

Background: Nerve injury induces Schwann cells to remove myelin coating and secrete factors that stimulate nerve regeneration.
Results: ChIP-seq was used to identify injury-regulated enhancers in peripheral nerve.
Conclusion: Dynamically regulated enhancers identify target sequences of injury-regulated transcription factors in Schwann cells.
Significance: These data elucidate transcription factor pathways that coordinate Schwann cell responses in peripheral nerve injury.
Keywords: ChIP Sequencing (ChIP-seq), Chromatin Immunoprecipitation (ChiP), Glial Cell, Histone Modification, Neurobiology, Schwann Cells
Abstract
Myelination of the peripheral nervous system is required for axonal function and long term stability. After peripheral nerve injury, Schwann cells transition from axon myelination to a demyelinated state that supports neuronal survival and ultimately remyelination of axons. Reprogramming of gene expression patterns during development and injury responses is shaped by the actions of distal regulatory elements that integrate the actions of multiple transcription factors. We used ChIP-seq to measure changes in histone H3K27 acetylation, a mark of active enhancers, to identify enhancers in myelinating rat peripheral nerve and their dynamics after demyelinating nerve injury. Analysis of injury-induced enhancers identified enriched motifs for c-Jun, a transcription factor required for Schwann cells to support nerve regeneration. We identify a c-Jun-bound enhancer in the gene for Runx2, a transcription factor induced after nerve injury, and we show that Runx2 is required for activation of other induced genes. In contrast, enhancers that lose H3K27ac after nerve injury are enriched for binding sites of the Sox10 and early growth response 2 (Egr2/Krox20) transcription factors, which are critical determinants of Schwann cell differentiation. Egr2 expression is lost after nerve injury, and many Egr2-binding sites lose H3K27ac after nerve injury. However, the majority of Egr2-bound enhancers retain H3K27ac, indicating that other transcription factors maintain active enhancer status after nerve injury. The global epigenomic changes in H3K27ac deposition pinpoint dynamic changes in enhancers that mediate the effects of transcription factors that control Schwann cell myelination and peripheral nervous system responses to nerve injury.
Introduction
Myelination of the peripheral nervous system is a vital aspect of nervous system development and function. The myelin sheath produced by Schwann cells is a specialized multilayer membrane essential for the rapid transmission of nerve impulses by axons. Impaired myelin structure and function are causative factors in a number of peripheral nerve disorders, such as Charcot-Marie-Tooth disease. Schwann cells not only produce and maintain the myelin sheath but also retain the developmental plasticity to break down myelin in response to injury, secrete molecules to support axonal survival, and then eventually remyelinate axons in regenerating nerves (1–3).
Induction and maintenance of Schwann cell myelination are mediated by several key transcription factors (4, 5). For example, early growth response 2 (also known as Krox20) is a C2H2 zinc finger transcription factor required for myelin formation and maintenance by Schwann cells (6–8), and it has been shown to regulate several major myelin genes (8, 9). Sox10 is a critical HMG box transcription factor for Schwann cell specification and development, and it is also required for the maintenance of peripheral nerve myelination (10). Egr2 and Sox10 activate myelin gene transcription through coordinate binding of distal and intronic enhancer sites in critical myelin genes such as connexin 32 (gap junction B1/Gjb1), myelin basic protein (Mbp), peripheral myelin protein 22 (Pmp22), and myelin protein zero (Mpz) (11–16).
The breakdown of axon-Schwann cell contact following nerve injury results in reduced expression of major myelin genes, which can be attributed in part to a rapid loss of Egr2 expression (17, 18). Several studies have highlighted the involvement of specific signaling pathways in mediating demyelination after nerve injury, such as p38, JNK, and ERK signaling (19–22). JNK and p38 signaling activate c-Jun, a basic leucine zipper transcription factor that is one of the major transcriptional determinants that trigger rapid demyelination in Schwann cells and induce a genetic program that supports axon regeneration (23). Gain of c-Jun and loss of Egr2 are therefore major contributors to altered gene expression in Schwann cells of injured nerve.
Gene expression changes during development and injury are shaped by the actions of enhancers that integrate the actions of multiple transcription factor inputs to regulate associated genes. These distal enhancer elements are integration hubs for signaling pathways activated by extracellular signals, and sequence analysis of active enhancers can be used to determine binding motifs for transcription factors that regulate enhancer activity (24). Recent genomics studies using chromatin immunoprecipitation (ChIP-seq) have offered tools to assess not only transcription factor binding but also enhancer activation status in specific tissues in a stage-dependent manner (25, 26). General characteristics of enhancers include open chromatin, histone H3K4 monomethylation, and recruitment of histone acetylases CBP/p300 (27). Recently, the chromatin mark for acetylated histone H3 lysine 27 (H3K27ac) has been identified as an epigenetic marker of actively engaged enhancer elements (28–30), and it allows differentiation from other nucleosome-depleted regions of chromatin, such as poised enhancers.
Substantial progress has been made in profiling gene expression changes and establishing the regulatory hierarchy and target genes of individual transcription factors during development and injury of peripheral nerve (9, 23, 31–33). Previous studies have elucidated combinatorial control of individual enhancers driving Schwann cell differentiation genes, such as Gjb1, Mbp, Mpz, Egr2, and Sox10 (4, 12, 14, 15, 34–40). However, genome-wide identification of the enhancers that form the substrate of the transcription factors' effects on gene expression is required to understand targeting mechanisms and to reveal the combinatorial control of genome-wide regulatory networks underlying myelin formation and the Schwann cell's response to nerve injury.
To identify active regulatory elements in Schwann cells, the chromatin signature H3K27ac was assessed in mature peripheral nerve tissue and after injury-induced demyelination. These data identified injury-regulated enhancers that mediate the effects of transcription factors that decline or increase after nerve injury. Furthermore, transcriptional regulators of Schwann cell differentiation were identified through analysis of enhancers targeted for epigenomic reprogramming that occurs after a demyelinating injury.
EXPERIMENTAL PROCEDURES
Nerve Injury Surgery
All animal experiments were performed according to protocols approved by the School of Veterinary Medicine, University of Wisconsin (Madison, WI). Male P25 Sprague-Dawley rats or adult male mice (Harlan Laboratory) were housed with normal light/dark cycles and ad libitum access to food and water. Prior to surgery, animals were anesthetized with isoflurane (Piramal Healthcare), and an injection of 5 mg/kg ketoprofen was given for analgesia. Under aseptic conditions, an incision (∼5 mm long) was made through the skin and sciatic nerve at the proximal lateral region of the femur. As a control, the contralateral leg also received a sham operation consisting of only a skin incision. The skin wound was sutured with rodent surgical staples, and the rats were caged for 72 h post-operation. The nerve tissue distal to the transection or sham site was isolated for use in expression and ChIP experiments.
Immunohistochemistry
Freshly dissected mouse sciatic nerve from sham and distal sections of injured nerve were fixed with 4% paraformaldehyde for 4 h at 4 °C and then immersed in PBS overnight at 4 °C. The nerve samples were embedded in Tissue-Tek OCT compound (Sakura Finetek) and cut into 10-μm cryostat sections. For fluorescence immunohistochemistry, the sections were blocked in 3% donkey serum, 0.2% Triton X-100 for 1 h at room temperature. Incubation with primary antibody was performed overnight at 4 °C in blocking solution, and secondary antibody incubation was performed at room temperature for 1 h. The primary antibodies used were rabbit anti-Runx2 (1:100; Santa Cruz Biotechnology, SC10758) and goat anti-Sox10 (1:200; R&D Systems AF2864). Secondary antibodies used were anti-rabbit Alexa 488 and anti-goat Alexa 568 (1:1000; Invitrogen, A21206 and A11057). Confocal images were taken using a Leica TCS LSI Macro microscope (Leica, Wetzlar, Germany).
ChIP
Freshly dissected rat sciatic nerve was minced in 1% formaldehyde for 10 min and then quenched for 10 min with glycine to a final concentration of 0.125 m. Samples were sequentially lysed in buffers LB1, LB2, and LB3 + 0.03% SDS (41). DNA was fragmented to an average size of 0.5–2 kb using 4× for 10 min Bioruptor (Diagenode) cycles on the medium setting. Each aliquot of sonicated chromatin (300 μg) was incubated overnight at 4 °C with 5 μg of antibody. A 10% aliquot was saved for input analysis. For histone antibody ChIP experiments, an 80-μl aliquot of protein G Dynabead (Invitrogen) slurry was added to each ChIP sample for 4 h at 4 °C. Immunoprecipitations were washed five times in RIPA buffer and then eluted at 65 °C in reverse cross-linking buffer (50 mm Tris, 10 mm EDTA, 1% SDS). For transcription factor antibody ChIP experiments, chromatin/antibody mixtures incubated in protein G-Sepharose beads (GE Healthcare) were used as described previously (12, 42), excluding herring sperm DNA from the blocking procedure. ChIP DNA was purified by phenol chloroform extraction and resuspended in 10 mm Tris, pH 8.0. Antibodies used in the study are as follows: H3K27ac (Active Motif, 37133), normal rabbit IgG (Millipore, 12-370), and c-Jun H-79 (Santa Cruz Biotechnology, SC-1694). Statistics were calculated using Student's t test. Error bars represent standard deviation, and asterisks denote p value (*, p ≤ 0.05; **, p ≤ 0.005). The samples were generated from independent chromatin pools (n = 3 for transcription factor ChIP, n = 2 or 3 for H3K27ac ChIP) and were analyzed using quantitative PCR primers listed in Table 1.
TABLE 1.
ChIP primers
Neg. is negative; TF is transcription factor.
| Gene | Primer | No. | Forward | Reverse |
|---|---|---|---|---|
| Hmga1 | rHMGA1-DB Ch1 | 1 | GTGGAGGGTGGGTTAAGGAT | TTCTGAGAGCCCAGTGAGGT |
| rmHMGA1-TFs Ch1 | 2 | CTGCTGAGTCACCCACACAC | GAGATGCCCTCCTCTTCCTC | |
| Shh | rShh-Jun Ch4 | 1 | GCCACTTACTGGCCTACCAT | TCAGACCGACCAAGCTCTTT |
| rShh-DB Ch1 | 2 | ATGTTAATGCAGGTGCCACA | GGGTTCCAGTGCTTTGTCAT | |
| Runx2 | rRunx2-TF Ch1 | 1 | TGCAGAGTTCTGCTCTCCAA | ACAAAAGGCTTGTGGTGAGG |
| rRunx2-TF Ch2 | 2 | GTGAGGATGGCAGATGTCCT | ACCTCATCAAAACCCCACAA | |
| mrRunx2-RUNX2 Ch4 | 3 | CCACCCAGCTGCTTGTACTT | TTCACATTCACTGCCCTCAG | |
| mrRunx2-RUNX2 Ch5 | 4 | AAGCCACAGTGGTAGGCAGT | TTGTTTGTGAGGCGAATGAA | |
| Gdnf | rGDNF-Jun Ch1 | 1 | CCATGAATCGGGAGTAGGAA | CCGGTCAAAGAGCACAAACT |
| Egr2 | rEGR2 pro- Ch1 | 1 | GAAAGTCTCGGAGAACCGGAA | GCAGCGATGGTAGCTCTGTCT |
| mrEGR2 MSE | 2 | TCCTGACCAGAAAGATTGTTA | TGCAGGATTTCAGCTTTGTGA | |
| Hmgcr | rmHMG pro-CRE Ch1 | 1 | TCGTGACGTAGGCCGTCAG | CCAATAAGGAAGGATCGTCCG |
| Mag | rMag Int Ch2 | 1 | ACAGGGATCTTTTGTATTGGCTACA | CGCAGCAGGCTTAGGGTG |
| Pmp22 | rPmp22 Ch24 | 1 | CCTTCCTCCTGTGCAGCATAAG | GGGCCTGGCGAAGCA |
| mrPmp22 − 90-kb Ch2 | 2 | TCACTCCCACTGGCTGGAA | AAATGCCCTTTGTCCGAACTC | |
| mrTEKT3 Ch1 (Neg.) | Neg. | AATACCAGCAGATCCGGAAGAC | TTGACTCGTTCTCCCAGGTTTT | |
| rPmp22 − Ch1 | 3 | AGGCCCGCCCTCCTGCACACG | TTTGCGTCGCTGCAGAAG |
ChIP-seq
Libraries were prepared from sham/injured P25 sciatic nerve H3K27ac ChIP samples and respective inputs, using the Illumina TruSeq ChIP sample preparation kit (Illumina Inc., San Diego). After library construction and amplification, quality and quantity were assessed using an Agilent DNA 1000 series chip assay (Agilent Technologies, Santa Clara, CA). The libraries were sequenced using Illumina HiSeq2000. Base calling was performed using the standard Illumina Pipeline. Reads were mapped to the Rattus norvegicus genome Rn5 using Bowtie to produce SAM files for further analysis. From the two biological replicates, we obtained 33,244,433 and 24,935,382 (sham) and 21,485,993 and 45,718,907 (injury) uniquely mapping reads. Input runs mapped 39,351,486 and 21,297,471 (sham) and 42,439,666 and 38,164,305 (injury) reads.
Peak Calling
MOSAiCS (43) was used to determine enriched binding regions for Sham_H3K27ac Injury_H3K27ac ChIP, respectively. For all data sets, BED files were generated with HOMER and subsequently uploaded to the UCSC genome browser for visualization purposes. Peak finding was performed using the R package MOSAiCS (MOdel-based one and two Sample Analysis and inference for ChIP-seq Data) controlled for a false discovery rate at 0.05. MOSAiCS implements a model-based approach where the background distribution for unbound regions takes into account systematic biases such as mappability and GC content, and the peak regions are described with a two-component negative binomial mixture model. Statistically different peaks were determined using DBChIP (44). Peak files were deposited in NCBI GEO under accession number GSE63103.
Functional Enrichment and Genome Ontology Analysis
DAVID (Database for Annotation, Visualization, and Integrated Discovery) is an integrated biological knowledge base and gene annotation tool set for the identification and clustering of statistically relevant functional and biological categories in large gene sets. Gene Ontology categories were clustered using high stringency, and categories with p values <0.05 and Benjamini <0.05 were chosen for analysis (45, 46).
Data Analysis
We used HOMER to analyze H3K27ac-bound regions to identify and compare motif signatures characterizing myelination-specific enhancers. Enhancer regions were compared with genomic fragments in 500-bp bins for motif enrichment. A heatmap matrix file was generated using HOMER, clustered with Cluster 3.0 using hierarchical average linkage with an uncentered correlation similarity metric, and was visualized using Java Tree View. Conversions between genome builds and genomes were calculated using the Liftover utility, and intersections between genomic regions were calculated on bed file tracks using the Table Browser utility; both programs are available in the UCSC Genome browser.
Schwann Cell Culture and siRNA Transfection
Primary rat Schwann cells were maintained on poly l-lysine-coated plates in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% bovine growth serum (Hyclone, Logan, UT), 0.02 μg/ml bovine pituitary extract (Sigma), and 2 μm forskolin (Sigma). Briefly, 24 h after seeding cells in 6- or 24-well plates, rat Schwann cells were transfected using Lipofectamine 3000 (Invitrogen, according to the manufacturer's protocol) with 30 pmol of either NC1 negative control siRNA (siControl, Integrated DNA Technologies catalog no. 51-01-14-03), c-Jun siRNA (siJun, IDT trifecta NM_053470), or Runx2 siRNA, (siRunx2, IDT trifecta NM_621835). The transfected cells were cultured in 1:1 serum-free DMEM/Ham's F-12, with insulin/transferrin/selenium supplement (Sigma). RNA was purified from cells 48 h after transfection using TriReagent (Ambion) and analyzed by quantitative reverse transcription (RT)-PCR using the StepOne Plus system (Applied Biosystems). Relative amounts of each gene were determined by the comparative CT method and normalized to 18 S rRNA (47). Statistics were calculated using Student's t test. Error bars represent standard deviation, and asterisks denote p value (*, p ≤ 0.05; **, p ≤ 0.005). The statistics were generated from independently generated samples in triplicate (n = 3) and were analyzed using primers listed in Table 2.
TABLE 2.
qRT-PCR primers
| Gene | Primer | Forward | Reverse |
|---|---|---|---|
| 18 S | 18 S rRNA | CGCCGCTAGAGGTGAAATTCT | CGAACCTCCGACTTTCGTTCT |
| c-Jun | rJun TM2 | GAGAGGAAGCGCATGAGGAAC | CCTTTTCCGGCACTTGGAG |
| Runx2 | rRunx2 TM | GCACCCAGCCCATAATAGAA | TGGAGATGTTGCTCTGTTCG |
| Shh | rShh TM | GCGGGCATCCACTGGTACT | TCGGACTTCAGCTGGACTTGA |
| Olig1 | rOlig1 TM | ACATCAAGGGTGTTGCCGAT | GACACCGGACTCTGGGCT T |
| GDNF | rGDNF TM | ACTGACTTGGGTTTGGGCTA | CCTGGCCTACCTTGTCACTT |
| HMGA1 | rHMGA1 TM2 | AGTGCAGGGGAAGCTTAGGT | GCTTGATGGGGGACATACAT |
| DRP2 | rDrp2 TM | GAGAAGATCCTGGCCCATTT | CCTCAGCTCTCCCTGAAGAA |
RESULTS
Identification of H3K27ac-marked Enhancers in Rat Sciatic Nerve
To examine genome-wide enhancer activity involved in Schwann cell myelination, we performed ChIP-seq mapping of H3K27ac-containing enhancers in rat sciatic nerve. Although H3K27ac ChIP-seq has been performed in several tissues (25, 26), the sciatic nerve is a good substrate for enhancer profiling because it is highly enriched in Schwann cells, which constitute ∼80–90% of the tissue. More than 50% of these cells are myelinating Schwann cells, whereas the remaining are nonmyelinating (Remak) Schwann cells (17, 18). In addition, the presence of neuronal nuclei in the spinal cord allows physical separation from Schwann cell nuclei in sciatic nerve. Based on these unique features, ChIP techniques applied to peripheral nerve allow analysis of dynamically regulated epigenetic changes in vivo.
We wished to not only profile active enhancers in peripheral nerve, but also to examine genome-wide changes in enhancers after injury. Therefore, ChIP-seq analysis was performed using sciatic nerve from P25 rats with either nerve transection or sham surgery (Fig. 1A). Two independent pools of cross-linked chromatin were made from sciatic nerve collected 72 h after injury (or sham surgery) and immunoprecipitated with an H3K27ac antibody validated for minimal histone mark cross-reactivity (48, 49). This time point was chosen based on published analyses of nerve injury gene expression (9, 18, 31) as most myelin genes are decreased to basal levels, and injury-induced genes are fully activated by this time.
FIGURE 1.
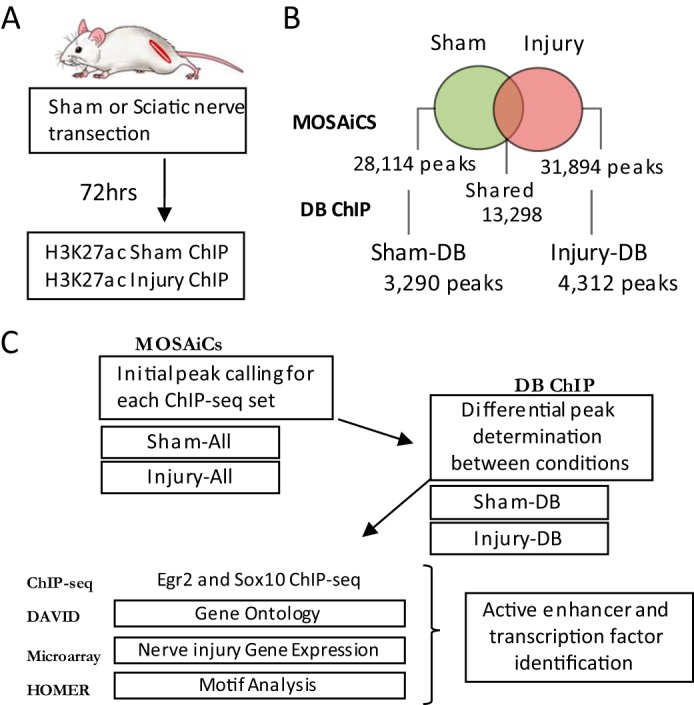
Peak calling and analysis of H3K27ac-marked active enhancers in myelinating nerve. A, ChIP-seq mapping of H3K27ac-containing enhancers was performed in vivo using distal nerve tissue from rat sciatic nerve 72 h post-nerve transection or sham surgery. B, Venn diagram illustrates the overlap between called peaks identified in sham and injury H3K27ac ChIP-seq runs and those peaks with statistically significant differential binding. C, workflow diagram and name designations for data sets. Total H3K27ac ChIP-seq peaks are called using MOSAiCs (Sham-All and Injury-All). Differentially bound peaks are determined using DB ChIP, which identifies peaks that are significantly changed between two conditions (Sham-DB and Injury-DB). Peak sequences were analyzed using Homer to identify enriched transcription factor motifs, and H3K27ac-marked enhancers were analyzed with respect to previous ChIP-seq data sets of transcription factor binding in peripheral nerve (Egr2 and Sox10). Genes linked to differential peaks were correlated with published nerve injury expression microarray data. DAVID was used to determine gene ontology groupings for genes determined to be regulated by DB sites.
Fig. 1C describes the process used to identify H3K27ac-enriched regions, determine differential presence of active enhancer marks between sham and injury conditions, and analyze differential sites using other available bioinformatics tools and data sets. ChIP-seq reads were mapped to the Rn5 genome, and peaks were called for each data set using MOSAiCS (43). A total of 28,114 peaks were called in the sham condition and 31,894 peaks in the nerve injury condition.
To identify peaks that were specific to sham or injured nerve, we employed DBChIP (44), which identifies differentially enriched peaks between the data sets. The core methodology of DBChIP relies on formally testing candidate peak regions for differential enrichment across multiple datasets. DBChIP first merges the lists of predicted binding locations from biological replicates by clustering nearby peaks into aggregated coordinates. Then, the hypothesis of nondifferential binding is tested at each consensus site to identify peaks that differentially enriched in the two data sets. The output from DBChIP is a list of p values quantifying evidence against equal enrichment and the corresponding fold changes. A major advantage of this method is that it eliminates spurious differential peaks that are slightly above threshold for peak calling in one data set and slightly below threshold in the comparison set.
Based on this analysis, many of the unique peaks were not statistically different in the two data sets, but DBChIP identified 4312 differentially bound H3K27ac peaks in the injured state (increased after nerve injury, designated InjuryDB) and 3290 peaks specific to the sham condition (decreased after nerve injury, designated ShamDB) (Fig. 1B). Because individual genes can be associated with multiple DB peaks, the total number of genes annotated within 50 kb of ShamDB peaks is 1909 and 2197 for InjuryDB. All differentially bound peaks were enriched >5-fold in one peak set relative to the other, with 75% exceeding 10-fold difference (listed in the supplemental Tables). The indicated number of shared peaks (13,298) reflects those peaks that were called in both data sets. To expand our bioinformatic analysis, we integrate several data sets with the information of active enhancer binding through H3K27ac. Together, this multidimensional analysis is used to shed light on the noncoding regulatory sites that underlie the gene expression networks that control peripheral nerve myelination.
In Vivo Analysis of Enhancer Dynamics after Nerve Injury
As examples of ShamDB and InjuryDB sites, Fig. 2 shows differential H3K27 acetylation in the Drp2 and Ngfr genes. Dystrophin-related protein 2 (Drp2) interacts with Periaxin to form the Cajal bands in myelinating Schwann cells (50), and its gene is one of the most down-regulated genes in a recent microarray analysis of nerve injury (23). A single enhancer marked by H3K27 acetylation was noted within an intron, which was dramatically reduced at 3 days post-injury. As shown previously (28–30), H3K27 acetylation often flanks the nucleosome-depleted core of enhancer sequences where transcription factors are bound. This pattern is apparent in the Drp2 gene, where a previously described Egr2-binding site (51) resides in a cleft between two peaks of H3K27 acetylation. Nerve growth factor receptor (Ngfr/p75) is induced in Schwann cells after nerve injury (52), and several InjuryDB H3K27ac peaks are observed in the promoter and upstream regions in the injury condition.
FIGURE 2.
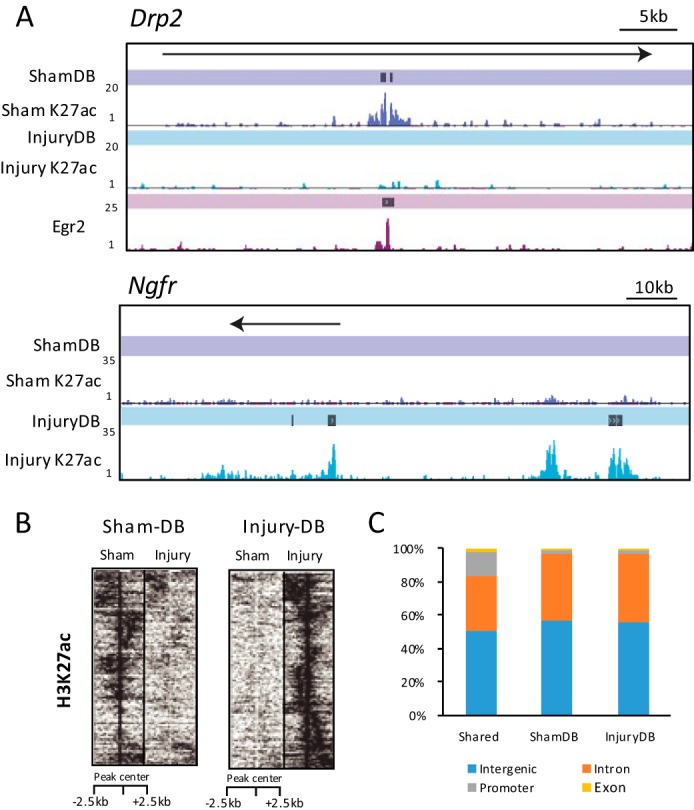
Dynamic regulation of H3K27ac-marked enhancer regions following nerve injury. A, H3K27 acetylation and DB tracks are shown for sham and injury conditions at Drp2 and Ngfr genes. The Egr2-binding peak track and a called peak within Drp2 is also shown. B, heatmap plot shows enrichment of H3K27ac sequencing tags aligned to a 5-kb window around DB peaks in sham and injury conditions. The height of the two heat maps was adjusted to be equal, even though they represent different numbers of genes. Although the heat map is centered on the DB peaks, there are sometimes adjoining regions within the 5-kb window that were also differentially regulated, and these were clustered together during heatmap generation. C, bar graph shows genomic distribution of differentially bound (DB) and shared peak sets for intronic, intergenic, and promoter (−1 kb to +0.88 kb around the transcription start site) classifications.
The changes in acetylation in the differentially enriched peaks are depicted in a heatmap plot of H3K27ac tag density in a 5-kb window around identified DB peaks in injured and sham nerve (Fig. 2B). Differentially bound enhancers are most commonly found in introns and intergenic regions between injury and sham conditions. Proximal enhancer regions around gene promoters are under-represented in the DB peak sets (p value ≤0.0001) relative to peaks shared in both H3K27ac sets (Fig. 2C). This is consistent with previous findings that H3K27ac effectively distinguishes active versus poised distal enhancer regions but does not effectively differentiate active promoters from poised/inactive ones (25).
To test the accuracy of the DB peak designation, several ShamDB peaks at selected sites were analyzed by qPCR2 analysis of independent ChIP samples (Fig. 3, A and B).These assays confirmed injury-sensitive H3K27ac levels at intronic ShamDB sites in the Drp2 gene (major peak in Fig. 2A) and myelin-associated glycoprotein (Mag, tracks shown in Fig. 5A). In addition, nerve injury reduced H3K27ac in the promoter region of the cholesterol biosynthetic gene, HMG-CoA reductase (Hmgcr), which is decreased after nerve injury (9). In addition, H3K27ac loss was confirmed on a downstream enhancer previously shown to activate Egr2 expression in myelinating Schwann cells (37, 53).
FIGURE 3.

Analysis of ShamDB peaks. A, H3K27ac profiles in sham and injury conditions are shown for ShamDB peaks in genes down-regulated during nerve injury: Egr2 and Hmgcr. B, in vivo ChIP H3K27ac assays were performed for sites with H3K27ac loss after nerve injury. Independent pools of chromatin from sham or injured sciatic nerve were immunoprecipitated using an anti-H3K27ac antibody and negative control antibody (IgG). ChIP samples were analyzed using quantitative PCR with the indicated primer sets, and percent recovery is calculated relative to input. Primer locations for the Drp2 and Mag genes are the single ShamDB peaks, shown in Figs. 2A and 5A. C, intersection of annotated sham or injury differentially bound peaks with genes induced or repressed 2-fold or greater after nerve injury (23). D, transcription factor-binding motifs generated from Sham-DB peaks. The percentage indicates the proportion of Sham-DB peaks with the indicated motif, and the p value shows the significance of motif association relative to a randomized set of peaks. *, p ≤ 0.05; **, p ≤ 0.005.
FIGURE 5.

Egr2 and Sox10 bind a small subset of developmentally important enhancers for Schwann cell myelination. A, Egr2 and Sox10 binding is shown relative to Sham-DB peaks for Sema5a, Mag, and Fa2h genes, which decrease after nerve injury. B, heatmap plot shows enrichment of H3K27ac sequencing tags aligned to a 5-kb window centered on Egr2- or Sox10-binding sites contained within DB enhancer sites. C and D, transcription factor-binding motifs generated from Egr2 and Sham-All (C) or Sox10 and Sham-All peaks (D). The percentage indicates proportion of Sham-All peaks with the indicated motif, and the p value shows the significance of motif association relative to a randomized set of peaks.
The DB peaks were annotated to the nearest transcription start site to determine whether the associated genes were regulated in nerve injury. Comparing the gene annotations to established recent microarray profiling of gene expression following nerve transection (23), we found ∼30% of genes identified as induced >2-fold after nerve injury had a nearby annotated InjuryDB peak. In contrast, InjuryDB peaks were associated with only ∼13% of genes expressed more highly in the sham condition (i.e. repressed after injury), whereas ShamDB peaks were associated with >30% of genes in this class (Fig. 3C). A limitation of this analysis is that enhancers often do not regulate the nearest gene, but the prevalence of differential peaks is generally correlated with increased and decreased gene sets in nerve injury.
Motif Analysis of Decommissioned Enhancers
Enhancers function as a platform for integration of signaling pathways and lineage-specific transcription factors and epigenetic states, culminating in highly specific gene expression programs. We therefore sought to use the sequences within ShamDB peaks to identify factors that bind to enhancers specific to the uninjured state. Accordingly, motif analysis was performed on ShamDB peaks to identify enriched sequences corresponding to binding sites of known transcription factors. Two of the top enriched sequences correspond to binding sites for Egr2 and Sox10, which are well established regulators of Schwann cell differentiation (4, 6–8, 10). Egr2 expression is lost after peripheral nerve injury (17, 18), although Sox10 expression is generally unchanged in published microarray studies of nerve injury (9, 23). Binding sites for TEAD (transcriptional enhancer activation domain containing) and NFI (nuclear factor I) sites were also enriched in ShamDB peaks (Fig. 3D). TEAD transcription factors do not currently have a known role in myelination or Schwann cell development, but they are targets of the hippo signaling pathway that modulates Tead interactions with YAP/TAZ coactivators (54). Interestingly, Tead transcription factors have recently been linked to merlin/neurofibromatosis 2 and Erbb2 signaling pathways (55), which are important for cell cycle control and differentiation in Schwann cells (56, 57).
Association of Egr2- and Sox10-bound Regions with Engaged Enhancers
The appearance of Egr2 motifs in the ShamDB peak sequences was anticipated based on its fundamental role in the myelination process (6–8), and its loss after nerve injury (17, 18). In previous work, we had generated genome-wide binding maps for transcription factors Egr2 and Sox10 in P15 rat sciatic nerve (51). Using these data sets, we tested whether Egr2 ChIP-seq and Sox10 ChIP-seq peaks fell within the H3K27ac-marked distal enhancer regions in uninjured nerve. After transferring peak locations to the Rn5 genome, we found that approximately one-third of Egr2 and Sox10 peaks overlapped with active enhancer regions marked by H3K27ac (37.3 and 34.75%, respectively, see Table 3). Our previous genomic analysis revealed that 5–10% of Egr2- and Sox10-binding sites colocalize (depending on spacing) in peripheral nerve (51), reflecting “composite elements” that have been identified in several myelin genes, including Gjb1, Pmp22, Mbp, Mag, and Mpz (11–16, 39). When sites of Egr2/Sox10 colocalization were examined in the context of active enhancer marks, the majority (73%) of the composite Egr2 and Sox10 peaks were marked with H3K27ac in the sham condition. The co-occurrence of these factors with active enhancers indicates that combinatorial Egr2/Sox10 binding is more highly indicative of actively engaged enhancers than either factor alone.
TABLE 3.
Peak overlap frequency between Egr2, Sox10, and H3K27ac overlapping regions
Values indicate number and overlap percentage between Egr2, Sox10, and differentially bound H3K27ac peaks (ShamDB or InjuryDB) or total H3K27ac peaks at each condition (ShamAll or InjuryAll). Percentages are calculated from fraction of all sites shared for each condition. TF is transcription factor.
| Egr2, Sox10, and H3K27ac overlapping regions | Peaks | TF | Relative to |
|---|---|---|---|
| % | |||
| Egr2-Sox10 composite sites | 618 | ||
| Egr2 and ShamAll | 2679 | 37.32 | Egr2 peaks |
| Sox10 and ShamAll | 2047 | 34.75 | Sox10 peaks |
| Egr2-Sox10 and ShamAll | 454 | 73.46 | Egr2/Sox10 peaks |
| Egr2 and ShamDB | 312 | 11.65 | Egr2/ Sham-All |
| Sox10 and ShamDB | 312 | 15.24 | Sox10/Sham-All |
| Egr2-Sox10 and ShamDB | 94 | 20.70 | Egr2/Sox10/Sham-All |
| Egr2 and InjuryAll | 1891 | 26.34 | Egr2 peaks |
| Sox10 and InjuryAll | 1281 | 21.75 | Sox10 peaks |
| Egr2-Sox10 and InjuryAll | 304 | 49.19 | Egr2/Sox10 peaks |
| Egr2 and InjuryDB | 44 | 2.33 | Egr2/Injury-All |
| Sox10 and InjuryDB | 51 | 2.49 | Sox10/Injury-All |
| Egr2-Sox10 and InjuryDB | 3 | 0.49 | Egr2/Sox10/Injury-All |
Consistent with the role of Egr2 and Sox10 in myelination, both Egr2- and Sox10-binding sites that overlap differential H3K27ac regions are enriched ∼6-fold more for sites deactivated in injury (ShamDB) versus sites that gain this enhancer histone mark (InjuryDB). The ∼300 sites overlapping ShamDB locations for each factor are listed in supplemental Tables S1 and S2.
Gene Ontology Analysis of Egr2 and Sox10 Enhancer Elements
To collectively assess the H3K27ac-marked binding sites for Egr2 and Sox10, we annotated each distal enhancer peak to the nearest transcription start site and calculated the enrichment of Gene Ontology terms for functional groupings. Egr2 peaks colocalizing with H3K27ac were associated with genes involved in axon ensheathment and regulation of locomotion. H3K27ac-marked Sox10 peaks were enriched near genes involved in the regulation of nerve impulse transmission and axon ensheathment (Fig. 4).
FIGURE 4.

Gene ontology for sham peaks colocalizing with Egr2/Sox10 binding. A, gene ontology cluster and Kegg pathway enrichment calculated for genes annotated to shared Egr2 and Sham-DB or Sox10 and Sham-DB peak regions. B, gene ontology cluster and Kegg pathway enrichment calculated for genes annotated to shared Egr2 and Sham-All or Sox10 and Sham-All peak regions. C, tag densities for H3K27ac, Egr2, and Sox10 across Lamb1, Itga6, and Dag1, genes important for extracellular matrix interactions and bound by Egr2 and Sox10 transcription factors.
Interestingly, Kegg pathway analysis identified focal adhesion and adherens junction categories. Lipid-rich myelin contains not only major myelin protein but also a rich assortment of anchoring junction proteins. Anchoring junctions such as gap junctions and adherens junctions are traditionally found to link two cells, but in peripheral myelin, they are found on opposing membrane layers. All these junction ontologies were enriched in the Sox10 peak sets. Further exploration of focal adhesion genes that encode proteins linking Schwann cells to the extracellular matrix identified several laminins and their integrin receptors. Schwann cell myelination requires the integration of laminin activities for process extension and morphogenesis (58–63). Laminins 411 and 211 (encoded by Lama4, Lama2, Lamb1, and Lamc1 genes) interact with integrin receptors, such as integrin α6 (Itga6), during the process of radial sorting (64, 65). All of these genes have Sox10-bound enhancers (see Lamb1 and Itga6 in Fig. 4C) and are regulated by Sox10 in siRNA studies of the S16 Schwann cell line (51). Another laminin receptor is dystroglycan (Dag1), which interacts with the actin cytoskeleton and is required for Cajal band formation (50, 58, 66). Dystroglycan also interacts with Periaxin and Drp2 (50), and all three of these genes have prominent Sox10-binding sites (Fig. 4C) and are regulated by Sox10 (51).
Nerve Injury Effects on Egr2-bound Enhancers
Consistent with the loss of Egr2 expression after nerve injury (17, 18), several Egr2-binding sites lose H3K27ac after nerve injury. An example is shown for the myelin-associated glycoprotein (Mag) gene (Fig. 5A), which was previously shown to have Egr2 and Sox10 binding within a conserved intronic region (39, 40, 42). These sites are flanked by H3K27ac, but the active histone mark is practically abolished at 3 days after nerve injury. Similar reductions in H3K27ac are observed in intronic regions of the Sema5a (semaphorin 5a) and Fa2h (fatty acid 2-hydroxylase) genes, where Egr2-binding sites are associated with a series of ShamDB peaks. The Fa2h and Sema5a genes are induced in myelinating Schwann cells in an Egr2-dependent manner (8) and are down-regulated after nerve injury (23). These data would suggest that Egr2 is required for H3K27 acetylation at these enhancers.
Based on these examples, we had hypothesized that most Egr2-bound enhancers would lose H3K27 acetylation after nerve injury. However, we surprisingly found that only a minor percentage of Egr2 sites with H3K27 acetylation (∼12%) lost acetylation after nerve injury (Table 3). Because it was possible that H3K27ac may be generally lost over all Egr2 sites, but only reach statistical significance at a minor subset of sites in the DBChIP analysis, we also calculated whether reads in all Egr2 peak regions (summed over the genome) were reduced after nerve injury, but we did not find a statistical difference of H3K27ac reads in aggregated Egr2 peak windows. Overall, ∼300 Egr2 peaks lost acetylation after nerve injury (Table 3), suggesting that other co-binding factors retain H3K27 acetylation at most Egr2-binding sites after nerve injury. The Egr2 peaks marked by H3K27ac were analyzed by motif finding, and we observed enrichment of several motifs besides Egr2 itself, including the TEAD and SOX motifs noted above (Fig. 5C). This analysis also revealed a cAMP-response element-binding protein-binding motif, which may reflect binding of cAMP-response element-binding protein family members that could mediate GPR126-cAMP signaling that is required for myelination (67, 68).
Because Sox10 mRNA expression is generally unchanged after nerve injury (9, 23), we anticipated that most of these sites would retain H3K27ac after nerve injury. Although this is true for most Sox10 sites, we did find that ∼15% of Sox10/H3K27ac sites lost H3K27 acetylation after nerve injury (Table 3). InjuryDB enhancers bound by Egr2 and Sox10 are depicted in the heatmap plot representation of transcription factor-bound DB sites (Fig. 5B). One of the interesting aspects of this analysis is that only ∼10% of the differentially bound sites that are lost after nerve injury were initially occupied by Egr2. This suggests that the nerve injury program in Schwann cells leads to displacement of other injury-sensitive transcription factors. Using the motifs found in ShamDB peaks (Fig. 3), the corresponding transcription factor genes were analyzed in nerve injury microarray studies to determine whether their levels declined after nerve injury. There were ∼2-fold reductions of Nur77 factors (another ShamDB enriched motif for Nr4a1 and Nr4a2) and in NFIA after nerve injury (23, 32). However, there was no observed decrease in TEAD family transcription factors after nerve injury, although the binding and activity of these factors are commonly modulated by post-translational modifications.
Analysis of Injury-induced Enhancers and the c-Jun Pathway
Analysis of transcription factor motifs enriched in InjuryDB peaks revealed cognate sites for several factors up-regulated in nerve injury (Fig. 6A), such as c-Jun. Identification of the c-Jun/AP-1-binding site provides an additional validation for the data set, as c-Jun coordinates important aspects of the Schwann cell's response to nerve injury (23, 69, 70). Microarray analysis showed 62 injury-induced genes were reduced >2-fold in c-Jun-deficient mice after nerve injury (23). We found that 21 of these genes had InjuryDB peaks. Interestingly, c-Jun-binding motifs were found in InjuryDB peaks proximal to several c-Jun target genes, including Runx2 and Gdnf, which are induced over 40-fold after peripheral nerve injury (9, 23). Glia cell-derived neurotropic factor (Gdnf) induction in Schwann cells plays a prominent role in supporting axons after nerve injury (70).
FIGURE 6.

Analysis of InjuryDB peaks. A, transcription factor-binding motifs generated from Injury-DB peaks. The p value shows the significance of motif association relative to a randomized set of peaks. B, histogram indicates motif frequency for c-Jun and Runx2 relative to the center of Injury DB peaks. C, immunohistochemistry was performed for Sox10 and Runx2 on sections from peripheral nerve in both sham and 3-day post-injury conditions. The expression of c-Jun target genes was determined by quantitative RT-PCR after siRNA-mediated reduction of c-Jun (D) or Runx 2 (E) in primary Schwann cells relative to a negative control siRNA. Error bars represent standard deviation and asterisks denote p value (* ≤ 0.05; ** ≤ 0.005). The statistics were generated from independently generated samples in triplicate (n = 3).
The induction of the Runx2 transcription factor (also known as Core-binding factor a1) after nerve injury is consistent with enrichment of the RUNX-binding motif in InjuryDB peaks (Fig. 6A). The RUNX-binding motif was found in injury-induced enhancers proximal to 13 of the c-Jun-dependent, injury-induced genes, including Runx2 itself, Olig1, and Shh. Although no role for Runx transcription factors has been shown in the peripheral nervous system development or injury, Runx2 has been previously found to physically interact and cooperatively bind with c-Jun on both activator protein-1 (AP-1) and RUNX-binding sites (71). RUNX-binding motifs were found in close proximity to c-Jun motif sites within the Runx2 gene. A histogram of c-Jun and Runx2 motif locations with respect to InjuryDB peak centers shows that the factor motifs are enriched at about 350 bp from the centers of H3K27ac peaks (Fig. 6B). Although Runx2 has been shown to be induced in a c-Jun-dependent manner (23), it has not been shown that Runx2 is induced in Schwann cells. Therefore, immunohistochemistry for Runx2 was performed in sham and 3-day injured sciatic nerve (Fig. 6C). Although there was some minimal staining in the sham condition, we see a significant increase in Runx2 staining, which was colocalized with Schwann cell expression of Sox10, indicating that the Runx2 induction occurs in Schwann cells in the peripheral nerve.
To test the involvement of c-Jun and Runx2 in activation of injury-associated genes, we performed siRNA analysis of these two transcription factors in primary rat Schwann cells to determine their effect on gene expression (Fig. 6, D and E). Previous experiments have shown that c-Jun is induced in primary Schwann cells cultured in serum-free medium (72). Interestingly, c-Jun siRNA caused Schwann cells to adopt a morphology similar to that described for primary Schwann cells from c-Jun null mice (23), in which the normal bipolar morphology was changed to a more flattened cell type lacking processes (data not shown). This was not observed with siRunx2.
As anticipated from analysis of c-Jun in nerve injury (23), we found that reduction of c-Jun expression resulted in down-regulation of several injury-induced genes: sonic hedgehog (Shh), Glial-derived neurotrophic factor (Gdnf), Olig1, Hmga1, and Runx2. Interestingly, down-regulation of Runx2 had no effect on c-Jun expression, but it did down-regulate Shh and Olig1, suggesting that c-Jun and Runx2 coordinately activate at least some injury-induced genes. Although c-Jun can alter cAMP-induced changes in differentiation markers (69, 72), c-Jun and Runx2 siRNA did not change Egr2 and Mpz levels in the basal medium conditions used for this assay (data not shown).
To explore whether the regulation of these genes are directly controlled by c-Jun, we took a closer look at the enhancers over these gene loci. Fig. 7A shows the location of induced enhancers in the Runx2 gene after nerve injury, which were validated using ChIP analysis of selected sites (Fig. 7C). Several enhancers contain consensus binding sites for the c-Jun and Runx2 transcription factors (Fig. 7B), and Runx2 binding has previously been observed in this locus (73). In addition, the ENCODE project has demonstrated binding of c-Jun at sites 1–3 at the homologous regions of the human RUNX2 locus by ChIP-seq (74).
FIGURE 7.
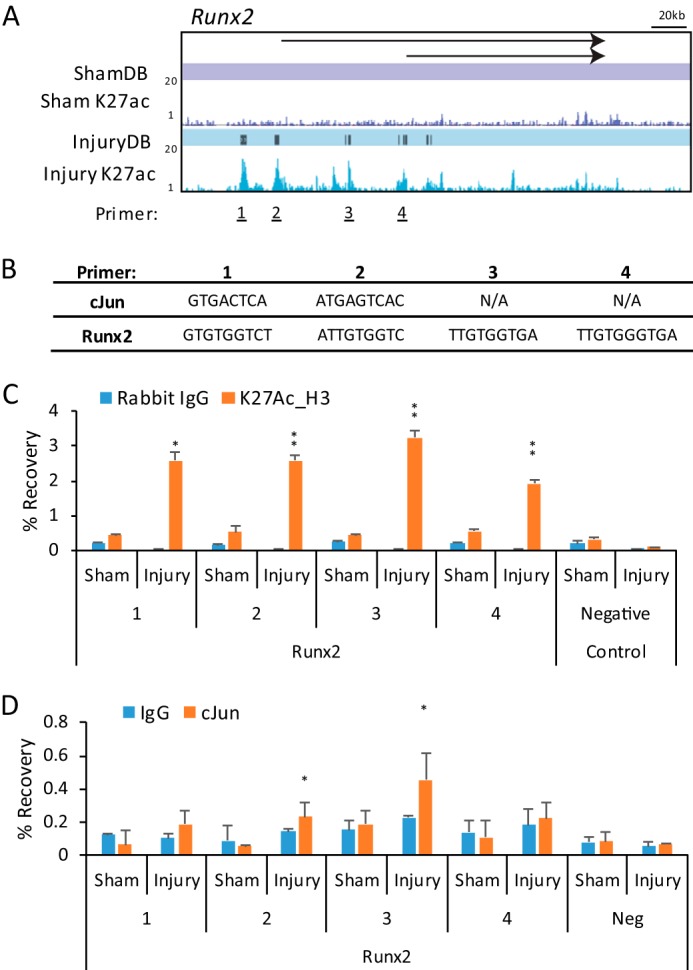
Injury-induced regulatory elements in the Runx2 gene. A, ChIP-seq profiles for H3K27ac with Sham-DB and Injury-DB peak designations are shown for the Runx2 gene. Primers for ChIP-qPCR validation are marked at InjuryDB peaks. B, c-Jun or Runx2-binding sites found at each Injury-DB peak are listed. C, chromatin from sham or injured sciatic nerve were immunoprecipitated using an anti-H3K27ac antibody and negative control antibody (IgG). ChIP samples were analyzed using quantitative PCR with the indicated primer sets, and percent recovery is calculated relative to input. D, similar ChIP assays were performed for c-Jun binding to the indicated sites within Runx2. Error bars represent standard deviation, and asterisks denote significant p value (* ≤ 0.05; ** ≤ 0.005).
To test for c-Jun binding at InjuryDB sites containing c-Jun consensus binding motifs, we performed ChIP in sham and injured nerves. By using a c-Jun antibody used previously in the ENCODE project (75), we examined Runx2 InjuryDB peaks containing either c-Jun or Runx2-binding motifs and found detectable binding of c-Jun at two sites in the Runx2 gene, which was specific to the injury condition (Fig. 7C). Interestingly, site 3 showed positive binding for c-Jun, although only a Runx2 consensus motif was found at this site, which may be due to cooperative binding between Runx2 and c-Jun. In addition, we also identified binding sites for c-Jun within injury-induced enhancers proximal to the Shh, Hmga1, and Gdnf genes as well, consistent with regulation of these genes by c-Jun after nerve injury (Fig. 8) (23). Overall, we found binding of c-Jun on six of the eight sites examined in our analysis.
FIGURE 8.

c-Jun binds a small subset of developmentally important enhancers for Schwann cell repair. A, ChIP-seq profiles for sham and injury conditions and InjuryDB peaks are shown at Shh, Hmga1, and Gdnf genes. B, in vivo ChIP-qPCR analysis of selected Injury-DB peaks was performed as described for Fig. 7 using an anti-H3K27ac antibody and negative control antibody (IgG). ChIP samples were analyzed using qPCR with the indicated primer sets, and percent recovery is calculated relative to input. C, ChIP assays for c-Jun binding was performed at the indicated primer locations in sham and injury chromatin. Error bars represent standard deviation, and asterisks denote significant p value (* ≤ 0.05; ** ≤ 0.005).
DISCUSSION
Recent studies have highlighted pathways that underlie the developmental plasticity of Schwann cells to break down myelin in response to injury and then eventually remyelinate axons. Injury-induced gene expression changes direct a mature Schwann cell to dissolve its basement membrane, resume proliferation, support axonal regrowth, direct the regenerating axon across the site of injury, and finally remyelinate (2, 3, 76). Interestingly, although demyelination is initiated by axonal cues, the age-dependent decline in the efficiency of axon regeneration has recently been attributed to alterations in the Schwann cell gene expression network rather than age-dependent effects on the axonal response (77).
Originally considered a dedifferentiation process, gene expression differences and functional analysis have shown that demyelinating Schwann cells are functionally distinct from immature Schwann cells (23). Because regulation of this process has important implications for the successful regeneration of axons, identification of key regulatory elements and the factors that bind them will help elucidate the connectivity of injury-induced transcription factors with their cognate regulatory elements. A key regulator of the Schwann cell's response to nerve injury is c-Jun, which drives demyelination and subsequent remyelination after nerve injury in Schwann cells. Recent work has identified c-Jun-dependent genes that are induced after nerve injury (23, 69). Accordingly, our analysis uncovered a number of injury-induced enhancers in c-Jun-dependent genes (such as Runx2, see Fig. 7D) that contain canonical binding sites for c-Jun/AP-1, and the c-Jun-binding motif is enriched in injury-induced enhancers.
Although c-Jun plays a profound role in the Schwann cell's response to nerve injury, many other transcription factors are induced. Some of them are c-Jun-dependent, such as Runx2, but others appear to be induced in a c-Jun-independent manner (23). Another pathway implicated in the Schwann cell's response to nerve injury is the ERK pathway. For example, induced activation of the Raf-ERK pathway results in induction of demyelination, and many of the genes induced by ERK activation (Mcp1/Ccl2, Cxcl10, Timp1, TGFβ1, and Megf10) appear to be distinct from c-jun-dependent genes (20, 22). In particular, Ccl2 is a critical chemokine involved in recruiting macrophages to peripheral nerves after injury and in a peripheral neuropathy induced by a myelin gene mutation (78). Interestingly, the InjuryDB sites are enriched in binding sites for the ETS family of transcription factors, which are commonly activated by ERK kinases. In Schwann cells, Ets1 is induced after nerve injury (9, 23), and Ets factors have been previously implicated in Schwann cell survival (79).
Our data sets allow us to not only detect injury-induced enhancers but also to determine the fate of Egr2-bound enhancers after nerve injury (17, 18, 80). Previous studies have indicated that c-Jun antagonizes Egr2 expression (69, 72). Egr2 is lost relatively soon after peripheral nerve injury (17, 18), and its expression is regained as Schwann cells resume myelination. We had anticipated that Egr2-bound enhancers would be selectively lost after nerve injury. However, we surprisingly found that most Egr2-bound enhancers were retained, and only about 10% of the Egr2-bound enhancers lost the H3K27ac mark upon injury. This may be a minimum estimate because the cutoffs used to select differentially bound peaks are somewhat conservative.
The maintenance of H3K27 acetylation on most Egr2-bound enhancers after injury could reflect compensatory binding of Egr2-related transcription factors that are induced after nerve injury, such as Egr1 (17). Alternatively, it is possible that the requisite histone acetylases are retained by other factors binding to the same enhancers, which may facilitate rebinding of Egr2 to these enhancers during remyelination. Our analysis highlighted other transcription factors whose binding motifs are enriched in injury-sensitive enhancers. These include the NF1, Nur77, and TEAD-binding motifs. As noted above, TEAD factors have been implicated in pathways that respond to neuregulin stimulation (55). Further studies will be required to elucidate the roles of these factors, because myelination depends on diverse transcription factors beyond Egr2 and Sox10 (81). It is anticipated that this enhancer analysis will help identify other transcription factors in Schwann cell development and their target enhancers.
As noted above, we also found that some ShamDB enhancers were colocalized with Sox10 sites. The loss of acetylation at such sites could reflect coordinated binding of Sox10 with factors like Egr2 and Nfatc3/c4 (40, 82) and potentially others that are lost after nerve injury. In addition, there may be competition with injury-induced Sox family members, such as Sox2 (3, 8), which may repress a subset of Sox10-occupied enhancers.
Using enhancer marks to identify regulatory regions has been used extensively in established cell lines and stem cells based on the ENCODE project and other efforts (24, 25, 28, 29), but our studies have identified active regulatory elements in vivo using sciatic nerve as a ChIP-seq substrate. To our knowledge, no previous study has profiled dynamic enhancer changes during an injury process. The ChIP-seq analysis showed clear enhancer changes in major myelin genes that decrease upon nerve injury (Mag and other myelin genes), as well as injury-induced genes, such as Gdnf and Ngfr (70). As such, profiling of H3K27ac reveals those enhancers that specifically decommissioned or activated after nerve injury. Although we speculate that enhancer changes are responsible for gene expression changes after injury, it remains quite possible that enhancers with constitutive H3K27ac may mediate changes in gene expression through other steps of transcriptional regulation besides H3K27 acetylation.
The H3K27 acetylation data were used to refine sites of Egr2 and Sox10 binding to those that bind to active enhancers. Approximately one-third of Egr2- and Sox10-binding sites are colocalized with active enhancers. Binding sites without H3K27ac may be active earlier in embryonic Schwann cell development, although we find relatively little overlap with neural crest enhancers (24). We have investigated whether these represent poised enhancers that could become active after nerve injury, but that is for the most part not the case (data not shown). Probably the most likely explanation is that Egr2 and Sox10 bind adventitiously to areas of open chromatin surrounding other types of elements in the genome (83). Our data also suggest that isolated binding of Egr2 or Sox10 is not sufficient to form an active enhancer region, because most clustered Egr2/Sox10-binding sites are positive for H3K27 acetylation. Moreover, most of the Egr2- and Sox10-bound enhancers surrounding major myelin genes are marked by H3K27ac, consistent with previous studies using H3K27ac as a mark of active enhancers (28, 29).
Although previous ChIP-seq studies of peripheral nerve reflected binding of Schwann cell-specific factors (51), it is acknowledged that analysis of histone modifications in peripheral nerve is complicated by the presence of non-Schwann cell types. Nonetheless, Schwann cells compose >80% of the nuclei in sciatic nerve (17, 18), and colocalization of enhancer marks with Egr2 and Sox10 indicates that these enhancers are derived from Schwann cells. Overall, the differentiation state of Schwann cells is highly dependent on axonal signals (84), so analysis of intact nerve is best able to capture epigenomic consequences of axon-initiated signaling pathways.
Histone acetylation is controlled by activity of both histone acetylases (principally CBP/p300 for H3K27ac) and deacetylases. Accordingly, ChIP-seq profiling of CBP/p300 has also been used to identify regulatory elements genome-wide (85), and previous studies have shown an interaction between Egr2 and CBP/p300 (12, 86). Histone deacetylase activity is required for Schwann cell differentiation (87, 88). After nerve injury, it is possible that there is targeted deacetylation of specific enhancer regions, although the complexes and factors required for targeted deacetylation in nerve injury have not been identified.
Identification of enhancers in peripheral nerve may also inform studies of the genetic basis of human peripheral neuropathy. Although substantial progress has been made in identifying causes of genetic diseases such as Charcot-Marie-Tooth disease (89), most of the successes have resulted from a focus on coding regions. In contrast, analysis of the vast majority of noncoding DNA has been hampered by a lack of functional annotation. However, noncoding regulatory elements constitute a significant percentage of the genome, and mutations and copy number variations affecting regulatory elements have been associated with a variety of genetic disorders (90–92). Only a few noncoding regulatory element mutations have been identified for hereditary peripheral neuropathies, including mutations of Sox10-binding sites in the Gjb1/connexin 32 promoter (16, 93) and recent identification of a mutation that is a candidate modifier of CMT1A (94). Until recently, technical limitations have limited mutational analysis to coding region mutations, but the exponential advances in sequencing technology have made it possible to identify tissue-specific regulatory elements that may harbor genetic lesions that cause or modify severity of peripheral nerve disorders.
Acknowledgments
We thank Marie Adams at the University of Wisconsin Biotechnology Center for performance and preliminary analysis of Illumina sequencing, Xiao-yu Liu and the Bioinformatics Resource Center for bioinformatic assistance, and Karla Knobel for assistance in microscopy. We also thank Tony Antonellis for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant NS075269 (to J. S.) and Core Grant HD03352.

This article contains supplemental Tables S1–S4.
The amino acid sequence of this protein can be accessed through NCBI Protein Database under NCBI accession number GSE63103.
- qPCR
- quantitative PCR.
REFERENCES
- 1. Brosius Lutz A., Barres B. A. (2014) Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev. Cell 28, 7–17 [DOI] [PubMed] [Google Scholar]
- 2. Patodia S., Raivich G. (2012) Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim H. A., Mindos T., Parkinson D. B. (2013) Plastic fantastic: Schwann cells and repair of the peripheral nervous system. Stem Cells Transl. Med. 2, 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Svaren J., Meijer D. (2008) The molecular machinery of myelin gene transcription in Schwann cells. Glia 56, 1541–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pereira J. A., Lebrun-Julien F., Suter U. (2012) Molecular mechanisms regulating myelination in the peripheral nervous system. Trends Neurosci. 35, 123–134 [DOI] [PubMed] [Google Scholar]
- 6. Topilko P., Schneider-Maunoury S., Levi G., Baron-Van Evercooren A., Chennoufi A. B., Seitanidou T., Babinet C., Charnay P. (1994) Krox-20 controls myelination in the peripheral nervous system. Nature 371, 796–799 [DOI] [PubMed] [Google Scholar]
- 7. Decker L., Desmarquet-Trin-Dinh C., Taillebourg E., Ghislain J., Vallat J. M., Charnay P. (2006) Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J. Neurosci. 26, 9771–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le N., Nagarajan R., Wang J. Y., Araki T., Schmidt R. E., Milbrandt J. (2005) Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc. Natl. Acad. Sci. U.S.A. 102, 2596–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nagarajan R., Le N., Mahoney H., Araki T., Milbrandt J. (2002) Deciphering peripheral nerve myelination by using Schwann cell expression profiling. Proc. Natl. Acad. Sci. U.S.A. 99, 8998–9003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Finzsch M., Schreiner S., Kichko T., Reeh P., Tamm E. R., Bösl M. R., Meijer D., Wegner M. (2010) Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J. Cell Biol. 189, 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. LeBlanc S. E., Jang S. W., Ward R. M., Wrabetz L., Svaren J. (2006) Direct regulation of myelin protein zero expression by the Egr2 transactivator. J. Biol. Chem. 281, 5453–5460 [DOI] [PubMed] [Google Scholar]
- 12. Jang S. W., Svaren J. (2009) Induction of myelin protein zero by early growth response 2 through upstream and intragenic elements. J. Biol. Chem. 284, 20111–20120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones E. A., Lopez-Anido C., Srinivasan R., Krueger C., Chang L. W., Nagarajan R., Svaren J. (2011) Regulation of the PMP22 gene through an intronic enhancer. J. Neurosci. 31, 4242–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones E. A., Brewer M. H., Srinivasan R., Krueger C., Sun G., Charney K. N., Keles S., Antonellis A., Svaren J. (2012) Distal enhancers upstream of the Charcot-Marie-Tooth type 1A disease gene PMP22. Hum. Mol. Genet. 21, 1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Denarier E., Forghani R., Farhadi H. F., Dib S., Dionne N., Friedman H. C., Lepage P., Hudson T. J., Drouin R., Peterson A. (2005) Functional organization of a Schwann cell enhancer. J. Neurosci. 25, 11210–11217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bondurand N., Girard M., Pingault V., Lemort N., Dubourg O., Goossens M. (2001) Human connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 10, 2783–2795 [DOI] [PubMed] [Google Scholar]
- 17. Topilko P., Levi G., Merlo G., Mantero S., Desmarquet C., Mancardi G., Charnay P. (1997) Differential regulation of the zinc finger genes Krox-20 and Krox-24 (Egr-1) suggests antagonistic roles in Schwann cells. J. Neurosci. Res. 50, 702–712 [DOI] [PubMed] [Google Scholar]
- 18. Zorick T. S., Syroid D. E., Arroyo E., Scherer S. S., Lemke G. (1996) The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in Schwann cell differentiation. Mol. Cell. Neurosci. 8, 129–145 [DOI] [PubMed] [Google Scholar]
- 19. Yang D. P., Kim J., Syed N., Tung Y. J., Bhaskaran A., Mindos T., Mirsky R., Jessen K. R., Maurel P., Parkinson D. B., Kim H. A. (2012) p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J. Neurosci. 32, 7158–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Napoli I., Noon L. A., Ribeiro S., Kerai A. P., Parrinello S., Rosenberg L. H., Collins M. J., Harrisingh M. C., White I. J., Woodhoo A., Lloyd A. C. (2012) A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73, 729–742 [DOI] [PubMed] [Google Scholar]
- 21. Harrisingh M. C., Perez-Nadales E., Parkinson D. B., Malcolm D. S., Mudge A. W., Lloyd A. C. (2004) The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 23, 3061–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shin Y. K., Jang S. Y., Park J. Y., Park S. Y., Lee H. J., Suh D. J., Park H. T. (2013) The Neuregulin-Rac-MKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia 61, 892–904 [DOI] [PubMed] [Google Scholar]
- 23. Arthur-Farraj P. J., Latouche M., Wilton D. K., Quintes S., Chabrol E., Banerjee A., Woodhoo A., Jenkins B., Rahman M., Turmaine M., Wicher G. K., Mitter R., Greensmith L., Behrens A., Raivich G., et al. (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S. A., Swigut T., Wysocka J. (2012) Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 11, 633–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nord A. S., Blow M. J., Attanasio C., Akiyama J. A., Holt A., Hosseini R., Phouanenavong S., Plajzer-Frick I., Shoukry M., Afzal V., Rubenstein J. L., Rubin E. M., Pennacchio L. A., Visel A. (2013) Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cotney J., Leng J., Oh S., DeMare L. E., Reilly S. K., Gerstein M. B., Noonan J. P. (2012) Chromatin state signatures associated with tissue-specific gene expression and enhancer activity in the embryonic limb. Genome Res. 22, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calo E., Wysocka J. (2013) Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S. A., Flynn R. A., Wysocka J. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heintzman N. D., Hon G. C., Hawkins R. D., Kheradpour P., Stark A., Harp L. F., Ye Z., Lee L. K., Stuart R. K., Ching C. W., Ching K. A., Antosiewicz-Bourget J. E., Liu H., Zhang X., Green R. D., et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barrette B., Calvo E., Vallières N., Lacroix S. (2010) Transcriptional profiling of the injured sciatic nerve of mice carrying the Wld(S) mutant gene: identification of genes involved in neuroprotection, neuroinflammation, and nerve regeneration. Brain Behav. Immun. 24, 1254–1267 [DOI] [PubMed] [Google Scholar]
- 32. Jiang N., Li H., Sun Y., Yin D., Zhao Q., Cui S., Yao D. (2014) Differential gene expression in proximal and distal nerve segments of rats with sciatic nerve injury during Wallerian degeneration. Neural Regen. Res. 9, 1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verheijen M. H., Chrast R., Burrola P., Lemke G. (2003) Local regulation of fat metabolism in peripheral nerves. Genes Dev. 17, 2450–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Werner T., Hammer A., Wahlbuhl M., Bösl M. R., Wegner M. (2007) Multiple conserved regulatory elements with overlapping functions determine Sox10 expression in mouse embryogenesis. Nucleic Acids Res. 35, 6526–6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reiprich S., Kriesch J., Schreiner S., Wegner M. (2010) Activation of Krox20 gene expression by Sox10 in myelinating Schwann cells. J. Neurochem. 112, 744–754 [DOI] [PubMed] [Google Scholar]
- 36. Fulton D. L., Denarier E., Friedman H. C., Wasserman W. W., Peterson A. C. (2011) Towards resolving the transcription factor network controlling myelin gene expression. Nucleic Acids Res. 39, 7974–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghislain J., Charnay P. (2006) Control of myelination in Schwann cells: a Krox20 cis-regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep. 7, 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jagalur N. B., Ghazvini M., Mandemakers W., Driegen S., Maas A., Jones E. A., Jaegle M., Grosveld F., Svaren J., Meijer D. (2011) Functional dissection of the oct6 Schwann cell enhancer reveals an essential role for dimeric sox10 binding. J. Neurosci. 31, 8585–8594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones E. A., Jang S. W., Mager G. M., Chang L.-W., Srinivasan R., Gokey N. G., Ward R. M., Nagarajan R., Svaren J. (2007) Interactions of Sox10 and Egr2 in myelin gene regulation. Neuron Glia Biol. 3, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. LeBlanc S. E., Ward R. M., Svaren J. (2007) Neuropathy-associated Egr2 mutants disrupt cooperative activation of myelin protein zero by Egr2 and Sox10. Mol. Cell. Biol. 27, 3521–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmidt D., Wilson M. D., Spyrou C., Brown G. D., Hadfield J., Odom D. T. (2009) ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 48, 240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jang S. W., LeBlanc S. E., Roopra A., Wrabetz L., Svaren J. (2006) In vivo detection of Egr2 binding to target genes during peripheral nerve myelination. J. Neurochem. 98, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 43. Sun G., Chung D., Liang K., Kelȩ S. (2013) Statistical analysis of ChIP-seq data with MOSAiCS. Methods Mol. Biol. 1038, 193–212 [DOI] [PubMed] [Google Scholar]
- 44. Liang K., Keles S. (2012) Detecting differential binding of transcription factors using ChIP-Seq. Bioinformatics 28, 121–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 46. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 48. Buecker C., Srinivasan R., Wu Z., Calo E., Acampora D., Faial T., Simeone A., Tan M., Swigut T., Wysocka J. (2014) Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14, 838–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Egelhofer T. A., Minoda A., Klugman S., Lee K., Kolasinska-Zwierz P., Alekseyenko A. A., Cheung M. S., Day D. S., Gadel S., Gorchakov A. A., Gu T., Kharchenko P. V., Kuan S., Latorre I., Linder-Basso D., et al. (2011) An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 18, 91–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sherman D. L., Wu L. M., Grove M., Gillespie C. S., Brophy P. J. (2012) Drp2 and periaxin form Cajal bands with dystroglycan but have distinct roles in Schwann cell growth. J. Neurosci. 32, 9419–9428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Srinivasan R., Sun G., Keles S., Jones E. A., Jang S. W., Krueger C., Moran J. J., Svaren J. (2012) Genome-wide analysis of EGR2/SOX10 binding in myelinating peripheral nerve. Nucleic Acids Res. 40, 6449–6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taniuchi M., Clark H. B., Johnson E. M. (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc. Natl. Acad. Sci. U.S.A. 83, 4094–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghislain J., Desmarquet-Trin-Dinh C., Jaegle M., Meijer D., Charnay P., Frain M. (2002) Characterisation of cis-acting sequences reveals a biphasic, axon-dependent regulation of Krox20 during Schwann cell development. Development 129, 155–166 [DOI] [PubMed] [Google Scholar]
- 54. Chen L., Loh P. G., Song H. (2010) Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway. Protein Cell 1, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Serrano I., McDonald P. C., Lock F., Muller W. J., Dedhar S. (2013) Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 4, 2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ramesh V. (2004) Merlin and the ERM proteins in Schwann cells, neurons and growth cones. Nat. Rev. Neurosci. 5, 462–470 [DOI] [PubMed] [Google Scholar]
- 57. Salzer J. L. (2012) Axonal regulation of Schwann cell ensheathment and myelination. J. Peripher. Nerv. Syst. 17, Suppl. 3, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Court F. A., Hewitt J. E., Davies K., Patton B. L., Uncini A., Wrabetz L., Feltri M. L. (2009) A laminin-2, dystroglycan, utrophin axis is required for compartmentalization and elongation of myelin segments. J. Neurosci. 29, 3908–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heller B. A., Ghidinelli M., Voelkl J., Einheber S., Smith R., Grund E., Morahan G., Chandler D., Kalaydjieva L., Giancotti F., King R. H., Fejes-Toth A. N., Fejes-Toth G., Feltri M. L., Lang F., Salzer J. L. (2014) Functionally distinct PI 3-kinase pathways regulate myelination in the peripheral nervous system. J. Cell Biol. 204, 1219–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu W. M., Feltri M. L., Wrabetz L., Strickland S., Chen Z. L. (2005) Schwann cell-specific ablation of laminin γ1 causes apoptosis and prevents proliferation. J. Neurosci. 25, 4463–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu W. M., Chen Z. L., North A. J., Strickland S. (2009) Laminin is required for Schwann cell morphogenesis. J. Cell Sci. 122, 929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McKee K. K., Yang D. H., Patel R., Chen Z. L., Strickland S., Takagi J., Sekiguchi K., Yurchenco P. D. (2012) Schwann cell myelination requires integration of laminin activities. J. Cell Sci. 125, 4609–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang D., Bierman J., Tarumi Y. S., Zhong Y. P., Rangwala R., Proctor T. M., Miyagoe-Suzuki Y., Takeda S., Miner J. H., Sherman L. S., Gold B. G., Patton B. L. (2005) Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J. Cell Biol. 168, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berti C., Bartesaghi L., Ghidinelli M., Zambroni D., Figlia G., Chen Z. L., Quattrini A., Wrabetz L., Feltri M. L. (2011) Non-redundant function of dystroglycan and β1 integrins in radial sorting of axons. Development 138, 4025–4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pellegatta M., De Arcangelis A., D'Urso A., Nodari A., Zambroni D., Ghidinelli M., Matafora V., Williamson C., Georges-Labouesse E., Kreidberg J., Mayer U., McKee K. K., Yurchenco P. D., Quattrini A., Wrabetz L., Feltri M. L. (2013) α6β1 and α7β1 integrins are required in Schwann cells to sort axons. J. Neurosci. 33, 17995–18007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nodari A., Previtali S. C., Dati G., Occhi S., Court F. A., Colombelli C., Zambroni D., Dina G., Del Carro U., Campbell K. P., Quattrini A., Wrabetz L., Feltri M. L. (2008) α6β4 integrin and dystroglycan cooperate to stabilize the myelin sheath. J. Neurosci. 28, 6714–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mogha A., Benesh A. E., Patra C., Engel F. B., Schöneberg T., Liebscher I., Monk K. R. (2013) Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J. Neurosci. 33, 17976–17985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arthur-Farraj P., Wanek K., Hantke J., Davis C. M., Jayakar A., Parkinson D. B., Mirsky R., Jessen K. R. (2011) Mouse schwann cells need both NRG1 and cyclic AMP to myelinate. Glia 59, 720–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parkinson D. B., Bhaskaran A., Arthur-Farraj P., Noon L. A., Woodhoo A., Lloyd A. C., Feltri M. L., Wrabetz L., Behrens A., Mirsky R., Jessen K. R. (2008) c-Jun is a negative regulator of myelination. J. Cell Biol. 181, 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fontana X., Hristova M., Da Costa C., Patodia S., Thei L., Makwana M., Spencer-Dene B., Latouche M., Mirsky R., Jessen K. R., Klein R., Raivich G., Behrens A. (2012) c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 198, 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. D'Alonzo R. C., Selvamurugan N., Karsenty G., Partridge N. C. (2002) Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J. Biol. Chem. 277, 816–822 [DOI] [PubMed] [Google Scholar]
- 72. Parkinson D. B., Bhaskaran A., Droggiti A., Dickinson S., D'Antonio M., Mirsky R., Jessen K. R. (2004) Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol. 164, 385–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barutcu A. R., Tai P. W., Wu H., Gordon J. A., Whitfield T. W., Dobson J. R., Imbalzano A. N., Lian J. B., van Wijnen A. J., Stein J. L., Stein G. S. (2014) The bone-specific Runx2-P1 promoter displays conserved three-dimensional chromatin structure with the syntenic Supt3h promoter. Nucleic Acids Res. 42, 10360–10372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gerstein M. B., Kundaje A., Hariharan M., Landt S. G., Yan K. K., Cheng C., Mu X. J., Khurana E., Rozowsky J., Alexander R., Min R., Alves P., Abyzov A., Addleman N., Bhardwaj N., et al. (2012) Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Raha D., Wang Z., Moqtaderi Z., Wu L., Zhong G., Gerstein M., Struhl K., Snyder M. (2010) Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. U.S.A. 107, 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Patodia S., Raivich G. (2012) Downstream effector molecules in successful peripheral nerve regeneration. Cell Tissue Res. 349, 15–26 [DOI] [PubMed] [Google Scholar]
- 77. Painter M. W., Brosius Lutz A., Cheng Y. C., Latremoliere A., Duong K., Miller C. M., Posada S., Cobos E. J., Zhang A. X., Wagers A. J., Havton L. A., Barres B., Omura T., Woolf C. J. (2014) Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 83, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fischer S., Weishaupt A., Troppmair J., Martini R. (2008) Increase of MCP-1 (CCL2) in myelin mutant Schwann cells is mediated by MEK-ERK signaling pathway. Glia 56, 836–843 [DOI] [PubMed] [Google Scholar]
- 79. Parkinson D. B., Langner K., Namini S. S., Jessen K. R., Mirsky R. (2002) β-Neuregulin and autocrine-mediated survival of Schwann cells requires activity of Ets family transcription factors. Mol. Cell. Neurosci. 20, 154–167 [DOI] [PubMed] [Google Scholar]
- 80. Stewart H. J. (1995) Expression of c-Jun, Jun B, Jun D and cAMP response element binding protein by Schwann cells and their precursors in vivo and in vitro. Eur. J. Neurosci. 7, 1366–1375 [DOI] [PubMed] [Google Scholar]
- 81. Meijer D., Svaren J. (2013) in Patterning and Cell Type Specification in the Developing CNS and PNS (Alvarez-Buylla A., Rowitch D., eds) pp. 759–770, Elsevier, New York [Google Scholar]
- 82. Kao S. C., Wu H., Xie J., Chang C. P., Ranish J. A., Graef I. A., Crabtree G. R. (2009) Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science 323, 651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Biggin M. D. (2011) Animal transcription networks as highly connected, quantitative continua. Dev. Cell 21, 611–626 [DOI] [PubMed] [Google Scholar]
- 84. Taveggia C., Feltri M. L., Wrabetz L. (2010) Signals to promote myelin formation and repair. Nat. Rev. Neurol. 6, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Visel A., Blow M. J., Li Z., Zhang T., Akiyama J. A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F., Afzal V., Ren B., Rubin E. M., Pennacchio L. A. (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Luciano R. L., Wilson A. C. (2003) HCF-1 functions as a coactivator for the zinc finger protein Krox20. J. Biol. Chem. 278, 51116–51124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jacob C., Christen C. N., Pereira J. A., Somandin C., Baggiolini A., Lötscher P., Ozçelik M., Tricaud N., Meijer D., Yamaguchi T., Matthias P., Suter U. (2011) HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat. Neurosci. 14, 429–436 [DOI] [PubMed] [Google Scholar]
- 88. Chen Y., Wang H., Yoon S. O., Xu X., Hottiger M. O., Svaren J., Nave K. A., Kim H. A., Olson E. N., Lu Q. R. (2011) HDAC-mediated deacetylation of NF-κB is critical for Schwann cell myelination. Nat. Neurosci. 14, 437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Timmerman V., Strickland A. V., Züchner S. (2014) Genetics of Charcot-Marie-Tooth (CMT) Disease within the frame of the human genome project success. Genes 5, 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schaub M. A., Boyle A. P., Kundaje A., Batzoglou S., Snyder M. (2012) Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kellis M., Wold B., Snyder M. P., Bernstein B. E., Kundaje A., Marinov G. K., Ward L. D., Birney E., Crawford G. E., Dekker J., Dunham I., Elnitski L. L., Farnham P. J., Feingold E. A., Gerstein M., et al. (2014) Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. U.S.A. 111, 6131–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Houlden H., Girard M., Cockerell C., Ingram D., Wood N. W., Goossens M., Walker R. W., Reilly M. M. (2004) Connexin 32 promoter P2 mutations: a mechanism of peripheral nerve dysfunction. Ann. Neurol. 56, 730–734 [DOI] [PubMed] [Google Scholar]
- 94. Brewer M. H., Ma K. H., Beecham G. W., Gopinath C., Baas F., Choi B. O., Reilly M. M., Shy M. E., Züchner S., Svaren J., Antonellis A. (2014) Haplotype-specific modulation of a SOX10/CREB response element at the Charcot-Marie-Tooth disease type 4C locus SH3TC2. Hum. Mol. Genet. 23, 5171–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]