CCR, conserved C-terminal catalytic region) and rAKAP18δ protein domains, with RII binding site (aa 301–314) and a unique N terminus (aa 1–26). B and C, His-tagged rAKAP18δ (100 ng) and 50 units of FLAG-tagged rhPDE3A1 were incubated for 30 min at 30 °C in phosphorylation buffer containing 200 μm ATP and 5 mm MgCl2 in the absence or presence of the indicated concentrations (units) of rPKAc (B) or in the absence or presence of 50 units of rPKAc (C). B and C, as described under “Methods,” proteins were immunoprecipitated (IP) with anti-His mAb magnetic beads, and immunoprecipitated proteins bound to the anti-His mAb magnetic beads were eluted by boiling in Laemmli SDS buffer (200 μl). Samples (15–20 μl) of eluted proteins as well as of reaction mixtures (Input, lower panels) were subjected to SDS-PAGE and Western immunoblotted (IB) with anti-FLAG-HRP, anti-AKAP18-His, anti-PKAc, and anti-phospho-PKA substrate (pPDE3A) antibodies as indicated. Similar amounts of AKAP18δ were immunoprecipitated in the control groups and in reactions incubated with rPKAc. C, as a control, rhPDE3A1 (50 units) was incubated with or without rPKAc (50 units) and with SF21 cell supernatants that contained or did not contain rAKAP18δ. The bar graph summarizes binding of rhPDE3A1-FLAG with rAKAP18δ-His in the absence (control) and presence of rPKAc. The band density of rhPDE3A1 was normalized to that of input rAKAP18δ and calculated as the amounts of rhPDE3A1 bound to rAKAP18δ in the absence (Control, C) or presence of rPKAc; there was an ∼9-fold increase in the association of rhPDE3A1 with AKAP18δ in the presence of rPKAc (*, p < 0.001). Shown are representative blots from three independent experiments.
Immunoprecipitated proteins bound to the three types of magnetic beads were washed (3×, buffer B) and then eluted by boiling (5 min) the beads in 200 μl of Laemmli SDS sample buffer (65 mm Tris, pH 6.8, 2.5% SDS, 10% glycerol, and 0.002% bromphenol blue with 100 mm β-mercaptoethanol and 100 mm DTT). After boiling, magnetic beads were separated from the eluted proteins by placing the tubes in a magnetic stand. Samples (15–20 μl) of eluted proteins were subjected to SDS-PAGE, electrotransferred to nitrocellulose membranes (Invitrogen), and immunoblotted sequentially with appropriate primary antibodies and HRP-labeled secondary antibodies or anti-FLAGM2-HRP antibody. Immunoreactive proteins were reacted with Supersignal® Westpico or Westfemto chemiluminescent reagents; signals were detected with Image reader LAS3000.
cAMP-dependent Phosphorylation of Endogenous PLB
Resuspended human SR fractions (20 μg of protein) were phosphorylated in buffer containing 50 mm HEPES, 5 mm MgCl2, 50 mm sucrose, 100 mm NaCl, 5 mm NaF, 10 mm PPi, 1 mm EDTA, pH 7.6. After incubation (2 min, 30 °C) in the absence or presence of cilostamide (1 μm) or rolipram (10 μm), 0.5 mm ATP and 0.1 μm cAMP were added as indicated, and incubations (50 μl final volume) were continued for 30 min. Reactions were terminated by the addition of Laemmli SDS sample buffer (25 μl). Samples were immediately boiled (5 min) and subjected to SDS-PAGE. Phosphorylation of PLB Ser-16, and endogenous PLB, PKAc, and PDE3A proteins were identified by Western immunoblotting (Fig. 2A).
FIGURE 2.

cAMP, PKA, and PDE3 inhibition increase SERCA2 activity and Ca2+ uptake in human SR fractions. A, as described under “Methods,” after incubation of SR fractions (20 μg) in the absence or presence of the indicated concentrations of ATP and cAMP without or with cilostamide (Cil) or rolipram (Rol), endogenous PLB, phosphorylated PLB, and β-actin were detected after SDS-PAGE and immunoblotting. Data are representative of three experiments. In these and other Western blots PLB is predominantly monomeric, most likely due to the heating of samples before electrophoresis under reducing conditions with Laemmli SDS sample buffer. B, bar graph summarizing pSer16PLB/PLB total ratios. Cilostamide significantly enhanced the effect of 0.1 μm cAMP on phosphorylation of PLB. *, p < 0.01 versus control (n = 3 independent experiments). C, PDE activity in human cardiac SR fractions was assayed as described under “Methods” and expressed as specific activity (pmol of cAMP hydrolyzed/min/mg). Results are presented as the mean ± S.E. (n = 3 preparations). PDE3 activity was determined as the cilostamide-sensitive fraction, and PDE4 activity was determined as the rolipram-sensitive fraction. PDE3 activity was significantly higher than PDE4 activity (*, p < 0.001). D, after incubation of SR fractions without or with the indicated concentrations of cAMP (0–10 μm), 45Ca2+ uptake was measured in the presence of 0.5 μm free Ca2+ as described under “Methods.” Results are presented as % increase due to cAMP, with basal Ca2+ uptake (9.4 ± 0.8 nmol/mg/min, n = 3) taken as 100%. cAMP significantly enhanced 45Ca2+ uptake (*, p < .01; **, p < 0.04). E, after incubation of SR fractions with or without cAMP (3 μm) in the presence or absence of cilostamide (1 μm), 45Ca2+ uptake (0.5 μm free Ca2+) was assayed as described under “Methods.” Results are presented as the mean ± S.E. (n = 3). Cilostamide significantly enhanced the effect of cAMP on 45Ca2+ uptake (*, p < .02). F, Ca2+ uptake (upper panel) and SERCA activity (lower panel) were assayed in reaction mixtures containing Mg2+ and ATP in the presence or absence of rPKAc as described under “Methods.” Results are presented as nmol (Ca2+ or Pi)/min/mg (mean ± S.E.) (n = 3). rPKAc significantly enhanced 45Ca2+ uptake and SERCA2 activity (*, p < .001).
Effect of cAMP and Recombinant PKA Catalytic Subunit (rPKAc) on 45Ca2+ Uptake or SERCA Activity in Human SR Vesicles
For experiments in Fig. 2, D–F, SR vesicle fractions (50∼100 μg), prepared as described above, were incubated (30 min, 30 °C) without or with the indicated concentrations of cAMP, 250 units of rPKAc, or 1.0 μm cilostamide (PDE3 inhibitor) in buffer containing 10 mm imidazole-HCl, pH 7.0, 0.1 μm okadaic acid, 10 mm MgCl2, 5 mm DTT, 0.5 mm ATP, and 1× phosphatase inhibitor mixture (1 rPKAc activity unit is defined as the amount of enzyme required to transfer 1 nmol of phosphate to PKA peptide (GRTGRRNSI)/min at 30 °C). Reaction mixtures were centrifuged (100,000 × g, 30 min, 4 °C), and the resulting pellets were resuspended in 0.3 m sucrose, 10 mm K-PIPES buffer containing 1× phosphatase inhibitor mixture, pH 7.0, for Ca2+ uptake and SERCA activity assays as described below.
Measurement of 45Ca2+ Uptake into SR Vesicles
For experiments in Fig. 2, D–F, oxalate-dependent Ca2+ uptake was quantified as described previously (19). As described above, after incubation (30 min, 30 °C) without or with various additions, SR vesicles were collected and resuspended in a sucrose/K-PIPES buffer. Then portions (50 μl, 10 μg of protein) were incubated at 37 °C for 1 min in 0.4 ml of Ca2+ uptake buffer consisting of 50 mm imidazole-HCl, pH 7.0, 100 mm KCl, 6 mm MgCl2, 10 mm NaN3 (to inhibit mitochondrial Ca2+ uptake), 10 mm potassium oxalate, 20 μm ruthenium red (to inhibit SR Ca2+ release), 0.1 mm EGTA, 45CaCl2 (10,000 dpm/nmol) (PerkinElmer Life Sciences, catalog #NEZ013), and unlabeled CaCl2 (0.5 μm free Ca2+). Uptake was initiated by adding 50 mm ATP (50 μl), and reactions were terminated by filtration through 0.45-μm Millipore filters. After washing four times with 4 ml of buffer containing 140 mm KCl, 10 mm NaCl, 2 mm MgCl2, 1 mm CaCl2, and 50 mm imidazole-HCl, pH 7.0, radioactivity retained on the filters was quantified by liquid scintillation counting. Free Ca2+ was calculated using a MAXCHELATOR program obtained at Stanford University.
SERCA Activity Assay
In Fig. 2F, Ca2+-ATPase activity in SR fractions was determined by measuring the amount of Pi released after the addition of ATP using the malachite green ATPase method (20). Briefly, as described above, after incubation (30 min, 30 °C) without or with 250 units of rPKAc, SR vesicles were isolated and suspended in sucrose/K-PIPES buffer. Then samples (50 μl, 10 μg of protein) were assayed for SERCA2 enzymatic activity in the presence or absence of thapsigargin (10 μm), as SERCA2 activity was determined as that portion of total activity inhibited by thapsigargin. The assay mixture (total volume, 125 μl) contained 0.125 m KCl, 20 mm imidazole, pH 7.0, 0.1 mm EGTA, 0.103 mm CaCl2, 5 mm MgCl2, 1 μm ionomycin, and 10 μg of SR. To initiate the reaction, 25 μl of substrate (1.25 mm ATP) was added to a final concentration of 0.25 mm. The mixture was incubated at room temperature for 3 min. The reaction was terminated by adding 25 μl of 250 g/liter TCA, vortexed quickly, and centrifuged (8000 × g, 3 min). Supernatants (20 μl) were added to 96-well plates followed by color reagent (100 μl/sample), which consisted of 54 mm ammonium molybdate and 0.73 mm malachite green. After 1 min sodium citrate (340 g/liter, 10 μl) was added with gentle shaking (room temperature for >20 min), after which plates were scanned at 650 nm. Pi was calculated by converting OD650 to nanomoles by means of a standard curve.
Gel Filtration Chromatography on Superose 6 Columns
Solubilized myocardial membranes (1.0 ml, 3 mg of total protein), prepared as described above, were applied to a Superose 6 HR 10/30 column (ÄKTA FPLC System, GE Healthcare) that was equilibrated and eluted with buffer C (buffer B without sucrose containing 150 mm NaCl and 1% Nonidet P-40) (Fig. 3A). Cytosolic fractions (1.0 ml, 3 mg of total protein), prepared as described above, were also applied to Superose 6 that was equilibrated and eluted with buffer D (buffer C, without Nonidet P-40) (Fig. 3B). Portions of the fractions (0.5 ml) were used for assay of PDE3 activity or SDS-PAGE/Western blotting (using anti-PDE3A-CT antibodies) (Fig. 3, A and B) as described above. In some experiments (Fig. 3C), portions of eluted HMW (H) and LMW (L) peaks from solubilized membrane fractions (M) and cytosol fractions (C) were pooled and concentrated. As described above (see “Immunoprecipitation and Immunoblotting”), fractions were cleared by incubation with 5 μg of rabbit non-immune IgG and then with 50 μl of Protein G magnetic beads for 30 min before placing the tubes into a magnetic stand to collect the beads against the side of the tube. As presented in Fig. 3C, cleared fractions were transferred to new tubes and incubated overnight at 4 °C with 10 μg of non-immune IgG (IgG lane) or with 10 μg of anti-PDE3A-CT (PDE3A lanes) followed by incubation (1 h, 4 °C) and immunoprecipitation with 50 μl of fresh Protein G magnetic beads before placing the tubes into a magnetic stand to collect the beads. Immunoprecipitated proteins bound to the magnetic beads were washed (3×, buffer B) and then eluted by boiling (5 min) in 200 μl of Laemmli SDS sample buffer. After boiling, magnetic beads were separated from the eluted proteins by placing the tubes in a magnetic stand. Samples of eluted proteins were subjected to SDS-PAGE and immunoblotting with anti-pPKA substrate or anti-PDE3A-CT (PDE3A) antibodies (Fig. 3C) as described above.
In Vitro Phosphorylation of Endogenous PDE3A
For experiments in Fig. 3D, solubilized membrane fractions (500 μg) were cleared with 5 μg of rabbit non-immune IgG and 50 μl of Protein G magnetic beads, and PDE3A was immunoprecipitated using 10 μg of anti-PDE3A-CT and 50 μl of Protein G magnetic beads as described above. Immunoprecipitated PDE3A bound to the magnetic beads was incubated for 1 h at 30 °C in phosphorylation buffer (50 mm HEPES, pH 7.5, 5 mm MgCl2, 100 mm NaCl, 1 mm EDTA, 10 mm PPi, 5 mm NaF, 0.1 μm okadaic acid) with 200 μm ATP or 200 μm ATP supplemented with [γ-32P]ATP (10 μCi per reaction; 3000 Ci/mmol stock) in the absence or presence of 250 units of rPKAc or rPKAc plus 10 μm PKAc inhibitor (PKI). After the incubation, tubes were placed in a magnetic stand to separate the beads from the reaction mixtures, and beads (with bound immunoprecipitated PDE3A) were washed (3×, buffer B).
As indicated in Fig. 3D (lower panel) immunoprecipitated PDE3A (not incubated with [γ-32P]ATP) was assayed for PDE3 activity using [3H]cAMP as substrate. The assays were terminated, and tubes were placed in a magnetic stand to separate the beads from the reaction mixtures. In the separated reaction mixtures, the product 5′-[3H]AMP was converted to [3H]adenosine, which was quantified as described in our published method (18).
As shown in Fig. 3D (upper panel), after incubation of immunoprecipitated PDE3A with rPKAc and [γ-32P]ATP, the beads were separated from the reaction mixtures and washed as described above. 32P-Labeled PDE3A bound to the magnetic beads was eluted by boiling the beads (5 min) in Laemmli SDS sample buffer and then by placing the tubes in a magnetic stand to separate the eluted 32P-labeled PDE3A from the magnetic beads. Samples of eluted 32P-labeled PDE3A were subjected to SDS-PAGE. γ-32P phosphorylation of PDE3A was detected by scanning the wet gels by phosphorimaging (GE Healthcare). Proteins on wet gels were then electrophoretically transferred to nitrocellulose membranes, and PDE3A was identified by Western blotting using anti-PDE3A antibody (Fig. 3D, middle panel). PKI blocked rPKAc-induced phosphorylation and activation of PDE3A.
In other experiments (Fig. 4) chromatography of solubilized myocardial membranes (3 mg) on Superose 6 was repeated 2 times and LMW fractions (#24–34, Fig. 3A) were pooled and concentrated via Centriprep YM-3 (centrifugal filter units with Ultracel-3 membranes (nominal molecular weight limit, >3 kDa)). The concentrated fractions were split, and after incubation for 1 h at 30 °C without (LMW control) or with (LMW + PKA) rPKAc in phosphorylation buffer containing 200 μm ATP and 5 mm MgCl2 were re-chromatographed on Superose 6 (Fig. 4). Portions of the fractions (0.5 ml) were used for assay of PDE3 activity or SDS-PAGE/Western blotting (using anti-PDE3A-CT and other indicated antibodies) as described above.
FIGURE 4.

Treatment of LMW fractions of solubilized human myocardial membranes with rPKAc induces the shift of PDE3A isoforms and components of the SERCA2 regulatory signalosome into HMW fractions during S6 gel filtration chromatography. Solubilized myocardial membranes (3 mg of protein, 1 ml) were subjected to chromatography on S6 columns as described in Fig. 3A and fractionated into LMW and HMW fractions. As described under “Methods,” LMW fractions were pooled from two different experiments (cf. Fig. 3A) and concentrated via Centriprep YM-3. Upper panels, pooled and concentrated Superose 6 LMW fractions were split, incubated (1 h, 30 °C) in phosphorylation buffer with 200 μm ATP and 5 mm MgCl2 in the absence or presence of rPKAc, and re-chromatographed on Superose 6. Portions (10 μl) of fractions (0.5 ml) were assayed for PDE3 activity (▴) (pmol of cAMP hydrolyzed/min/0.5 ml) and protein content (milliabsorption units (mAU)) 280 nm) (●). Molecular mass standards: 1, thyroglobulin (670 kDa); 2, γ-globulin (158 kDa); 3, ovalbumin (44 kDa); 4, myoglobin (17 kDa); 5, vitamin B12 (1.35 kDa). Bottom panels, portions (20 μl) of the indicated fractions were subjected to SDS/PAGE and immunoblotted (IB) with the indicated antibodies. Representative results from one of two independent experiments are shown. Cav3, caveolin 3.
Co-immunoprecipitation of PDE3A with Components of SERCA2 Regulatory Signalosomes from Pooled LMW Fractions after Incubation with or without rPKAc
For these studies (Fig. 5), pooled LMW Superose 6 fractions from solubilized myocardial membranes (two experiments) were prepared as described above (cf. Figs. 3A and 4). These LMW fractions were concentrated and split into three fractions that were then incubated (1 h, 30 °C) in phosphorylation buffer containing 200 μm ATP and 5 mm MgCl2, without (IgG, Control) or with (PKA-C) rPKAc. To study co-immunoprecipitation of PDE3A with components of the SERCA2/AKAP18 signalosome (Fig. 5), at the completion of these reactions the three LMW fractions were cleared with rabbit non-immune IgG (5 μg)and Protein G magnetic beads (50 μl) as described above and then incubated (overnight, 4 °C) with non-immune IgG (10 μg) (IgG) or anti-PDE3A-CT antibody (10 μg) (control, PKA-C) before incubation (1 h, 4 °C) with Protein G magnetic beads. Tubes were placed in a magnetic stand to separate the beads from the reaction mixtures. Immunoprecipitated proteins bound to Protein G magnetic beads were washed and eluted as described above, and portions of eluted samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with indicated antibodies (Fig. 5). Total membrane proteins (10 μg, input) were loaded on gels as controls.
Co-immunoprecipitation of FLAG-tagged Recombinant Human PDE3A (rhPDE3A) Variants and rSERCA2 after Incubation with or without rPKAc
FLAG-tagged rhPDE3A1 (open reading frame, accession number NP_000912) and its phosphorylation site mutants (rhPDE3A1-S292A/S293A (P1), rhPDE3A1-S312A (P2), rhPDE3A1-S428A (P3), rhPDE3A1-S438A (P4), and rhPDE3A1-S292A/S293A/S312A/S438A (P5) (Fig. 6B) were synthesized by Genscript, Inc (Piscataway, NJ) and subcloned into pAcSG2 baculoviral expression vector. FLAG-tagged rhPDE3A variants and phosphorylation site mutants were expressed in SF21 cells ((10–20) × 106 SF21 cells/75-cm2 flask) as described (21, 22). SF21 cells were harvested, sedimented (10 min, 1000 × g, 4 °C), washed twice with ice-cold PBS, resuspended, homogenized (in a Dounce homogenizer) in buffer B, and sonicated on ice (2 × 20 s pulses, output 2, 40% cycle) with a Sonifier cell disruptor 350 (Branson Sonic Power Co., Danbury, CT). After incubation/rotation in buffer B containing 1% (v/v) Nonidet P40, the sonicated homogenates were centrifuged (20 min, 4 °C, 12,000 × g). The supernatants containing rhPDE3A variants were used for experiments.
Purified human rSERCA2 (full-length ORF (accession number NP_001672.1, 1–997 aa)) protein with a GST tag at its N terminus was purchased from Abnova (Taiwan). rSERCA2 and FLAG-tagged rhPDE3A variants (usually 50 arbitrary units, 1 unit being the amount of PDE3 that hydrolyzed 1 pmol of cAMP/min at 30 °C) were incubated (30 min, 30 °C) in phosphorylation buffer containing 5 mm MgCl2, 200 μm ATP with or without 50 units of rPKAc (final volume, 200 μl). Reactions were stopped by the dilution of reaction mixtures to 1.0 ml with buffer B containing 1 mg/ml SF21 cell supernatants and cleared as described above (see “Immunoprecipitation and Immunoblotting”) by incubation with 5 μg of mouse non-immune IgG and then with 50 μl of Protein G magnetic beads. To block nonspecific binding of proteins to anti-FLAG M2 magnetic beads, ∼1 ml of anti-FLAG M2 magnetic beads was incubated (overnight, 4° C) with 5 ml (1 mg/ml) of Sf21 cell supernatants (prepared by homogenization of cells in buffer B containing 1% Nonidet P-40 and centrifugation (12,000 × g, 20 min)).
Treated anti-FLAG M2 magnetic beads were placed in a magnetic stand to separate the beads from Sf21 cell supernatants and washed (PBS, 3×). These washed magnetic beads were used for immunoprecipitation of recombinant proteins (25 μl, 2 h, 4 °C) from the cleared reaction mixtures. Tubes were placed in a magnetic stand to separate the beads from the reaction mixtures, which were removed. Immunoprecipitated proteins bound to the anti-FLAG-M2 magnetic beads were washed and eluted by boiling in Laemmli SDS sample buffer (200 μl). As described above, tubes were placed in a magnetic stand to separate the beads from the eluted proteins, and samples of eluted proteins as well as of SERCA2 (reaction mixture input protein) were subjected to SDS-PAGE and Western immunoblotting with the indicated antibodies as described above.
Co-immunoprecipitation of His-tagged Recombinant Rat AKAP18δ (rAKAP18δ) with FLAG-tagged rhPDE3A1 after Incubation with or without rPKAc
His-tagged rAKAP18δ open reading frame (accession number NP_001001801, (4)) was synthesized by Genscript, Inc. (Piscataway, NJ), subcloned into pAcSG2 baculoviral expression vector, and expressed in SF21 cells (21). A comparison of the translated sequences of human AKAP18γ (accession number NP_057461 and AK300587; 348-aa residues) and rat AKAP18δ (4) (accession number Q6JP77; 353 aa residues) indicate that 8 of the first 12 aa residues of human AKAP18γ and rat AKAP18δ are identical and that the proteins are homologous (∼74% aa identity). SF21 cell supernatants, containing His-tagged rAKAP18δ, were prepared as described above. FLAG-tagged rhPDE3A1 (usually 50 arbitrary units) was incubated (30 min, 30 °C) with rAKAP18δ (50 ng) in phosphorylation buffer containing 2 mm DTT, 5 mm MgCl2, and 200 μm ATP (final volume, 300 μl) with or without different concentrations of rPKAc (Fig. 7B) and with or without 50 units of rPKAc (Fig. 7C). As a control, in Fig. 7C, rhPDE3A1 (50 units) was incubated with or without rPKAc (50 units) and with SF21 cell supernatants that contained or did not contain rAKAP18δ. Reactions were stopped by dilution of reaction mixtures to 1.0 ml with buffer B containing 1 mg/ml SF21 cell supernatants and cleared by incubation with 5 μg of mouse non-immune IgG and then with 50 μl of Protein G magnetic beads as described above. Anti-His mAb magnetic beads incubated with SF21 cell supernatants as described above for anti-FLAG M2 magnetic beads were used for immunoprecipitation of recombinant proteins (25 μl, 2 h, 4 °C). Tubes were placed in a magnetic stand to separate the beads from the reaction mixtures, which were removed. Immunoprecipitated proteins bound to anti-His mAb magnetic beads were washed and eluted by boiling in Laemmli SDS sample buffer (200 μl). As described above, samples of eluted immunoprecipitated proteins as well as of reaction mixtures (“input proteins”) were subjected to SDS-PAGE and immunoblotted with indicated antibodies.
RESULTS
PDE3A Co-localizes with PLB, SERCA2, and AKAP18 in the Z-bands of Human Cardiac Myocytes
The intracellular distribution of PDE3A in cryostat sections of normal human left ventricle was examined by immunostaining with peptide affinity-purified anti-PDE3A-CT antibodies (Fig. 1). Consistent with our previous studies (2, 23), co-staining with anti-PDE3A-CT and anti-desmin antibodies demonstrated that PDE3A is distributed in a striated pattern in the Z-lines of human cardiac myocytes (Fig. 1, A and B), with little co-localization with myomesin (M-line marker protein) (Fig. 1B). PDE3A co-localized with PLB, SERCA2, and an AKAP18 variant (detected by antibodies to human AKAP18 (Abcam: Ab 30987)) (Fig. 1, A and B).
FIGURE 1.

PDE3A, SERCA2, PLB, and AKAP18 co-localize in the Z-bands in normal human myocardium. A, cryostat sections of normal human left ventricle were prepared and, as described under “Methods,” permeabilized, blocked, and then incubated in blocking buffer with rabbit anti-PDE3A-CT, anti-desmin, anti-SERCA2, anti-PLB, anti-AKAP18, and anti-myomesin primary antibodies followed by incubation with Alexa Fluor® 488- or 594-conjugated anti-mouse or anti-rabbit secondary antibodies. Signals were detected with a Zeiss LSM510 laser scanning confocal microscope. From the left: first column, red fluorescent staining for marker proteins: desmin, SERCA2, myomesin, PLB, and AKAP18; second column, green fluorescence staining for PDE3A; third column, DAPI staining of nuclei (blue); fourth column, merged images of PDE3A, marker proteins, and DAPI indicate that PDE3A exhibits a striated pattern and co-localizes with desmin, SERCA2, PLB, and AKAP18. B, merged images from stacks of 10–15 sections (with 1-μm intervals) reveal colocalization of PDE3A with desmin, SERCA2, PLB, and AKAP18 but not with myomesin (labeling M-line Red). X-Y (center), above X-Z (top), and Y-Z (right) planes are at the indicated positions. Representative images from three independent experiments are shown.
PDE3 Inhibition Potentiates cAMP-dependent Phosphorylation of PLB and Its Stimulation of Ca2+ Uptake in Human Myocardial Membranes
The effects of PDE3 inhibition on PLB phosphorylation and Ca2+ uptake were examined in SR fractions prepared from human left ventricular myocardium. At 0.1 μm cAMP, phosphorylation of PLB at Ser-16 by endogenous PKA was stimulated by the PDE3-selective inhibitor cilostamide (Fig. 2, A and B). Because PDE4 has also been found to co-immunoprecipitate with PLB in subcellular preparations from human myocardium (16), we tested the effect of the PDE4-specific inhibitor rolipram on PLB phosphorylation and found no effect (Fig. 2, A and B). This result corresponds to the lower amount of rolipram-sensitive cAMP hydrolytic activity relative to cilostamide-sensitive cAMP hydrolytic activity in these SR preparations (Fig. 2C). cAMP increased ATP-dependent, oxalate-supported Ca2+ uptake (Fig. 2D), and this effect was potentiated by cilostamide (Fig. 2E). The addition of rPKAc stimulated both Ca2+ uptake and SERCA2 activity (Fig. 2F). These results indicate that PDE3 has a specific role in regulating phosphorylation of PLB by cAMP/PKA and the consequent stimulation of Ca2+ uptake in human myocardium.
Gel Filtration Chromatography of PDE3A Isoforms from Human Myocardium
In previous studies we showed that the phosphorylation of rPDE3A isoforms in transfected HEK293 cells promotes their interactions with other proteins (15). To address whether phosphorylation might promote the integration of PDE3A into multiprotein SERCA2 signalosomes in cardiac myocytes, we analyzed cytosolic and Nonidet P-40-solubilized membrane proteins from human left ventricular myocardium by Superose 6 gel filtration chromatography (Fig. 3). As seen in Fig. 3A, PDE3 activity in solubilized myocardial membrane proteins was recovered in distinct HMW (∼3000 kDa) and LMW (∼700 kDa) peaks. Western blotting with anti-PDE3A antibodies indicated that the HMW and LMW peaks contained all three PDE3A isoforms. In contrast, upon Superose 6 chromatography of cytosolic fractions, from which PDE3A1 is absent, PDE3A2 and PDE3A3 eluted in a single LMW peak (Fig. 3B). As seen in Fig. 3C, immunoprecipitation of PDE3A from pooled HMW or LMW peaks of solubilized membranes followed by Western blotting with anti-phospho PKA substrate and anti-PDE3A-CT antibodies demonstrated that endogenous PDE3A1 in HMW peaks was the most highly PKA-phosphorylated PDE3A isoform. These results suggested that phosphorylation of endogenous PDE3A1 by PKA may be involved in its incorporation into HMW multiprotein complexes.
To confirm these findings, studies of the phosphorylation of endogenous PDE3A by rPKAc in the absence or presence of [γ-32P]ATP or unlabeled ATP were carried out in vitro using immunoprecipitated PDE3A from solubilized myocardial membranes. As seen in Fig. 3D (upper panels), after immunoprecipitated PDE3A was incubated with [γ-32P]ATP in the absence (Ctrl) or presence of rPKAc or rPKAc plus PKI peptide, phosphorimaging of wet gels after SDS-PAGE combined with Western blotting demonstrated that both PDE3A1 and PDE3A2 could be markedly phosphorylated by rPKAc, with little if any increase in the phosphorylation of PDE3A3. PKI blocked phosphorylation of PDE3A1 and PDE3A2. As seen in Fig. 3D (lower panel), phosphorylation of immunoprecipitated PDE3A by rPKAc was accompanied by stimulation of PDE3A hydrolytic activity (35% compared with controls, p < 0.01). PKI blocked rPKAc-induced activation of PDE3A.
PDE3A Associates Phosphorylation-dependently with PLB, SERCA2, and AKAP18
We examined the effects of phosphorylation by rPKAc on the interaction of endogenous PDE3A with PLB, SERCA2, and AKAP18 (Figs. 4 and 5). Pooled and concentrated membrane LMW Superose 6 fractions (analogous to fractions 24–34 in Fig. 3A) were divided and incubated in phosphorylation buffer containing ATP and MgCl2 in the absence or presence of rPKAc and then re-chromatographed on Superose 6 (S6) columns (Fig. 4) or immunoprecipitated with anti-PDE3A-CT antibody (Fig. 5). As seen in Fig. 4, PDE3 activity in control-pooled membrane LMW fractions exhibited an apparent molecular mass of 700 kDa during re-chromatography on S6 columns (Fig. 4, LMW Control). After incubation with rPKAc, however, the apparent molecular mass of most of the eluted PDE3A1 and PDE3A2 was shifted to from LMW fractions to ≥3000 kDa (Fig. 4, LMW + PKA). Most of the PDE3A3, which was not phosphorylated or only weakly phosphorylated by rPKAc (Fig. 3), remained in the LMW peak. This indicates that phosphorylation by rPKAc (cf. Fig. 3D) correlates with the shift in elution of PDE3A1 and PDE3A2. As also seen in Fig. 4, shifts from LMW to the HMW fractions were observed as well for AKAP18, PLB, PKA-RII, and PKA catalytic subunits as well as PP1 and caveolin 3.
As seen in Fig. 5, incubation of pooled and concentrated membrane LMW fractions with rPKAc increased the co-immunoprecipitation of PDE3A with AKAP18 (not AKAP-LBC), SERCA2, PP2A, PP1, and cav3 but reduced the co-immunoprecipitation of PLB. These results suggest that phosphorylation of membrane-associated PDE3A promotes its integration into signalosomes containing AKAP18, SERCA2, and other proteins involved in the regulation of Ca2+ transients and myocardial contractility by cAMP. The decreased association of PLB with these proteins after phosphorylation by PKA is consistent with the idea that the integration of PLB into this complex is dependent upon its interactions with SERCA2 and AKPA18 and that these interactions are reduced by phosphorylation of PLB by PKA at Ser-16 (2, 3, 24).
In Fig. 4, the shift of the PDE3A3 isoform from LMW to HMW fractions after incubation with PKA cannot be explained by phosphorylation of PDE3A3, as it is not a substrate for PKA (cf. Fig. 3). Although PDE3A2 can be readily phosphorylated by rPKAc in vitro (Figs. 3D and 5) and this phosphorylation is correlated with its shift from LMW to HMW fractions (Fig. 4), endogenous HMW PDE3A2 is less highly phosphorylated than PDE3A1. It is possible that PDE3A3 forms heteromeric complexes with PDE3A1 and/or PDE3A2 and that phosphorylation of multiple proteins contributes to the incorporation of PDE3A isoforms into the endogenous SERCA2 regulatory signalosome. In addition, phosphorylation at more than one site in PDE3A isoforms may contribute to their shift in migration, as our previous work indicates that PDE3A isoforms interact directly or indirectly with a large number of other proteins (15).
Because murine PDE3A1 is a component of a SERCA2 regulatory signalosome in murine myocardium (2, 3) and because endogenous PDE3A1 is the most highly PKA-phosphorylated isoform in human myocardium (Fig. 3C), we studied its phosphorylation-dependent interactions with SERCA2 and AKAP18δ in more detail using recombinant forms of these proteins. The co-immunoprecipitation of both proteins with rhPDE3A1 was increased by phosphorylation of rhPDE3A1 by rPKAc (Figs. 6C and 7).
To gain insight into molecular mechanisms contributing to the phosphorylation-dependent interaction of rhPDE3A1 and rSERCA2, we generated PDE3A1 constructs with C- and N-terminal deletions and serine-to-alanine mutations at PKA sites in the PDE3A1 N terminus. As seen in Fig. 6C, rSERCA2 also co-immunoprecipitated with phosphorylated rhPDE3A-RD. rPDE3A-RD contains only the N-terminal portion of PDE3A1 (aa 146–484), including both its unique N-terminal extension and some of its shared sequence with PDE3A2). SERCA2 did not co-immunoprecipitate with rhPDE3A-Δ510, which is a recombinant form from which the first 510 aa of the PDE3A open reading frame were deleted and which is not phosphorylated by rPKAc. This indicated that interactions with rSERCA2 involved the N terminus of rhPDE3A1.
With at least four PKA consensus sites (RRX(S/T)) in the rhPDE3A1 N terminus, we inserted serine-to-alanine mutations in these sites: S292A/S293A (P1), S312A (P2), S428A (P3), S438A (P4), and S292A/S293A/S312A/S438A (P5). As seen in Fig. 6D, P1 and P5 mutations markedly diminished PKA-stimulated interactions with rSERCA2. PDE3A1 mutants P2 (S312A) and P3 (S428A) showed slightly decreased phosphorylation-dependent interactions with rSERCA2, whereas the PDE3A1 P4 mutation (S438A) had no effect (Fig. 6). These data indicate that Ser-292/Ser-293 is the major PKA site regulating the interaction of rhPDE3A1 with rSERCA2.
DISCUSSION
PDEs have critical roles in the compartmentation of cAMP signaling (25–29). A number of studies involving many different PDEs have demonstrated that these enzymes are recruited to specific intracellular multiprotein complexes (signalosomes) through protein-protein interactions and that as a result of this localization individual PDEs are able to regulate specific cAMP-mediated signaling pathways with great precision (25–29). Understanding the mechanisms by which PDEs are localized to signalosomes and the consequences of this localization is especially important in cardiac muscle, where inhibitors of the PDE3 family of enzymes are used to increase myocardial contractility in patients with heart failure. In earlier work we showed that inotropic responses to PDE3 inhibition in mice, which correlated with increases in intracellular Ca2+ transients, are attributable specifically to isoforms in the PDE3A subfamily and that PDE3A1 is a component of a murine SERCA2-regulatory signalosome (1–3). In the experiments described here, we have confirmed that human PDE3A1 is part of a similar SERCA2-, PLB- and AKAP18-containing signalosome localized to sarcomeric Z-bands in human cardiac myocytes. We also showed that the integration of PDE3A isoforms into this signalosome is phosphorylation-dependent and that the interaction of PDE3A1 with SERCA2 is dependent upon the phosphorylation of PDE3A1 at Ser-292/Ser-293, a sequence in its unique N-terminal extension.
Although both PDE3 and PDE4 have been found to co-immunoprecipitate with PLB in complexes from mouse and human myocardium (16), our studies show that PDE3-selective inhibition (but not PDE4 inhibition) potentiates the phosphorylation of PLB by endogenous PKA and stimulation of SERCA2 activity and Ca2+ uptake in SR-enriched vesicles prepared from human myocardium. This most likely reflects the higher amount of PDE3 activity relative to PDE4 activity in SR preparations from human myocardium. Taken together, our observations provide evidence for the physiologic and therapeutic importance of the association of PDE3A with the SERCA2-regulatory signalosome.
As noted above, previous studies of PDE3B showed a role for N-terminal phosphorylation in integrating adipocyte PDE3B into signalosomes in response to insulin or β3-adrenergic receptor agonists (5, 6, 10). N-terminal phosphorylation in response to PKA and PKC activation has also been shown to regulate the interactions of PDE3A isoforms with 14-3-3 and with other (as-yet-unidentified) proteins (15). The interactions with 14-3-3 were consequences of the phosphorylation of two sites in the N-terminal sequence common to both PDE3A1 and PDE3A2, Ser-312 and Ser-428; there was no evidence for the involvement of any other phosphorylation sites in regulating these protein-protein interactions. Our new results show that PDE3A1 interacts directly with both SERCA2 and AKAP18 in a phosphorylation-dependent manner. The interaction with SERCA2 is dependent upon the phosphorylation of Ser-292/Ser-293, a site unique to PDE3A1 that was not known to have a role in regulating protein-protein interactions. The fact that PDE3A2, which lacks this site, also interacts phosphorylation-dependently with SERCA2 indicates that phosphorylation of other sites in its sequence is responsible for this interaction and/or that the lack of the unique N terminus in PDE3A2 alters its phosphorylation and/or interactions with other signaling proteins. These findings add significantly to our understanding of the molecular mechanisms through which PDE3A1 is localized to intracellular signalosomes that control intracellular Ca2+ handling and suggest further that the interactions of each PDE3A isoform with SERCA2 can be selectively targeted.
As noted earlier, the sequences of PDE3A1 and PDE3A2 are identical save for the presence in the former of a unique 154-amino acid N-terminal extension. Understanding how this extension affects the function of PDE3A1 is, therefore, central to understanding the unique roles of the two isoforms. A growing body of evidence indicates that several molecular mechanisms are involved. The hydrophobic loops within this sequence restrict PDE3A1 distribution to cellular and intracellular membranes. Within these membranes, the protein-protein interactions of each isoform are clearly different; the 5-hydroxytryptamine receptor, for example, binds to PDE3A1 but not to PDE3A2 (30). The N-terminal extension also affects the tertiary structure of the downstream sequence; its presence converts Ser-428 from being a strong PKC site in PDE3A2 to a weaker one in PDE3A1. This contributes to the selective regulation of these isoforms by preferential phosphorylation of PDE3A1 at the protein binding site Ser-312 by PKA and preferential phosphorylation of PDE3A2 by PKC at the protein binding site Ser-428 (15). Because phosphorylation promotes the association of each isoform with different proteins, this is likely to be a factor in the two isoforms having distinct interactomes. In addition, phosphorylation by PKB of the Ser-292/Ser-293 site, which is found within the N-terminal extension unique to PDE3A1, stimulates cAMP hydrolytic activity, providing another mechanism by which PDE3A1 can be selectively regulated (31).
Our results showing that the interaction of PDE3A1 with SERCA2 is dependent upon the phosphorylation of Ser-292/Ser-293 reveal a new and important mechanism whereby the unique N-terminal extension of PDE3A1 regulates its function. The amino acid sequence surrounding this site, aa 288–294, is RRRRSSS, and all three serines can be phosphorylated in vitro under different conditions (31); hence, it is likely this site can be phosphorylated by different kinases activated in response to different upstream signals. Phosphorylation at Ser-292/Ser-293 may promote direct interactions of PDE3A1-binding proteins such as SERCA2 with this site. Alternatively, phosphorylation at Ser-292/Ser-293 may influence the conformation of PDE3A1 allosterically to promote binding to a different protein-protein interface. In either case, our results suggest that an agent capable of binding to PDE3A1 in its Ser-292/Ser-293-phosphorylated conformation may be able to inhibit its protein-protein interactions without affecting those of PDE3A2, thereby selectively inhibiting the integration of PDE3A1 into specific signalosomes.
Our findings add to the understanding of the mechanisms whereby PDE3A, a component of a SERCA2-containing signalosome, regulates cAMP-mediated changes in contractility in cardiac myocytes. Phosphorylation of PLB on Ser-16 causes its dissociation from SERCA2, relieving its inhibition of SERCA2 activity and increasing Ca2+ transport into the SR (24, 32, 33). These actions increase the amplitude of intracellular Ca2+ transients, which are attenuated in dilated cardiomyopathy (34). PKA-catalyzed phosphorylation of PLB at Ser-16, induced by isoproterenol, forskolin, or isobutylmethylxanthine, correlated with increased cardiac relaxation (24, 35). The interaction of PLB with AKAP18 is also reduced by phosphorylation of Ser-16 (3). Phosphorylation of PDE3A1 at Ser-292/Ser-293, which correlates with its recruitment into the SERCA2 regulatory signalosome, would tend to counteract this effect by reducing the concentration of cAMP in the proximity of this complex (Fig. 8). This combination of stimulation and inhibition of SERCA2 activity in response to PKA activation through separate mechanisms may permit a greater degree of “fine-tuning” of SR Ca2+ handling in response to β-adrenergic receptor agonists. It may also explain the synergism observed when PDE3 inhibitors are used in combination with β-adrenergic receptor agonists as inotropic agents. Because PP2A and PP1 are thought to be the principal phosphatases responsible for dephosphorylation of PKA substrates and PLB (36), the presence of PP1 and PP2A in the complex would be expected to catalyze the dephosphorylation of PDE3A, PLB, and other PKA substrates and return the SERCA2 complex to its basal state. Of note, AKAP18 associates with inhibitor I and thereby can control PP1 activity (37).
FIGURE 8.
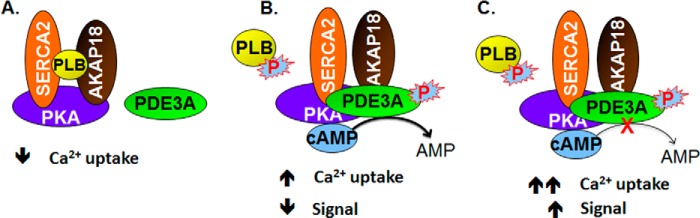
Model of the regulation of SERCA2 activity by cAMP and the AKAP18 and PLB-containing signalosome. A, components of the AKAP18/SERCA2/PLB signalosome are shown. B, in the absence of cAMP, SERCA2 was inhibited by its interaction with PLB. Activation of PKA by cAMP resulted in the phosphorylation of PLB and PDE3A (and, most likely, other molecules in the signalosome). The former dissociates from SERCA2, increasing SERCA2 activity, but the integration of phosphorylated PDE3A into the signalosome limits this effect by increasing hydrolysis of cAMP. PP1 and PP2A in the signalosome would be expected to catalyze the dephosphorylation of PDE3A, PLB, and other PKA substrates and return the SERCA2 complex to its basal state. C, PDE3 inhibition potentiates the effect of cAMP on SERCA2.
Our results may have therapeutic implications. Selective inhibition of the PDE3A isoforms associated with SERCA2 might allow the inotropic benefits of stimulating Ca2+ transport into the sarcoplasmic reticulum without the harmful effects of global inhibition of PDE3 activity (2, 38). Currently available PDE3 inhibitors, however, have little selectivity for PDE3A versus PDE3B isoforms, whose catalytic domains are similar, and no selectivity for individual PDE3A isoforms, whose catalytic domains are identical (39). Blocking the integration of PDE3A1 into SERCA2 regulatory complexes either by blocking its phosphorylation or by blocking the interactions of phosphorylated PDE3A1 with SERCA2 may be a way of targeting PDE3 activity in a specific microdomain in cardiac myocytes to stimulate SR Ca2+ uptake and produce inotropic actions without the adverse consequences that accompany diffuse increases in intracellular cAMP content (29, 38).
Acknowledgments
We acknowledge the professional skills and advice from Dr. Christian A. Combs (Light Microscopy Core Facility, NHLBI, National Institutes of Health (NIH)), Angel Aponte (Proteomics Core Facility, NHLBI), and Dr. Zu-Xi Yu (Pathology Core Facility, NHLBI).
This work was supported, in whole or in part, by National Institutes of Health NHLBI Intramural program. This work was also supported by medical research funds from the United States Department of Veterans Affairs and Foundation Leducq Grant 06CVD02 cAMP and an American Heart Association grant-in-aid (to M. M.); by Swedish Research Council Project 3362 (to E. D.); and by Deutsche Forschungsgemeinschaft Grant KL1415-4/2, the Else Kröner-Fresenius-Stiftung (2013_A145), and the German-Israeli Foundation (I-1210-286.13/2012) (to E. K. and F. G.).
- PDE3
- cyclic nucleotide phosphodiesterase 3
- AKAP
- A-kinase anchoring protein
- rAKAP
- recombinant AKAP
- HMW
- high molecular weight
- LMW
- low molecular weight
- SR
- sarcoplasmic reticulum
- signalosome
- macromolecular regulatory complex
- PKAc
- PKA catalytic subunit
- rPKAc
- recombinant PKAc
- PLB
- phospholamban
- PP2A
- protein phosphatase 2A
- PP1
- protein phosphatase 1
- rhPDE3A
- recombinant human PDE3A
- SERCA2
- sarcoplasmic reticulum Ca2+ ATPase 2
- rSERCA2
- recombinant SERCA2
- rAKAP18
- recombinant AKAP18
- CT domain
- C-terminal domain
- S6
- Superose 6
- aa
- amino acid(s).
REFERENCES
- 1. Sun B., Li H., Shakur Y., Hensley J., Hockman S., Kambayashi J., Manganiello V. C., Liu Y. (2007) Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 19, 1765–1771 [DOI] [PubMed] [Google Scholar]
- 2. Beca S., Ahmad F., Shen W., Liu J., Makary S., Polidovitch N., Sun J., Hockman S., Chung Y. W., Movsesian M., Murphy E., Manganiello V., Backx P. H. (2013) Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ. Res. 112, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lygren B., Carlson C. R., Santamaria K., Lissandron V., McSorley T., Litzenberg J., Lorenz D., Wiesner B., Rosenthal W., Zaccolo M., Taskén K., Klussmann E. (2007) AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. Embo. Rep. 8, 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stefan E., Wiesner B., Baillie G. S., Mollajew R., Henn V., Lorenz D., Furkert J., Santamaria K., Nedvetsky P., Hundsrucker C., Beyermann M., Krause E., Pohl P., Gall I., MacIntyre A. N., Bachmann S., Houslay M. D., Rosenthal W., Klussmann E. (2007) Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 18, 199–212 [DOI] [PubMed] [Google Scholar]
- 5. Ahmad F., Lindh R., Tang Y., Weston M., Degerman E., Manganiello V. C. (2007) Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem. J. 404, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmad F., Lindh R., Tang Y., Ruishalme I., Ost A., Sahachartsiri B., Strålfors P., Degerman E., Manganiello V. C. (2009) Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the β3-adrenergic receptor agonist CL316243: effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem. J. 424, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Onuma H., Osawa H., Yamada K., Ogura T., Tanabe F., Granner D. K., Makino H. (2002) Identification of the insulin-regulated interaction of phosphodiesterase 3B with 14-3-3 β protein. Diabetes 51, 3362–3367 [DOI] [PubMed] [Google Scholar]
- 8. Palmer D., Jimmo S. L., Raymond D. R., Wilson L. S., Carter R. L., Maurice D. H. (2007) Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J. Biol. Chem. 282, 9411–9419 [DOI] [PubMed] [Google Scholar]
- 9. Nilsson R., Ahmad F., Swärd K., Andersson U., Weston M., Manganiello V., Degerman E. (2006) Plasma membrane cyclic nucleotide phosphodiesterase 3B (PDE3B) is associated with caveolae in primary adipocytes. Cell. Signal. 18, 1713–1721 [DOI] [PubMed] [Google Scholar]
- 10. Rondinone C. M., Carvalho E., Rahn T., Manganiello V. C., Degerman E., Smith U. P. (2000) Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J. Biol. Chem. 275, 10093–10098 [DOI] [PubMed] [Google Scholar]
- 11. Perino A., Ghigo A., Ferrero E., Morello F., Santulli G., Baillie G. S., Damilano F., Dunlop A. J., Pawson C., Walser R., Levi R., Altruda F., Silengo L., Langeberg L. K., Neubauer G., Heymans S., Lembo G., Wymann M. P., Wetzker R., Houslay M. D., Iaccarino G., Scott J. D., Hirsch E. (2011) Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell. 42, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilson L. S., Baillie G. S., Pritchard L. M., Umana B., Terrin A., Zaccolo M., Houslay M. D., Maurice D. H. (2011) A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J. Biol. Chem. 286, 16285–16296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pozuelo Rubio M., Campbell D. G., Morrice N. A., Mackintosh C. (2005) Phosphodiesterase 3A binds to 14-3-3 proteins in response to PMA-induced phosphorylation of Ser-428. Biochem. J. 392, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunter R. W., Mackintosh C., Hers I. (2009) Protein kinase C-mediated phosphorylation and activation of PDE3A regulate cAMP levels in human platelets. J. Biol. Chem. 284, 12339–12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vandeput F., Szabo-Fresnais N., Ahmad F., Kho C., Lee A., Krall J., Dunlop A., Hazel M. W., Wohlschlegel J. A., Hajjar R. J., Houslay M. D., Manganiello V. C., Movsesian M. A. (2013) Selective regulation of cyclic nucleotide phosphodiesterase PDE3A isoforms. Proc. Natl. Acad. Sci. U.S.A. 110, 19778–19783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richter W., Xie M., Scheitrum C., Krall J., Movsesian M. A., Conti M. (2011) Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 106, 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Babick A. P., Cantor E. J., Babick J. T., Takeda N., Dhalla N. S., Netticadan T. (2004) Cardiac contractile dysfunction in J2N-k cardiomyopathic hamsters is associated with impaired SR function and regulation. Am. J. Physiol. Cell Physiol. 287, C1202–C1208 [DOI] [PubMed] [Google Scholar]
- 18. Manganiello V., Vaughan M. (1973) An effect of insulin on cyclic adenosine 3′:5′-monophosphate phosphodiesterase activity in fat cells. J. Biol. Chem. 248, 7164–7170 [PubMed] [Google Scholar]
- 19. Mishra S., Sabbah H. N., Rastogi S., Imai M., Gupta R. C. (2005) Reduced sarcoplasmic reticulum Ca2+ uptake and increased Na+-Ca2+ exchanger expression in left ventricle myocardium of dogs with progression of heart failure. Heart Vessels 20, 23–32 [DOI] [PubMed] [Google Scholar]
- 20. Kirchgesser M., Dahlmann N. (1990) A colorimetric assay for the determination of acid nucleoside triphosphatase activity. J. Clin. Chem. Clin. Biochem. 28, 407–411 [DOI] [PubMed] [Google Scholar]
- 21. Wechsler J., Choi Y. H., Krall J., Ahmad F., Manganiello V. C., Movsesian M. A. (2002) Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J. Biol. Chem. 277, 38072–38078 [DOI] [PubMed] [Google Scholar]
- 22. Shakur Y., Takeda K., Kenan Y., Yu Z. X., Rena G., Brandt D., Houslay M. D., Degerman E., Ferrans V. J., Manganiello V. C. (2000) Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3): Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J. Biol. Chem. 275, 38749–38761 [DOI] [PubMed] [Google Scholar]
- 23. Vandeput F., Wolda S. L., Krall J., Hambleton R., Uher L., McCaw K. N., Radwanski P. B., Florio V., Movsesian M. A. (2007) Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J. Biol. Chem. 282, 32749–32757 [DOI] [PubMed] [Google Scholar]
- 24. MacLennan D. H., Kranias E. G. (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577 [DOI] [PubMed] [Google Scholar]
- 25. Yan C., Miller C. L., Abe J. (2007) Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ. Res. 100, 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mongillo M., McSorley T., Evellin S., Sood A., Lissandron V., Terrin A., Huston E., Hannawacker A., Lohse M. J., Pozzan T., Houslay M. D., Zaccolo M. (2004) Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 95, 67–75 [DOI] [PubMed] [Google Scholar]
- 27. Houslay M. D., Baillie G. S., Maurice D. H. (2007) cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 100, 950–966 [DOI] [PubMed] [Google Scholar]
- 28. Zaccolo M. (2009) cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 158, 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maurice D. H., Ke H., Ahmad F., Wang Y., Chung J., Manganiello V. C. (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 13, 290–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weninger S., Van Craenenbroeck K., Cameron R. T., Vandeput F., Movsesian M. A., Baillie G. S., Lefebvre R. A. (2014) Phosphodiesterase 4 interacts with the 5-HT4(b) receptor to regulate cAMP signaling. Cell. Signal. 26, 2573–2582 [DOI] [PubMed] [Google Scholar]
- 31. Han S. J., Vaccari S., Nedachi T., Andersen C. B., Kovacina K. S., Roth R. A., Conti M. (2006) Protein kinase B/Akt phosphorylation of PDE3A and its role in mammalian oocyte maturation. EMBO J. 25, 5716–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simmerman H. K., Jones L. R. (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 78, 921–947 [DOI] [PubMed] [Google Scholar]
- 33. Sande J. B., Sjaastad I., Hoen I. B., Bøkenes J., Tønnessen T., Holt E., Lunde P. K., Christensen G. (2002) Reduced level of serine 16-phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc. Res. 53, 382–391 [DOI] [PubMed] [Google Scholar]
- 34. Kawase Y., Hajjar R. J. (2008) The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat. Clin. Pract. Cardiovasc. Med. 5, 554–565 [DOI] [PubMed] [Google Scholar]
- 35. Kuschel M., Karczewski P., Hempel P., Schlegel W. P., Krause E. G., Bartel S. (1999) Ser16 prevails over Thr17 phospholamban phosphorylation in the β-adrenergic regulation of cardiac relaxation. Am. J. Physiol. 276, H1625–H1633 [DOI] [PubMed] [Google Scholar]
- 36. Schwoerer A. P., Neuber C., Schmechel A., Melnychenko I., Mearini G., Boknik P., Kirchhefer U., Schmitz W., Ehmke H., Eschenhagen T., El-Armouche A. (2008) Mechanical unloading of the rat heart involves marked changes in the protein kinase-phosphatase balance. J. Mol. Cell Cardiol. 45, 846–852 [DOI] [PubMed] [Google Scholar]
- 37. Singh A., Redden J. M., Kapiloff M. S., Dodge-Kafka K. L. (2011) The large isoforms of A-kinase anchoring protein 18 mediate the phosphorylation of inhibitor-1 by protein kinase A and the inhibition of protein phosphatase 1 activity. Mol. Pharmacol. 79, 533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Movsesian M. A. (2003) PDE3 inhibition in dilated cardiomyopathy: reasons to reconsider. J. Card. Fail. 9, 475–480 [DOI] [PubMed] [Google Scholar]
- 39. Hambleton R., Krall J., Tikishvili E., Honeggar M., Ahmad F., Manganiello V. C., Movsesian M. A. (2005) Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J. Biol. Chem. 280, 39168–39174 [DOI] [PubMed] [Google Scholar]