

Background: Akt signaling is activated by G protein-coupled receptors (GPCRs) via the PI3K-mTORC2 signaling cascade, but mechanistic insight is lacking.
Results: GPCR signaling promotes lysosomal and ESCRT-dependent degradation of DEPTOR, an antagonist of mTORC2, that regulates Akt activation and signaling.
Conclusion: ESCRTs regulate signaling by promoting degradation of a cytosolic antagonist of signaling.
Significance: This study begins to identify the signaling pathways governing the physiological roles of ESCRTs and Akt.
Keywords: Adrenergic Receptor, Akt PKB, CXC Chemokine Receptor Type 4 (CXCR-4), Endosomal Sorting Complexes Required for Transport (ESCRT), Epidermal Growth Factor Receptor (EGFR), G Protein-coupled Receptor (GPCR), Heterotrimeric G Protein, Insulin Receptor, Lysosome, Mammalian Target of Rapamycin (mTOR)
Abstract
G protein-coupled receptor (GPCR) signaling mediates many cellular functions, including cell survival, proliferation, and cell motility. Many of these processes are mediated by GPCR-promoted activation of Akt signaling by mammalian target of rapamycin complex 2 (mTORC2) and the phosphatidylinositol 3-kinase (PI3K)/phosphoinositide-dependent kinase 1 (PDK1) pathway. However, the molecular mechanisms by which GPCRs govern Akt activation by these kinases remain poorly understood. Here, we show that the endosomal sorting complex required for transport (ESCRT) pathway mediates Akt signaling promoted by the chemokine receptor CXCR4. Pharmacological inhibition of heterotrimeric G protein Gαi or PI3K signaling and siRNA targeting ESCRTs blocks CXCR4-promoted degradation of DEPTOR, an endogenous antagonist of mTORC2 activity. Depletion of ESCRTs by siRNA leads to increased levels of DEPTOR and attenuated CXCR4-promoted Akt activation and signaling, consistent with decreased mTORC2 activity. In addition, ESCRTs likely have a broad role in Akt signaling because ESCRT depletion also attenuates receptor tyrosine kinase-promoted Akt activation and signaling. Our data reveal a novel role for the ESCRT pathway in promoting intracellular signaling, which may begin to identify the signal transduction pathways that are important in the physiological roles of ESCRTs and Akt.
Introduction
Signaling by the CXC chemokine receptor 4 (CXCR4) promoted by its cognate ligand CXCL12 is important during development and in the adult (1–4). CXCR4 signaling has also been linked to several pathologies, including WHIM (warts, hypogammaglobulinemia, infections, myelokathexis) syndrome and cancer progression (5, 6). Yet, despite the importance in health and disease, the molecular mechanisms governing CXCR4 signaling remain poorly understood. CXCR4 belongs to the superfamily of G protein-coupled receptors (GPCRs)3 and couples with the heterotrimeric guanine nucleotide-binding protein Gαi and the associated dimer consisting of β and γ (Gβγ) (7). Typical of GPCRs, activation of CXCR4 by CXCL12 promotes release of GDP from the Gα subunit and binding to GTP, which in turn leads to the release of the Gβγ heterodimer. The GTP-bound Gαi and the released Gβγ activate various effector molecules and signaling pathways (7). Two notable signaling pathways activated by CXCR4 are the phosphatidylinositol 3-kinase (PI3K)-Akt and mitogen-activated protein kinase, such as extracellular signal-regulated kinases-1 and -2 (ERK-1/2), signaling pathways. Akt and ERK-1/2 signaling play important roles in CXCR4-promoted cell survival and migration (7). We recently reported that CXCR4-promoted ERK-1/2 activation is spatially restricted to caveolae and is mediated by the E3 ubiquitin ligase AIP4 and the adaptor protein STAM1 (8). However, the molecular mechanisms mediating CXCR4-promoted Akt signaling remain poorly understood.
Akt is a serine/threonine kinase belonging to the AGC family of protein kinases (9). Upon activation, Akt phosphorylates a diverse array of proteins involved in cell survival, growth, proliferation, cell metabolism, and cell motility (9). Despite the importance of Akt signaling in health and disease, the mechanisms governing its activation by signaling receptors remain poorly understood. GPCR-promoted Akt activation is typically mediated by the Gβγ heterodimer, which binds directly to PI3Kβ (10, 11) or PI3Kγ (12). This interaction is generally considered to be confined to the inner leaflet of the plasma membrane where PI3K catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) to phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) (13). PtdIns(3,4,5)P3 serves as a docking site for the pleckstrin homology domains of PDK1 (PtdIns(3,4,5)P3-dependent protein kinase) and Akt, leading to their plasma membrane recruitment and PDK1 phosphorylation of Akt at threonine residue 308 (Thr-308 of Akt1). Akt is also phosphorylated by mTORC2 (mechanistic or mammalian target of rapamycin complex 2) at serine residue 473 (Ser-473 of Akt1) (14). How GPCR signaling leads to activation of mTORC2 remains virtually unknown.
mTORC2 is a multiprotein complex consisting of mTOR, Rictor, mSin1, mLst8, and DEPTOR plus other recently defined subunits (15). mTOR, mLst8, and DEPTOR are also found in a distinct multisubunit kinase complex referred to as mTORC1, which also consists of Raptor and PRAS40 (15). DEPTOR (DEP-domain containing mTOR-interacting protein) negatively regulates the activity of mTORC2 and mTORC1 (15). DEP (Dishevelled, EGL-10, pleckstrin) domains have been linked to regulation of GPCR signaling (16). DEPTOR interacts with mTOR and inhibits mTOR kinase activity, thereby serving as an endogenous inhibitor of both mTORC1 and mTORC2 (17). DEPTOR is ubiquitinated and is degraded by the proteasome following serum treatment of cells, leading to increased mTORC2 and mTORC1 activity (17–20). DEPTOR regulation of mTORC1 and mTORC2 is critical, as perturbations in this regulation have been linked to aberrant Akt signaling in several cancers (17, 21). Despite this, the molecular mechanisms that regulate DEPTOR in cells remain poorly understood.
In addition to the proteasome, the lysosome is the major degradative compartment in cells (22). Targeting proteins for lysosomal degradation can occur via multiple mechanisms, including the ESCRT pathway (23). The ESCRT pathway consists of four distinct protein complexes (ESCRT-0-III) plus the disassembly VPS4 complex (24, 25). These complexes act in a coordinated manner on the limiting membrane of endosomes to promote the formation of intraluminal vesicles (ILVs) leading to the creation of the multivesicular body (MVB) (26). Mature MVBs then fuse with lysosomes where their contents are degraded (27). ESCRTs also target ubiquitinated transmembrane cargo into ILVs by virtue of their ability to interact with ubiquitin moieties on the cargo via the ubiquitin binding domains present in several ESCRT subunits (28–30). Ubiquitinated cell signaling receptors are well defined ESCRT-dependent cargo and are targeted into ILVs for eventual lysosomal degradation (30–32). As such, the ESCRT pathway typically mediates down-regulation of receptor signaling (33), but recently this pathway has also been linked to positive regulation of cell signaling (34–36). For example, glycogen synthase kinase 3β (GSK-3β), which serves as an endogenous antagonist of the canonical Wnt/β-catenin signaling pathway, is targeted into ILVs by ESCRTs. GSK-3β is normally active and free in the cytoplasm to phosphorylate β-catenin, thereby promoting its proteasomal degradation (34–36). However, upon activation of Wnt signaling, GSK-3β is sequestered into ILVs of MVBs via the ESCRT pathway, thereby preventing its interaction with β-catenin, which leads to β-catenin stabilization and long lasting signaling (34–36). To our knowledge, whether DEPTOR is sequestered in MVBs and/or degraded by lysosomes remains unknown.
Here, we show that activation of the chemokine receptor CXCR4 and α2-adrenergic receptors, prototypical GPCRs, promotes degradation of DEPTOR. We show that GPCR-promoted DEPTOR degradation is blocked by lysomotrophic agents and by siRNA directed against ESCRTs. ESCRT depletion leads to increased levels of DEPTOR and attenuation of Akt activation by mTORC2. ESCRT depletion also attenuates receptor tyrosine kinase (RTK)-promoted Akt activation and signaling. Our findings indicate that ESCRTs regulate receptor-promoted DEPTOR degradation and Akt signaling.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, siRNA, DNA Constructs
HeLa cells were from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Hyclone Laboratories (Logan, UT)) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories). Antibodies directed against pAkt-S473 (catalog no. 9271), Akt (catalog no. 2967), GSK-3β (catalog no. 12456), pFoxO1-T24/FoxO3a-T32 (catalog no. 9464), FoxO3a (catalog no. 2497), TSC2 (4308), pTSC2-T1462 (catalog no. 3617), Rictor (catalog no. 2140), p-p70 S6 kinase-T389 (catalog no. 9234), p70 S6 kinase (catalog no. 9202), DEPTOR (catalog no. 11816), and mTOR (catalog no. 2972) were from Cell Signaling Technologies (Danvers, MA). Antibodies directed against STAM1 (catalog no. 12434-1-AP) and UBAP1 (catalog no. 12385-1-AP) were from ProteinTech Group (Chicago, IL). The antibody directed against SIN1 (catalog no. 05-1044) was from Millipore (Billerica, MA). Antibodies directed against pGSK3-S9 (catalog no. Ab75814) and Akt1 (catalog no. Ab32038, E45) were from Abcam (Cambridge, MA). The anti-ERK-1/2 (catalog no. M8159), anti-Tsg101 (catalog no. T5701), anti-Rictor (catalog no. SAB5300210), anti-FLAG (catalog no. F3165), and anti-tubulin (catalog no. T5293) antibodies were from Sigma. The antibody directed against CXCR4 (2B11) was from BD Biosciences. The anti-actin antibody was from MP Biomedicals (Aurora, OH). The anti-Myc antibody was from Covance (catalog no. MMS-150R). Horseradish peroxidase-conjugated secondary antibodies were from Vector Laboratories (Burlingame, CA). Stromal cell-derived factor-1α (SDF-1α; CXCL12) and epidermal growth factor (EGF) were from PeproTech (Rocky Hill, NJ). Insulin was from Gemini Bio-Products (West Sacramento, CA). Norepinephrine, dimethyl sulfoxide (DMSO), cycloheximide, chloroquine, wortmannin, and 3-methyladenine were from Sigma. Gallein and pertussis toxin were from R & D Systems (Minneapolis, MN). Lactacystin was from BioVision (Milpitas, CA).
siRNA-directed against luciferase (catalog no. P-002099-01-20), Tsg101 (catalog no. M-003549-01-0005), and STAM1 (catalog no. D-011423-01) was from Dharmacon RNA Technologies (Lafayette, CO). siRNA directed against UBAP1 (catalog no. HSC.RNAI.N001171201.12), Vps22 (catalog no. HSC.RNAI.N007241.12.2), CHMP4C (catalog no. HSC.RNAIN152284.12.3), and RICTOR (catalog no. HSC.RNAI.N152756.12.4) was from Integrated DNA Technologies (Coralville, IA). pLKO human shRNA1 DEPTOR (Addgene plasmid catalog no. 21335), pRK5 FLAG human DEPTOR (Addgene plasmid catalog no. 21334), and myc-Raptor (Addgene plasmid catalog no. 1859) were gifts from David Sabatini (17, 37).
CXCR4 and DEPTOR Degradation Assays
CXCL12-promoted degradation of endogenous CXCR4 in HeLa cells was assessed by immunoblot analysis, as described previously (38–40). siRNA transfections were as described previously (38–40). For DEPTOR degradation, HeLa cells (300,000) were passaged onto a 6-well plate. The following day, cells were washed with DMEM containing 20 mm HEPES and then serum-starved in the same media for 3 h. Cells were then incubated with the same media containing 50 μg/ml cycloheximide for 15 min at 37 °C, followed by treatment with vehicle (0.1% BSA in PBS) or 10 nm CXCL12 for various times. To examine the effect of inhibitors on CXCR4-promoted DEPTOR degradation, cells were washed with warm DMEM supplemented with 20 mm HEPES, then treated with the same media containing 50 μg/ml cycloheximide for 15 min, followed by pretreatment with dimethyl sulfoxide (DMSO), 200 μm chloroquine, 10 μm lactacystin, 50 ng/ml pertussis toxin, 50 μm gallein, 50–100 nm wortmannin, and 0.5–5 mm 3-methyladenine for 1 h. Cells were then treated with vehicle or 10 nm CXCL12 for 3 h in the continued presence of the inhibitors. Cells were washed once with ice-cold PBS and collected in 300 μl of 2× sample buffer (8% SDS, 10% glycerol, 0.7 m β-mercaptoethanol, 37.5 mm Tris-HCl, pH 6.5, 0.003% bromphenol blue). Equal amounts of samples were analyzed by 10% SDS-PAGE and immunoblotting. DEPTOR degradation was determined by densitometric analysis of similar exposures of film across multiple experiments.
Signaling Experiments
HeLa and BAEC cells were transfected with siRNA (50 nm final concentration) directed against individual subunits of ESCRT-0 (STAM1), ESCRT-I (UBAP1 or Tsg101), ESCRT-II (Vps22), ESCRT-III (CHMP4C), or control siRNA (luciferase or GAPDH) siRNA. After 48 h, 300,000 cells were plated onto 6-well dishes, and the next day cells were washed once with warm DMEM containing 20 mm HEPES and serum-starved in the same media for 3 h. Cells were treated for 5 min with 10 nm CXCL12, 100 ng/ml EGF, 10 μm norepinephrine, 50–200 nm insulin or vehicle (0.1% BSA in PBS). Cells were washed once with PBS and collected in 300 μl of 2× sample buffer. Equal amounts of samples were analyzed by 10% SDS-PAGE and immunoblotting with antibodies directed against phosphorylated and total levels of several signaling molecules. The phosphorylation status of Akt at Ser-473 was quantitated by densitometric analysis of similar exposures of film across multiple experiments.
Co-immunoprecipitation Assay
HeLa cells (300,000) grown in 6-well plates were transfected with 50 nm siRNA directed against luciferase or STAM1, using Lipofectamine 3000 (Invitrogen). In some experiments, 24 h later cells were also transfected with FLAG-DEPTOR and myc-Raptor. Forty eight h after treatment with siRNA, cells were washed once with ice-cold PBS, and lysed in 300 μl of immunoprecipitation buffer (40 mm HEPES, pH 7.4, 0.3% CHAPS, 2 mm EDTA, 50 mm sodium fluoride, 10 mm sodium pyrophosphate decahydrate, 10 mm β-glycerophosphate, and protease inhibitors (10 μg/ml each of aprotinin, pepstatin A, and leupeptin; Roche Applied Science)). Lysates were cleared by centrifugation at 13,000 rpm for 20 min at 4 °C using a 5417r-Eppendorf microcentrifuge. The cleared supernatants were collected, and protein concentrations were determined using the BCA protein assay kit (Pierce). A total of 300–400 μg of supernatant in a volume of 200–300 μl was incubated with either anti-mouse IgG, anti-Rictor, or anti-Myc mouse monoclonal antibodies for 12–16 h. Samples were then incubated with 20 μl of a 50% slurry of protein G-agarose (Roche Applied Science) for 1 h at 4 °C while rocking. Bound proteins were eluted in nonreducing 2× sample buffer, followed by 10% SDS-PAGE and immunoblotting. For data in Fig. 6E, HeLa cells were serum-starved for 3 h and treated with vehicle for 30 min or CXCL12 for 5 or 30 min, followed by immunoprecipitation of endogenous Rictor, as described above.
FIGURE 6.

DEPTOR depletion enhances and DEPTOR overexpression attenuates CXCR4-promoted Akt activation and signaling. A and B, role of ESCRTs in regulating the association of DEPTOR with mTORC2 and mTORC1. HeLa cells were treated with siRNA directed against STAM1 or luciferase (A), followed by transfection with FLAG-tagged DEPTOR and with Myc-tagged Raptor or empty vector (pcDNA) (B). Endogenous Rictor (A) or myc-Raptor (B) were immunoprecipitated (IP) and analyzed for endogenous DEPTOR (A) or FLAG-DEPTOR (B), respectively, and the indicated proteins. Representative blots from three independent experiments are shown. The asterisks in A indicate nonspecific bands. C and D, role of DEPTOR in regulating Akt activation promoted by CXCR4. HeLa cells were transfected FLAG-DEPTOR or shRNA targeting DEPTOR, followed by serum starvation and stimulation with CXCL12. Cell lysates were analyzed for the indicated proteins and their phosphorylation status. Data are from three independent experiments. E, role of CXCR4 activation on DEPTOR association with mTORC2. HeLa cells were stimulated with CXCL12 for 5 or 30 min or vehicle, followed by immunoprecipitation of endogenous Rictor and immunoblotting for DEPTOR and mTOR. Representative blots from two independent experiments are shown. Ctrl, control.
Statistical Analysis
Data are represented as the mean ± S.E. of at least three experiments or determinations. All statistical tests were done using GraphPad Prism 6.2b for Mac OS X (GraphPad Software, San Diego). Student's t test was used to compare the difference between two groups; one-way analysis of variance (ANOVA) was used to compare the difference between three or more groups, and two-way ANOVA was used to compare the difference between different groups under different treatment conditions. ANOVA was followed by Tukey's or Bonferroni's post hoc test. A probability (p) value of <0.05 was considered significant. Specific values are provided in the figure panels or in the figure legends.
RESULTS
GPCR Activation Promotes Lysosomal Degradation of DEPTOR
To examine whether GPCRs regulate DEPTOR degradation, we treated HeLa cells with CXCL12, the cognate ligand for CXCR4, a prototypical GPCR, and analyzed DEPTOR levels by immunoblotting. As shown in Fig. 1A, treatment with CXCL12 promoted time-dependent degradation of endogenous DEPTOR, whereas the levels of Akt or other mTORC2 subunits mTOR, Rictor, and Sin1 were not changed. DEPTOR degradation was noticeable after 15–30 min of agonist treatment (Fig. 1A), albeit not statistically significant (Fig. 1B), but nevertheless indicating a very rapid mechanism of degradation. Endogenous CXCR4 was also degraded (Fig. 1A), as we have shown previously (38, 40, 41).
FIGURE 1.
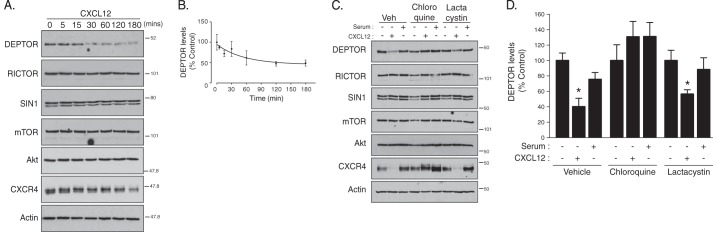
CXCR4-promoted lysosomal degradation of DEPTOR. A, CXCR4 activation induces degradation of DEPTOR. HeLa cells were treated with vehicle (PBS + 0.1% BSA) or CXCL12 for the indicated times, as described under “Experimental Procedures.” Equal amounts of whole cell lysates were immunoblotted for DEPTOR and the indicated proteins. B, immunoblots were analyzed by densitometry. Graph shows the average amount of DEPTOR remaining normalized to actin at each CXCL12 time point compared with vehicle (0 time point; set to 100%) from four independent experiments. Error bars represent the mean ± S.E. C, analysis of DEPTOR degradation in the presence of lysosomal or proteasomal inhibitors. HeLa cells were pretreated with vehicle (Veh), chloroquine, or lactacystin, followed by treatment with vehicle, CXCL12, or serum (10% FBS) for 3 h, as described under “Experimental Procedures.” Equal amounts of whole cell lysates were immunoblotted for DEPTOR and the indicated proteins. D, quantification of DEPTOR levels by densitometry. DEPTOR levels were normalized to actin, and the bars represent the average amount of DEPTOR remaining after CXCL12 or serum treatment compared with vehicle (set to 100%) under each inhibitor condition. Data are from 4 to 7 independent experiments, and error bars represent mean ± S.E. Data were analyzed by a two-way ANOVA followed by Tukey's post hoc test. *, data are significant from control (p < 0.05).
To determine whether the proteasome mediates CXCR4-promoted DEPTOR degradation, cells were pretreated with the selective proteasomal inhibitor lactacystin (42). In addition, we also pretreated cells with the lysosomotropic agent chloroquine, which increases endo-lysosomal pH, thereby inhibiting the function of lysosomal hydrolases (43). As shown in Fig. 1C, CXCR4-promoted DEPTOR degradation was completely blocked in chloroquine-treated cells but not in lactacystin-treated cells (Fig. 1, C and quantification in D). CXCR4 is known to be degraded in lysosomes (38), and accordingly, CXCR4 degradation was also blocked by chloroquine but not lactacystin (Fig. 1C). In contrast, serum treatment for 3 h did not promote significant degradation of DEPTOR (Fig. 1, C and quantification in D). These data suggest that lysosomes but not the proteasome mediates CXCR4-induced DEPTOR degradation.
The ESCRT pathway targets ubiquitinated transmembrane proteins, such as CXCR4, into ILVs of MVBs for eventual degradation in lysosomes (40, 44). ESCRTs have also been implicated in targeting cytosolic proteins into ILVs of MVBs (34, 35). We next examined whether DEPTOR degradation is regulated by ESCRTs. To explore this, we first focused on ESCRT-I. ESCRT-I is composed of Tsg101, Vps28, Vps37, and Mvb12 or UBAP1, and each is stoichiometrically incorporated into ESCRT-I, forming a stable heterotetrameric complex (45). Loss of a single ESCRT-I subunit destabilizes the complex and leads to degradation of the other subunits (46). Tsg101 is a core component of ESCRT-I, and it has been shown to mediate sorting of CXCR4 to lysosomes (47). There are at least four predicted Mvb12 isoforms in mammalian cells, and at least three of them (Mvb12A, Mvb12B, and UBAP1) can assemble into distinct ESCRT-I complexes with discrete functions (45, 46). ESCRT-I complexes that contain UBAP1 are involved in lysosomal degradation of transmembrane receptors, such as EGFR, whereas those that contain Mvb12 are involved in cytokinesis, another cellular process that also requires ESCRTs (46). Similar to EGFR, siRNA targeting UBAP1, but not Mvb12A or Mvb12B, significantly attenuated agonist-induced degradation of CXCR4 compared with control (Fig. 2, A–D), indicating for the first time that ESCRT-I complexes mainly containing UBAP1 mediate CXCR4 lysosomal degradation.
FIGURE 2.

Depletion of ESCRT-I subunit UBAP1 attenuates CXCR4 and DEPTOR degradation. A–F, HeLa cells treated with siRNA directed against UBAP1, Mvb12A, Mvb12B, or luciferase (Ctrl) were stimulated with vehicle (minus symbol) or CXCL12 (plus symbol) for 3 h, as described under “Experimental Procedures.” Equal amounts of whole cell lysates were immunoblotted for CXCR4 (A and C) or DEPTOR (E) and the indicated proteins. Bars represent the average amount of CXCR4 degraded compared with vehicle control (B and D) or DEPTOR remaining (F) after each treatment condition, compared with vehicle and luciferase siRNA-transfected cells (set to 100%). CXCR4 (B and D) and DEPTOR (F) levels were normalized to actin. Data are from 3 (B and D) or 4 (F) independent experiments. Error bars (B, D, and F) represent mean ± S.E. Data were analyzed by a Student's t test (B), one-way ANOVA (D), or two-way ANOVA (F). CXCR4 degraded in UBAP1 siRNA-treated cells is significantly different from control cells +(p = 0.003).
We next examined whether UBAP1 regulates DEPTOR degradation. DEPTOR levels were significantly increased in HeLa cells in which UBAP1 was depleted by siRNA, as compared with control (Fig. 2, E and quantification in F). In addition, CXCR4-promoted DEPTOR degradation was blocked in UBAP1-depleted cells (Fig. 2, E and quantification in F). These data indicate that UBAP1 and by extension ESCRTs regulate DEPTOR degradation. Given that ESCRT depletion leads to elevated DEPTOR levels, these data suggest that the ESCRT pathway regulates mTORC2 activity.
ESCRTs Regulate mTORC2-dependent Phosphorylation of Akt at Ser-473
To examine the activity of mTORC2, we determined the phosphorylation status of Akt at serine residue 473 (pAkt-S473), an mTORC2 phosphorylation site, by immunoblotting with a phospho-specific antibody. HeLa cells were serum-starved for 3 h and then treated for various times with CXCL12. As shown in Fig. 3A, CXCL12 induced a rapid and transient increase in phosphorylation of Akt at Ser-473 that was maximal at 5 min but remained somewhat elevated for up to 60 min of agonist treatment. Phosphorylation of Ser-473 was mediated by mTORC2, as depletion of Rictor, a defining subunit of mTORC2 (37), attenuated phosphorylation of Ser-473 in HeLa cells treated with CXCL12 (Fig. 3B).
FIGURE 3.

Depletion of mTORC2 subunit Rictor attenuates phosphorylation of Akt at Ser-473 upon CXCR4 activation. A, time course of Akt activation promoted by stimulation of CXCR4. HeLa cells were treated with vehicle (PBS + 0.1% BSA) or CXCL12 for the indicated times, as described under “Experimental Procedures.” Samples were immunoblotted using anti-phosphospecific antibodies at Ser-473 (pAkt-S473) or Thr-308 (pAkt-T308) and total Akt. Immunoblots were analyzed by densitometry. Graph shows the average pAkt-S473 levels normalized to total Akt at each time point compared with control (5-min time point; set to 100%). Data are from three independent experiments, and error bars represent mean ± S.E. B, depletion of Rictor attenuates CXCR4-promoted phosphorylation of Akt at Ser-473. HeLa cells treated with siRNA directed against Rictor or luciferase (Ctrl) were serum-starved for 3 h followed by treatment with vehicle (minus symbol) or CXCL12 (plus symbol) for 5 min, as described under “Experimental Procedures.” Equal amounts of whole cell lysates were immunoblotted for the indicated proteins and their phosphorylation status. Representative blots from three independent experiments are shown. Arrow points to pAkt-T308 band. The asterisk represents nonspecific bands.
We next examined the role of ESCRTs on mTORC2 activity in HeLa cells treated with siRNA directed against a subunit in ESCRT-0 (STAM1), ESCRT-I (UBAP1), ESCRT-II (Vps22), or ESCRT-III (CHMP4C). As shown in Fig. 4, A–D, CXCR4-promoted phosphorylation of Akt at Ser-473 was attenuated in cells depleted of a subunit from each ESCRT complex. DEPTOR levels were increased in cells depleted of all ESCRTs (Fig. 4, A, B, or D), except ESCRT-II (Fig. 4C), consistent for a role of ESCRTs in regulating DEPTOR degradation. Importantly, ESCRTs do not have a global effect on signaling because CXCR4-promoted phosphorylation of ERK-1/2 was not affected in cells treated with siRNA targeting ESCRT-I (Fig. 4B), ESCRT-II (Fig. 4C), or ESCRT-III (Fig. 4D). In contrast, depletion of ESCRT-0 subunit STAM1 attenuated CXCR4-promoted ERK-1/2 activation, which we have previously shown likely occurs via a discrete pool of STAM1 that is localized to caveolae, possibly unrelated to its functional role as part of ESCRT-0 (Fig. 4A) (8). A role for ESCRTs in regulating mTORC2 activity is not unique to HeLa cells because siRNA-mediated depletion of Tsg101, a core subunit of ESCRT-I (25), also significantly attenuated CXCR4-induced phosphorylation of Akt at Ser-473 in BAEC (Fig. 5, A and quantification in B), further confirming a role for ESCRTs in regulating mTORC2 activity. However, DEPTOR levels were not changed, as determined by immunoblotting, suggesting that in addition to modulating DEPTOR levels (Fig. 5A), ESCRTs may regulate Akt activation via an additional mechanism. CXCL12 treatment of BAEC cells displayed similar time-dependent phosphorylation of Akt at Ser-473 (Fig. 5C), as compared with HeLa cells (Fig. 3A).
FIGURE 4.
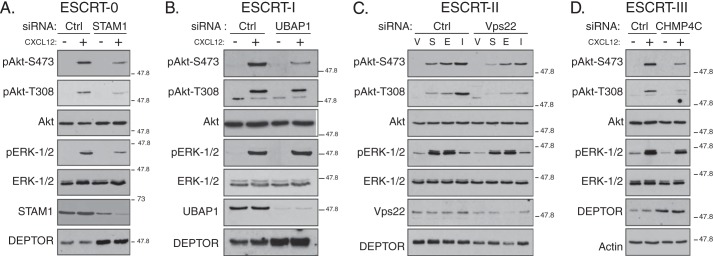
Depletion of subunits from ESCRT-0, -I, -II, or -III attenuates phosphorylation of Akt upon CXCR4 activation. A–D, depletion of individual subunits from each of the ESCRT complexes attenuates CXCR4-promoted Akt activation. HeLa cells treated with siRNA directed against STAM1 (ESCRT-0), UBAP1 (ESCRT-I), Vps22 (ESCRT-II), CHMP4C (ESCRT-III), or luciferase (Ctrl) were serum-starved for 3 h followed by treatment with vehicle (minus symbol) or CXCL12 (plus symbol) for 5 min, as described under “Experimental Procedures.” Equal amounts of whole cell lysates were immunoblotted for the indicated proteins and their phosphorylation status. Representative blots from four independent experiments are shown. V, vehicle; S, CXCL12; E, EGF; and I, insulin.
FIGURE 5.

Depletion of ESCRT-I subunit Tsg101 attenuates CXCR4-promoted phosphorylation of Akt at Ser-473. A, BAEC treated with siRNA directed against Tsg101 or GAPDH (Ctrl) were serum-starved in DMEM containing 20 mm HEPES for 3 h, followed by treatment with vehicle (minus symbol) or CXCL12 (plus symbol) for 10 min. Equal amounts of whole cell lysates were immunoblotted for pAkt-S473 and the indicated proteins. Representative immunoblots from three independent experiments are shown. The asterisk likely represents a nonspecific band. B, densitometric analysis of pAkt-S473 levels. pAkt-S473 levels were normalized to total Akt levels, and bars represent the average levels of pAkt-S473 compared with control (Ctrl) and CXCL12-treated cells (set to 100%) from three independent experiments. The error bars represent mean ± S.E. Data were analyzed by a two-way ANOVA followed by Bonferroni's post hoc test (p < 0.05). C, time course of Akt activation promoted by CXCR4 in BAEC cells. Cells were treated with vehicle (time 0) or CXCL12 for the indicated times, and equal amounts of whole cell lysates were immunoblotted for pAkt-S473 and total Akt. Immunoblots were analyzed by densitometry, and the graph shows the average pAkt-S473 levels normalized to total Akt at each time point compared with control (10-min time point; set to 100%) from three independent experiments. Error bars represent S.E.
Because DEPTOR can antagonize mTOR activity, we next examined whether ESCRT depletion by siRNA also leads to an increase in the amount of DEPTOR associated with mTOR by co-immunoprecipitation with Raptor or Rictor, which are found in mTORC1 and mTORC2, respectively (15). As shown in Fig. 6A, there was a greater amount of DEPTOR in STAM1-depleted cells compared with control, and this correlated with a greater amount of DEPTOR in Rictor immunoprecipitates (Fig. 6A). There was no change in the amount of mTOR associated with Rictor (Fig. 6A). In contrast, although FLAG-DEPTOR levels were increased in STAM1-depleted cells, there was no change in the amount of FLAG-DEPTOR associated with myc-Raptor when compared with control (Fig. 6B). Taken together, these data suggest that ESCRTs regulate DEPTOR associated with mTORC2, but not mTORC1, and they further suggest that ESCRTs regulate mTORC2 activity by regulating DEPTOR degradation.
To confirm that DEPTOR does indeed regulate CXCR4-promoted mTORC2 activity, we next examined Akt activation in cells overexpressing FLAG-tagged DEPTOR or in cells transfected with shRNA targeting DEPTOR. Overexpression of FLAG-DEPTOR attenuated whereas DEPTOR depletion enhanced phosphorylation of Akt at Ser-473 (Fig. 6, C and D, respectively). CXCR4-promoted ERK-1/2 activation was not impacted by DEPTOR overexpression (Fig. 6C) or depletion (Fig. 6D). Therefore, DEPTOR regulates CXCR4-promoted Akt activation.
Our data indicate that the ESCRT pathway regulates CXCR4-promoted Akt activation, likely via promoting degradation of DEPTOR. However, Akt is maximally activated within 5 min of CXCL12 treatment (Fig. 2A), whereas DEPTOR is only noticeably degraded after 30 min of CXCL12 treatment (Fig. 1A). It is likely that upon CXCR4 activation DEPTOR becomes dissociated from mTORC2 before it is sequestered into ILVs of MVBs and noticeably degraded in lysosomes. To examine this, we treated HeLa cells with vehicle or CXCL12 for 5 or 30 min, followed by immunoprecipitation of endogenous Rictor and immunoblotting to detect DEPTOR. As shown in Fig. 6E, there was less DEPTOR associated with Rictor after treatment with CXCL12 for 5 or 30 min, compared with vehicle. This is consistent with the idea that CXCR4 activation promotes DEPTOR dissociation from mTORC2, which is likely required for its incorporation into ILVs and maximal Akt activation following receptor activation.
ESCRTs Regulate Akt Signaling
Phosphorylation of Ser-473 is necessary for full activation of Akt and hence Akt signaling (14, 49). Next, we determined whether ESCRTs regulate Akt signaling by examining the phosphorylation status of Akt substrates FoxO1/O3a, TSC2, and GSK-3β (49, 50). CXCR4-promoted phosphorylation of FoxO1/O3a at Akt sites Thr-24/Thr-32 was markedly attenuated in cells treated with siRNA directed against a subunit in each of the ESCRTs compared with control siRNA (Fig. 7, A–D). CXCR4-promoted phosphorylation of TSC2 and GSK-3β at Akt sites Thr-1462 and Ser-9, respectively, was attenuated by ESCRT depletion, although modestly (Fig. 7, A–D). Consistent for a role in Akt signaling, DEPTOR overexpression attenuated (Fig. 6C) and DEPTOR depletion enhanced (Fig. 6D) CXCR4-promoted phosphorylation of FoxO1/O3a at Akt sites Thr-24/Thr-32. Taken together, our data indicate that ESCRTs govern Akt signaling, likely by regulating the levels of DEPTOR and thereby controlling the activity of mTORC2.
FIGURE 7.
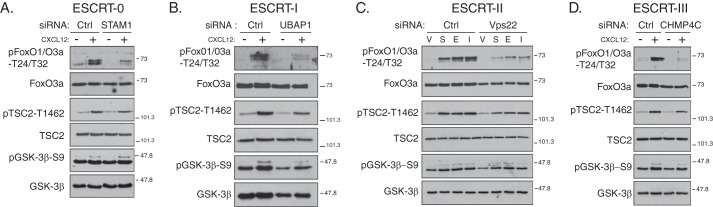
Depletion of subunits from ESCRT-0, -I, -II, or -III attenuates CXCR4-promoted Akt signaling. A–D, depletion of individual subunits from each of the ESCRT complexes attenuates CXCR4-promoted Akt activation. HeLa cells treated with siRNA directed against STAM1 (ESCRT-0), UBAP1 (ESCRT-I), Vps22 (ESCRT-II), CHMP4C (ESCRT-III), or luciferase (Ctrl) were serum-starved for 3 h followed by treatment with vehicle (minus symbol) or CXCL12 (plus symbol) for 5 min. Equal amounts of whole cell lysates were immunoblotted for the indicated proteins and their phosphorylation status. Samples are from the experiments described in Fig. 4. Representative blots from four independent experiments are shown. V, vehicle; S, CXCL12; E, EGF; and I, insulin.
ESCRTs Have a Broad Role in Akt Signaling
Because ESCRTs regulate DEPTOR levels in cells, it is possible that ESCRTs have a broad role in Akt signaling. HeLa cells endogenously express α2-adrenergic receptors and similar to CXCR4 are coupled with Gαi heterotrimeric G proteins (51). HeLa cells also express epidermal growth factor and insulin receptors, two well studied RTKs that readily promote Akt signaling (49, 50). Next, we explored whether ESCRTs regulate Akt signaling induced by activation of these receptors. To accomplish this, HeLa cells were treated with siRNA directed against ESCRT-I subunit UBAP1, followed by serum starvation and stimulation with the adrenergic receptor agonist norepinephrine (10 μm), EGF (100 ng/ml), or insulin (50 nm) for 5 min. Depletion of UBAP1 attenuated Akt phosphorylation at Ser-473 promoted by norepinephrine (Fig. 8A), EGF (Fig. 8B), or insulin (Fig. 8C). Activation of EKR-1/2 by norepinephrine, EGF, or insulin was not impacted by depletion of UBAP1 (Fig. 8, A–C, respectively). In addition, depletion of ESCRT-II subunit Vps22 also attenuated Akt phosphorylation at Ser-473, but not activation of ERK-1/2, promoted by EGF or insulin (see Fig. 4C).
FIGURE 8.

Depletion of ESCRT-I subunit UBAP1 attenuates Akt activation and signaling promoted by GPCRs and RTKs. A–C, HeLa cells transfected with siRNA directed against ESCRT-I subunit UBAP1 or luciferase (Ctrl) were treated with vehicle (0.1% BSA in PBS), 10 μm norepinephrine (A, norepi), 100 ng/ml EGF (B), or 50 nm insulin (C) for 5 min. Equal amounts of whole cell lysates were analyzed by immunoblotting for the indicated proteins and their phosphorylation status. Representative blots from four independent experiments are shown. D and E, role of GPCRs and RTKs on DEPTOR degradation. HeLa cells were treated with vehicle (VEH), CXCL12, norepinephrine (norepi), EGF, or insulin for 3 h, and DEPTOR levels were determined by immunoblotting. E, quantification of DEPTOR levels. Bars represent the average amount of DEPTOR remaining normalized to actin compared with vehicle (VEH; set to 100%) from five independent experiments. Error bars represent S.E. Data were analyzed by a one-way ANOVA followed by Bonferroni's post hoc test (VEH versus CXCL12 or norepinephrine, p = 0.00072, as indicated by the brackets).
To determine whether Akt signaling was impacted by ESCRT depletion, we examined the phosphorylation status of Akt substrates FoxO1/O3a, TSC2, and GSK-3β. Similar to CXCL12, norepinephrine, EGF, or insulin promoted phosphorylation of FoxO1/O3a at Thr-24/Thr-32 was attenuated in UBAP1-depleted cells compared with control, whereas phosphorylation of TSC2 at Thr-1462 and GSK-3β at Ser-9 was not noticeably impacted (Fig. 8, A–C, respectively). Similar results for EGF and insulin were observed in cells depleted of Vps22 (see Fig. 4C).
We next examined whether activation of α2-adrenergic receptors, EGFR, or the insulin receptor promoted DEPTOR degradation. HeLa cells were treated with norepinephrine, EGF, or insulin for 3 h, and DEPTOR levels were determined by immunoblotting. Norepinephrine, but not EGF or insulin, promoted DEPTOR degradation (Fig. 8, D and quantification in E), suggesting that GPCRs, but not RTKs, regulate DEPTOR degradation, at least under our experimental conditions.
DEPTOR is also a component of mTORC1, and DEPTOR levels associated with mTORC1 were not changed in STAM1-depleted cells (Fig. 6B), suggesting that mTORC1 signaling is not directly regulated by ESCRTs. To address this, we examined the phosphorylation status of p70S6 kinase (p70S6K), which is directly phosphorylated by mTORC1 at Thr-389 (17). As shown in Fig. 9A, CXCR4-promoted phosphorylation of p70S6K at Thr-389 was not affected in cells treated with siRNA directed against ESCRT-I subunit UBAP1. Similarly, norepinephrine- (Fig. 9B), EGF- (Fig. 9C), or insulin (Fig. 9D)-promoted phosphorylation of p70S6K at Thr-389 was not affected by depletion of UPAB1. These data suggest that ESCRTs do not impact mTORC1 activity.
FIGURE 9.

Depletion of ESCRT-I subunit UBAP1 does not have an effect on GPCR and RTK-promoted mTORC1 activity. A–D, HeLa cells were treated with siRNA directed against ESCRT-I subunit UBAP1 and stimulated with CXCL12 (A), norepinephrine (B), EGF (C), or insulin (D) for 5 min. Equal amounts of whole cell lysates were immunoblotted for p70S6K and its phosphorylation state at Thr-389, an mTORC1 phosphorylation site. Samples are from the experiment described in Fig. 4. Representative blots from four independent experiments are shown.
GPCR-promoted DEPTOR Degradation Is G Protein- and PI3K-dependent
To gain further insight into the mechanism driving GPCR-promoted DEPTOR degradation, we next examined whether this occurs via balanced G protein-dependent and G protein-independent pathways (52). CXCR4 couples with the pertussis toxin-sensitive Gαi heterotrimeric G protein (7, 53), and as shown in Fig. 10A, CXCL12-induced degradation of DEPTOR was completely blocked in HeLa cells treated with pertussis toxin, indicating that G protein signaling is necessary for CXCR4-promoted DEPTOR degradation. In addition, pertussis toxin completely blocked CXCR4-promoted phosphorylation of Akt at Ser-473 or Thr-308 and phosphorylation of FoxO1/O3a at Thr-24/Thr-32 (Fig. 10B). Pertussis toxin did not have an impact on EGFR-promoted phosphorylation of Akt at Ser-473 or Thr-308 or phosphorylation of FoxO1/O3a at Thr-24/Thr-32 (Fig. 10B). These data indicate that CXCR4-promoted DEPTOR degradation and Akt signaling occurs via G protein-dependent signaling.
FIGURE 10.

Role of G protein and PI3K signaling in DEPTOR degradation and Akt signaling promoted by CXCR4. A, role of G protein and PI3K signaling on DEPTOR degradation. HeLa cells were pretreated with DMSO, pertussis toxin (PTX), wortmannin, or 3-MA, followed by treatment with vehicle (VEH) (minus symbol) or CXCL12 (plus symbol) for 3 h. Whole cell lysates were immunoblotted for DEPTOR, CXCR4, and actin. Representative blots from three independent experiments are shown. B–D, role of G protein and PI3K signaling on Akt activation and signaling. HeLa cells were pretreated with pertussis toxin (B, PTX), gallein (C), and varying concentrations of wortmannin or 3-methyladenine (D), followed by treatment with vehicle (VEH), CXCL12, or EGF for 5 min. Equal amounts of whole cell lysates were immunoblotted for the indicated proteins and their phosphorylation status. Arrow represents pAkt-T308 and asterisks represent nonspecific bands (B–D). Representative blots from three independent experiments are shown.
Typically, GPCR-promoted Akt signaling occurs via the released Gβγ heterodimer, which binds directly to and activates either PI3Kβ or PI3Kγ leading to the production of PtdIns(3,4,5)P3 at the plasma membrane (10, 12) and recruitment of PDK1 and Akt, thereby enabling phosphorylation of Akt at Thr-308 by PDK1 (54). Consistent with this, gallein, a pharmacological inhibitor of Gβγ (55), attenuated CXCR4-promoted Akt phosphorylation at Thr-308 but not phosphorylation promoted by EGFR (Fig. 10C). Gallein also attenuated phosphorylation of Akt at Ser-473, possibly because it prevents reassociation of α- and βγ-subunits. It is also possible that phosphorylation of Thr-308 by PDK1 and Ser-473 by mTORC2 are mechanistically linked (54, 56). In contrast, inhibition of Ser-473 phosphorylation by Rictor depletion did not have an impact on phosphorylation of Thr-308 (see Fig. 3B). Importantly, phosphorylation of Ser-473 is necessary for Akt signaling because in Rictor-depleted cells phosphorylation of FoxO1/O3a was also attenuated, compared with control (see Fig. 3B). Gallein also attenuated CXCR4-promoted phosphorylation of FoxO1/O3a (Fig. 10C). These data indicate that Gβγ are required for CXCR4-promoted Akt signaling likely via PI3K signaling.
To confirm that PI3K is involved in CXCR4-promoted Akt signaling, we incubated cells with wortmannin, a nonselective inhibitor of several PI3K isoforms (57). As shown in Fig. 10D, CXCR4-promoted phosphorylation of Akt at Thr-308 and phosphorylation of FoxO1/O3a at Thr-24/Thr-32 was dose-dependently blocked in cells treated with wortmannin, compared with control. We have recently shown that wortmannin (100 nm) reduces the amount of phosphatidylinositol 3-phosphate (PtdIns3P) on early endosomes in HeLa cells, assessed by using a fluorescent based-reporter of PtdIns3P levels (40), consistent with the fact that wortmannin also inhibits the class III PI3K Vps34 (58). Vps34 catalyzes the formation of PtdIns3P on the cytosolic leaflet of endosomal membranes where it plays a role in the ESCRT pathway (59, 60). Vps34 can also be found in another discrete complex that has an essential role in autophagy (61). To examine Vps34, we treated cells with varying doses of 3-methyladenine (3-MA), a somewhat selective inhibitor of Vps34 (62). As shown in Fig. 10D, 3-MA dose-dependently inhibited CXCR4-promoted phosphorylation of Akt at Thr-308 or Ser-473 and phosphorylation of FoxO1/O3a at Thr-24/Thr-32. At higher doses (5 and 0.5 mm) 3-MA likely also inhibits the class I PI3K catalytic subunits p110β and p110γ (62). In addition, wortmannin or 3-MA, blocked CXCR4-promoted degradation of DEPTOR, indicating that PI3K signaling drives DEPTOR degradation (Fig. 10A). In contrast, CXCR4 degradation was only modestly inhibited by pertussis toxin, wortmannin, or 3-MA treatments, likely because the remaining PtdIns3P (63) is still able to efficiently support CXCR4 sorting for degradation, at least during the 3-h treatment time used in this experiment (Fig. 10A). Overall, these data suggest that class I and class III PI3Ks mediate Akt signaling and DEPTOR degradation induced by CXCR4.
DISCUSSION
We show here that the canonical ESCRT pathway mediates CXCR4-promoted DEPTOR degradation. Depletion of ESCRTs leads to elevated levels of DEPTOR (Fig. 2D), and because DEPTOR is a natural endogenous antagonist of mTOR kinase activity (17), this likely negatively impacts mTORC2 activity and thereby inhibits Akt activation and signaling promoted by GPCRs (Figs. 4, A–D, and 8A) and RTKs (Fig. 8, B and C). In addition, acute CXCR4 (<30 min) activation also promotes DEPTOR degradation, and this requires G protein and PI3K signaling (Fig. 10A). This suggests that Akt signaling is linked to DEPTOR degradation, but whether this is linked to activation of mTORC2 remains to be clearly defined. Phosphorylation of Akt at Ser-473 was maximal at 5 min in the presence of CXCL12 (Fig. 3A), which does not necessarily coincide with the time of DEPTOR degradation (Fig. 1B). However, treatment of cells with CXCL12 for 5 min led to dissociation of DEPTOR from mTORC2 (Fig. 6E). This is consistent with the idea that DEPTOR is somehow displaced from the mTORC2 complex and then is likely incorporated into ILVs of MVBs before it is degraded in lysosomes. This will need to be examined using high resolution microscopy and more sensitive biochemical strategies to conclusively determine whether DEPTOR is targeted into ILVs at these times points. Our data suggest a model in which DEPTOR degradation or sequestration into ILVs serves as a mechanism to promote mTORC2 activity and thereby facilitate Akt activation and signaling. DEPTOR associated with mTORC1 does not seem to be impacted by ESCRTs (Figs. 6B and 9), which may be linked to the fact that activated mTORC1 is found on the surface of lysosomes (64). Our working model is depicted in Fig. 11. Our study confirms and extends the notion that ESCRTs can serve as positive regulators of signaling and extends our knowledge about the cellular and molecular mechanisms that govern mTORC2 and Akt signaling.
FIGURE 11.

Proposed model for the role of ESCRTs in regulating GPCR-promoted Akt activation. Binding to agonist (black circle) leads to GPCR-promoted activation of G protein signaling, whereby GDP is exchanged for GTP leading to an active Gα subunit (Gα-GTP) and dissociation of the Gβγ heterodimer. Free Gβγ activates PI3K, which leads to the recruitment of PDK1 and Akt to the plasma membrane where Akt is phosphorylated at Thr-308 by PDK1 and by mTORC2 at Ser-473. Gβγ and PI3K signaling drive DEPTOR degradation likely by promoting DEPTOR targeting into the ILV of the MVBs)via the ESCRT pathway. MVBs eventually fuse with lysosomes where DEPTOR degradation occurs. DEPTOR degradation may facilitate phosphorylation of Akt at Ser-473 by mTORC2. GPCRs can also be targeted into ILVs via ubiquitination and direct interactions with ESCRTs. How DEPTOR is targeted into ILVs of MVBs remains to be determined.
DEPTOR is a cytosolic protein, but it is targeted for ESCRT- and lysosome-dependent degradation. Typically, ESCRTs target ubiquitinated transmembrane proteins for entry into ILVs of MVBs for eventual degradation in lysosomes (65). Cytosolic factors and even those that are peripherally associated with endosomal membranes are typically excluded from ILVs, such as the ESCRT proteins themselves (66). Because ubiquitination is required for CXCR4 entry into the ESCRT pathway and for its degradation (44, 67), it is possible that DEPTOR interacts with ubiquitinated CXCR4 directly or indirectly to be targeted into ILVs. For example, GSK-3β appears to be sequestered into ILVs as a large molecular complex with transmembrane proteins (34–36). An argument against this idea is that PI3K inhibition completely blocked DEPTOR degradation, while only modestly attenuating CXCR4 degradation (Fig. 10A). Alternatively, DEPTOR could enter ILVs via a receptor-independent mechanism (68), although it would still require G protein and PI3K signaling. In this case, DEPTOR could interact with an unknown factor that enters ILVs (68, 69). Other possibilities are likely, but in any case, other mTORC2 subunits would be excluded from ILVs; because of the mTORC2 subunits we examined, DEPTOR was the only one degraded upon GPCR activation (Fig. 1A) or impacted by ESCRT depletion (Fig. 2E). Further work will be required to precisely define the mechanism by which DEPTOR is targeted for lysosomal degradation.
In addition to regulating DEPTOR levels and phosphorylation of Akt at Ser-473 by mTORC2, ESCRTs also regulate phosphorylation of Akt at Thr-308. Phosphorylation of Thr-308 promoted by CXCR4 (Fig. 4, A–D) was also attenuated in ESCRT-depleted cells. PI3K catalyzes the formation of PtdIns(3,4,5)P3 from PtdIns(4,5)P2 at the plasma membrane, which recruits PDK1 and Akt via their respective pleckstrin homology domains (13). ESCRTs likely do not regulate PDK1 activity because it is constitutively activated (54). It is possible that ESCRT depletion could alter the localization of PDK1, Akt, or some other factor that regulates the phosphorylation status of Akt, but this remains to be examined. The effect of ESCRT depletion on Akt activation or signaling is likely not because of an indirect effect linked to other roles that ESCRTs have in other membrane-related functions, such as cytokinesis, viral budding, or plasma membrane repair, because to the best of our knowledge ESCRT-0 and ESCRT-II are not involved in these processes (70–73). Further work will be required to thoroughly understand how ESCRTs govern full activation of Akt.
We also show that Akt signaling is impacted by ESCRTs. In ESCRT-depleted cells phosphorylation of Akt substrates FoxO1/3a was substantially impaired, whereas phosphorylation of other Akt substrates GSK-3β and TSC2 was only moderately impaired (Figs. 7, A–D, and 8, A–C). These data are consistent with other studies that have shown that silencing of mTORC2 phosphorylation of Akt at Ser-473 impacts phosphorylation of Thr-24/Thr-32 on FoxO1/O3a, but not phosphorylation of Thr-1462 on TSC2 or Ser-9 on GSK-3β (49, 50). It is possible that phosphorylation of these sites is less sensitive to perturbations in Akt activity or a redundant kinase is able to phosphorylate these proteins when Akt activity is attenuated or absent (15). Alternatively, differences in subcellular localization may account for differences in Akt-mediated phosphorylation of its substrates (74). Taken together, our data indicate that ESCRTs govern Akt signaling, likely by regulating the levels of DEPTOR and thereby controlling the activity of mTORC2.
ESCRTs may play a general role in GPCR-promoted Akt signaling. For example, in addition to CXCL12, stimulation with norepinephrine also promotes DEPTOR degradation and Akt signaling via ESCRTs (Fig. 8, A, D, and E). Whether this applies to other GPCRs remains to be determined, but it may not extend to all GPCRs. For example, in contrast to CXCR4, activation of the angiotensin II type 1 receptor by angiotensin II does not activate Akt via mTORC2 (75). This suggests that angiotensin II type 1 receptor-promoted Akt signaling may occur via an ESCRT-independent mechanism. Therefore, it is possible that ESCRTs mediate Akt signaling of only a subset of GPCRs.
ESCRT-dependent Akt signaling is not limited to GPCRs. We also show that ESCRTs mediate Akt signaling induced by EGF or insulin (Fig. 8, B and C, respectively). However, our study highlights an important distinction between GPCRs and RTKs in that GPCR activation promotes rapid degradation of DEPTOR, whereas activation of EGF or insulin receptors does not (Fig. 8, D and quantification in E). Serum treatment of cells has been shown to promote DEPTOR degradation, but it occurs over long treatment times (4–24 h) and via the proteasome (17). In contrast, CXCR4 promotes DEPTOR degradation very rapidly (<30 min of CXCL12 treatment), and it occurs via lysosomes (Fig. 1). Serum (10% FBS) treatment, at least under our experimental conditions, did not promote DEPTOR degradation (Fig. 1C). Because ESCRT depletion leads to an overall increase in the cellular complement of DEPTOR, it is likely that Akt signaling induced by cell signaling receptors in general will be impacted. Alternatively, RTKs could selectively promote DEPTOR sequestration into ILVs without leading to its degradation, similar to what occurs with GSK-3β (34). Whether growth factor or RTK-driven DEPTOR sequestration into ILVs occurs will require further investigation to determine.
Akt signaling is involved in several cellular processes, including cell survival and cell motility (9). Phosphorylation of FoxO1/O3a transcription factors by Akt prevents them from translocating to the nucleus and enhancing the expression of genes that promote apoptosis, thereby facilitating cell survival (76–78). Interestingly, ESCRT-0 or ESCRT-I knock-out mice die early during embryogenesis, and the embryos show a high degree of cellular death in part due to apoptosis (33), which is consistent with a decrease in Akt signaling. We recently showed that siRNA targeting ESCRT-0 subunit STAM1 does not affect CXCR4-induced proliferation or survival of HeLa cells (8). The role of ESCRTs on cell survival remains to be examined further, but it is also possible that ESCRTs have other roles. ESCRTs have been linked to cell polarity and controlling focal adhesion dynamics (79, 80). Akt signaling has been linked to CXCR4-induced directed migration of HeLa cells (81), and directed cell migration is an important developmental and pathophysiological function of CXCR4 (3, 4, 6, 48, 82). Therefore, it is possible that ESCRTs and Akt signaling have important physiological roles in directed cell migration. In conclusion, our findings begin to explain the signal transduction pathways important in the physiological roles of ESCRTs and Akt.
Acknowledgments
We thank Debra Wyatt for technical assistance and Rohit Malik for help with experiments at the beginning of the project. We are especially grateful to JoAnn Trejo (University of California at San Diego) for kindly and generously sharing reagents. We thank Amy Rosenfeld for critically reading an early version of the manuscript and Tarun Patel for generous support.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM106727 (to A. M.).
- GPCR
- G protein-coupled receptor
- ESCRT
- endosomal sorting complex required for transport
- mTOR
- mammalian target of rapamycin
- RTK
- receptor tyrosine kinase
- MVB
- multivesicular body
- ILV
- intraluminal vesicle
- ANOVA
- analysis of variance
- BAEC
- bovine aortic endothelial cell
- PtdIns3P
- phosphatidylinositol 3-phosphate
- PtdIns(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PtdIns(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- 3-MA
- 3-methyladenine
- EGFR
- EGF receptor.
REFERENCES
- 1. Agarwal U., Ghalayini W., Dong F., Weber K., Zou Y. R., Rabbany S. Y., Rafii S., Penn M. S. (2010) Role of cardiac myocyte CXCR4 expression in development and left ventricular remodeling after acute myocardial infarction. Circ. Res. 107, 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ceradini D. J., Kulkarni A. R., Callaghan M. J., Tepper O. M., Bastidas N., Kleinman M. E., Capla J. M., Galiano R. D., Levine J. P., Gurtner G. C. (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858–864 [DOI] [PubMed] [Google Scholar]
- 3. Ma Q., Jones D., Borghesani P. R., Segal R. A., Nagasawa T., Kishimoto T., Bronson R. T., Springer T. A. (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9448–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zou Y. R., Kottmann A. H., Kuroda M., Taniuchi I., Littman D. R. (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393, 595–599 [DOI] [PubMed] [Google Scholar]
- 5. Hernandez P. A., Gorlin R. J., Lukens J. N., Taniuchi S., Bohinjec J., Francois F., Klotman M. E., Diaz G. A. (2003) Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 34, 70–74 [DOI] [PubMed] [Google Scholar]
- 6. Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verástegui E., Zlotnik A. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 [DOI] [PubMed] [Google Scholar]
- 7. Busillo J. M., Benovic J. L. (2007) Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malik R., Soh U. J., Trejo J., Marchese A. (2012) Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J. Biol. Chem. 287, 9013–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 10. Dbouk H. A., Vadas O., Shymanets A., Burke J. E., Salamon R. S., Khalil B. D., Barrett M. O., Waldo G. L., Surve C., Hsueh C., Perisic O., Harteneck C., Shepherd P. R., Harden T. K., Smrcka A. V., et al. (2012) G protein-coupled receptor-mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Sci. Signal. 5, ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guillermet-Guibert J., Bjorklof K., Salpekar A., Gonella C., Ramadani F., Bilancio A., Meek S., Smith A. J., Okkenhaug K., Vanhaesebroeck B. (2008) The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc. Natl. Acad. Sci. U.S.A. 105, 8292–8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leopoldt D., Hanck T., Exner T., Maier U., Wetzker R., Nürnberg B. (1998) Gβγ stimulates phosphoinositide 3-kinase-γ by direct interaction with two domains of the catalytic p110 subunit. J. Biol. Chem. 273, 7024–7029 [DOI] [PubMed] [Google Scholar]
- 13. Hirsch E., Katanaev V. L., Garlanda C., Azzolino O., Pirola L., Silengo L., Sozzani S., Mantovani A., Altruda F., Wymann M. P. (2000) Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science 287, 1049–1053 [DOI] [PubMed] [Google Scholar]
- 14. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 15. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ballon D. R., Flanary P. L., Gladue D. P., Konopka J. B., Dohlman H. G., Thorner J. (2006) DEP-domain-mediated regulation of GPCR signaling responses. Cell 126, 1079–1093 [DOI] [PubMed] [Google Scholar]
- 17. Peterson T. R., Laplante M., Thoreen C. C., Sancak Y., Kang S. A., Kuehl W. M., Gray N. S., Sabatini D. M. (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Y., Xiong X., Sun Y. (2011) DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao D., Inuzuka H., Tan M.-K., Fukushima H., Locasale J. W., Liu P., Wan L., Zhai B., Chin Y. R., Shaik S., Lyssiotis C. A., Gygi S. P., Toker A., Cantley L. C., Asara J. M., Harper J. W., Wei W. (2011) mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan S., Skaar J. R., Kuchay S., Toschi A., Kanarek N., Ben-Neriah Y., Pagano M. (2011) mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol. Cell 44, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Efeyan A., Sabatini D. M. (2010) mTOR and cancer: many loops in one pathway. Curr. Opin. Cell Biol. 22, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ciechanover A. (2013) Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg. Med. Chem. 21, 3400–3410 [DOI] [PubMed] [Google Scholar]
- 23. Luzio J. P., Parkinson M. D., Gray S. R., Bright N. A. (2009) The delivery of endocytosed cargo to lysosomes. Biochem. Soc. Trans. 37, 1019–1021 [DOI] [PubMed] [Google Scholar]
- 24. Henne W. M., Stenmark H., Emr S. D. (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henne W. M., Buchkovich N. J., Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 26. Hurley J. H., Hanson P. I. (2010) Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gruenberg J., Stenmark H. (2004) The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5, 317–323 [DOI] [PubMed] [Google Scholar]
- 28. Raiborg C., Stenmark H. (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452 [DOI] [PubMed] [Google Scholar]
- 29. Clague M. J., Liu H., Urbé S. (2012) Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 23, 457–467 [DOI] [PubMed] [Google Scholar]
- 30. Hurley J. H., Stenmark H. (2011) Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu. Rev. Biophys. 40, 119–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchese A., Trejo J. (2013) Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 25, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hislop J. N., von Zastrow M. (2011) Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic 12, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodahl L. M., Stuffers S., Lobert V. H., Stenmark H. (2009) The role of ESCRT proteins in attenuation of cell signalling. Biochem. Soc. Trans. 37, 137–142 [DOI] [PubMed] [Google Scholar]
- 34. Taelman V. F., Dobrowolski R., Plouhinec J. L., Fuentealba L. C., Vorwald P. P., Gumper I., Sabatini D. D., De Robertis E. M. (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobrowolski R., De Robertis E. M. (2012) Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol. 13, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dobrowolski R., Vick P., Ploper D., Gumper I., Snitkin H., Sabatini D. D., De Robertis E. M. (2012) Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2, 1316–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarbassov D. D., Ali S. M., Kim D.-H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 38. Marchese A., Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 [DOI] [PubMed] [Google Scholar]
- 39. Bhandari D., Trejo J., Benovic J. L., Marchese A. (2007) Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 282, 36971–36979 [DOI] [PubMed] [Google Scholar]
- 40. Holleman J., Marchese A. (2014) The ubiquitin ligase Deltex-3L regulates endosomal sorting of the G protein-coupled receptor CXCR4. Mol. Biol. Cell 25, 1892–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malik R., Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fenteany G., Standaert R. F., Lane W. S., Choi S., Corey E. J., Schreiber S. L. (1995) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268, 726–731 [DOI] [PubMed] [Google Scholar]
- 43. Seglen P. O. (1983) Inhibitors of lysosomal function. Methods Enzymol. 96, 737–764 [DOI] [PubMed] [Google Scholar]
- 44. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 [DOI] [PubMed] [Google Scholar]
- 45. Hurley J. H. (2010) The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45, 463–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stefani F., Zhang L., Taylor S., Donovan J., Rollinson S., Doyotte A., Brownhill K., Bennion J., Pickering-Brown S., Woodman P. (2011) UBAP1 is a component of an endosome-specific ESCRT-I complex that is essential for MVB sorting. Curr. Biol. 21, 1245–1250 [DOI] [PubMed] [Google Scholar]
- 47. Valiathan R. R., Resh M. D. (2008) Differential control of CXCR4 and CD4 downregulation by HIV-1 Gag. Virol. J. 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tachibana K., Hirota S., Iizasa H., Yoshida H., Kawabata K., Kataoka Y., Kitamura Y., Matsushima K., Yoshida N., Nishikawa S., Kishimoto T., Nagasawa T. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594 [DOI] [PubMed] [Google Scholar]
- 49. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 50. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 51. Gibson S. K., Gilman A. G. (2006) Giα and Gβ subunits both define selectivity of G protein activation by α2-adrenergic receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 53. Katada T. (2012) The inhibitory G protein G(i) identified as pertussis toxin-catalyzed ADP-ribosylation. Biol. Pharm. Bull. 35, 2103–2111 [DOI] [PubMed] [Google Scholar]
- 54. Mora A., Komander D., van Aalten D. M., Alessi D. R. (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161–170 [DOI] [PubMed] [Google Scholar]
- 55. Bonacci T. M., Mathews J. L., Yuan C., Lehmann D. M., Malik S., Wu D., Font J. L., Bidlack J. M., Smrcka A. V. (2006) Differential targeting of Gβγ-subunit signaling with small molecules. Science 312, 443–446 [DOI] [PubMed] [Google Scholar]
- 56. Robles-Molina E., Dionisio-Vicuña M., Guzmán-Hernández M. L., Reyes-Cruz G., Vázquez-Prado J. (2014) Gβγ interacts with mTOR and promotes its activation. Biochem. Biophys. Res. Commun. 444, 218–223 [DOI] [PubMed] [Google Scholar]
- 57. Brunn G. J., Williams J., Sabers C., Wiederrecht G., Lawrence J. C., Jr., Abraham R. T. (1996) Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 15, 5256–5267 [PMC free article] [PubMed] [Google Scholar]
- 58. Volinia S., Dhand R., Vanhaesebroeck B., MacDougall L. K., Stein R., Zvelebil M. J., Domin J., Panaretou C., Waterfield M. D. (1995) A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 14, 3339–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Raiborg C., Bremnes B., Mehlum A., Gillooly D. J., D'Arrigo A., Stang E., Stenmark H. (2001) FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J. Cell Sci. 114, 2255–2263 [DOI] [PubMed] [Google Scholar]
- 60. Raiborg C., Schink K. O., Stenmark H. (2013) Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J. 280, 2730–2742 [DOI] [PubMed] [Google Scholar]
- 61. Simonsen A., Tooze S. A. (2009) Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 186, 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miller S., Tavshanjian B., Oleksy A., Perisic O., Houseman B. T., Shokat K. M., Williams R. L. (2010) Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 327, 1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shpetner H., Joly M., Hartley D., Corvera S. (1996) Potential sites of PI-3 kinase function in the endocytic pathway revealed by the PI-3 kinase inhibitor, wortmannin. J. Cell Biol. 132, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Efeyan A., Zoncu R., Sabatini D. M. (2012) Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shields S. B., Piper R. C. (2011) How ubiquitin functions with ESCRTs. Traffic 12, 1306–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hurley J. H., Boura E., Carlson L. A., Różycki B. (2010) Membrane budding. Cell 143, 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marchese A. (2014) Endocytic trafficking of chemokine receptors. Curr. Opin. Cell Biol. 27, 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mageswaran S. K., Dixon M. G., Curtiss M., Keener J. P., Babst M. (2014) Binding to any ESCRT can mediate ubiquitin-independent cargo sorting. Traffic 15, 212–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Malerød L., Pedersen N. M., Sem Wegner C. E., Lobert V. H., Leithe E., Brech A., Rivedal E., Liestøl K., Stenmark H. (2011) Cargo-dependent degradation of ESCRT-I as a feedback mechanism to modulate endosomal sorting. Traffic 12, 1211–1226 [DOI] [PubMed] [Google Scholar]
- 70. Pornillos O., Higginson D. S., Stray K. M., Fisher R. D., Garrus J. E., Payne M., He G. P., Wang H. E., Morham S. G., Sundquist W. I. (2003) HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 162, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Langelier C., von Schwedler U. K., Fisher R. D., De Domenico I., White P. L., Hill C. P., Kaplan J., Ward D., Sundquist W. I. (2006) Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 80, 9465–9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jimenez A. J., Maiuri P., Lafaurie-Janvore J., Divoux S., Piel M., Perez F. (2014) ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- 73. Morita E., Sandrin V., Chung H.-Y., Morham S. G., Gygi S. P., Rodesch C. K., Sundquist W. I. (2007) Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26, 4215–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schenck A., Goto-Silva L., Collinet C., Rhinn M., Giner A., Habermann B., Brand M., Zerial M. (2008) The endosomal protein appl1 mediates akt substrate specificity and cell survival in vertebrate development. Cell 133, 486–497 [DOI] [PubMed] [Google Scholar]
- 75. Nazarewicz R. R., Salazar G., Patrushev N., San Martin A., Hilenski L., Xiong S., Alexander R. W. (2011) Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J. Biol. Chem. 286, 2886–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Greer E. L., Brunet A. (2008) FOXO transcription factors in ageing and cancer. Acta Physiologica 192, 19–28 [DOI] [PubMed] [Google Scholar]
- 77. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Delgado-Martín C., Escribano C., Pablos J. L., Riol-Blanco L., Rodríguez-Fernández J. L. (2011) Chemokine CXCL12 uses CXCR4 and a signaling core formed by bifunctional Akt, extracellular signal-regulated kinase (ERK)1/2, and mammalian target of rapamycin complex 1 (mTORC1) proteins to control chemotaxis and survival simultaneously in mature dendritic cells. J. Biol. Chem. 286, 37222–37236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lobert V. H., Stenmark H. (2012) The ESCRT machinery mediates polarization of fibroblasts through regulation of myosin light chain. J. Cell Sci. 125, 29–36 [DOI] [PubMed] [Google Scholar]
- 80. Tu C., Ortega-Cava C. F., Winograd P., Stanton M. J., Reddi A. L., Dodge I., Arya R., Dimri M., Clubb R. J., Naramura M., Wagner K. U., Band V., Band H. (2010) Endosomal-sorting complexes required for transport (ESCRT) pathway-dependent endosomal traffic regulates the localization of active Src at focal adhesions. Proc. Natl. Acad. Sci. U.S.A. 107, 16107–16112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peng S. B., Peek V., Zhai Y., Paul D. C., Lou Q., Xia X., Eessalu T., Kohn W., Tang S. (2005) Akt activation, but not extracellular signal-regulated kinase activation, is required for SDF-1α/CXCR4-mediated migration of epithelioid carcinoma cells. Mol. Cancer Res. 3, 227–236 [DOI] [PubMed] [Google Scholar]
- 82. Li Y. M., Pan Y., Wei Y., Cheng X., Zhou B. P., Tan M., Zhou X., Xia W., Hortobagyi G. N., Yu D., Hung M. C. (2004) Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6, 459–469 [DOI] [PubMed] [Google Scholar]