

Background: AMPK, which monitors cellular energy levels, is dysregulated in most cancers.
Results: AMPK suppresses PLD activity, and PLD suppresses AMPK via mTOR.
Conclusion: PLD, which is elevated in many cancers, negatively regulates AMPK signals via mTOR.
Significance: This study implicates PLD and its metabolite phosphatidic acid as an integral part of energy input to mTOR.
Keywords: AMP-activated Protein Kinase (AMPK), Mammalian Target of Rapamycin (mTOR), Phosphatidic Acid, Phospholipase D (PLD), Tuberous Sclerosis Complex (TSC)
Abstract
AMP-activated protein kinase (AMPK), a critical sensor of energy sufficiency, acts as central metabolic switch in cell metabolism. Once activated by low energy status, AMPK phosphorylates key regulatory substrates and turns off anabolic biosynthetic pathways. In contrast, the mammalian/mechanistic target of rapamycin (mTOR) is active when there are sufficient nutrients for anabolic reactions. A critical factor regulating mTOR is phosphatidic acid (PA), a central metabolite of membrane lipid biosynthesis and the product of the phospholipase D (PLD)-catalyzed hydrolysis of phosphatidylcholine. PLD is a downstream target of the GTPase Rheb, which is turned off in response to AMPK via the tuberous sclerosis complex. Although many studies have linked AMPK with mTOR, very little is known about the connection between AMPK and PLD. In this report, we provide evidence for reciprocal regulation of PLD by AMPK and regulation of AMPK by PLD and PA. Suppression of AMPK activity led to an increase in PLD activity, and conversely, activation of AMPK suppressed PLD activity. Suppression of PLD activity resulted in elevated AMPK activity. Exogenously supplied PA abolished the inhibitory effects of elevated AMPK activity on mTOR signaling. In contrast, exogenously supplied PA could not overcome the effect AMPK activation if either mTOR or Raptor was suppressed, indicating that the inhibitory effects of PLD and PA on AMPK activity are mediated by mTOR. These data suggest a reciprocal feedback mechanism involving AMPK and the PLD/mTOR signaling node in cancer cells with therapeutic implications.
Introduction
AMP-activated protein kinase (AMPK)3 is a critical signaling node that responds to cellular energy levels and alters cell metabolism accordingly (1, 2). Altered metabolism has become a hallmark of cancer (3), and accordingly, AMPK is suppressed in many cancers (2). The best characterized mechanism for suppressing AMPK activity in human cancers is through mutations to the gene encoding liver kinase B1 (LKB1) (4). LKB1 had previously been reported to be a tumor suppressor associated with an inherited tumor susceptibility known as Peutz-Jeghers syndrome (5, 6). The discovery of the connection between LKB1 and AMPK (7–9) clearly implicated altered metabolism in cancer. In response to elevated AMP/ATP ratios, AMPK binds AMP and is then phosphorylated by LKB1. This leads to activated AMPK, suppression of anabolic metabolism, and a concomitant shift to catabolic events that generate ATP to restore energy homeostasis (2, 10).
In addition to causing a shift from anabolic to catabolic metabolism, AMPK also stimulates p53-dependent G1 cell cycle arrest (11, 12). The LKB1/AMPK pathway suppresses mammalian/mechanistic target of rapamycin (mTOR) signaling (13, 14). Suppression of mTOR also results in G1 cell cycle arrest (15). A key downstream target of AMPK is the tuberous sclerosis complex (TSC), consisting of TSC1 and TSC2. The TSC has a GTPase-activating protein (GAP) activity for the GTPase Ras homolog enriched in brain (Rheb) (16). AMPK phosphorylates TSC2 and activates its GAP activity, resulting in suppressed Rheb due to the hydrolysis of bound GTP to GDP. Rheb is a key regulator of mTOR complex 1 (mTORC1) (17). Thus mechanistically, activating AMPK could cause cell cycle arrest by suppressing mTOR. Of interest is that although GTP-bound Rheb is required for mTOR activation, Rheb binds mTOR in a GTP-independent manner (18). However, Rheb binds to and activates phospholipase D1 (PLD1) in a GTP-dependent manner (19). PLD generates the metabolite phosphatidic acid (PA), which is required for the stability and activity of the mTOR complexes mTORC1 and mTORC2 (20, 21). PLD activity is required for the stimulation of mTOR by growth factors (22) and amino acids (23, 24). Thus, a key role for Rheb could be the generation of PA needed for mTOR activity. PLD activity is commonly elevated in human cancer cells and, like mTOR, promotes cell cycle progression and survival (22, 25).
Although the negative impact of AMPK on mTOR has been previously reported (13), much less is known about the impact of mTOR on AMPK signals. We report here that AMPK suppresses PLD activity in human cancer cells with elevated PLD activity and that PLD and PA suppress AMPK in an mTOR-dependent manner. This study reveals a means for cancer cells to suppress AMPK to promote cell proliferation.
EXPERIMENTAL PROCEDURES
Cells, Cell Culture Conditions, and Transfection
The MDA-MB-231, Calu-1, and BJ-hTERT cells used in this study were obtained from American Type Culture Collection. All cells were maintained in DMEM (Sigma-Aldrich D6429) supplemented with 10% fetal bovine serum (Sigma) and penicillin/streptomycin (Sigma-Aldrich). Transfections were performed using PolyFect (Qiagen) for plasmid transfection and Lipofectamine RNAiMAX (Invitrogen) for siRNA transfection according to the manufacturers' instructions.
Materials
Reagents were obtained from the following sources. 12-O-Tetradecanoylphorbol-13-acetate (TPA) and antibodies against phospho-AMPK (Thr-172), AMPK, HA tag, S6 kinase, phospho-S6 kinase (Thr-389), eIF4E-binding protein 1 (4E-BP1), phospho-4E-BP1 (Thr-37/Thr-46), acetyl-CoA carboxylase (ACC), phospho-ACC (Ser-79), LKB1, Rheb, TSC2, phospho-TSC2 (Ser-1387), mTOR, Raptor, UNC-51-like kinase-1 (ULK1), phosphor-ULK1 (Ser-555/Ser-757), and actin were obtained from Cell Signaling. Glucose-free DMEM (11966-025) was from Invitrogen. Dialyzed fetal bovine serum (F0392) was obtained from Sigma-Aldrich. siRNAs targeting LKB1 (sc-35816), AMPK (sc-45312), Raptor (sc-44069), and mTOR (sc-35409) were obtained from Santa Cruz Biotechnology, and siRNAs targeting Rheb (M-009692-02-0005), PLD1 (M-009413-00-0010), and PLD2 (M-005064-01-0010) were obtained from Dharmacon. 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and compound C were obtained from Tocris Bioscience. PLD inhibitors for PLD1 (VU0379595) and PLD2 (VU0285655-1), 1-palmitoyl-2-oleoyl-PA, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (PS) in chloroform were purchased from Avanti Polar Lipids.
Plasmids
Plasmids for transient transfections were obtained from the following sources. The pcDNA3.1 control plasmid was obtained from Invitrogen. The plasmid expression vectors for HA-tagged catalytically inactive PLD1 and PLD2 (pCGN-PLD1-K898R and pCGN-PLD2-K758R) were generous gifts from Dr. Michael Frohman (State University of New York, Stony Brook, NY).
siRNA
Cells were plated on 6-well plates at 30% confluence in medium containing 10% serum. After 24 h, cells were transfected with siRNA at 100 nm using Lipofectamine RNAiMAX according to the manufacturer's instructions. After 6 h, the medium was changed to fresh medium containing 10% serum, and 48 h later, cells were lysed and analyzed by Western blotting.
Western Blot Analysis
Cell lysates were collected using M-PER (Thermo Scientific 78501), and proteins were separated on denaturing SDS-polyacrylamide gels. Electrophoresed proteins were transferred to nitrocellulose membrane. After transfer, membranes were blocked in isotonic solution containing 5% nonfat dry milk in PBS. Membranes were incubated with primary antibodies as indicated, and depending on the origin of the primary antibody, either HRP-conjugated anti-mouse or anti-rabbit IgG was used for detection using the ECL system (Pierce). The relative protein levels of AMPK phosphorylation were normalized to AMPK and quantified using LI-COR Image Studio software.
Preparation of PA and PS
Immediately before use, the appropriate amount of PA or PS was dried under nitrogen and resuspended by vortexing briefly in Dulbecco's PBS (SAFC Biosciences 59321C). The lipid suspension was then sonicated in a water bath sonicator for 5 min. The resulting PA suspension was immediately added to cell culture to a final concentration of 300 μm. Cells were lysed after 45 min of phospholipid stimulation.
PLD Activity
PLD activity was determined by accumulation of the transphosphatidylation product [3H]phosphatidylbutanol as described previously (21). Lipid membranes were labeled with [3H]myristic acid (60 Ci/mmol, 1.5 μCi/ml; PerkinElmer Life Sciences) for 4 h. 1-BtOH was added for 20 min before lipids were collected. Lipids were extracted and separated by TLC along with a phosphatidylbutanol standard (Enzo Life Sciences BML-ST401-0050). The phosphatidylbutanol fraction was identified by co-migration with standards, and the level of the PLD product [3H]phosphatidylbutanol was determined by scintillation counting after scraping the phosphatidylbutanol band from the TLC plates.
RESULTS
Activation of AMPK Suppresses PLD Activity
To investigate a connection between PLD and AMPK activities, we examined the effect of glucose deprivation, which is known to elevate AMPK activity, on PLD activity. We used two K-Ras-driven human cancer cell lines, MDA-MB-231 breast and Calu-1 lung, which we reported previously to have elevated PLD activity (26, 27). As expected, glucose deprivation resulted in elevated phosphorylation of AMPK at the LKB1 site at Thr-172 in both MDA-MB-231 and Calu-1 cells (Fig. 1A). As shown in Fig. 1A, glucose deprivation also led to suppression of PLD activity in both MDA-MB-231 and Calu-1 cells. These data reveal a correlation between elevated AMPK activity and suppressed PLD activity.
FIGURE 1.

Activation of AMPK suppresses PLD activity. A, MDA-MB-231 and Calu-1 cells were plated at 80% confluence in 60-mm plates in DMEM containing 10% serum. 24 h later, the cells were shifted to either complete DMEM or DMEM lacking glucose. Both complete DMEM and DMEM without glucose contained 10% dialyzed fetal bovine serum. After 4 h, cells were harvested, and lysates were prepared and used for Western blot analysis of the levels of phospho-AMPK (P-AMPK), AMPK, and actin. The relative levels of AMPK phosphorylation were normalized to total AMPK and quantified using LI-COR Image Studio software. For PLD activity assay, [3H]myristic acid was added for 4 h in fresh medium to label lipids. 1-BtOH was added for 20 min, and the amount of the PLD-catalyzed transphosphatidylation product phosphatidylbutanol was determined as described under “Experimental Procedures.” Values were normalized to the levels of PLD activity in controls, which were given a value of 100%. B, MDA-MB-231 cells were plated as described for A, and 24 h later, AICAR (2 mm) was added for the indicated times. Cells were harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC (P-ACC), ACC, and actin were determined by Western blot analysis. C, MDA-MB-231 and Calu-1 cells were plated as described for A, and AICAR was added for 45 min. The cells were then harvested, and the PLD activity was determined as described for A. D, MDA-MB-231 cells were plated as described for B. Where indicated, cells were treated with AICAR and/or PA (300 μm) for 45 min. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for B. E, MDA-MB-231 cells were plated as described for B. Where indicated, cells were treated with AICAR and/or PS (300 μm) for 45 min. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for B. F, BJ-hTERT cells were plated as described for D. Where indicated, cells were treated with AICAR and/or PA (300 μm) for 4 h. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for D. G, MDA-MB-231 cells were plated as described for D. Where indicated, cells were treated with AICAR and/or TPA (100 nm) for 24 h. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for D. Error bars for PLD assays represent S.D. for at least two independent experiments. The Western blots shown are representative of experiments repeated at least two times. The statistical significance (p value) was determined by Student's two-tailed paired t test. *, p ≤ 0.01 compared with the control for all values. The relative levels of AMPK and ACC phosphorylation were normalized to total AMPK and total ACC, respectively, and quantified as described for A.
AMPK can be activated by AICAR, a cell-permeable nucleoside that is metabolically converted to 5-aminoimidazole-4-carboxamide ribonucleoside monophosphate or ZMP by adenosine kinase. ZMP mimics the allosteric effects of AMP on the AMPK system (28). As shown in Fig. 1B, AICAR treatment activated AMPK as indicated by increased levels of phosphorylated AMPK at the LKB1 site. Also shown is the AICAR-induced increase in the level of phosphorylation of ACC, a substrate of AMPK that regulates the synthesis of fatty acids. We next examined the effect of AICAR on PLD activity, and as shown in Fig. 1C, AICAR suppressed the PLD activity in both MDA-MB-231 and Calu-1 cells. PLD catalyzes the hydrolysis of phosphatidylcholine to generate PA. We therefore examined the effect of AICAR on MDA-MB-231 cells treated with PA. Of significance, the AICAR-induced increase in the phosphorylation of both AMPK and ACC was suppressed by PA treatment (Fig. 1D). In contrast, PS did not suppress AMP activation (Fig. 1E). We also examined the effect AICAR on the non-cancerous human fibroblast cell line BJ-hTERT. As shown in Fig. 1F, AICAR treatment also led to increased phosphorylation of AMPK and ACC, and the increased phosphorylation was suppressed by PA, indicating that the observed effects are not restricted to cancer cells. To further establish that the effect of exogenously added PA was not an artifact of the exogenous addition of PA, we investigated the effect of stimulating endogenous PA production with the phorbol ester TPA, which potently activates PLD (29). As shown in Fig. 1G, like exogenously supplied PA, TPA also suppressed the AICAR-induced increases in AMPK and ACC phosphorylation in MDA-MB-231 cells. These data demonstrate that elevated AMPK activity suppresses PLD activity and suggest that PLD-generated PA prevents AMPK activation.
Inhibiting AMPK Increases PLD Activity in a Rheb-dependent Manner
The data in Fig. 1 indicate that AMPK suppresses PLD activity. We therefore investigated the effect of inhibiting AMPK on PLD activity. We first examined the effect of compound C, a pharmacological inhibitor of AMPK. As shown in Fig. 2A, compound C inhibited the phosphorylation of both AMPK and ACC. Compound C also increased the PLD activity in both MDA-MB-231 and Calu-1 cells (Fig. 2A). We also suppressed the expression of AMPK and LKB1 using siRNA. As shown in Fig. 2B, suppression of either AMPK or LKB1 expression elevated PLD activity in both MDA-MB-231 and Calu-1 cells. Thus, activation of AMPK suppresses PLD activity, and suppression of AMPK elevates PLD activity.
FIGURE 2.
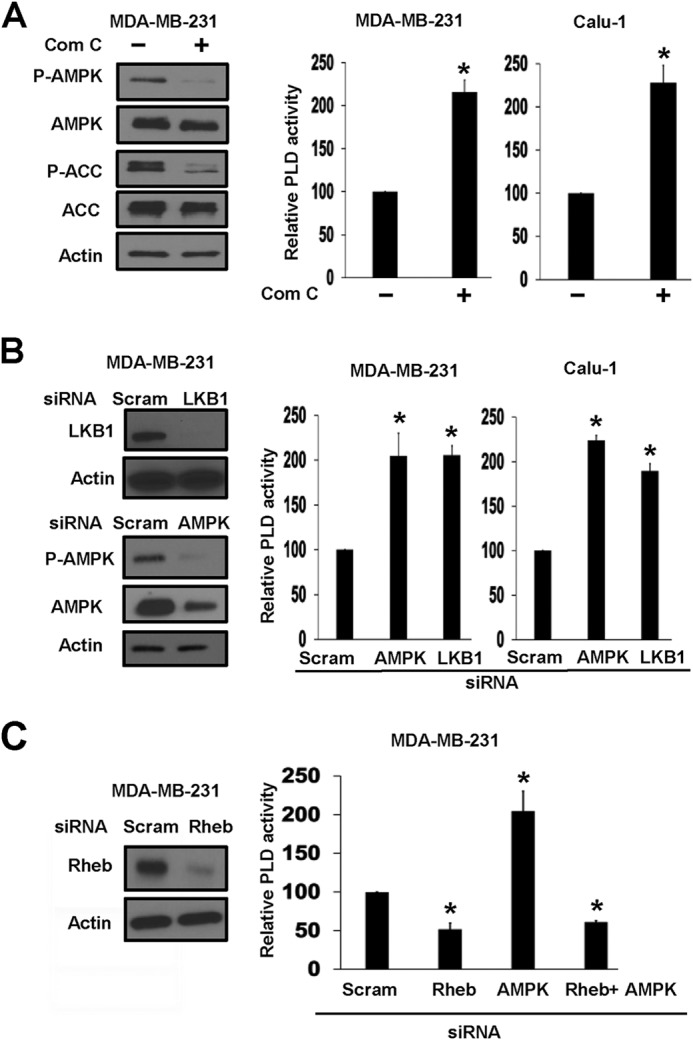
Inhibiting AMPK increases PLD activity in a Rheb-dependent manner. A, MDA-MB-231 cells were plated as described for Fig. 1B and then treated with compound C (Com C; 20 μm) for 45 min. The cells were harvested, and the levels of phospho-AMPK (P-AMPK), AMPK, phospho-ACC (P-ACC), ACC, and actin were determined by Western blot analysis. For PLD activity assay, MDA-MB-231 and Calu-1 cells were plated and treated with compound C for 45 min, and the relative PLD activity was then determined as described for Fig. 1C. B and C, MDA-MB-231 cells were plated overnight in 6-well plates at 30% confluence. The cells were then transfected with siRNAs for scrambled (scram) control siRNA, LKB1, or AMPK (B) or Rheb (C) as indicated. 6 h later, the cells were treated with fresh medium containing 10% serum for an additional 48 h. The cells were harvested, and the levels of LKB1, phospho-AMPK, AMPK (B), Rheb (C), and actin were determined by Western blot analysis. For PLD activity assays, Calu-1 (B) and MDA-MB-231 (B and C) cells were plated and transfected with siRNAs as described above. After 48 h of siRNA transfection, cells were harvested, and PLD activity was evaluated as described for A. Values were normalized to the control scrambled siRNAs, which were given a value of 100%. Error bars for PLD assays represent S.D. for at least two independent experiments. The Western blots shown are representative of experiments repeated at least two times. The statistical significance (p value) was determined by Student's two-tailed paired t test. *, p ≤ 0.01 compared with the control.
AMPK phosphorylates TSC2 and activates its GAP activity for Rheb (16), resulting in the suppression of Rheb. Rheb binds to and activates PLD1 in a GTP-dependent manner (19). We therefore examined whether the elevated PLD activity observed in response to suppression of AMPK was dependent on Rheb. We stimulated PLD activity in MDA-MB-231 cells by suppressing AMPK expression with AMPK siRNA as shown in Fig. 2B and then examined the effect of suppressing Rheb expression on PLD activity. As shown in Fig. 2C, suppressing Rheb expression with siRNA prevented the induction of PLD activity observed with AMPK knockdown with siRNA. These data demonstrate that the increased PLD activity observed with AMPK inhibition was dependent upon Rheb.
Inhibition of PLD Activates AMPK
PLD catalyzes the hydrolysis of phosphatidylcholine to generate choline and PA. PA is required for mTOR complex stability (21) and activity (20). Because mTOR is sensitive to the energy status of the cell, we evaluated the effect of suppressing PLD activity on AMPK. We first examined the effect of PLD1 and PLD2 siRNAs on AMPK activity as measured by the level of phosphorylated AMPK and ACC. As shown in Fig. 3A, introduction of PLD1 and PLD2 siRNAs led to the suppression of PLD activity and elevated both AMPK and ACC phosphorylation levels. Similar results were obtained using catalytically inactive mutants of PLD1 and PLD2 (Fig. 3B) that act as dominant-negative mutants (21) or pharmacological inhibitors of PLD1 and PLD2 (Fig. 3C). Importantly, the effect of the PLD inhibitors on the phosphorylation of AMPK and ACC could be suppressed by exogenously supplied PA in both MDA-MB-231 and Calu-1 cells (Fig. 3, D and E), indicating that the effect of the inhibitors was due to preventing PA production. Thus, there appears to be reciprocal regulation of PLD by AMPK and of AMPK by PLD.
FIGURE 3.

Inhibition of PLD activates AMPK. A, MDA-MB-231 cells were plated as described for Fig. 2B and transfected with PLD1 and PLD2 siRNAs or a scrambled (scram) control siRNA as indicated. 6 h later, the cells were treated with fresh medium containing 10% serum for an additional 72 h. The cells were then harvested, and PLD activity was evaluated as described under “Experimental Procedures”. Values were normalized to the control scrambled siRNAs, which were given a value of 100%. For Western blot analysis, cells were harvested at 72 h, and the levels of phospho-AMPK (P-AMPK), AMPK, phospho-ACC (P-ACC), ACC, and actin were determined. B, MDA-MB-231 cells were plated as described for A and transfected with vectors expressing catalytically inactive dominant-negative (DN) mutant forms of PLD1 and PLD2. The parental vector pcDNA3.1 was used as control (Con). 48 h later, the cells were harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, and ACC were determined as described for A. Expression of PLD mutants was evaluated by probing the blots for HA tags on the PLD mutants. Blots were also probed for actin as loading controls. C, MDA-MB-231 cells were plated, the PLD1 and PLD2 inhibitors (PLDi; 10 μm each) were added for 1 h, and the relative PLD activity was then determined as described for Fig. 1C. The concentrations used were based on a previous study in which we used these inhibitors to block mTOR (24). For Western blot analysis, MDA-MB-231 cells were plated and treated with the PLD inhibitors for the indicated times. Cells were harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for A. D, MDA-MB-231 cells were plated as described for C and treated with the PLD inhibitors and/or PA for 45 min as indicated. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for A. E, Calu-1 cells were plated, and the PLD inhibitors (10 μm each) were added for 1 h. The relative PLD activity was then determined as described for Fig. 1C. For Western blot analysis, Calu-1 cells were plated as described for C and treated with the PLD inhibitors and/or PA for 45 min as indicated. The cells were then harvested, and the levels of phospho-AMPK, AMPK, phospho-ACC, ACC, and actin were determined as described for A. The Western blot data are representative of experiments repeated at least two times. Error bars for PLD assays represent S.D. for at least two independent experiments. The statistical significance (p value) was determined by Student's two-tailed paired t test. *, p ≤ 0.01 compared with the control. The relative levels of AMPK and ACC phosphorylation were normalized to total AMPK and total ACC, respectively, and quantified as described for Fig. 1A.
Effects of PLD on AMPK Are Mediated by mTORC1
The data presented in Figs. 1–3 reveal a reciprocal regulation of AMPK and PLD whereby suppression of AMPK activates PLD, and suppression of PLD activates AMPK. A critical target of PLD and PA is mTOR (25), which, like PLD, is dependent upon Rheb (16, 18). It was previously reported that rapamycin can increase phosphorylation of AMPK and ACC (30). We therefore examined whether suppression of mTORC1 would, like that of PLD, elevate AMPK activity. We treated MDA-MB-231 cells with siRNAs for both mTOR and the mTORC1 companion protein Raptor and examined the levels of AMPK and ACC phosphorylation. As shown in Fig. 4A, suppressing the expression of either mTOR or Raptor led to substantial increases in both AMPK and ACC phosphorylation. We next evaluated the effect of PA on AICAR-induced phosphorylation of TSC and the mTOR substrates S6 kinase and 4E-BP1. As expected, AICAR induced phosphorylation of TSC at the AMPK site at Ser-1387 and suppressed phosphorylation of the mTORC1 substrates S6 kinase and 4E-BP1 (Fig. 4B, left panels). Importantly, the effect of AICAR was reversed if cells were provided with PA, indicating that the effect of AICAR was the result of suppressing PLD activation. However, if the expression of either mTOR (Fig. 4B, middle panels) or Raptor (right panels) was suppressed with siRNA, PA was unable to reverse the effect of AICAR. These data demonstrate that the presence of PA overcomes the effects of AMPK activation, implicating PLD, and, importantly, that suppression of mTORC1 overcomes the ability of PA to suppress the effect of AICAR. The inability of PA to overcome the effect of AICAR in the absence of mTOR or Raptor indicates that the effect of AICAR on mTORC1 is due to the suppression of PLD-stimulated PA production. The inability of PA to rescue the inhibitory effect of AICAR on TSC2 phosphorylation in the absence of either mTOR or Raptor supports the hypothesis that the effect of PA is dependent on mTOR.
FIGURE 4.
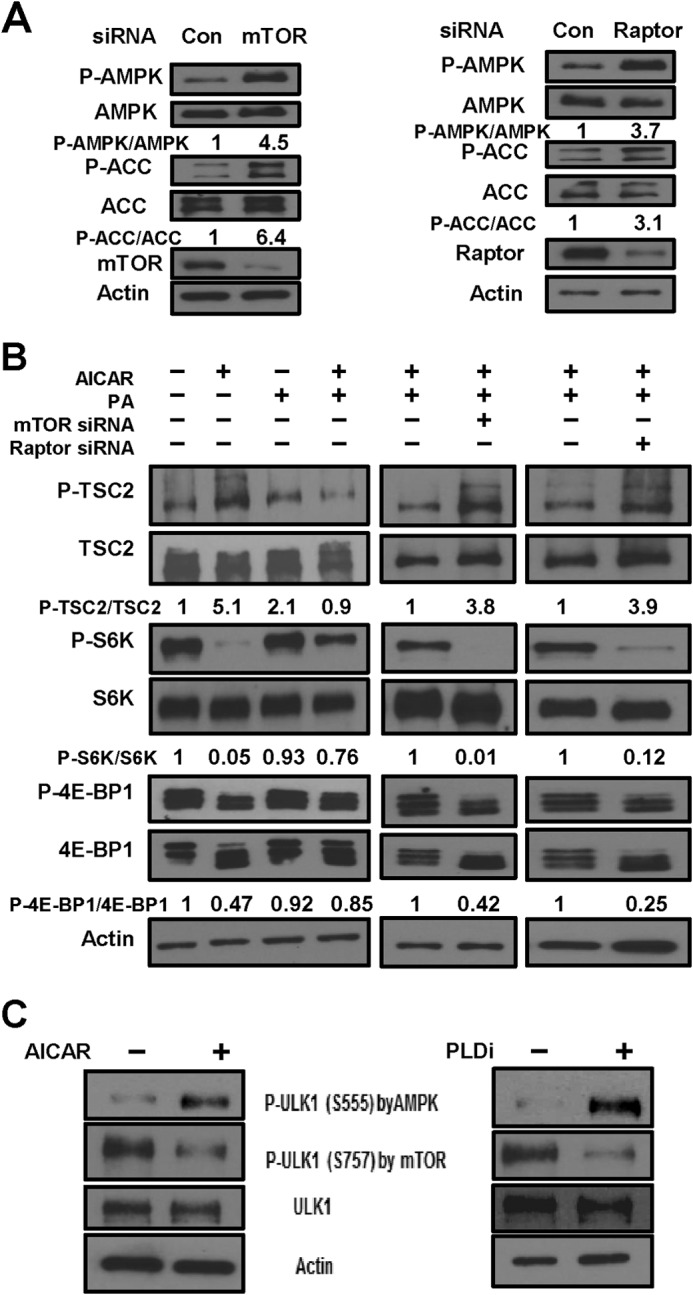
Effects of PLD on AMPK are mediated by mTORC1. A, MDA-MB-231 cells were plated as described for Fig. 3A and transfected with mTOR, Raptor, or scrambled control (Con) siRNA as indicated. After 72 h of transfection, cells were harvested and analyzed for phospho-AMPK (P-AMPK), AMPK, phospho-ACC (P-ACC), ACC, mTOR, Raptor, and actin by Western blot analysis. The relative levels of AMPK and ACC phosphorylation were normalized to total AMPK and total ACC, respectively, and quantified as described for Fig. 1A. B, MDA-MB-231 cells were plated and transfected with mTOR or Raptor siRNA as described for A. After 72 h, cells were treated with AICAR and/or PA as indicated for 45 min. The cells were then harvested, and the levels of phospho-TSC2 (P-TSC2), TSC2, phospho-S6 kinase (P-S6K), S6K, phospho-4E-BP1 (P-4E-BP1), 4E-BP1, and actin were determined as described for A. The relative levels of TSC2, S6K, and 4E-BP1 phosphorylation were normalized to total TSC2, total S6K, and total 4E-BP1, respectively, and quantified as described for Fig. 1A. C, MDA-MB-231 cells were plated as described for Fig. 1B and, where indicated, treated with the PLD inhibitors (PLDi) or AICAR for 1 h. The cells were then harvested, and the levels of phospho-ULK1 (P-ULK1; Ser-555 (AMPK site) and Ser-757 (mTOR site)), ULK1, and actin were determined by Western blotting. The data shown are representative of experiments repeated at least two times.
We next evaluated the effect of PLD inhibition on autophagy as a readout for mTOR suppression. Autophagy is regulated by several kinases, including AMPK, a positive regulator of autophagy (31). Treatment with either AICAR or PLD inhibitors induced phosphorylation of ULK1 Ser-555, a site mediated by activated AMPK (Fig. 4C). In contrast, both treatments suppressed the phosphorylation of ULK1 at Ser-757, which is mediated by mTORC1. These data suggest that inhibition of PLD either directly with PLD inhibitors or indirectly with AICAR results in autophagy by inducing AMPK-ULK1 phosphorylation at Ser-555 and suppressing mTOR-ULK1 phosphorylation at Ser-757. These data are consistent with a recent report by Min and co-workers (32), who reported that PLD inhibition enhances autophagic flux via ULK1. These data reveal that activation of AMPK or suppression of PLD contributes to activation of the autophagic kinase ULK1.
DISCUSSION
The level of nutrients and energy in cells is carefully monitored. Two critical signaling nodes for nutrient and energy sensing are AMPK and mTORC1, with AMPK being activated in response to energy deprivation and mTOR being activated by nutritional sufficiency. An emerging theme in cancer is that these signaling nodes are dysregulated, with AMPK being suppressed and mTOR being activated during tumorigenesis (33). One of the more common tumor suppressor genes is LKB1, which contributes to the suppression of AMPK when lost by mutation (34), underscoring the importance of AMPK suppression in tumorigenesis. In this study, we have provided evidence for reciprocal regulation of AMPK and mTOR involving PLD and its metabolite PA. Suppression of PLD activity increased the phosphorylation of AMPK at the LKB1 site at Thr-172 and of ACC at the AMPK site at Ser-79. Exogenously supplied PA suppressed phosphorylation of AMPK and ACC in an mTORC1-dependent manner. These data indicate that the elevated PLD activity commonly observed in human cancer cells (22, 25) contributes to the suppression of AMPK via activation of mTOR.
The suppression of AMPK in response to elevated PLD activity in cancer cells provides a positive feedback loop for sustaining elevated PLD and mTORC1 activity. This keeps the TSC GAP activity suppressed, and thus, Rheb remains GTP-bound and capable of activating PLD1. This is likely the case in cancer cells, where PLD activity is commonly elevated (22). However, the reverse could also be true, and elevated AMPK could suppress PLD activity, which would lead to less PA and an inactive mTOR and, as a consequence, prevent the suppression of AMPK. In this case, there is a positive feedback loop that favors AMPK. This is shown schematically in Fig. 5. It is not clear at this point what determines which loop will predominate: the one that favors AMPK or the one that favors PLD and mTOR. However, this is an important point with implications for tumorigenesis and possible therapeutic options.
FIGURE 5.
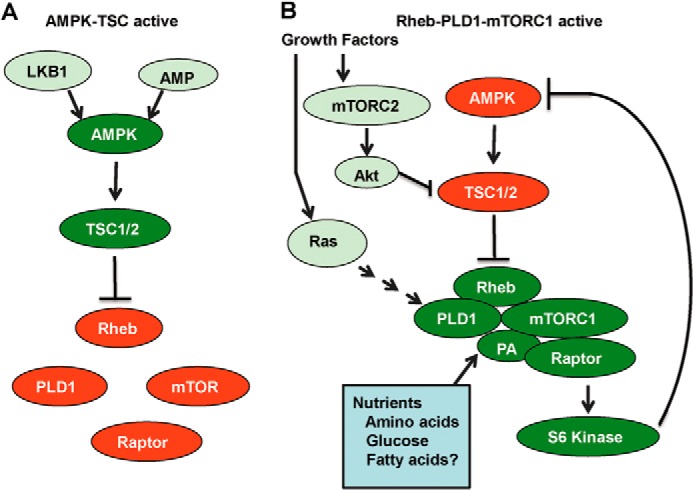
Model for reciprocal regulation of AMPK and PLD. Two scenarios are presented for the regulation of PLD by AMPK and the regulation of AMPK by PLD. A, AMPK is activated by the presence AMP and phosphorylation by LKB1. AMPK then phosphorylates TSC, which stimulates the GAP activity of TSC for Rheb, resulting in the hydrolysis of bound GTP to GDP and inactivation of Rheb. As a consequence, PLD is not activated (19), and the PA needed for stabilization of mTORC1 (21) is not generated, so mTORC1 is inactive. B, Rheb is GTP-bound and promotes the production of PA by PLD and the stabilization of mTORC1. Active mTORC1 then causes suppression of AMPK in a manner that is likely dependent on S6 kinase in that rapamycin has been reported to activate AMPK (30) at concentrations that suppress S6 kinase (38). Other signals promoted by oncogenic stimuli contribute to the activation of this pathway, such as mTORC2-Akt, which suppresses TSC (16), and Ras, which leads to PLD activation (27). It is also important to note that mTOR is an integrator on nutrient and growth factor signals (54), both of which are required for cell cycle progression and proliferation. mTOR requires amino acids (55), glucose (24), and possibly lipids (51) for activity.
The regulation of mTOR by AMPK has two separate pathways, one of which goes through phosphorylation of TSC2 and activation of the GAP activity for Rheb (13, 14). However, AICAR can still suppress mTOR in TSC2−/− cells, which led to the discovery that AMPK can also suppress mTORC1 by direct phosphorylation of the mTORC1 substrate-recognizing subunit Raptor at Ser-792 (35, 36). These findings indicate that the relationship between AMPK and mTOR is complex and that there may be multiple connections between the two signaling nodes that monitor nutrient and energy sufficiency. It is not clear how PLD and mTOR suppress AMPK; however, it was reported previously that rapamycin can stimulate the phosphorylation of AMPK at the LKB1 site at Thr-172 and of ACC at the AMPK site at Ser-79 (30). This can be achieved at low nanomolar concentrations of rapamycin that suppress S6 kinase phosphorylation but that have no effect on 4E-BP1 phosphorylation (37, 38), suggesting a role for S6 kinase.
In contrast with cancer and other proliferating cells, AMPK stimulates glucose uptake when ATP levels are reduced (39). Interestingly, it has been reported that AMPK activates PLD1 in muscle cells and that PLD activation is required for glucose uptake (40, 41). This apparently occurs via direct phosphorylation of PLD1 by AMPK at Ser-505 and requires ERK (40). Whether PA impacted on mTOR in these studies was not clear, but it has been reported that PLD can stimulate the mTOR-dependent expression of hypoxia-inducible factors 1α and 2α (42, 43), promoting glucose uptake and glycolysis (44). These studies indicate that AMPK can both suppress and activate PLD1 depending upon metabolic need.
With regard to the PLD isoform involved, we used strategies that inhibit both PLD1 and PLD2. This was based on our previous observation that suppression of both PLD1 and PLD2 is more effective in suppressing mTOR than suppression of either isoform by itself (21, 24), indicating that PLD1 and PLD2 work together. Clearly, PLD1 is more strongly implicated in the signals coming through Rheb (Fig. 5). However, Rheb impacts on mTORC1 primarily on the lysosomal membrane (45). PLD1 goes to the lysosomal membrane in response to amino acids (46); however, PLD2, which exists primarily on the plasma membrane, also moves to the lysosomes via endocytosis and the endosomes, which was implicated by our study, demonstrating that both PLD1 and PLD2 are involved in EGF receptor endocytosis (47). Thus, it is possible that PA generated by both PLD1 and PLD2 may be involved via convergence at the lysosomal membrane.
In this study, we have provided evidence for a reciprocal regulation of PLD and mTOR by AMPK and of AMPK by PLD and mTOR. A theme that has emerged in cancer is that it is critical to keep AMPK activity suppressed, as evidenced by the frequency of LKB1 mutations in cancer (4). There are two means for AMPK to suppress mTOR: indirectly through phosphorylation of TSC (13, 14) and directly through phosphorylation of Raptor (35, 36). It has been speculated that the most commonly dysregulated signals in human cancer are those that lead to elevated mTOR activity (48, 49). In this regard, it is significant that PLD activity is elevated in a wide variety of human cancers and cancer cell lines (22, 25). The finding here that PLD suppresses AMPK in an mTOR-dependent manner is consistent with a central role for PLD and its metabolite PA in signals involved in the regulation of nutrient and energy status. PA occupies a central spot in membrane phospholipid biosynthesis and is therefore an ideal indicator of lipid sufficiency. It could also indicate sufficient glucose and glutamine, which are key nutrients that are incorporated into PA (50). We have proposed that the PA requirement of mTOR evolved as a means to sense the presence of sufficient materials for membrane synthesis to complement the ability to sense amino acids (50, 51). Consistent with this hypothesis, the enzymes involved in the de novo synthesis of PA stimulate mTOR (52, 53), indicating that the PA requirement of mTOR is related to the ability to synthesis membrane phospholipids. Given that AMPK is an important mediator of energy homeostasis, it is not surprising that AMPK regulates the level of PA, a metabolite at the center of membrane lipid biosynthesis and a critical factor for mTOR activity.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-CA046677 and R01-CA179542 from NCI and by National Institutes of Health Research Centers in Minority Institutions Award RR-03039 from the National Center for Research Resources (to the Department of Biological Sciences, Hunter College of the City University of New York).
- AMPK
- AMP-activated protein kinase
- LKB
- liver kinase B
- mTOR
- mammalian/mechanistic target of rapamycin
- mTORC
- mTOR complex
- TSC
- tuberous sclerosis complex
- GAP
- GTPase-activating protein
- Rheb
- Ras homolog enriched in brain
- PLD
- phospholipase D
- PA
- phosphatidic acid
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- 4E-BP
- eIF4E-binding protein
- ACC
- acetyl-CoA carboxylase
- ULK
- UNC-51-like kinase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- PS
- phosphatidylserine.
REFERENCES
- 1. Burkewitz K., Zhang Y., Mair W. B. (2014) AMPK at the nexus of energetics and aging. Cell Metab. 20, 10–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardie D. G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 4. Hardie D. G., Alessi D. R. (2013) LKB1 and AMPK and the cancer-metabolism link–ten years after. BMC Biol. 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., Toro T., Bodmer W., Olschwang S., Olsen A. S., Stratton M. R., de la Chapelle A., Aaltonen L. A. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 [DOI] [PubMed] [Google Scholar]
- 6. Jenne D. E., Reimann H., Nezu J., Friedel W., Loff S., Jeschke R., Müller O., Back W., Zimmer M. (1998) Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 18, 38–43 [DOI] [PubMed] [Google Scholar]
- 7. Shaw R. J., Kosmatka M., Bardeesy N., Hurley R. L., Witters L. A., DePinho R. A., Cantley L. C. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U.S.A. 101, 3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hawley S. A., Boudeau J., Reid J. L., Mustard K. J., Udd L., Mäkelä T. P., Alessi D. R., Hardie D. G. (2003) Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 10. Rehman G., Shehzad A., Khan A. L., Hamayun M. (2014) Role of AMP-activated protein kinase in cancer therapy. Arch. Pharm. 347, 457–468 [DOI] [PubMed] [Google Scholar]
- 11. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 12. Imamura K., Ogura T., Kishimoto A., Kaminishi M., Esumi H. (2001) Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287, 562–567 [DOI] [PubMed] [Google Scholar]
- 13. Shaw R. J., Bardeesy N., Manning B. D., Lopez L., Kosmatka M., DePinho R. A., Cantley L. C. (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99 [DOI] [PubMed] [Google Scholar]
- 14. Corradetti M. N., Inoki K., Bardeesy N., DePinho R. A., Guan K. L. (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18, 1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saqcena M., Menon D., Patel D., Mukhopadhyay S., Chow V., Foster D. A. (2013) Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS ONE 8, e74157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang J., Manning B. D. (2009) A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 37, 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Avruch J., Long X., Ortiz-Vega S., Rapley J., Papageorgiou A., Dai N. (2009) Amino acid regulation of TOR complex 1. Am. J. Physiol. Endocrinol. Metab. 296, E592–E602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. (2005) Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713 [DOI] [PubMed] [Google Scholar]
- 19. Sun Y., Fang Y., Yoon M. S., Zhang C., Roccio M., Zwartkruis F. J., Armstrong M., Brown H. A., Chen J. (2008) Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc. Natl. Acad. Sci. U.S.A. 105, 8286–8291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 21. Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foster D. A., Xu L. (2003) Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. 1, 789–800 [PubMed] [Google Scholar]
- 23. Yoon M. S., Sun Y., Arauz E., Jiang Y., Chen J. (2011) Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J. Biol. Chem. 286, 29568–29574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu L., Salloum D., Medlin P. S., Saqcena M., Yellen P., Perrella B., Foster D. A. (2011) Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 286, 25477–25486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Foster D. A. (2009) Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim. Biophys. Acta 1791, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng Y., Rodrik V., Toschi A., Shi M., Hui L., Shen Y., Foster D. A. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281, 15862–15868 [DOI] [PubMed] [Google Scholar]
- 27. Shi M., Zheng Y., Garcia A., Xu L., Foster D. A. (2007) Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Lett. 258, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corton J. M., Gillespie J. G., Hawley S. A., Hardie D. G. (1995) 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 29. Song J., Foster D. A. (1993) v-Src activates a unique phospholipase D activity that can be distinguished from the phospholipase D activity activated by phorbol esters. Biochem. J. 294, 711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Habib S. L., Kasinath B. S., Arya R. R., Vexler S., Velagapudi C. (2010) Novel mechanism of reducing tumourigenesis: upregulation of the DNA repair enzyme OGG1 by rapamycin-mediated AMPK activation and mTOR inhibition. Eur. J. Cancer 46, 2806–2820 [DOI] [PubMed] [Google Scholar]
- 31. Wong P. M., Puente C., Ganley I. G., Jiang X. (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jang Y. H., Choi K. Y., Min D. S. (2014) Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ. 21, 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Inoki K., Kim J., Guan K. L. (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 52, 381–400 [DOI] [PubMed] [Google Scholar]
- 34. Shackelford D. B. (2013) Unravelling the connection between metabolism and tumorigenesis through studies of the liver kinase B1 tumour suppressor. J. Carcinog. 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J. W., Park S., Takahashi Y., Wang H. G. (2010) The association of AMPK with ULK1 regulates autophagy. PLoS ONE 5, e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yellen P., Chatterjee A., Preda A., Foster D. A. (2013) Inhibition of S6 kinase suppresses the apoptotic effect of eIF4E ablation by inducing TGF-β-dependent G1 cell cycle arrest. Cancer Lett. 333, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yellen P., Saqcena M., Salloum D., Feng J., Preda A., Xu L., Rodrik-Outmezguine V., Foster D. A. (2011) High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 10, 3948–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shirwany N. A., Zou M. H. (2014) AMPK: a cellular metabolic and redox sensor. A minireview. Front. Biosci. 19, 447–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J. H., Park J. M., Yea K., Kim H. W., Suh P. G., Ryu S. H. (2010) Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLoS ONE 5, e9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen H. C., Bandyopadhyay G., Sajan M. P., Kanoh Y., Standaert M., Farese R. V., Jr., Farese R. V. (2002) Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-β-d-riboside (AICAR)-stimulated glucose transport. J. Biol. Chem. 277, 23554–23562 [DOI] [PubMed] [Google Scholar]
- 42. Toschi A., Lee E., Gadir N., Ohh M., Foster D. A. (2008) Differential dependence of hypoxia-inducible factors 1α and 2α on mTORC1 and mTORC2. J. Biol. Chem. 283, 34495–34499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Toschi A., Edelstein J., Rockwell P., Ohh M., Foster D. A. (2008) HIFα expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene 27, 2746–2753 [DOI] [PubMed] [Google Scholar]
- 44. Semenza G. L. (2013) HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 123, 3664–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoon M. S., Du G., Backer J. M., Frohman M. A., Chen J. (2011) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 195, 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shen Y., Xu L., Foster D. A. (2001) Role for phospholipase D in receptor-mediated endocytosis. Mol. Cell. Biol. 21, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blagosklonny M. V. (2011) Molecular damage in cancer: an argument for mTOR-driven aging. Aging 3, 1130–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ward P. S., Thompson C. B. (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster D. A., Salloum D., Menon D., Frias M. (2014) Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mTOR. J. Biol. Chem. 289, 22583–22588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Foster D. A. (2013) Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol. Metab. 24, 272–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Avila-Flores A., Santos T., Rincón E., Mérida I. (2005) Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem. 280, 10091–10099 [DOI] [PubMed] [Google Scholar]
- 53. Blaskovich M. A., Yendluri V., Lawrence H. R., Lawrence N. J., Sebti S. M., Springett G. M. (2013) Lysophosphatidic acid acyltransferase β regulates mTOR signaling. PLoS ONE 8, e78632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fingar D. C., Blenis J. (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171 [DOI] [PubMed] [Google Scholar]
- 55. Efeyan A., Zoncu R., Sabatini D. M. (2012) Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]