

Background: The neurotoxin 1-methyl-4-phenylpyridinium (MPP+) kills dopaminergic neurons by a variety of mechanisms.
Results: MPP+ affects dopamine (DA) vesicular storage, plasma membrane transport, and catabolic breakdown, leading to accumulation of cytosolic DA and neurotoxicity.
Conclusion: Alterations in DA homeostasis account for ∼30% of MPP+-mediated toxicity.
Significance: Comprehensive analysis of the effects of MPP+ helps to understand the mechanisms underlying the development of Parkinson disease.
Keywords: Dopamine, Mouse, Neurotoxin, Neurotransmitter, Parkinson Disease, Vesicles, MPTP, MPP+
Abstract
1-Methyl-4-phenylpyridinium (MPP+), the active metabolite of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, selectively kills dopaminergic neurons in vivo and in vitro via a variety of toxic mechanisms, including mitochondrial dysfunction, generation of peroxynitrite, induction of apoptosis, and oxidative stress due to disruption of vesicular dopamine (DA) storage. To investigate the effects of acute MPP+ exposure on neuronal DA homeostasis, we measured stimulation-dependent DA release and non-exocytotic DA efflux from mouse striatal slices and extracellular, intracellular, and cytosolic DA (DAcyt) levels in cultured mouse ventral midbrain neurons. In acute striatal slices, MPP+ exposure gradually decreased stimulation-dependent DA release, followed by massive DA efflux that was dependent on MPP+ concentration, temperature, and DA uptake transporter activity. Similarly, in mouse midbrain neuronal cultures, MPP+ depleted vesicular DA storage accompanied by an elevation of cytosolic and extracellular DA levels. In neuronal cell bodies, increased DAcyt was not due to transmitter leakage from synaptic vesicles but rather to competitive MPP+-dependent inhibition of monoamine oxidase activity. Accordingly, monoamine oxidase blockers pargyline and l-deprenyl had no effect on DAcyt levels in MPP+-treated cells and produced only a moderate effect on the survival of dopaminergic neurons treated with the toxin. In contrast, depletion of intracellular DA by blocking neurotransmitter synthesis resulted in ∼30% reduction of MPP+-mediated toxicity, whereas overexpression of VMAT2 completely rescued dopaminergic neurons. These results demonstrate the utility of comprehensive analysis of DA metabolism using various electrochemical methods and reveal the complexity of the effects of MPP+ on neuronal DA homeostasis and neurotoxicity.
Introduction
The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)4 was first identified to induce parkinsonism in humans (1, 2) and has since been widely used to produce Parkinson disease-like degeneration in animal models (3, 4). Following systemic administration, MPTP crosses the blood-brain barrier by lipophilic diffusion and is metabolized by astroglial monoamine oxidase (MAO) B to 1-methyl-4-phenyl-2,3-dihydropyridinium, which undergoes spontaneous oxidation to the active toxin 1-methyl-4-phenylpyridinium (MPP+) (5, 6). MPP+ has been shown to exit astroglia via OCT3 (organic cation transporter 3) (7) or the extraneuronal monoamine transporter (8) and to accumulate in dopaminergic neurons via dopamine uptake transporter (DAT)-mediated uptake (9, 10). In the neuronal cytosol, the inside-negative mitochondrial membrane potential drives the uptake of MPP+ into these organelles, where the toxin inhibits complex I of the electron transport chain, leading to ATP depletion and generation of reactive oxygen species (11–14). With additional mechanisms, including generation of peroxynitrite (15), inhibition of peroxidase Prx2 (16), and destabilization of microtubules (17–19), cumulative stress induced by MPP+ leads to neuronal apoptosis (4, 20, 21).
Several theories address the highly selective pattern of neurodegeneration observed in animals exposed to MPTP. The ability of the toxin to accumulate specifically in dopamine (DA) neurons via DAT-mediated uptake can target them for MPTP-mediated toxicity. Alternatively, dopaminergic cells may be more sensitive to MPP+ due to intrinsic factors, including poorly myelinated long and highly branched axons (22), cell-selective expression of the Cav1.3 (23) and GirK2 (24, 25) channels, or activation of KATP channels (26) or DLP1 (dynamin-like protein 1) (27). DA itself may be a risk factor that renders cells vulnerable to stress (28, 29), although the contribution of DA homeostasis in MPP+-induced toxicity remains controversial (30). Following DAT-mediated uptake, VMAT2 (vesicular monoamine transporter 2) accumulates MPP+ in synaptic vesicles (31, 32), leading to a redistribution of stored DA to the cytosol. This is followed by DAT-mediated reverse transport of DA (11), which is thought to be responsible for massive release of DA into the extracellular space after administration of MPTP in vivo (33–35). However, many of the proposed effects of MPP+ have never been demonstrated experimentally.
We studied the time and concentration dependences of the alterations in DA metabolic pools in MPP+-treated acute striatal slices and primary cultures of midbrain dopaminergic neurons. Our findings indicate that MPP+ affects DA vesicular storage, DAT-mediated transport, and catabolic breakdown, leading to the accumulation of DAcyt and neurotoxicity.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were used for slice preparations, and ventral midbrain primary cultures were used for neurotoxicity and HPLC experiments. For intracellular patch electrochemistry (IPE), cultures were generated from transgenic mice that express GFP under the control of the rat tyrosine hydroxylase (TH) promoter (TH-GFP+/−) (36). VMAT2 overexpression experiments were performed on ventral midbrain neurons from Sprague-Dawley rats. All animals were used in accordance with the National Institutes of Health guidelines for the use of live animals, and the animal protocols were approved by the Institutional Animal Care and Use Committee of Columbia University.
Acute Striatal Slice Preparation and Measurement of DA Release by Fast-scan Cyclic Voltammetry
7–9-week-old mice were decapitated, and 300-μm coronal slices that contained cortex and striatum were cut on a Leica VT1200 vibratome (Leica Biosystems Nussloch GmbH, Nussloch, Germany) in ice-cold cutting saline containing 125 mm NaCl, 2.5 mm KCl, 26 mm NaHCO3, 0.3 mm KH2PO4, 3.3 mm MgSO4, 0.8 mm NaH2PO4, and 10 mm glucose (pH 7.2–7.4, 292–296 mosmol/liter). Slices were allowed to recover for 1–2 h at 37 °C in oxygenated (95% O2, 5% CO2) recording saline containing 125 mm NaCl, 2.5 mm KCl, 26 mm NaHCO3, 0.3 mm KH2PO4, 2.4 mm CaCl2, 1.3 mm MgSO4, 0.8 mm NaH2PO4, and 10 mm glucose (pH 7.2–7.4, 292–296 mosmol/liter). Fast-scan cyclic voltammetry recordings were performed with 5-μm cylinder carbon fiber electrodes (CFEs) positioned at the dorsolateral striatum ∼50 μm below the exposed surface. Striatal slices were electrically stimulated using a bipolar stainless steel electrode placed at a distance of ∼100 μm from the recording electrode. Square pulses of 0.4-ms duration were produced by an ISO-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Stimulus magnitude was selected by plotting a current-response curve and selecting the minimum value that produced the maximum response.
Triangular voltage ramps from a holding potential of −450 mV to +800 mV over 8.5 ms (scan rate of 295 mV/ms) were applied to the CFEs at 100-ms intervals. Current was recorded with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA) filtered with a 10-kHz low-pass Bessel filter and digitized at 25 kHz (ITC-18 board, InstruTECH, Great Neck, NY). Triangular wave generation and data acquisition were controlled by a locally written computer routine in IGOR Pro (WaveMetrics, Lake Oswego, OR). Background-subtracted cyclic voltammograms obtained in DA solutions of known concentration served to calibrate the electrodes and to identify released DA.
Cell Cultures
Ventral midbrain dopaminergic neurons from postnatal day 0–2 mice were dissected, dissociated, and plated on a monolayer of cortical astrocytes at a plating density of ∼100,000 cells/cm2 as described (37, 38). Experiments were conducted 7–14 days post-plating.
Adenoviral Vector Construction and Transfection
HA-tagged VMAT2 cDNA was first subcloned into the shuttle vector pTet-EF and then co-infected with donor virus DNA (ψ5) into HEK293 cells expressing Cre recombinase, and the resultant VMAT2-HA-harboring adenovirus was purified and stored at ∼8000 pfu/μl as described previously (39). Rat primary ventral midbrain cultures were incubated with 1 μl of VMAT2 adenovirus diluted in 100 μl of medium for 5 h, and 2 ml of medium was added to each dish; ψ5 alone was used as a negative (empty virus) control. As described previously (39), overexpression of VMAT2 was confirmed by immunostaining for both VMAT2 and the HA epitope placed in the luminal loop between transmembrane domains 1 and 2, which does not interfere with transporter activity or subcellular localization. Transfection efficiency reached 80–90% with expression of VMAT2 protein observed in both dopaminergic and non-dopaminergic neurons (40). Overexpression of VMAT2 in midbrain neurons increased the overall transporter levels measured by Western blotting and enhanced the depolarization-evoked release of DA and the total intracellular DA by ∼2-fold relative to control cells as assessed by HPLC (41).
HPLC Measurements of DA and 3,4-Dihydroxyphenylacetic Acid (DOPAC) Concentrations
Whole-cell (cytosolic + vesicular) DA and DOPAC levels were determined by HPLC with electrochemical detection as described (42, 43). Molar amounts of metabolites were calculated from areas under HPLC peaks using calibration curves and normalized to protein concentrations in each sample. Incubations with MPP+ were performed at room temperature.
Measurements of DAcyt by IPE
Measurements of neuronal DAcyt were performed as described previously (40). Briefly, a specially designed electrode holder allowed us to house a 5-μm polyethylene-coated CFE inside the glass patch pipette. Voltage ramps from a holding potential of −450 mV to +800 mV over 8.5 ms (scan rate of 295 mV/ms) were applied to the CFE at 100-ms intervals using a subroutine locally written in IGOR Pro. Subtraction voltammograms were generated, and DA concentration at the maximum of the oxidation wave was calculated using calibration curves generated for CFEs with different detection surface areas (44). The initial DA concentration in the cellular cytosol was calculated using the cell body volume and the volume of the pipette tip estimated from photographs taken before each recording. Glass patch pipettes were back-filled with the same extracellular saline as the bath solutions, which reduced the background current drifts because of the exchange of ions between the patch pipette and the bath. The saline contained 118.6 mm NaCl, 3 mm KCl, 2.7 mm Na-HEPES, 3.3 mm HEPES, 1.2 mm MgCl2·6H2O, 2 mm CaCl2, and 10 mm glucose (pH 7.2–7.4, 250–255 mosmol/liter). All drugs and inhibitors were present in the bath and in the patch pipette.
The sensitivity of IPE to DA depends on multiple factors, including the signal-to-noise ratio, the exposed area of the CFE, the geometry of the cell, and the proximity of the cell to the CFE (44). Because the concentration of DAcyt was below the detection limits in untreated dopaminergic neurons, cultures were preincubated with 100 μm l-3,4-dihydroxyphenylalanine (l-DOPA) before and during the recordings. The presence of l-DOPA increased the baseline oxidation current but did not otherwise interfere with IPE measurements. Within each experiment, the same CFE was used for measurements from experimental and control groups of cells. Incubations with all drugs were done at 37 °C, and l-DOPA was always added 30 min before the start of the recordings. IPE measurements were performed at room temperature.
Neurotoxicity Assays
Cells were preincubated with VMAT2-HA-harboring adenovirus for 1 day and with various DA metabolism inhibitors for the times indicated before the application of MPP+. Following a 2-day incubation with the toxin, immunostaining of 4% paraformaldehyde-fixed cultures was performed using mouse anti-TH antibodies (1:1000; Chemicon, Temecula, CA), followed by secondary antibodies conjugated with Alexa Fluor 488 (1:200; Molecular Probes, Eugene, OR). Images were acquired on a conventional fluorescence microscopy setup (Axiovert 100 microscope, Carl Zeiss MicroImaging, Inc. Thornwood, NY) equipped with Zeiss AxioCam MRm camera and FITC and rhodamine filter sets (Chroma Technology, Bellows Falls, VT). The total number of immunoreactive neurons in a culture dish was tallied and analyzed by counting the number of immunoreactive cells in 20 fields of view at ×200 magnification (Plan-Neofluar ×20 objective, ∼0.8-mm2 viewing field) and taking the average as a representative for each dish (40). The counts were performed by an observer blinded to the experimental treatments.
In Vitro MAO Activity Assay
Measurements of the activity of recombinant MAO-A in the presence of various competitive and non-competitive inhibitors were performed using a luminescent MAO-Glo assay kit (Promega, Madison, WI) according to the manufacturer's instructions.
Data Analysis
Statistical analysis was performed with Prism 4 (GraphPad Software, La Jolla, CA) using one-way analysis of variance (ANOVA), followed by Tukey's post-hoc test for comparisons across multiple groups or two-way ANOVA with the Bonferroni post-test for the paired data. In some cases, data in each experiment performed on sister cultures were normalized to values in control samples and pooled for statistical analysis.
RESULTS
Effects of MPP+ on DA Release from Mouse Striatal Slices
MPP+ is a substrate for both DAT and VMAT2, leading to its accumulation inside dopaminergic synaptic vesicles and leakage of stored transmitter (9, 11, 45). To investigate the effects of MPP+ on vesicular DA homeostasis, we measured changes in stimulation-dependent and stimulation-independent DA release from acute dorsal striatal slices treated with 10 and 50 μm MPP+ (Fig. 1). Every 2 min, the slices were stimulated by an electrical pulse that elicited exocytotic DA release from striatal terminals. Evoked release of DA can be maintained for several hours in control slices (data not shown); however, MPP+ decreased the amplitude (Imax) of DA spikes within 20–40 min after the start of the perfusion in a concentration-dependent manner (Fig. 1, A, B, and F), consistent with the depletion of vesicular neurotransmitter storage. The release signals also became wider (Fig. 1, E and H, and Table 1), confirming that MPP+ inhibited DAT-mediated reuptake of released DA.
FIGURE 1.

Cyclic voltammetry recordings of DA release from striatal slices exposed to MPP+. A–C, representative traces from slices exposed to 10 μm MPP+ (A) or 50 μm MPP+ (B and C) at the indicated temperatures. Perfusion of MPP+ was started at time 0. Stimulation-dependent DA release evoked by electrical stimulation every 2 min is visible as sharp spikes, whereas a much taller and wider stimulation-independent DA overflow is observed later. C, the DAT blocker nomifensine (10 μm) was perfused at the indicated times when the depletion of evoked DA release had reached its maximum. D, representative peaks of DA release in slices treated with 10 or 50 μm MPP+ for 20 min at 37 °C. E, same signals as in D normalized to their maximum current to more clearly indicate the differences in t½ (dotted line). F and G, exposure to MPP+ resulted in a time-dependent reduction of the amplitude (Imax) of evoked DA release. This effect reached its maximum faster in slices treated with higher MPP+ doses and exhibited different kinetics at 37 °C (F) and 24 °C (G). H, changes in the half-width (t½, where t½ indicates width of a peak at half of its height of evoked DA release events at 6 and 20 min after the start of 10 μm (green) or 50 μm (magenta) MPP+ treatment at 24 or 37 °C. See Table 1 for statistical analysis.
TABLE 1.
Changes in parameters of stimulation-dependent DA release from striatal slices treated with 10 or 50 μm MPP+ at 37 or 24 °C
Values are normalized to parameters of release events before MPP+ application and represent mean ± S.E. The number of independent recordings is given in parentheses. Average Imax and t½ from untreated slices were 1.7 ± 0.4 μm and 340 ± 25 ms at 37 °C (n = 10) and 3.0 ± 0.6 μm and 690 ± 120 ms at 24 °C (n = 10), respectively.
| Imax | t½ | |
|---|---|---|
| % | % | |
| 37 °C | ||
| 10 μm MPP+ (n = 6) | ||
| 6 min | 82 ± 9a | 102 ± 3 |
| 20 min | 49 ± 7a | 129 ± 11a |
| 50 μm MPP+ (n = 4) | ||
| 6 min | 80 ± 5a | 118 ± 6a |
| 20 min | 11 ± 4a | 137 ± 10a |
| 24 °C | ||
| 10 μm MPP+ (n = 4) | ||
| 6 min | 110 ± 2a,b | 110 ± 2a |
| 20 min | 77 ± 10a,b | 145 ± 14a |
| 50 μm MPP+ (n = 6) | ||
| 6 min | 112 ± 8a,b | 134 ± 6a |
| 20 min | 28 ± 8a | 166 ± 11a,b |
a p < 0.05 from corresponding untreated controls by two-way ANOVA.
b p < 0.05 from the corresponding time point at 37 °C by two-way ANOVA.
MPP+-mediated effects on evoked DA release demonstrated a steep temperature dependence. First, both the Imax and t½ of stimulation-dependent release signals were ∼2-fold smaller at 37 °C than at 24 °C (1.7 ± 0.4 μm and 340 ± 25 ms (n = 10) versus 3.0 ± 0.6 μm and 690 ± 120 ms (n = 10), respectively; p < 0.05 by t test for both pairs of values). Second, in slices incubated with MPP+ at 24 °C, evoked DA release decreased at a slower rate than at 37 °C (Fig. 1, F and G). Third, at 24 °C, there was an increase in the Imax of DA release spikes over the first 10 min of drug exposure (Fig. 1G and Table 1). These data indicate that, consistent with other findings (46), DAT-mediated uptake of both DA and MPP+ was decreased at lower temperature, thus leading to a substantially delayed effect of MPP+ on vesicular DA depletion.
Once the depletion of evoked DA release had reached its maximum, a far greater stimulation-independent peak of DA overflow appeared. This peak was larger and occurred earlier when a higher concentration of MPP+ was used (Fig. 1, A and B, and Table 2). Surprisingly, DA efflux was never observed in slices incubated with MPP+ at 24 °C, but began shortly after the temperature was increased to 37 °C (Fig. 1B). Because the effects of MPP+ were similar to those reported for amphetamine exposure (Ref. 47; also see “Discussion”), we investigated the possibility that the delayed stimulation-independent peak of DA overflow was caused by reverse transport via DAT (48–50). We exploited the temperature dependence of MPP+-mediated DA overflow to confirm its reliance on reverse transport by applying the DAT inhibitor nomifensine (10 μm) immediately prior to changing the temperature from 24 to 37 °C. This protocol ensured that MPP+ entered dopaminergic terminals, as evident from the toxin-induced decrease in stimulation-dependent DA release (Fig. 1C). In agreement with the DAT-dependent mechanism of DA efflux, nomifensine prevented DA overflow from MPP+-treated slices (Fig. 1C and Table 2).
TABLE 2.
Parameters of stimulation-independent DA efflux from striatal slices treated with 10 or 50 μm MPP+ at 37 or 24 °C
Nomifensine was added at 10 μm 30 min after the start of MPP+ incubation when slices were still at 24 °C. Values represent mean ± S.E. The number of independent recordings is given in parentheses. NA, not applicable.
| Imax | Rise timea | Time with MPP+b | |
|---|---|---|---|
| μm | min | min | |
| 37 °C | |||
| 10 μm MPP+ (n = 10) | 8.5 ± 2.8 | 5.8 ± 0.5 | 48 ± 7 |
| 50 μm MPP+ (n = 15) | 13.5 ± 1.7c | 5.5 ± 0.5 | 29 ± 3c |
| 24 °C for 60–120 min, then 37 °Cd | |||
| 10 μm MPP+ (n = 8) | 27.2 ± 5.0e | 6.4 ± 0.7 | 18 ± 3e |
| 50 μm MPP+ (n = 8) | 29.8 ± 3.3e | 7.7 ± 0.6 | 14 ± 2e |
| 50 μm MPP+ with Nomifensine (n = 3) | 6.2 ± 1.1f | NAg | 19 ± 1g |
a Rise time was calculated as duration from the first detectable DA overflow signal to its maximum.
b Time from the start of MPP+ perfusion to the maximum of DA efflux.
c p < 0.05 from 10 μm MPP+ at 37 °C by one-way ANOVA.
d Efflux was never observed in slices incubated with MPP+ at 24 °C (Fig. 1B).
e p < 0.05 with both 10 and 50 μm MPP+ at 37 °C by one-way ANOVA.
f p < 0.05 with 50 μm MPP+ at both 37 and 24 °C by one-way ANOVA.
g There was no well defined peak of DA efflux in the presence of nomifensine (Fig. 1C).
Effects of MPP+ on DA Homeostasis in Mouse Primary Dopaminergic Neuronal Cultures
We next employed cultured mouse neurons, which, in contrast to striatal slices, have both the terminals and the cell bodies exposed to MPP+ during treatment. First, we verified the effects of the toxin on total intracellular (cytosolic + vesicular) and extracellular DA and DOPAC levels measured by HPLC. Consistent with previous reports (11), MPP+ caused a time-dependent decrease in intracellular DA levels while increasing the extracellular DA concentration (Fig. 2). Because >90% of the intracellular DA is stored inside the vesicles (40, 44, 51), these data further confirm that MPP+ redistributes DA from vesicles to the cytosol, followed by DA extrusion from neurons via reverse transport by DAT. In contrast to the slice data, however, MPP+-mediated efflux of DA was observed at room temperature.
FIGURE 2.
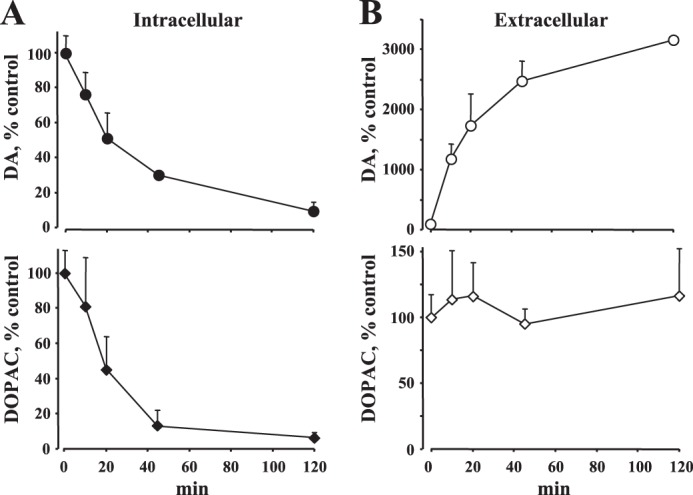
Changes in total cellular DA and DOPAC levels. HPLC measurements of intracellular (A) and extracellular (B) DA and DOPAC concentrations in cultured midbrain neurons exposed to 50 μm MPP+ at room temperature. Note the similarity of the time course of changes in DA pools in cultured cells and in the striatal slice (see Fig. 1).
We then measured the concentration of DAcyt in neuronal cell bodies using IPE (44). To differentiate DA from other intracellular metabolites, we used a cyclic voltammetric mode of detection (52) that generates a signature current-voltage dependence for oxidizable compounds (Fig. 3A). As ventral midbrain cultures contain different neuronal populations, including dopaminergic, GABAergic, and glutamatergic cells, we employed cultures from transgenic mice that overexpress enhanced GFP under the control of the TH promoter (36). As we have shown previously, DAcyt in untreated cultured mouse neurons is below the detection limits of IPE, which is ∼50 nm (40). However, in midbrain neurons treated with 50 μm MPP+ for 2 h, we could detect DAcyt at an average level of 100 ± 30 nm (n = 14 cells).
FIGURE 3.

Effect of MPP+ on DAcyt homeostasis. A, oxidation profile of MPP+ shows that the presence of the toxin did not interfere with cyclic voltammetry measurements of DA. B, MPP+ (10 μm, 1 h) induced a large increase in DAcyt, which was prevented by the DAT inhibitor nomifensine (Nmf; 5 μm, 15-min pretreatment). *, p < 0.01 compared with all other groups by one-way ANOVA. Ctrl, control. C, time course of changes in DAcyt following cell treatment with 10 or 50 μm MPP+. The curves are statistically different by two-way ANOVA (p < 0.01). D, DAcyt in neurons exposed to 10 μm MPP+ for 1 h in the presence and absence of the VMAT2 inhibitor reserpine (Res; 2 μm, 60-min pretreatment) or the MAO inhibitor pargyline (PGL; 10 μm, 15-min pretreatment). In B–D, cells were pretreated with 100 μm l-DOPA for 1 h before the recordings to elevate DAcyt to levels detectable by IPE. Incubations with all drugs were done at 37 °C, whereas recordings were performed at room temperature. E, an in vitro assay of MAO-A activity performed at room temperature using a chemiluminescent substrate shows that MPP+ (IC50 = 1 μm) is a more potent enzyme inhibitor than pargyline (IC50 = 5.5 μm) or DA (IC50 = 150 μm). F, the Lineweaver-Burk plot demonstrates that MPP+ is a competitive inhibitor of MAO.
Pretreatment of cultures with the DA precursor l-DOPA (100 μm, 30 min; the same treatment was used in all other IPE experiments) elevated DAcyt to ∼25 μm. Addition of MPP+ (10 μm, added 30 min before l-DOPA) to these cells further increased DAcyt, a response that could be completely prevented by nomifensine, which inhibited DAT-mediated uptake of MPP+ into the cells (Fig. 3B). A time course of changes induced by MPP+ showed that DAcyt increased sharply during the first 2 h of treatment, followed by a plateau and a slow decrease in DA levels over the next 24 h (Fig. 3C).
Several mechanisms could account for the increased DAcyt, including activation of DA synthesis via TH or aromatic l-amino acid decarboxylase, inhibition of DA degradation by MAO, or redistribution of DA from vesicles to the cytosol. In a previous study of midbrain neurons, we reported that inhibition of VMAT2 does not significantly affect DAcyt in neuronal somas (40). We speculated that there are too few vesicles in neuronal cell bodies to cause any significant impact on DAcyt by VMAT2-mediated uptake of the transmitter, although the influence of disrupted DA storage could be different in neuronal terminals, where the ratio of vesicular to cytosolic DA is much larger (53). Consistent with this, the VMAT2 uptake blocker reserpine did not affect DAcyt in neurons treated with MPP+ (Fig. 3D), ruling out the leakage of vesicular DA as a cause of increased DAcyt. We noted, however, that in MPP+-treated cells, intracellular DOPAC levels measured by HPLC did not increase, as would be expected with elevated DAcyt (Fig. 2). It is therefore possible that MPP+ is a MAO inhibitor. Indeed, measurements of DAcyt in MPP+-treated neurons in the presence of pargyline demonstrated that whereas the MAO blocker significantly increased DAcyt levels when applied alone, the effect was not additive to that produced by MPP+ (Fig. 3D). We then directly assessed the ability of MPP+ to inhibit MAO using an in vitro bioluminescence assay kit. MPP+ inhibited MAO-A activity with an IC50 of ∼1 μm, which was more potent than either the non-competitive inhibitor pargyline or the competitive inhibitor/substrate DA (Fig. 3E). Analysis of the kinetic parameters of MAO-A inhibition indicated that neurotoxin is a competitive inhibitor of the enzyme (Fig. 3F). Thus, the data point to MAO inhibition as the main cause of increased DAcyt in MPP+-treated cells.
Role of DA in MPP+-induced Toxicity
Mesencephalic dopaminergic neurons treated with MPP+ for 2 h showed morphological abnormalities, including a disrupted nuclear envelope and swollen neurites (Fig. 4, A and B), followed by MPP+ concentration-dependent cell death after 2 days of exposure (Fig. 4C). As disruption of DA homeostasis is one of several insults by which the toxin is suggested to compromise neuronal survival, we explored whether protection could be achieved by preventing the up-regulation of DAcyt. Pretreatment of cultures with the TH inhibitor α-methyltyrosine for 48 h depleted total intracellular DA content by ∼70% (from 280 ± 33 to 67 ± 27 pmol/mg of protein (n = 3), p < 0.05 by t test) as assessed by HPLC measurements. DA depletion also provided partial protection of midbrain neurons from MPP+-mediated toxicity, increasing cell survival from 40 ± 2 to 62 ± 1% (Fig. 4E). We previously demonstrated that overexpression of VMAT2 effectively protects DA midbrain neurons from l-DOPA-induced toxicity (40). Similarly, virus-mediated overexpression of VMAT2 completely rescued dopaminergic neurons from MPP+ (Fig. 4F).
FIGURE 4.

MPP+-induced toxicity in cultured midbrain dopaminergic neurons. A and B, representative images of cultured midbrain neuronal somas with proximal dendrites (left panels) and axons (right panels) of control neurons (A) and those exposed to 10 μm MPP+ (B). After 2 h at 37 °C (same conditions for experiments in E–G), cells were fixed and immunostained for TH. Note the disruption of the nuclear envelope and the abnormal morphology of neuronal processes in MPP+-treated cultures. C, exposure to MPP+ resulted in dose-dependent death of TH-positive ventral midbrain neurons. D, correlation between DAcyt dose, calculated as areas under the curves in Fig. 3C, and neurotoxicity. The dashed line represents the linear fit of the data. E, rescue of neurons from 10 μm MPP+-mediated toxicity by the TH inhibitor α-methyltyrosine (αMT; 500 μm, added 48 h before MPP+). *, p < 0.05 by one-way ANOVA against MPP+ only. F, neuroprotection of TH+ neurons in rat midbrain cultures infected with adenovirus carrying VMAT2 or empty vector for 1 day and then exposed to varying concentrations of MPP+ for 2 days. *, p < 0.05 versus other groups by two-way ANOVA. G, effect of pargyline (PGL), l-deprenyl (DPN), and propargylamine (PPGA) (all at 10 μm, added 1 h before MPP+) on the survival of dopaminergic neurons exposed to 10 μm MPP+. None of the compounds were neurotoxic when applied alone. *, p < 0.05 compared with control (Ctrl) and MPP+ alone by one-way ANOVA.
It has been hypothesized that blockade of neuronal MAO decreases DA-mediated toxicity by reducing the production of its reactive catabolic products, 3,4-dihydroxyphenylacetaldehyde and H2O2. We therefore investigated whether the MAO blockers pargyline (IC50 = 5.5 μm) (Fig. 3E) and l-deprenyl (IC50 = 0.8 μm) (54) can protect cells from MPP+. Although these inhibitors slightly increased cell survival in neurotoxin-treated cultures (Fig. 4G), the effect was comparable with that produced by N-propargylamine, their structural analog, which is a much less potent MAO inhibitor (IC50 = 28 μm) (55).
DISCUSSION
Although MPTP provides a model of Parkinson disease-like degeneration that selectively targets substantia nigra dopaminergic neurons, the contribution of different toxicity pathways in achieving the specificity of MPP+ for the subset of neurons has been a subject of discussion (17, 30, 56–58). Our study provides a detailed analysis of DA homeostasis in MPP+-treated neurons, demonstrating the complexity of the mechanisms of MPP+ action and the utility of comprehensive analysis of DA metabolism using various electrochemical techniques. MPP+ induced major alterations in vesicular, cytosolic, and extracellular DA pools, which accounted for up to 30% of its toxicity.
Effects of MPP+ on Stimulation-dependent and Stimulation-independent Release of DA
To our knowledge, this is the first study of the effects of MPP+ on stimulation-dependent and stimulation-independent DA release in striatal slices. The data demonstrate that MPP+ induced three consecutive effects: inhibition of DAT activity, followed by depletion of vesicular DA, followed by reverse transport of DA via DAT. Inhibition of DAT-mediated DA reuptake was noticeable almost immediately after the start of MPP+ perfusion, as evident from the increased t½ of evoked DA release events (Table 1), in agreement with previous reports (59, 60). Next, MPP+ completely inhibited evoked DA releases within 20–40 min of exposure, depending on the concentration of the toxin and temperature (Fig. 1). This is consistent with the depletion of vesicular DA storage that results from competitive inhibition of DA uptake by VMAT2 and the disruption of the electrochemical gradient inside synaptic vesicles (61). At 24 °C, the differences between the effects of MPP+ on DAT function and vesicular DA storage were particularly pronounced, as there was a larger increase in the t½ of release events (Fig. 1H), and the decrease in Imax was preceded by its temporal increase (Fig. 1G), similar to the effects of the DAT inhibitor nomifensine (47). This time course confirms that MPP+ has an immediate effect on the plasma transporter, whereas vesicular DA depletion only occurs following DAT- and VMAT2-mediated transport of the toxin, both of which are temperature-sensitive (46, 62).
Surprisingly, the magnitude of DAT inhibition by MPP+ was very small compared with the effect of another DAT competitive inhibitor, amphetamine. A modest ∼50% increase in the t½ of evoked DA spikes was observed in the presence of MPP+, whereas amphetamine produces a 5–7-fold increase in t½ within 5 min of exposure (47). This difference is puzzling considering that both drugs inhibit evoked DA release with similar kinetics, thus apparently accumulating at comparable intracellular concentrations.
Often coinciding with complete vesicular DA depletion, a large stimulation-independent DA overflow was observed. The presence of this peak depended on DAT activity, confirming that it results from DA reverse transport via DAT, similar to DA overflow described for amphetamine (47). Another similarity to amphetamine exposure5 was that MPP+-induced DA efflux was undetectable at 24 °C, arguing against the idea that this peak occurs as a result of cytotoxicity and demonstrating that both normal DAT activity and reversal are temperature-dependent. In contrast to slice data, DA efflux occurred at room temperature when dopaminergic neuronal cultures were exposed to MPP+ (Fig. 2), in agreement with previous reports (11).
DA efflux was larger and occurred earlier when slices were incubated with higher MPP+ concentrations at 37 °C. When, however, DA efflux was induced by a temperature switch following a 1–2-h preincubation at 24 °C, the parameters of DA overflow in slices exposed to 10 and 50 μm MPP+ were no longer different (Table 2). Evidently, the effect of MPP+ on intracellular DA pools had reached its maximum at both MPP+ concentrations during the preincubation, and the switch to 37 °C was only required to initiate DA efflux.
As discussed above, the effects of MPP+ were similar to those caused by amphetamine; both compounds inhibit DAT, deplete vesicular DA, and induce DAT-mediated efflux of the transmitter, albeit with different potencies and time kinetics. However, these similarities are at odds with the differences in neurotoxicity caused by these drugs; whereas MPP+ produces severe nigrostriatal neurodegeneration within several days of exposure, amphetamine, even when chronically abused at high doses, affects only dopaminergic terminals, with little effect on the survival of neurons (63, 64). Additional studies will be needed to address the difference in MPP+ and amphetamine effects.
Effects of MPP+ on DAcyt and Neurotoxicity
A long-standing hypothesis of neurodegeneration in Parkinson disease postulates that the buildup of DAcyt and associated oxyradical stress make dopaminergic neurons more susceptible (11, 29, 65–69). l-DOPA and exogenously added DA can be toxic to dopaminergic neurons in vitro (28, 40, 70–75), and several reports confirm that a buildup of DAcyt is sufficient to induce progressive neurodegeneration in rodents (67, 76, 77).
MPP+ has been suspected to increase DAcyt based on indirect evidence, such as HPLC measurements of cellular DA contents (11) and an increase in 5-cysteinyl-DA in animals treated with MPTP (78). Using IPE, a technique that can directly measure DAcyt, we confirmed that MPP+ exposure increased DAcyt in both the absence and presence of the DA precursor l-DOPA. DAcyt increased sharply during the first 2 h of exposure and then decreased slowly over the next 24 h. A higher concentration of MPP+ produced a larger elevation of DAcyt, leading to an ∼25% difference in DAcyt exposure dose as calculated from the area under the curves shown in Fig. 3C (1500 ± 103 and 1870 ± 150 μm DA per h for 10 and 50 μm MPP+, respectively). We have shown previously that the DAcyt dose is proportional to neurodegeneration induced by l-DOPA in the same culture system (40). This seems to also be the case for MPP+-induced degeneration, as there was a linear correlation between DAcyt dose and neurotoxicity (Fig. 4D), suggesting an explanation for the difference in cell death caused by increasing MPP+ levels. It should be noted that DAcyt measurements were performed in the presence of l-DOPA to overcome the detection limitations of cyclic voltammetry and therefore might not represent DA concentrations found in MPP+-treated cells. However, l-DOPA treatment would only augment differences in untreated cells, provided that these differences are downstream from the conversion of l-DOPA to DA.
Although the kinetics of MPP+-induced DAcyt increase and vesicular DA depletion were similar, in neuronal somas, the blockade of MAO rather than redistribution of stored DA was the main reason for the DAcyt increase. Consistent with MPP+ being a potent MAO blocker, the commonly used MAO inhibitor pargyline, which increased neuronal cytosolic transmitter levels when applied alone, had no additional effect on DAcyt in the presence of MPP+ (Fig. 3D). It is therefore unlikely that MAO inhibitors can provide neuroprotection from MPP+-mediated toxicity by further affecting MAO activity. Although we indeed observed that pargyline and l-deprenyl had small but significant effects on the survival of primary dopaminergic neurons exposed to MPP+, their structural analog N-propargylamine provided similar protection (Fig. 4G). This suggests that the presence of the propargylamine moiety on pargyline and l-deprenyl may rescue cells by other mechanisms, including preservation of the mitochondrial membrane and attenuation of pro-apoptotic molecular markers (79–83). It is also possible that other MAO inhibitors, such as rasagiline, have a different mechanism of action.
In agreement with a previous report (11), pharmacological depletion of DA levels by ∼70% provided partial protection against MPP+-mediated toxicity, although it is impossible to know to what extent DAcyt levels were affected by the TH blockade. In contrast, overexpression of VMAT2, which has been shown to increase vesicular DA uptake (41) and decrease DAcyt (40), completely rescued dopaminergic neurons from MPP+, similar to neuroprotection from l-DOPA toxicity in the same culture system (40). This is in agreement with the detoxifying nature of the transporter, which was cloned for its ability to protect cells from MPP+-mediated toxicity by removing it from the cytosol into the vesicles (45, 77).
The role for DA in genuine Parkinson disease pathogenesis is likely to differ in important ways from that in the MPTP toxin model, but because this neurotoxin is widely used in studies of possible therapeutic interventions, it is important to understand its toxicity mechanisms. In agreement with previous findings (30), our data show that the presence of DA by itself does not fully explain the selective vulnerability of catecholaminergic neurons to MPP+ exposure, implicating the involvement of other mechanisms, including inhibition of complex I, generation of reactive oxygen species and peroxynitrite, destabilization of microtubules, and cell-selective expression of some proteins and channels. However, as an additional stress factor that becomes more important in substantia nigra and locus coeruleus neurons, which rely on Ca2+-driven pacemaking activity (23, 40, 84), DA may shift the balance from cell survival to cell death.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS075222 from NINDS (to E. V. M.) and the NINDS Morris K. Udall Center of Excellence Research Grant (to D. S.). This work was also supported by the Parkinson's Disease and JPB Foundations (to D. S.) and by a fellowship from the Finnish Cultural Foundation (to A. P.).
S. J. Choi, unpublished data.
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MAO
- monoamine oxidase
- MPP+
- 1-methyl-4-phenylpyridinium
- DAT
- dopamine uptake transporter
- DA
- dopamine
- DAcyt
- cytosolic DA
- IPE
- intracellular patch electrochemistry
- TH
- tyrosine hydroxylase
- CFE
- carbon fiber electrode
- DOPAC
- 3,4-dihydroxyphenylacetic acid
- l-DOPA
- l-3,4-dihydroxyphenylalanine
- ANOVA
- analysis of variance
- VMAT2
- vesicular monoamine transporter 2.
REFERENCES
- 1. Langston J. W., Ballard P., Tetrud J. W., Irwin I. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 [DOI] [PubMed] [Google Scholar]
- 2. Davis G. C., Williams A. C., Markey S. P., Ebert M. H., Caine E. D., Reichert C. M., Kopin I. J. (1979) Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1, 249–254 [DOI] [PubMed] [Google Scholar]
- 3. Jackson-Lewis V., Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson's disease. Nat. Protoc. 2, 141–151 [DOI] [PubMed] [Google Scholar]
- 4. Bové J., Perier C. (2012) Neurotoxin-based models of Parkinson's disease. Neuroscience 211, 51–76 [DOI] [PubMed] [Google Scholar]
- 5. Ransom B. R., Kunis D. M., Irwin I., Langston J. W. (1987) Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+. Neurosci. Lett. 75, 323–328 [DOI] [PubMed] [Google Scholar]
- 6. Brooks W. J., Jarvis M. F., Wagner G. C. (1989) Astrocytes as a primary locus for the conversion MPTP into MPP+. J. Neural Transm. 76, 1–12 [DOI] [PubMed] [Google Scholar]
- 7. Cui M., Aras R., Christian W. V., Rappold P. M., Hatwar M., Panza J., Jackson-Lewis V., Javitch J. A., Ballatori N., Przedborski S., Tieu K. (2009) The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 8043–8048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Inazu M., Takeda H., Matsumiya T. (2003) Expression and functional characterization of the extraneuronal monoamine transporter in normal human astrocytes. J. Neurochem. 84, 43–52 [DOI] [PubMed] [Google Scholar]
- 9. Javitch J. A., D'Amato R. J., Strittmatter S. M., Snyder S. H. (1985) Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. U.S.A. 82, 2173–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bezard E., Gross C. E., Fournier M. C., Dovero S., Bloch B., Jaber M. (1999) Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol. 155, 268–273 [DOI] [PubMed] [Google Scholar]
- 11. Lotharius J., O'Malley K. L. (2000) The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J. Biol. Chem. 275, 38581–38588 [DOI] [PubMed] [Google Scholar]
- 12. Rossetti Z. L., Sotgiu A., Sharp D. E., Hadjiconstantinou M., Neff N. H. (1988) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and free radicals in vitro. Biochem. Pharmacol. 37, 4573–4574 [DOI] [PubMed] [Google Scholar]
- 13. Nicklas W. J., Vyas I., Heikkila R. E. (1985) Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenylpyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 36, 2503–2508 [DOI] [PubMed] [Google Scholar]
- 14. Schapira A. H. (1994) Evidence for mitochondrial dysfunction in Parkinson's disease–a critical appraisal. Mov. Disord. 9, 125–138 [DOI] [PubMed] [Google Scholar]
- 15. Przedborski S., Jackson-Lewis V., Yokoyama R., Shibata T., Dawson V. L., Dawson T. M. (1996) Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 93, 4565–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qu D., Rashidian J., Mount M. P., Aleyasin H., Parsanejad M., Lira A., Haque E., Zhang Y., Callaghan S., Daigle M., Rousseaux M. W., Slack R. S., Albert P. R., Vincent I., Woulfe J. M., Park D. S. (2007) Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron 55, 37–52 [DOI] [PubMed] [Google Scholar]
- 17. Burke R. E., O'Malley K. (2013) Axon degeneration in Parkinson's disease. Exp. Neurol. 246, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qureshi H. Y., Paudel H. K. (2011) Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and α-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem. 286, 5055–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cappelletti G., Maggioni M. G., Maci R. (1999) Influence of MPP+ on the state of tubulin polymerisation in NGF-differentiated PC12 cells. J. Neurosci. Res. 56, 28–35 [DOI] [PubMed] [Google Scholar]
- 20. Karunakaran S., Saeed U., Mishra M., Valli R. K., Joshi S. D., Meka D. P., Seth P., Ravindranath V. (2008) Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J. Neurosci. 28, 12500–12509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vila M., Przedborski S. (2003) Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 365–375 [DOI] [PubMed] [Google Scholar]
- 22. Matsuda W., Furuta T., Nakamura K. C., Hioki H., Fujiyama F., Arai R., Kaneko T. (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan C. S., Guzman J. N., Ilijic E., Mercer J. N., Rick C., Tkatch T., Meredith G. E., Surmeier D. J. (2007) 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 24. Chung C. Y., Seo H., Sonntag K. C., Brooks A., Lin L., Isacson O. (2005) Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum. Mol. Genet. 14, 1709–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lüscher C., Slesinger P. A. (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liss B., Haeckel O., Wildmann J., Miki T., Seino S., Roeper J. (2005) K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 8, 1742–1751 [DOI] [PubMed] [Google Scholar]
- 27. Wang X., Su B., Liu W., He X., Gao Y., Castellani R. J., Perry G., Smith M. A., Zhu X. (2011) DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: implications for Parkinson's disease. Aging Cell 10, 807–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hastings T. G., Berman S. B. (1999) Dopamine-induced toxicity and quinone modification of proteins: implications for Parkinson's disease. in Role of Catechol Quinone Species in Cellular Toxicity (Creveling C. R., ed) pp. 69–89, F. P. Graham Publishing, Inc., Johnson City, TN [Google Scholar]
- 29. Sulzer D., Surmeier D. J. (2013) Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov. Disord. 28, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hasbani D. M., Perez F. A., Palmiter R. D., O'Malley K. L. (2005) Dopamine depletion does not protect against acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in vivo. J. Neurosci. 25, 9428–9433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Speciale S. G., Liang C. L., Sonsalla P. K., Edwards R. H., German D. C. (1998) The neurotoxin 1-methyl-4-phenylpyridinium is sequestered within neurons that contain the vesicular monoamine transporter. Neuroscience 84, 1177–1185 [DOI] [PubMed] [Google Scholar]
- 32. Moriyama Y., Amakatsu K., Futai M. (1993) Uptake of the neurotoxin, 4-methylphenylpyridinium, into chromaffin granules and synaptic vesicles: a proton gradient drives its uptake through monoamine transporter. Arch. Biochem. Biophys. 305, 271–277 [DOI] [PubMed] [Google Scholar]
- 33. Lau Y. S., Fung Y. K., Trobough K. L., Cashman J. R., Wilson J. A. (1991) Depletion of striatal dopamine by the N-oxide of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurotoxicology 12, 189–199 [PubMed] [Google Scholar]
- 34. Sirinathsinghji D. J., Heavens R. P., McBride C. S. (1988) Dopamine-releasing action of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 1-methyl-4-phenylpyridine (MPP+) in the neostriatum of the rat as demonstrated in vivo by the push-pull perfusion technique: dependence on sodium but not calcium ions. Brain Res. 443, 101–116 [DOI] [PubMed] [Google Scholar]
- 35. Serra P. A., Pluchino S., Marchetti B., Desole M. S., Miele E. (2008) The MPTP mouse model: cues on DA release and neural stem cell restorative role. Parkinsonism Relat. Disord. 14, Suppl. 2, S189–S193 [DOI] [PubMed] [Google Scholar]
- 36. Sawamoto K., Nakao N., Kobayashi K., Matsushita N., Takahashi H., Kakishita K., Yamamoto A., Yoshizaki T., Terashima T., Murakami F., Itakura T., Okano H. (2001) Visualization, direct isolation, and transplantation of midbrain dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 6423–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burke R. E., Antonelli M., Sulzer D. (1998) Glial cell line-derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture. J. Neurochem. 71, 517–525 [DOI] [PubMed] [Google Scholar]
- 38. Rayport S., Sulzer D., Shi W. X., Sawasdikosol S., Monaco J., Batson D., Rajendran G. (1992) Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J. Neurosci. 12, 4264–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krantz D. E., Peter D., Liu Y., Edwards R. H. (1997) Phosphorylation of a vesicular monoamine transporter by casein kinase II. J. Biol. Chem. 272, 6752–6759 [DOI] [PubMed] [Google Scholar]
- 40. Mosharov E. V., Larsen K. E., Kanter E., Phillips K. A., Wilson K., Schmitz Y., Krantz D. E., Kobayashi K., Edwards R. H., Sulzer D. (2009) Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pothos E. N., Larsen K. E., Krantz D. E., Liu Y., Haycock J. W., Setlik W., Gershon M. D., Edwards R. H., Sulzer D. (2000) Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J. Neurosci. 20, 7297–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feigin A., Fukuda M., Dhawan V., Przedborski S., Jackson-Lewis V., Mentis M. J., Moeller J. R., Eidelberg D. (2001) Metabolic correlates of levodopa response in Parkinson's disease. Neurology 57, 2083–2088 [DOI] [PubMed] [Google Scholar]
- 43. Larsen K. E., Fon E. A., Hastings T. G., Edwards R. H., Sulzer D. (2002) Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 22, 8951–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mosharov E. V., Gong L. W., Khanna B., Sulzer D., Lindau M. (2003) Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J. Neurosci. 23, 5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Y., Peter D., Roghani A., Schuldiner S., Privé G. G., Eisenberg D., Brecha N., Edwards R. H. (1992) A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70, 539–551 [DOI] [PubMed] [Google Scholar]
- 46. Xie T., McCann U. D., Kim S., Yuan J., Ricaurte G. A. (2000) Effect of temperature on dopamine transporter function and intracellular accumulation of methamphetamine: implications for methamphetamine-induced dopaminergic neurotoxicity. J. Neurosci. 20, 7838–7845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmitz Y., Lee C. J., Schmauss C., Gonon F., Sulzer D. (2001) Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J. Neurosci. 21, 5916–5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sulzer D., Maidment N. T., Rayport S. (1993) Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J. Neurochem. 60, 527–535 [DOI] [PubMed] [Google Scholar]
- 49. Sulzer D., Chen T. K., Lau Y. Y., Kristensen H., Rayport S., Ewing A. (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J. Neurosci. 15, 4102–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jones S. R., Gainetdinov R. R., Wightman R. M., Caron M. G. (1998) Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 18, 1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chien J. B., Wallingford R. A., Ewing A. G. (1990) Estimation of free dopamine in the cytoplasm of the giant dopamine cell of Planorbis corneus by voltammetry and capillary electrophoresis. J. Neurochem. 54, 633–638 [DOI] [PubMed] [Google Scholar]
- 52. Michael D. J., Wightman R. M. (1999) Electrochemical monitoring of biogenic amine neurotransmission in real time. J. Pharm. Biomed. Anal. 19, 33–46 [DOI] [PubMed] [Google Scholar]
- 53. Mosharov E. V. (2014) Regulation of DA homeostasis and role of VMAT2 in DA-induced neurodegeneration. in Handbook of Neurotoxicity (Kostrzewa R., ed) pp. 973–994, Springer, New York [Google Scholar]
- 54. Youdim M. B., Gross A., Finberg J. P. (2001) Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol. 132, 500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yi H., Maruyama W., Akao Y., Takahashi T., Iwasa K., Youdim M. B., Naoi M. (2006) N-Propargylamine protects SH-SY5Y cells from apoptosis induced by an endogenous neurotoxin, N-methyl(R)salsolinol, through stabilization of mitochondrial membrane and induction of anti-apoptotic Bcl-2. J. Neural Transm. 113, 21–32 [DOI] [PubMed] [Google Scholar]
- 56. Jackson-Lewis V., Smeyne R. J. (2005) MPTP and SNpc DA neuronal vulnerability: role of dopamine, superoxide and nitric oxide in neurotoxicity. Neurotox. Res. 7, 193–202 [DOI] [PubMed] [Google Scholar]
- 57. Dauer W., Przedborski S. (2003) Parkinson's disease: mechanisms and models. Neuron 39, 889–909 [DOI] [PubMed] [Google Scholar]
- 58. Choi W. S., Kruse S. E., Palmiter R. D., Xia Z. (2008) Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. U.S.A. 105, 15136–15141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huguet F., Page G., Morel P., Tallineau C., Piriou A. (1997) MPTP toxicity in rat striatal slices: dopamine uptake alteration does not appear to be related to lipid peroxidation. Toxicology 122, 93–99 [DOI] [PubMed] [Google Scholar]
- 60. Barc S., Page G., Fauconneau B., Barrier L., Huguet F. (2001) A new in vitro approach for investigating the MPTP effect on DA uptake. Neurochem. Int. 38, 243–248 [DOI] [PubMed] [Google Scholar]
- 61. Sulzer D., Pothos E. N. (2000) Regulation of quantal size by presynaptic mechanisms. Rev. Neurosci. 11, 159–212 [DOI] [PubMed] [Google Scholar]
- 62. Volz T. J., Hanson G. R., Fleckenstein A. E. (2006) Measurement of kinetically resolved vesicular dopamine uptake and efflux using rotating disk electrode voltammetry. J. Neurosci. Methods 155, 109–115 [DOI] [PubMed] [Google Scholar]
- 63. Chang L., Alicata D., Ernst T., Volkow N. (2007) Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction 102, 16–32 [DOI] [PubMed] [Google Scholar]
- 64. Moszczynska A., Fitzmaurice P., Ang L., Kalasinsky K. S., Schmunk G. A., Peretti F. J., Aiken S. S., Wickham D. J., Kish S. J. (2004) Why is parkinsonism not a feature of human methamphetamine users? Brain 127, 363–370 [DOI] [PubMed] [Google Scholar]
- 65. Edwards R. H. (1993) Neural degeneration and the transport of neurotransmitters. Ann. Neurol. 34, 638–645 [DOI] [PubMed] [Google Scholar]
- 66. Gainetdinov R. R., Fumagalli F., Wang Y. M., Jones S. R., Levey A. I., Miller G. W., Caron M. G. (1998) Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J. Neurochem. 70, 1973–1978 [DOI] [PubMed] [Google Scholar]
- 67. Caudle W. M., Richardson J. R., Wang M. Z., Taylor T. N., Guillot T. S., McCormack A. L., Colebrooke R. E., Di Monte D. A., Emson P. C., Miller G. W. (2007) Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Uhl G. R. (1998) Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease. Ann. Neurol. 43, 555–560 [DOI] [PubMed] [Google Scholar]
- 69. Pifl C., Rajput A., Reither H., Blesa J., Cavada C., Obeso J. A., Rajput A. H., Hornykiewicz O. (2014) Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 34, 8210–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sulzer D., Zecca L. (2000) Intraneuronal dopamine-quinone synthesis: a review. Neurotox. Res. 1, 181–195 [DOI] [PubMed] [Google Scholar]
- 71. Pardo B., Mena M. A., Casarejos M. J., Paíno C. L., De Yébenes J. G. (1995) Toxic effects of l-DOPA on mesencephalic cell cultures: protection with antioxidants. Brain Res. 682, 133–143 [DOI] [PubMed] [Google Scholar]
- 72. Mytilineou C., Han S. K., Cohen G. (1993) Toxic and protective effects of l-DOPA on mesencephalic cell cultures. J. Neurochem. 61, 1470–1478 [DOI] [PubMed] [Google Scholar]
- 73. Keller J. N., Hanni K. B., Markesbery W. R. (2000) Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech. Ageing Dev. 113, 61–70 [DOI] [PubMed] [Google Scholar]
- 74. Montine T. J., Farris D. B., Graham D. G. (1995) Covalent crosslinking of neurofilament proteins by oxidized catechols as a potential mechanism of Lewy body formation. J. Neuropathol. Exp. Neurol. 54, 311–319 [DOI] [PubMed] [Google Scholar]
- 75. Whitehead R. E., Ferrer J. V., Javitch J. A., Justice J. B. (2001) Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem. 76, 1242–1251 [DOI] [PubMed] [Google Scholar]
- 76. Chen L., Ding Y., Cagniard B., Van Laar A. D., Mortimer A., Chi W., Hastings T. G., Kang U. J., Zhuang X. (2008) Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lohr K. M., Bernstein A. I., Stout K. A., Dunn A. R., Lazo C. R., Alter S. P., Wang M., Li Y., Fan X., Hess E. J., Yi H., Vecchio L. M., Goldstein D. S., Guillot T. S., Salahpour A., Miller G. W. (2014) Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 9977–9982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Teismann P., Tieu K., Choi D. K., Wu D. C., Naini A., Hunot S., Vila M., Jackson-Lewis V., Przedborski S. (2003) Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 100, 5473–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gerlach M., Riederer P., Youdim M. B. (1992) The molecular pharmacology of l-deprenyl. Eur. J. Pharmacol. 226, 97–108 [DOI] [PubMed] [Google Scholar]
- 80. Jenner P., Langston J. W. (2011) Explaining ADAGIO: a critical review of the biological basis for the clinical effects of rasagiline. Mov. Disord. 26, 2316–2323 [DOI] [PubMed] [Google Scholar]
- 81. Naoi M., Maruyama W. (2009) Functional mechanism of neuroprotection by inhibitors of type B monoamine oxidase in Parkinson's disease. Expert Rev. Neurother. 9, 1233–1250 [DOI] [PubMed] [Google Scholar]
- 82. Sharma S. K., Carlson E. C., Ebadi M. (2003) Neuroprotective actions of Selegiline in inhibiting 1-methyl-4-phenylpyridinium ion (MPP+)-induced apoptosis in SK-N-SH neurons. J. Neurocytol. 32, 329–343 [DOI] [PubMed] [Google Scholar]
- 83. Le W., Jankovic J., Xie W., Kong R., Appel S. H. (1997) (−)-Deprenyl protection of 1-methyl-4 phenylpyridium ion (MPP+)-induced apoptosis independent of MAO-B inhibition. Neurosci. Lett. 224, 197–200 [DOI] [PubMed] [Google Scholar]
- 84. Williams J. T., North R. A., Shefner S. A., Nishi S., Egan T. M. (1984) Membrane properties of rat locus coeruleus neurones. Neuroscience 13, 137–156 [DOI] [PubMed] [Google Scholar]