

Background: Single residue substitutions in sarcomeric proteins cause most inherited cardiomyopathies.
Results: Mutant α-tropomyosins cause multiple functional alterations in actin affinity and Ca2+ sensitivity.
Conclusion: Mutants follow distinct mechanisms to change Ca2+ sensitivity.
Significance: Fluorescence assays to measure changes in troponin C conformation may provide a simple platform for preliminary high throughput screening of modulatory small molecules to treat inherited cardiomyopathies.
Keywords: Actin, Cardiac Hypertrophy, Cardiomyopathy, Myosin, Tropomyosin, Regulated Thin Filament
Abstract
The most frequent known causes of primary cardiomyopathies are mutations in the genes encoding sarcomeric proteins. Among those are 30 single-residue mutations in TPM1, the gene encoding α-tropomyosin. We examined seven mutant tropomyosins, E62Q, D84N, I172T, L185R, S215L, D230N, and M281T, that were chosen based on their clinical severity and locations along the molecule. The goal of our study was to determine how the biochemical characteristics of each of these mutant proteins are altered, which in turn could provide a structural rationale for treatment of the cardiomyopathies they produce. Measurements of Ca2+ sensitivity of human β-cardiac myosin ATPase activity are consistent with the hypothesis that hypertrophic cardiomyopathies are hypersensitive to Ca2+ activation, and dilated cardiomyopathies are hyposensitive. We also report correlations between ATPase activity at maximum Ca2+ concentrations and conformational changes in TnC measured using a fluorescent probe, which provide evidence that different substitutions perturb the structure of the regulatory complex in different ways. Moreover, we observed changes in protein stability and protein-protein interactions in these mutants. Our results suggest multiple mechanistic pathways to hypertrophic and dilated cardiomyopathies. Finally, we examined a computationally designed mutant, E181K, that is hypersensitive, confirming predictions derived from in silico structural analysis.
Introduction
Approximately half of all inherited primary cardiomyopathies have been associated with single-residue substitutions in sarcomeric proteins (1–3). Hypertrophic cardiomyopathy (HCM)9 and dilated cardiomyopathy (DCM) are the two major types, characterized by left ventricle hypertrophy and thinning of the cardiac walls, respectively (4). In general, HCM-associated troponin (Tn) and tropomyosin (Tm) mutants are hypersensitive in assays measuring myosin enzymatic activity as a function of Ca2+ concentration (5–8). Conversely, DCM-associated mutants are usually hyposensitive in the same assays (5).
At the sarcomeric level, contractility is brought about by Ca2+ regulation of the cross-bridge cycle. Cross-bridges form between myosin heads protruding from bipolar thick filaments and the regulated thin filament (RTF). The RTF is composed of F-actin, Tm, and the Tn complex and responds to Ca2+ with a series of conformational changes. The most widely accepted model linking Ca2+ and muscle contraction is the three-state model (9, 10). In the absence of Ca2+, Tm physically occludes the myosin-binding sites on actin to produce the blocked state. Muscle contraction is triggered by the influx of Ca2+ into the sarcoplasm, where it binds to TnC and initiates a sequence of conformational changes through TnI and TnT. The ensuing conformational shift of TnT and its interaction with Tm (11) results in azimuthal movement of Tm and partial uncovering of the myosin-binding sites of actin. This is called the closed state. Myosin binding to F-actin stimulates the power stroke, associated with phosphate release, causing movement of thin filaments with respect to the myosin heads, and sarcomere contraction. ADP is released from the myosin head in the subsequent steps (12, 13). The binding of myosin further displaces Tm, producing the open state. Dissociation of Ca2+ from TnC and the ATP-induced dissociation of myosin from actin produce sarcomere relaxation. Thus, muscle contraction is brought about by coordination between Ca2+-induced changes in the thin filament and the ATPase cycle of myosin. The components of the RTF harbor almost 100 cardiomyopathy-associated single-residue substitutions (14), making the RTF a crucial system to study. In this study we focus on Tm, because its interactions with actin, Tn, and myosin define the states of the RTF.
α-Tropomyosin, encoded by the TPM1 locus, is a 284-amino acid residue, 33-kDa α-helical coiled-coil protein that associates with actin filaments as a homodimer. The Tm sequence has characteristic heptad repeats (Fig. 1) stabilized by hydrophobic core residues at the a and d positions, with predominantly hydrophilic residues at the b, c, and f positions on the surface. Flexibility is imparted by kinks and alanine clusters and is important for regulatory functions. The Tm sequence also can be visualized as seven periods, each divided into an N-terminal α band and C-terminal β band. The α bands are involved in interaction with actin and the β bands in interaction with the myosin head (15, 16). The head-to-tail association of Tm dimers forms a continuous super-helical cable that matches the contours of F-actin and imparts cooperativity to regulatory interactions (17, 18). This overlap region also interacts with TnT to control the state of the RTF.
FIGURE 1.

Tropomyosin mutants. A, distribution of the mutants shown on the Tm dimer. The backbone is shown in black, HCM mutants in red, DCM mutants in blue, and the conserved-site mutant in green. This color scheme is followed throughout. B, positions of mutants relative to the α-helical coiled-coil heptad repeat. C, representative SDS-PAGE of purified WT and mutant Tms. Lane M, molecular mass marker.
Of 30 identified mutations in human TPM1 (19), four (E40K, E54K, D175N, and E180G) have been characterized more than the others. E40K and E54K are DCM-causing mutants that decrease Ca2+ sensitivity in ATPase assays and lower tension at high Ca2+ concentrations in myofiber experiments, whereas the HCM-causing mutants D175N and E180G show opposite changes to the DCM mutants (19). Beyond these binary tendencies, the detailed causes of hypercontractility of HCM-causing mutants (20) and hypocontractility of DCM-causing mutants (21) are only partially understood. Although multiple sources of mammalian Tm and other sarcomeric proteins have been used previously (22, 23), we focus here on the human cardiac sarcomeric proteins. We report here the characterization of five HCM-causing (E62Q, I172T, L185R, S215L, and M281T) and two DCM-causing (D84N and D230N) human cardiac α-Tm mutants (24–30). In addition to being relatively less characterized previously, these mutants have significant clinical severity. Clinical data on the mutants are summarized in Table 1.
TABLE 1.
Tropomyosin mutants used in this study
The abbreviations used are as follows: NCCM, noncompaction cardiomyopathy; SCD, sudden cardiac death; HF, heart failure.
| Mutant | Period (exon) | Heptad repeat position | Phenotype | No. of families affected | Clinical manifestation | Ref. |
|---|---|---|---|---|---|---|
| E62Q | 2 (2) | f | HCM | 1 | Reported as cases of HCM in the age group 15–69 years, early onset leading to SCD at 15 years, variable manifestation upon late onset | 25 |
| D84N | 3 (3) | g | DCM | 1 | Age of diagnosis ranges from 5 months to 52 years; symptoms include DCM and NCCM; SCD reported as early as 5 years | 69 |
| I172T | 5 (5) | d | HCM | 1 | Diagnosed as HCM at 36 years of age with family history of SCD at 53 years | 26 |
| E181K | 5 (5) | f | NA | 0 | Predicted to be HCM from structural and in silico analysis | 32 |
| L185R | 5 (5) | c | HCM | 2 | Two cases of SCD reported at 6 and 8 years of age in a family with a history of HCM | 26 |
| S215L | 6 (7) | e | HCM | 2 | Reported in children under 15 years with HCM; independently reported as a compound heterozygous mutation with HCM-like symptoms in South Indian population | 29, 70 |
| D230N | 6 (7) | f | DCM | 2 | Presents with DCM; ranging from HF and SCD upon early onset to asymptomatic left ventricular dysfunction in cases of late onset | 30 |
| M281T | 7 (9) | a | HCM | 3 | Reported as cases of familial HCM with onset late into adulthood | 26 |
These substitutions are located along the length of the Tm coiled-coil rod (Fig. 1A). I172T and M281T are located at the hydrophobic a and d positions of the heptad repeat (the dimer interface; Fig. 1B) and may affect the stability of the dimer. M281T may affect the head-to-tail overlap between Tm dimers. The other substitutions are on the outside surface of the coiled coil, regions that may alter interactions with actin, myosin, or the Tn complex.
On the basis of sequence and structural analyses and considerations of charged-residue clusters (31, 32), Glu-181 was identified as an evolutionarily conserved residue that should play an important role because of close interactions with actin (32). The residue is located in the C-terminal half of Tm period 5 and is likely to function to stabilize the blocked and closed states via electrostatic interactions with actin. This leads to the prediction that a charge reversal will displace Tm from the blocking position and lead to hypersensitivity. To test this hypothesis, we created E181K and analyzed it along with the seven mutants known to cause cardiomyopathy.
Overall, our suite of assays suggested a surprising degree of mechanistic heterogeneity in that alterations in Ca2+ sensitivity could not be attributed to any single parameter of Tm function. Hence, we propose that a more fine-grained categorization of mechanisms could lead to better small molecule amelioration of disease.
EXPERIMENTAL PROCEDURES
DNA, Plasmids, and Mutagenesis
Plasmids encoding human cardiac isoforms of α-Tm with an Ala-Ser N-terminal dipeptide, TnC, TnT, and TnI were kindly provided by James D. Potter, University of Miami. The plasmid encoding the triple mutant of TnC (C35S, T53C, C84S; hereafter TnC3) was constructed as described (33). The mutations were introduced by standard site-directed mutagenesis. All mutants were confirmed by DNA sequencing.
Protein Purification
Escherichia coli Rosetta cells were transformed, selected with ampicillin and chloramphenicol, grown in terrific broth at 37 °C to an A600 of ∼1, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 25 °C, and pelleted. WT and mutant Tms were purified as described by Monteiro et al. (34), with the following modifications. The lysate was boiled and centrifuged. The supernatant was subjected to ammonium sulfate precipitation; the Tm was in the 40% ammonium sulfate saturation supernatant and the 70% pellet. The pellet was dialyzed into 20 mm Tris, 1 mm EDTA, 100 mm NaCl, 1 mm DTT, pH 7.5, at 4 °C and purified over DE52 or Q-Sepharose. Tm was eluted using a gradient from 0.1 to 1 m NaCl. The fractions containing Tm were pooled and dialyzed into equilibration buffer (5 mm sodium phosphate, 1 m NaCl, 1 mm DTT) for hydroxyapatite chromatography. The Tm was eluted using an increasing gradient of phosphate up to 400 mm. Positive fractions were run on a gel to confirm purity, and the pure concentrated fractions were pooled. These were dialyzed into storage buffer (20 mm imidazole, 300 mm KCl, 1 mm DTT, pH 7.0) and frozen in liquid nitrogen. Mutant Tm yields were generally lower than yields for WT Tm. For M281T, the lysate was incubated at lower temperature (80 °C) for only 7 min, likely reducing protein loss due to denaturation. Troponins were purified using published methods (33, 35–37). Harvested cells were resuspended in a buffer containing 6 m urea and lysed by sonication. The lysate was clarified by ultracentrifugation, and troponins were purified by column chromatography (33). Actin was purified from chicken pectoral muscle acetone powder (38). Human β-cardiac myosin subfragment 1 (S1), containing the ATPase head domain and the essential light chain, was expressed and purified from C2C12 cells (33, 39). Representative purifications and reconstitution into regulated thin filaments (RTFs) (35–37) are shown in Figs. 1C and 5, A and B.
FIGURE 5.

Ca2+ sensitivity. A and B, representative SDS-PAGE of proteins used for in vitro assays. Lane M, molecular mass marker. A, purified human β-cardiac S1 from C2C12 cells, actin from chicken skeletal muscle, human α Tm from E. coli, and human Tn from E. coli. B, purified Tns and reconstitution of components into regulated thin filaments. C, DCM mutants D84N and D230N (blue) shift the pCa50 of RTF-stimulated ATPase activity to the right relative to WT (black). D, HCM mutants (red) and predicted HCM mutant E181K (green) shift the pCa50 to the left. E, DCM mutants D84N and D230N (blue) do not significantly change the ANS fluorescence response to Ca2+ relative to WT (black). F, all HCM mutants (except I172T) have increased changes in ANS fluorescence relative to WT. Values are in Tables 3 and 4. All graphs were best fit to mean ± S.E. from ≥3 experiments. The maximum S.E. for ATPase was 11 units and for ANS assay was 4 units of the y axis.
Labeling of TnC3 with ANS and NBD
TnC3 was purified as described above and dialyzed into 50 mm Tris, 100 mm KCl, 1 mm EGTA, pH 7.5, at 4 °C. The protein concentration was estimated by the Bradford assay. A 2 mm stock solution of 2-(4′-(iodoacetamido)-ANS was prepared in DMSO. A 2-fold molar excess of 2-(4′-(iodoacetamido)-ANS was added to 3 ml of TnC3 protein, mixed thoroughly, wrapped in foil, and kept on ice for 2 h. The labeling reaction was terminated by adding a 10-fold molar excess of DTT. The labeled protein was dialyzed into 20 mm imidazole, 1 m KCl, 1 mm MgCl2, 1 mm DTT pH 7.5, at 4 °C. The labeling efficiency was measured using an extinction coefficient of 24,900 m−1cm−1 at 325 nm for ANS and by calculating the TnC3 concentration by the Bradford assay. Labeling efficiency was estimated to be 95% and separately confirmed by mass spectrometry (33). For labeling the TnC3 with NBD, a 2 mm stock solution of N-((2-(iodoacetoxy)ethyl)-N-methyl)-NBD was prepared in dimethylformamide. A 3-fold molar excess of N-((2-(iodoacetoxy)ethyl)-N-methyl)-NBD was added to 2 ml of TnC3 protein, mixed thoroughly, wrapped in foil, and kept on ice for 4 h. Labeling was terminated with a 10-fold molar excess of DTT. The labeled protein was dialyzed into 20 mm imidazole, 1 m KCl, 1 mm MgCl2, 1 mm DTT, pH 7.5, at 4 °C. Labeling efficiency was measured using an extinction coefficient of 23,000 m−1 cm−1 at 472 nm for NBD. Labeling efficiency was estimated to be 80%.
Reconstituting the Tn Complex, with or without ANS
TnI, TnT, and TnC (or TnC3-ANS or -NBD) were mixed in a 1.3:1.3:1 molar ratio in 20 m imidazole, 1 m KCl, 1 mm MgCl2, 1 mm DTT, pH 7.5, and incubated on ice for 1 h. The complex was dialyzed against decreasing concentrations of KCl in the same buffer, twice each as follows: 1, 0.7, 0.5, 0.3, 0.1, and 0.01 m. Finally, the complex was clarified by centrifugation at 16,000 × g and dialyzed into 200 mm HEPES, 10 mm KCl, 1 mm MgCl2, and 1 mm DTT, pH 7.5, and flash-frozen. NBD-labeled complex was dialyzed into 20 mm HEPES, 100 mm KCl, 2 mm MgCl2, 0.5 mm EGTA, 1 mm DTT, pH 7.5, at 4 °C and flash-frozen.
Measuring Tm Thermal Stability by Circular Dichroism
A 1.5 μm solution of Tm in 500 mm KCl, 20 mm potassium phosphate, pH 7, 5 mm MgCl2, 1 mm DTT was heated from 5 to 70 °C at 0.5 °C/min, and ellipticity was measured at 222 nm. The protein was then incubated at 70 °C for 5 min and cooled to 5 °C. The process was repeated twice for each sample. The normalized ellipticity signal was plotted as a function of temperature to understand changes in thermal stability.
Actin-Tropomyosin Cosedimentation Assay
The affinity of WT and mutant Tm for F-actin was measured as described (40) using the buffer system described by Coulton et al. (41). Briefly, F-actin and Tm were dialyzed into 20 mm MOPS, 100 mm KCl, 5 mm MgCl2, 1 mm DTT, pH 7. Tm was clarified by centrifugation at 375,000 × g for 30 min before the experiment. Increasing amounts of Tm (0.2 to 2.4 μm) were added to a constant concentration of F-actin (7 μm). Following incubation at 23 °C, reactions were ultracentrifuged in a TLA100 rotor at 375,000 × g for 20 min at 20 °C. Supernatants were separated and analyzed by SDS-PAGE. Standard concentrations of Tm were electrophoresed in parallel. All the gels were stained with Coomassie Blue and destained together. Nonsaturated gel images were acquired using an ImageQuant LAS 4000 (GE Healthcare) and the concentration of free Tm was calculated by densitometric analysis using ImageJ. The pellets were resuspended in the same volume of buffer as the initial reaction and separated by SDS-PAGE. These gels were also stained-destained and analyzed to determine fractional saturation of F-actin with Tm (θ). The graph was fit to Hill Equation 1 of the form
to obtain estimates of the dissociation constant (Kd) and cooperativity of the interaction (h). For the DCM mutants D84N and D230N, additional assays were carried out in a high salt buffer (200 mm KCl) with 0.4 to 8 μm Tm added to a constant concentration (5 μm) of F-actin.
Troponin-Tropomyosin Interaction
The binding of Tm to Tn was monitored by fluorescence of NBD in 20 mm HEPES, 100 mm KCl, 2 mm MgCl2, 0.5 mm EGTA, 1 mm DTT, pH 7.5, plus or minus 600 μm CaCl2. Tn-NBD and Tm were dialyzed into this buffer, and 200 nm of the Tn complex was incubated with increasing concentrations of Tm from 20 to 600 nm in 250 μl. 200 μl of each reaction was used to measure the fluorescence intensity of NBD in top reading mode using a Tecan Infinite M200Pro with excitation of 480 ± 2 nm and emission at 535 ± 5 nm, with integration time of 2 ms and gain of 100. Data were averaged over 25 reads for each well. The binding in absence of Ca2+ led to a decrease in fluorescence, although in the presence of Ca2+ there was an increase in fluorescence. The fluorescence intensities were normalized on a scale of 0 to 1 using ΔF/ΔFmax and plotted as a function of the total Tm concentration. These data were fit to a rectangular hyperbolic Equation 2 of the form
ANS Assay
The method was modified from Davis et al. (42). RTFs were reconstituted using G-actin, Tm, and Tn complex containing TnC3-ANS. G-actin concentration was determined using absorbance at 290 nm (ϵ = 26,600 m−1) in G-buffer (2 mm Tris, 0.2 mm CaCl2, 0.2 mm ATP, 2 mm DTT, pH 8). Tm was dialyzed into a buffer of 200 mm HEPES, 300 mm KCl, 4 mm MgCl2, 1 mm DTT, pH 7.5, and its concentration was determined by Bradford assay following denaturation in guanidine-HCl. BSA standards used were also denatured. The concentration of the Tn complex was estimated by Bradford assay. G-actin, Tm, and Tn for the RTF were mixed in a molar ratio of 7:1:0.7. This ratio ensured that there was no free Tn in the mixture (33). The concentration of F-actin was 7 μm. Typically, batches of 500 μl of RTF were prepared.
10× pCa buffer (40 mm nitrilotriacetic acid, 20 mm EGTA, pH 7.5 at 25 °C) and 5× base buffer (1 m HEPES, 50 mm KCl, 20 mm MgCl2) were filtered. G-actin was aliquoted and mixed with 50 μl of 10× pCa buffer and 100 μl of 5× base buffer. ATP and DTT were added. After allowing 30 min for the F-actin to polymerize on ice, Tm was added, mixed, and incubated a further 30 min on ice for stabilization. ANS-labeled Tn complex was added, and the volume was adjusted using water. The final KCl concentration varied between 15 and 30 mm.
For the assay, 200 μl of RTF was aliquoted in duplicate wells in a clear, flat-bottom 96-well plate (Corning Glass). Ca2+ concentration was increased stepwise by adding small volumes of 20 mm CaCl2 to the RTF. Each duplicate control well received an equal volume of KCl, reflecting the volume of CaCl2 added to the experimental well. The volume of CaCl2 to be added was calculated using a pCa calculator (43). ANS fluorescence was measured in top reading mode using a Tecan Infinite M200Pro with excitation of 325 ± 4.5 nm and emission at 450 ± 10 nm, with an integration time of 2 ms and a gain of 136. Data were averaged over 36 reads for each well for each Ca2+ addition. Following CaCl2 addition, the plate was shaken for 2 min before reading the fluorescence. Fluorescence readings for RTF with KCl addition were subtracted from readings for the same RTF with corresponding CaCl2 addition. From these subtracted readings, we derived F0 (basal fluorescence by averaging the first three data points). For each Tm mutant, the fluorescence values were normalized using (F − F0)/F0. The normalized data were fit with Equation 3 in SigmaPlot to get nH and pCa50.
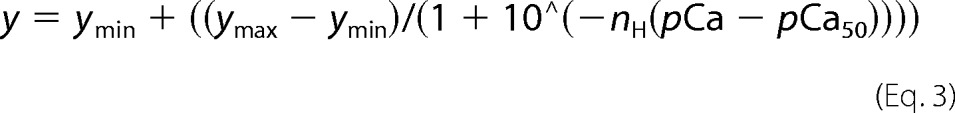 |
In the ANS assay with human β-cardiac S1, the ratio of S1 to actin was maintained at 0.1:1. All other details were identical.
ANS Quenching by Acrylamide
RTFs incorporating WT or mutant Tm were reconstituted as for the ANS assay, and the fluorescence of ANS was measured identically at pCa 5. Acrylamide was added to the reaction buffer in steps from 0 to 700 mm, whereas an equal volume of pCa 5 buffer lacking acrylamide was added to controls. The difference in fluorescence intensity was converted to the fractional change in fluorescence and modeled using the quenching sphere of action using Equation 4,
where F0 is the fluorescence in absence of quencher; F is the fluorescence upon addition of quencher; [Q] is the molar concentration of acrylamide, and V is the volume of the sphere surrounding the fluorophore. Within this volume the probability of quenching is unity (44). The Stern-Volmer coefficient for each mutant is equal to the slope obtained by fitting the data to a straight line with a y-intercept of 1.
ATPase Assays
Actin, Tm, Tn complex, and freshly prepared myosin S1 were dialyzed into buffer containing 20 mm imidazole, pH 7.5, 10 mm KCl, 3 mm MgCl2, 1 mm DTT, pH 7.5. RTFs were formed by incubating F-actin, Tm, and Tn complex in the molar ratio of 7:2:3 (excess Tm and Tn were used to ensure no unregulated actin remained) just before the assay. The 10× pCa buffers were prepared with 20 mm EGTA, 40 mm nitrilotriacetic acid, and varying concentrations of CaCl2. Free Ca2+ concentrations were calculated using the pCa calculator developed by Dweck et al. (43). Assays contained 7 μm actin, 2 μm Tm, 3 μm Tn, 0.1 μm human β-cardiac S1 and the appropriate 1× pCa buffer in a 96-well plate. Reactions were incubated at 30 °C, and the reaction was started by addition of 2 mm ATP. The ATPase activity of S1 was measured using the colorimetric Fiske-Subbarow method (45). For all assays, controls with S1 in the absence of actin provided baseline activity (typically 0.05 s−1) that was subtracted from all actin-activated measurements. For pCa curves, the RTF-activated S1 ATPase activity was monitored as a function of increasing Ca2+ concentrations. The data were fit to the modified Hill equation (Equation 3) using MATLAB to calculate the pCa50, Hill coefficient (nH), maximum activity (ymax), and minimum activity (ymin). For each S1 preparation, activities with the mutant Tm RTFs were scaled to average maximal activity with WT RTFs. For the ATPase inhibition assay, 7 μm actin, 2 μm Tm, and 0.1 μm human β-cardiac S1 was used at pCa 9 with increasing concentrations of Tn complex (0–5 μm).
RESULTS
Tropomyosin Mutants Are More Thermostable
Because the thermal stability of Tm structure is affected in some mutants (46–48), we monitored the thermal stability of the Tm mutants in the presence of DTT (Fig. 2). WT Tm shows two characteristic transitions that are confirmed by the first-order differential (data not shown). All of the mutant Tms stabilize the second transition relative to WT. Thus, most of the mutants have small effects on stability of the protein.
FIGURE 2.

Thermal stability of Tm mutants. Thermal denaturation profiles of WT Tm (black) and mutants in the presence of 1 mm DTT observed by change in circular dichroism at 222 nm. Signal change was normalized to 1.0 for the plateau.
HCM- and DCM-causing Tm Mutants Show Small Alterations in Their Binding Affinities for Actin and Tn Complex Relative to Wild Type Tm
To test for functional differences between WT and mutant Tms, we first examined their binding affinity with actin (40, 41). With the exception of E62Q and I172T, the mutations altered the Kd values (Fig. 3 and Table 2) relative to WT. Some (L185R, S215L, and D230N) also altered the cooperativity of binding in low salt buffer (Fig. 3G and Table 2), although D84N affected cooperativity in high salt buffer (Fig. 3I and Table 2). Ser-215 and Asp-230 are in the sixth quasi-repeat of Tm, the deletion of which is known to weaken actin binding (15). Accordingly, the largest change in the Kd value observed was for the DCM-causing mutant D230N, which had a nearly 5-fold higher binding affinity for actin than WT (Table 2 and Fig. 2, D and F). The validity of this observation was further tested in the presence of high salt. The Kd value for binding of actin to WT Tm shifted from ∼0.2 μm in the presence of 100 mm KCl to ∼2 μm in 200 mm KCl. Between these low and high salt conditions, the Kd value for D230N changed from ∼0.04 to ∼1.1 μm. A similar change in Kd values between low and high salt conditions was observed for the other DCM mutant D84N. S215L stood out among HCM mutants with an ∼2-fold decrease in affinity for actin and an ∼4-fold increase in cooperativity coefficient (Fig. 3, E–G).
FIGURE 3.

Actin-Tm binding. A–C, SDS-PAGE of representative actin-WT Tm cosedimentation assay. A, pellets. B, supernatants. C, standards for quantitation. D–G, binding of Tm to actin. D, DCM mutant D230N (blue); E, HCM mutant S215L (red) relative to WT control (black). F, dissociation constants; G, cooperativity of the binding (mean ± S.E.) for mutants relative to WT Tm in 100 mm KCl (*, p < 0.05 by t test). Values are in Table 2. H and I, dissociation constants and cooperativity of the binding (mean ± S.E.) for DCM mutants, D84N, and D230N relative to WT at 200 mm KCl.
TABLE 2.
Actin-tropomyosin cosedimentation assay
Values are mean ± S.E. from nine independent assays. Values with asterisks significantly differ from WT controls with p < 0.05 using Student's t test.
| Tm | Disease phenotype | Kd | Hill coefficient, nH |
|---|---|---|---|
| μm | |||
| WT (high salt) | 0.21 ± 0.020 (2.26 ± 0.110) | 2.2 ± 0.4 (3.08 ± 0.3) | |
| E62Q | HCM | 0.20 ± 0.010 | 1.9 ± 0.2 |
| D84N (high salt) | DCM | 0.10 ± 0.004* (1.39 ± 0.020)* | 2.0 ± 0.2 (5.9 ± 0.7)* |
| I172T | HCM | 0.22 ± 0.008 | 2.2 ± 0.2 |
| E181K | Predicted HCM | 0.27 ± 0.010* | 2.5 ± 0.3 |
| L185R | HCM | 0.13 ± 0.030* | 3.3 ± 0.3* |
| S215L | HCM | 0.36 ± 0.002* | 7.9 ± 0.4* |
| D230N (high salt) | DCM | 0.04 ± 0.005* (1.07 ± 0.050)* | 1.1 ± 0.1* (2.6 ± 0.2) |
| M281T | HCM | 0.14 ± 0.003* | 2.6 ± 0.1 |
Binding of Tm to the Tn complex in the absence of actin and in the presence and absence of Ca2+ was monitored by the change in fluorescence intensity of Tn complex containing TnC3-NBD. In the presence of Ca2+, the fluorescence increased with increasing Tm (data not shown). WT Tm binds to Tn with a Kapp of ∼50 ± 20 nm. In the absence of Ca2+, NBD fluorescence decreased as Tm concentration was increased. WT Tm has a Kapp of 130 ± 10 nm. In both the conditions, all the mutants had a similar affinity for Tn as WT Tm (data not shown). We did not detect any changes in the interaction with free Tn in the absence of actin.
Because Tm interacts with both actin and Tn and the Tm mutants affected these interactions independently, we examined how changes in the Tm-actin interaction affect Tn function. Actin-Tm-stimulated ATPase activity of myosin S1 is inhibited at pCa 9 by the binding of the Tn complex. All the Tm mutants except E181K and L185R were indistinguishable from WT in their inhibition of ATPase activity. These mutants showed a decreased inhibition of ATPase relative to WT controls (Fig. 4).
FIGURE 4.

Inhibition of myosin ATPase activity. Effect of TPM1 mutations on inhibition of myosin S1 ATPase activity by interaction of Tn with actin-Tm complexes in the absence of Ca2+. Data (mean ± S.E.) from three experiments.
All Mutants Show Altered Ca2+ Sensitivity in the Six-component Reconstituted Regulated System
The ability of Tm and Tn to inhibit the actin-myosin interaction at low Ca2+ concentrations but not at higher Ca2+ concentrations has been used as an in vitro correlate of sarcomere function. We therefore examined activation of human β-cardiac S1 ATPase by the RTF as a function of Ca2+ concentration. Representative purifications and reconstitution of RTFs are shown in Fig. 5, A and B. The DCM mutants D84N and D230N showed a decreased sensitivity to Ca2+ relative to WT, shifting the curve to the right (Fig. 5C, blue curves), although all of the HCM mutants showed an increased sensitivity to Ca2+, shifting the curve to the left (Fig. 5D, red curves). For the DCM mutants, the change in pCa50 (ΔpCa50) was −0.16 and −0.22 for D84N and D230N, respectively. For the HCM mutant I172T, the ΔpCa50 was +0.06 (Fig. 5D and Table 3). However, none of these three mutants produced a significant change in the maximal activation of S1 ATPase activity (Table 3). The other HCM mutants significantly increased both Ca2+ sensitivity and maximal activation of ATPase activity. D84N, L185R, and S215L reduced the Hill coefficient relative to WT (Fig. 5D and Table 3).
TABLE 3.
Thin filament activated ATPase assays
Values are mean ± S.E. from 4 to 6 independent assays. Statistical significance was calculated using Student's t test.
| Tm | Disease phenotype | pCa50 | ΔpCa50 from WT | % maximum activity | % minimum activity | Hill coefficient, nH |
|---|---|---|---|---|---|---|
| WT | 6.33 ± 0.09 | 104 ± 2 | 8 ± 1 | 1.63 ± 0.10 | ||
| E62Q | HCM | 6.39 ± 0.06a | +0.06 | 131 ± 7a | 8 ± 1 | 1.39 ± 0.16 |
| D84N | DCM | 6.17 ± 0.03b | −0.16 | 82 ± 1a | 2 ± 1 | 1.40 ± 0.1b |
| I172T | HCM | 6.51 ± 0.07a | +0.18 | 109 ± 11 | 14 ± 3 | 1.98 ± 0.44 |
| E181K | Predicted HCM | 6.78 ± 0.02a | +0.45 | 150 ± 6b | 23 ± 3b | 2.12 ± 0.53 |
| L185R | HCM | 7.26 ± 0.02b | +0.93 | 224 ± 5c | 32 ± 8a | 1.77 ± 0.19a |
| S215L | HCM | 6.49 ± 0.13a | +0.16 | 152 ± 5a | 21 ± 3a | 1.11 ± 0.07a |
| D230N | DCM | 6.11 ± 0.16a | −0.22 | 86 ± 7 | 10 ± 3 | 1.85 ± 0.36 |
| M281T | HCM | 6.63 ± 0.02a | +0.30 | 132 ± 5a | 9 ± 1 | 1.76 ± 0.18 |
a p < 0.05.
b p < 0.01.
c p < 0.005.
Interestingly, our in silico analysis predicted that the E181K substitution would destabilize the closed state and increase the time spent in the myosin-bound open state (32). Consistent with those predictions, we observed hypersensitivity, with reduced pCa50 and greater S1 ATPase activity at pCa 4 than WT Tm. These are typical of HCM mutants. Furthermore, the extent of the increase in Ca2+ sensitivity was greater than that of most of the HCM mutants in this study.
Enhanced Conformational Changes in TnC Correlate with the Increased Actin-activated ATPase above pCa 6
The finding that E181K and the HCM Tm mutants studied here (except I172T) had elevated ATPase activity at higher Ca2+ concentrations prompted us to assay for correlated structural changes in Tn. We therefore labeled TnC3 with a fluorescent reporter (ANS) that is sensitive to conformational changes in TnC (42, 49). With the exception of I172T, RTFs containing the HCM mutant Tms displayed a greater fluorescence increase between pCa 8 and pCa 4 (Fig. 5, E and F, and Table 4) relative to WT RTFs. These increases correlated well with the increased activation of ATPase (Fig. 6C) and to a lesser extent with change in the pCa50 of the ATPase activity (Fig. 5, C and D). The DCM mutants D84N and D230N slightly decreased ATPase activity at high Ca2+ concentrations (Fig. 5C) but did not have a significant effect on the magnitude of the ANS fluorescence change (Fig. 5E).
TABLE 4.
ANS fluorescence assay for thin filament conformational change
Values are mean ± S.E. from three independent assays.
| Tm | Phenotype | pCa50 | Maximum fluorescence ymax | Hill coefficient, nH | Stern-Volmer quenching constant, KSV |
|---|---|---|---|---|---|
| WT | 6.04 ± 0.05 | 10.46 ± 0.29 | 1.42 ± 0.35 | 0.0003 | |
| E62Q | HCM | 5.91 ± 0.11 | 18.58 ± 1.98a | 1.50 ± 0.31 | 0.0002 |
| D84N | DCM | 6.34 ± 0.11 | 10.10 ± 0.51 | 1.82 ± 0.12 | NDb |
| I172T | HCM | 5.92 ± 0.06 | 10.18 ± 2.40 | 1.03 ± 0.21 | 0.0002 |
| E181K | Predicted HCM | 5.88 ± 0.08 | 20.59 ± 2.09a | 0.77 ± 0.07 | 0.0002 |
| L185R | HCM | 6.19 ± 0.02a | 20.32 ± 2.42a | 1.30 ± 0.13 | 0.0002 |
| S215L | HCM | 5.95 ± 0.02 | 24.09 ± 3.67a | 0.75 ± 0.08 | nd |
| D230N | DCM | 5.99 ± 0.12 | 9.24 ± 0.36a | 0.81 ± 0.06 | 0.0003 |
| M281T | HCM | 6.00 ± 0.08 | 13.10 ± 0.46a | 0.93 ± 0.15 | 0.0003 |
a Values are significantly different from WT controls using Student's t test: p < 0.05.
b ND, not determined.
FIGURE 6.
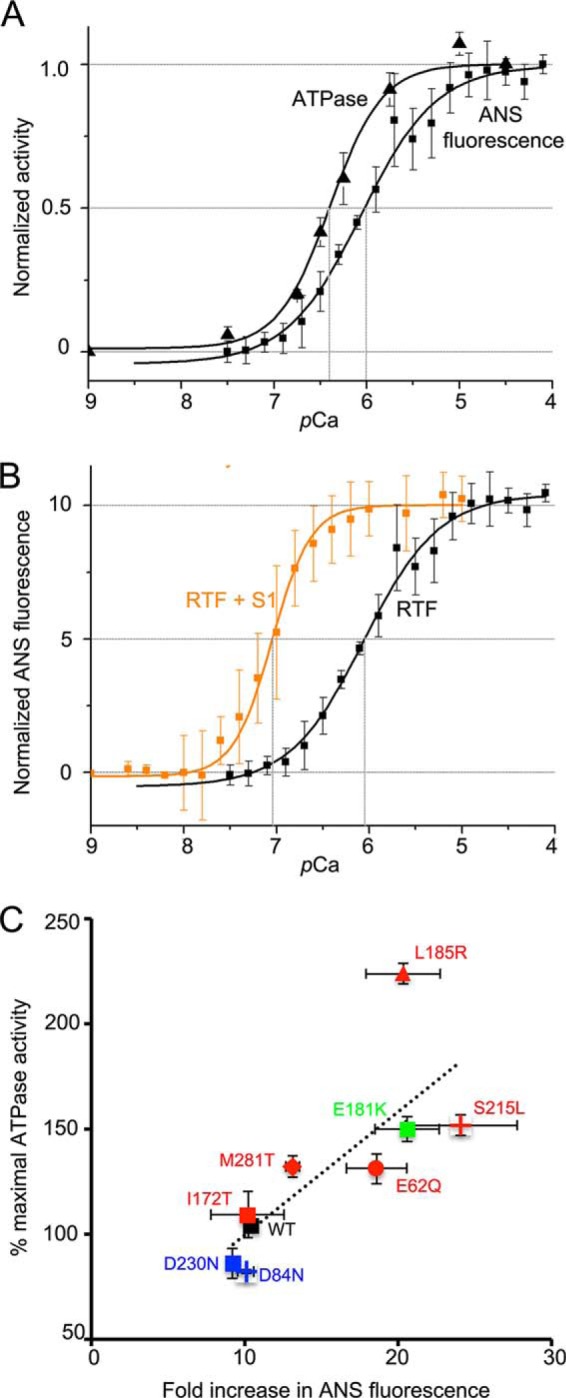
RTF activation. A, increase in ATPase activity and increase in ANS fluorescence from RTF, normalized, and plotted as a function of Ca2+ on the same axes. ATPase activity has a lower pCa50 and greater cooperativity. B, increase in ANS fluorescence as a function of Ca2+ concentration in the absence (black) or presence (orange) of S1. In the presence of S1, pCa50 is greatly reduced and cooperativity is greater. C, correlation between ATPase activity at pCa 4.5 (WT = 100%) and the fold change in ANS fluorescence for each Tm ((ymax − ymin)/ymin). Change in ATPase activity relative WT is correlated to the fold change in ANS fluorescence (Pearson's correlation coefficient r = 0.76).
To independently characterize the nature of the conformational changes induced by Tm mutants, we performed quenching of the TnC3-ANS signal with acrylamide. Although D230N and M281T were indistinguishable from WT, the other mutants had altered Stern-Volmer quenching constants, consistent with conformational differences in TnC in the presence of some mutants (Table 4).
Role of Myosin in Thin Filament Activation
For WT Tm, the ATPase assay yields a significantly lower pCa50 and greater cooperativity than the ANS fluorescence assay (Fig. 6A). To test whether the presence of myosin S1 is responsible for this difference, we monitored the ANS fluorescence change in RTFs at a 0.1 ratio of S1 to actin in the absence of ATP. This addition of S1 lowered pCa50 by almost 1 unit and significantly increased the cooperativity of activation (Fig. 6B). Thus, myosin potentiates thin filament activation by Ca2+.
DISCUSSION
Tropomyosin coordinates Ca2+-mediated changes in the structure of the Tn complex with myosin-mediated displacement of the actin filament. It has been observed that cardiomyopathy-causing mutations in Tm retrogradely affect the Ca2+ sensitivity (19), likely by inducing conformational changes in the Tn complex (50). In addition, Tm is a critical element in the cooperative binding of myosin to actin. We therefore reasoned that studying relatively unexplored Tm mutants as a group could yield novel insights into mechanisms that regulate cardiac contraction.
Because of the significant biochemical differences between myosins derived from different tissues and animal species (33), we have used human β-cardiac S1. The use of non-human sources of proteins could be one of the reasons for differing biochemical parameters with the same mutant. TnT mutants that showed no change in sliding velocity when tested with human β-cardiac myosin showed significant changes when tested with non-human myosins (33). The D175N HCM Tm mutant produced no change in Ca2+ sensitivity when assayed in rat or bovine contexts (51, 52), but it increased Ca2+ sensitivity when assayed using rabbit fast skeletal heavy meromyosin (53). When the R453C HCM mutant was produced in mouse α-cardiac MHC (54) and human β-cardiac MHC (55), there was a similar increase in single-molecule force production. However, the R453C substitution in mouse α-cardiac MHC did not change filament sliding velocity, although the same substitution in human β-cardiac S1 caused a reduction in filament sliding velocity; these have different implications for ensemble force. The use of all-human systems whenever feasible should reduce the likelihood of such complications with the mutants we report here.
Thermostability and Affinity for Actin
Our thermal stability data suggest possible explanations for some of our other observations. Other groups (56, 57) have demonstrated a relationship between thermostability and flexibility. We found that under reducing conditions, all mutants were more thermostable than WT. This may indicate reduced flexibility along the length of the molecule, which in turn could produce the dramatic changes in binding affinity for actin that we observed with the D84N, D230N, L185R, and M281T mutants. The increased affinity of D84N and D230N for actin provides a ready, but not necessarily sufficient, explanation for DCM. The stabilization of the Tm-actin interaction should make it less likely to shift the Tm into the active states, desensitizing the system. This explanation makes L185R and M281T interesting subjects for further mechanistic investigations.
The other mutant that showed a significantly altered affinity for actin (S215L) might do so because it alters the charge of a residue lying in an actin-interacting region so that it may weaken binding. Therefore, it is likely to be displaced more readily from the closed state to the open state. This alone could be sufficient to cause the hypersensitivity to Ca2+ we observed in the ATPase and ANS assays. However, S215L is the only HCM mutant for which we observed a large decrease in affinity for actin. In the cases of the other mutants, affinity for myosin and relative stabilization or destabilization of blocked, closed, or open states might contribute to pathogenicity.
Ca2+ Regulation of the RTF in the Absence and Presence of Myosin
As one of our long term goals is to identify therapeutic small molecule effectors of the cardiac contractile system, we sought to compare and contrast the effects of mutant Tms by two commonly used measurements of Ca2+ sensitivity, myosin ATPase activity and fluorescent assays for changes in Tn conformation. The pCa50 shifts observed in the ATPase assays (Fig. 5, C and D, and Table 3) were consistent with the binary paradigm that HCM mutants confer hypersensitivity and DCM mutants confer hyposensitivity to Ca2+ (5–8). As it is more complete and does not require extensive modifications (mutagenesis or labeling) of any of the six components, the ATPase assay remains the assay of choice to identify small molecule modulators.
The variation in fluorescence magnitudes observed in our ANS assays suggests two conclusions. First, TnC can assume multiple conformational states, each presumably having different affinity constants for Ca2+ binding. This is consistent with observations that proteins of the EF-hand family, of which TnC is a member, display a multitude of unique conformational changes together constituting a conformational continuum (58). In the context of the three-state model (9, 10), Ca2+ binding to TnC causes the transition from a blocked to a closed state. In the ANS assay, we are monitoring this transition using an increase in fluorescence as the readout. The variation suggests that the closed state could be a collection of multiple substrates, supporting earlier work (59). Second, we know that Ca2+ binding to TnC causes structural changes that are transmitted to Tm through TnI and TnT. Therefore, the differential fluorescence changes may represent Tm changing the manner in which structural transitions occur in TnC. This could be viewed as a retrograde aspect of bi-directional signal transmission, as has been suggested previously (50).
The pCa50 and cooperativity values from our ATPase and ANS fluorescence assays using RTFs containing WT Tm were consistent with those of previous studies (24, 33), but were different between the two assays (Tables 3 and 4). These differences in WT parameters (Fig. 6A) may reflect the role of myosin binding in induction of a further cooperative binding of myosin and a transition of the Tm out of its blocked state (60–62). This interpretation is supported by the observation that addition of a similar ratio of tight-binding myosin led to a further reduction in pCa50 and a further increase in cooperativity (Fig. 6B). Our data are consistent with our previous observations (33) that cooperativity is increased by the addition of a low ratio of cycling S1 to the RTF.
Which enzymatic parameter of myosin might the Tm mutants alter to dramatically increase ATPase activity at high Ca2+ concentrations? The inclusion of Tm-Tn and Ca2+ can be viewed as a change from the actomyosin Michaelis-Menten model to a cooperative Monod-Wyman-Changeux model (63), in which the important parameters are K0.5 and Vmax. Physically, K0.5 is a description of the affinity of myosin for the RTF. It has been proposed that Tm affects the K0.5 of the system. Therefore Tm mutants, particularly L185R in this study, may alter the rate of closure of the 50-kDa cleft in myosin (63–65), hence changing K0.5.
Correlation between Maximal ATPase Activity and ANS Fluorescence at Maximal Ca2+
In contrast with the ATPase results (Table 3), the pCa50 shifts observed in our ANS assays (Fig. 5, E and F) were greatly attenuated (Table 4), with only one mutant significantly different from WT. One potential caveat is that the three substitutions in TnC3 used to allow labeling by ANS and NBD may be altering its function. However, in skinned trabeculae both WT TnC and TnC3 responded identically to Ca2+ (42), suggesting that neither the three substitutions nor the label alter its function. Although pCa50 shifts were poorly correlated, we did note a much better correlation between maximal ATPase activity and the magnitude of the ANS fluorescence change at high Ca2+. This correlation held for all mutants (Fig. 6C, Pearson's r = 0.76).
At present, measuring Ca2+ sensitivity using ATPase activity provides us maximum information and consistency with the binary hypothesis of cardiomyopathy. However, there is one scenario in which the ANS fluorescence assay would have high utility: as an initial screen for small molecules for attenuating cardiomyopathic mechanisms by attenuating changes in RTF conformation. We hope to use the ANS assay as a primary tool to identify potentially interesting candidate compounds using limited amounts of protein and reagents. This type of fluorescence assay can also be easily automated for high throughput screening. Interesting candidates will then have to be more rigorously tested using ATPase, load-dependent, and other functional assays.
In addition to the mutant Tms found in patients, we also included a mutant E181K that we predicted would cause HCM. By modeling actin-Tm interactions, our earlier in silico analysis led us to predict that the E181K substitution would destabilize the closed state and stabilize a myosin-bound state, causing hypersensitivity (32). Consistent with these predictions, here we observed that E181K interacted weakly with actin relative to WT tropomyosin. This may lead to increased displacement of the mutant tropomyosin and produce greater sensitivity in the ATPase and ANS assays. Further validation of the hypothesis comes from recent results published after we began our studies; the same E181K substitution in β-Tm (encoded by TPM2) was reported to cause distal arthrogryposis (66, 67) through a gain-of-function mechanism (68).
In conclusion, TPM1 mutations cause differences in protein stability, actin binding, and Tn conformation. All of these differences could converge to change the Ca2+ dependence of myosin activity. Our data suggest multiple mechanistic routes to a very limited set of cardiac pathologies. If consistent and supported by additional assays (particularly load-based) and additional mutants, our observations could be useful in developing small molecule therapies.
Acknowledgments
We thank Shirley Sutton and Kathy Ruppel for help in human β-cardiac S1 preparation and Sadie Ingle, Mary Willard, and other members of the Spudich laboratory for their help and input. We thank James Potter for the generous gift of Tn expression plasmids and purification protocols. We also thank Jayant Udgaonkar, Nilesh Aghera, Jogender Singh, Pooja Malhotra, and Roumita Moulick for access to and help with mass spectrometry and circular dichroism spectroscopy and K. VijayRaghavan for tireless support of this project. We thank Satyaki Sarkar for help with the clinical literature. We thank Minhaj Sirajuddin for reviewing the manuscript.
Note Added in Proof
After submission of this manuscript, Orzechowski et al. (Orzechowki, M., Fischer, S., Moore, J. R., Lehman, W., and Farman, G. P. (2014) Arch. Biochem. Biophys. 564, 89–99) published a study of energy landscapes of tropomyosin mutants. Their calculations for the D84N mutant correlate well with our data, while the lack of correlation for the D230N mutant may be a consequence of their focus only on Tm-actin interactions in the immediate vicinity of the substitution.
This work was supported, in whole or in part, by National Institutes of Health Grants GM33289 and HL117138 (to J. A. S.). This work was also supported by intramural grants from inStem (Department of Biotechnology, Government of India), the Shanta Wadhwani Centre for Cardiac and Neural Research, and program Grant RGP0054/2009-C from the Human Frontiers Science Program (to R. S. and J. A. S.).
- HCM
- hypertrophic cardiomyopathy
- DCM
- dilated cardiomyopathy
- Tm
- tropomyosin
- Tn
- troponin
- TnC
- troponin C
- TnI
- troponin I
- TnT
- troponin T
- ANS
- anilino-naphtahlene-6-sulfonic acid
- NBD
- 7-nitrobenz-2-oxa-1,3-diazole
- S1
- myosin subfragment 1
- RTF
- regulated thin filament
- MHC
- myosin heavy chain.
REFERENCES
- 1. Harvey P. A., Leinwand L. A. (2011) The cell biology of disease: cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maron B. J., Nichols P. F., 3rd, Pickle L. W., Wesley Y. E., Mulvihill J. J. (1984) Patterns of inheritance in hypertrophic cardiomyopathy: assessment by M-mode and two-dimensional echocardiography. Am. J. Cardiol. 53, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 3. Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T., Bild D. E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 92, 785–789 [DOI] [PubMed] [Google Scholar]
- 4. Watkins H., Ashrafian H., Redwood C. (2011) Inherited cardiomyopathies. N. Engl. J. Med. 364, 1643–1656 [DOI] [PubMed] [Google Scholar]
- 5. Alves M. L., Gaffin R. D., Wolska B. M. (2010) Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J. Mol. Cell. Cardiol. 48, 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robinson P., Griffiths P. J., Watkins H., Redwood C. S. (2007) Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 7. Dyer E. C., Jacques A. M., Hoskins A. C., Ward D. G., Gallon C. E., Messer A. E., Kaski J. P., Burch M., Kentish J. C., Marston S. B. (2009) Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2, 456–464 [DOI] [PubMed] [Google Scholar]
- 8. Sheehan K. A., Arteaga G. M., Hinken A. C., Dias F. A., Ribeiro C., Wieczorek D. F., Solaro R. J., Wolska B. M. (2011) Functional effects of a tropomyosin mutation linked to FHC contribute to maladaptation during acidosis. J. Mol. Cell. Cardiol. 50, 442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKillop D. F., Geeves M. A. (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65, 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tobacman L. S., Butters C. A. (2000) A new model of cooperative myosin-thin filament binding. J. Biol. Chem. 275, 27587–27593 [DOI] [PubMed] [Google Scholar]
- 11. Murakami K., Stewart M., Nozawa K., Tomii K., Kudou N., Igarashi N., Shirakihara Y., Wakatsuki S., Yasunaga T., Wakabayashi T. (2008) Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl. Acad. Sci. U.S.A. 105, 7200–7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murphy C. T., Rock R. S., Spudich J. A. (2001) A myosin II mutation uncouples ATPase activity from motility and shortens step size. Nat. Cell Biol. 3, 311–315 [DOI] [PubMed] [Google Scholar]
- 13. Kraft T., Mählmann E., Mattei T., Brenner B. (2005) Initiation of the power stroke in muscle: insights from the phosphate analog AlF4. Proc. Natl. Acad. Sci. U.S.A. 102, 13861–13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tardiff J. C. (2011) Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 108, 765–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hitchcock-DeGregori S. E., Song Y., Greenfield N. J. (2002) Functions of tropomyosin's periodic repeats. Biochemistry 41, 15036–15044 [DOI] [PubMed] [Google Scholar]
- 16. Hitchcock-DeGregori S. E., Singh A. (2010) What makes tropomyosin an actin binding protein? A perspective. J. Struct. Biol. 170, 319–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greenfield N. J., Palm T., Hitchcock-DeGregori S. E. (2002) Structure and interactions of the carboxyl terminus of striated muscle α-tropomyosin: it is important to be flexible. Biophys. J. 83, 2754–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corrêa F., Farah C. S. (2007) Different effects of trifluoroethanol and glycerol on the stability of tropomyosin helices and the head-to-tail complex. Biophys. J. 92, 2463–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Redwood C., Robinson P. (2013) α-Tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 34, 285–294 [DOI] [PubMed] [Google Scholar]
- 20. Come P. C., Bulkley B. H., Goodman Z. D., Hutchins G. M., Pitt B., Fortuin N. J. (1977) Hypercontractile cardiac states simulating hypertrophic cardiomyopathy. Circulation 55, 901–908 [DOI] [PubMed] [Google Scholar]
- 21. Sanoudou D., Vafiadaki E., Arvanitis D. A., Kranias E., Kontrogianni-Konstantopoulos A. (2005) Array lessons from the heart: focus on the genome and transcriptome of cardiomyopathies. Physiol. Genomics 21, 131–143 [DOI] [PubMed] [Google Scholar]
- 22. Borovikov Y. S., Karpicheva O. E., Chudakova G. A., Robinson P., Redwood C. S. (2009) Dilated cardiomyopathy mutations in α-tropomyosin inhibit its movement during the ATPase cycle. Biochem. Biophys. Res. Commun. 381, 403–406 [DOI] [PubMed] [Google Scholar]
- 23. Mirza M., Robinson P., Kremneva E., Copeland O., Nikolaeva O., Watkins H., Levitsky D., Redwood C., El-Mezgueldi M., Marston S. (2007) The effect of mutations in α-tropomyosin (E40K and E54K) that cause familial dilated cardiomyopathy on the regulatory mechanism of cardiac muscle thin filaments. J. Biol. Chem. 282, 13487–13497 [DOI] [PubMed] [Google Scholar]
- 24. Chang A. N., Harada K., Ackerman M. J., Potter J. D. (2005) Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in α-tropomyosin. J. Biol. Chem. 280, 34343–34349 [DOI] [PubMed] [Google Scholar]
- 25. Jongbloed R. J., Marcelis C. L., Doevendans P. A., Schmeitz-Mulkens J. M., Van Dockum W. G., Geraedts J. P., Smeets H. J. (2003) Variable clinical manifestation of a novel missense mutation in the α-tropomyosin (TPM1) gene in familial hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 41, 981–986 [DOI] [PubMed] [Google Scholar]
- 26. Van Driest S. L., Ellsworth E. G., Ommen S. R., Tajik A. J., Gersh B. J., Ackerman M. J. (2003) Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108, 445–451 [DOI] [PubMed] [Google Scholar]
- 27. Van Driest S. L., Will M. L., Atkins D. L., Ackerman M. J. (2002) A novel TPM1 mutation in a family with hypertrophic cardiomyopathy and sudden cardiac death in childhood. Am. J. Cardiol. 90, 1123–1127 [DOI] [PubMed] [Google Scholar]
- 28. Makhoul M., Ackerman M. J., Atkins D. L., Law I. H. (2011) Clinical spectrum in a family with tropomyosin-mediated hypertrophic cardiomyopathy and sudden death in childhood. Pediatr. Cardiol. 32, 215–220 [DOI] [PubMed] [Google Scholar]
- 29. Nallari P., Rani D. S., Calambur N., Thangaraj K. (2010) Genetic variations in α-tropomyosin gene in HCM and DCM patients from India. Environ. Mol. Mutagenesis 51, 722 [Google Scholar]
- 30. Lakdawala N. K., Dellefave L., Redwood C. S., Sparks E., Cirino A. L., Depalma S., Colan S. D., Funke B., Zimmerman R. S., Robinson P., Watkins H., Seidman C. E., Seidman J. G., McNally E. M., Ho C. Y. (2010) Familial dilated cardiomyopathy caused by an α-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 55, 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barua B., Pamula M. C., Hitchcock-DeGregori S. E. (2011) Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc. Natl. Acad. Sci. U.S.A. 108, 10150–10155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Margaret Sunitha S., Mercer J. A., Spudich J. A., Sowdhamini R. (2012) Integrative structural modelling of the cardiac thin filament: energetics at the interface and conservation patterns reveal a spotlight on period 2 of tropomyosin. Bioinform. Biol. Insights 6, 203–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sommese R. F., Nag S., Sutton S., Miller S. M., Spudich J. A., Ruppel K. M. (2013) Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin. PLoS One 8, e83403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monteiro P. B., Lataro R. C., Ferro J. A., Reinach Fde C. (1994) Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 269, 10461–10466 [PubMed] [Google Scholar]
- 35. Gomes A. V., Guzman G., Zhao J., Potter J. D. (2002) Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J. Biol. Chem. 277, 35341–35349 [DOI] [PubMed] [Google Scholar]
- 36. Szczesna D., Guzman G., Miller T., Zhao J., Farokhi K., Ellemberger H., Potter J. D. (1996) The role of the four Ca2+ binding sites of troponin C in the regulation of skeletal muscle contraction. J. Biol. Chem. 271, 8381–8386 [DOI] [PubMed] [Google Scholar]
- 37. Sheng Z., Pan B. S., Miller T. E., Potter J. D. (1992) Isolation, expression, and mutation of a rabbit skeletal muscle cDNA clone for troponin I. The role of the NH2 terminus of fast skeletal muscle troponin I in its biological activity. J. Biol. Chem. 267, 25407–25413 [PubMed] [Google Scholar]
- 38. Pardee J. D., Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 [DOI] [PubMed] [Google Scholar]
- 39. Resnicow D. I., Deacon J. C., Warrick H. M., Spudich J. A., Leinwand L. A. (2010) Functional diversity among a family of human skeletal muscle myosin motors. Proc. Natl. Acad. Sci. U.S.A. 107, 1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Golitsina N., An Y., Greenfield N. J., Thierfelder L., Iizuka K., Seidman J. G., Seidman C. E., Lehrer S. S., Hitchcock-DeGregori S. E. (1997) Effects of two familial hypertrophic cardiomyopathy-causing mutations on α-tropomyosin structure and function. Biochemistry 36, 4637–4642 [DOI] [PubMed] [Google Scholar]
- 41. Coulton A. T., Koka K., Lehrer S. S., Geeves M. A. (2008) Role of the head-to-tail overlap region in smooth and skeletal muscle β-tropomyosin. Biochemistry 47, 388–397 [DOI] [PubMed] [Google Scholar]
- 42. Davis J. P., Norman C., Kobayashi T., Solaro R. J., Swartz D. R., Tikunova S. B. (2007) Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 92, 3195–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dweck D., Reyes-Alfonso A., Jr., Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 [DOI] [PubMed] [Google Scholar]
- 44. Zhou Z., Li K. L., Rieck D., Ouyang Y., Chandra M., Dong W. J. (2012) Structural dynamics of C-domain of cardiac troponin I protein in reconstituted thin filament. J. Biol. Chem. 287, 7661–7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trybus K. M. (2000) Biochemical studies of myosin. Methods 22, 327–335 [DOI] [PubMed] [Google Scholar]
- 46. Kalyva A., Schmidtmann A., Geeves M. A. (2012) In vitro formation and characterization of the skeletal muscle α β-tropomyosin heterodimers. Biochemistry 51, 6388–6399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Janco M., Kalyva A., Scellini B., Piroddi N., Tesi C., Poggesi C., Geeves M. A. (2012) α-Tropomyosin with a D175N or E180G mutation in only one chain differs from tropomyosin with mutations in both chains. Biochemistry 51, 9880–9890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X. E., Suphamungmee W., Janco M., Geeves M. A., Marston S. B., Fischer S., Lehman W. (2012) The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 424, 493–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson J. D., Collins J. H., Robertson S. P., Potter J. D. (1980) A fluorescent probe study of Ca2+ binding to the Ca2+-specific sites of cardiac troponin and troponin C. J. Biol. Chem. 255, 9635–9640 [PubMed] [Google Scholar]
- 50. Michele D. E., Coutu P., Metzger J. M. (2002) Divergent abnormal muscle relaxation by hypertrophic cardiomyopathy and nemaline myopathy mutant tropomyosins. Physiol. Genomics 9, 103–111 [DOI] [PubMed] [Google Scholar]
- 51. Michele D. E., Albayya F. P., Metzger J. M. (1999) Direct, convergent hypersensitivity of calcium-activated force generation produced by hypertrophic cardiomyopathy mutant α-tropomyosins in adult cardiac myocytes. Nat. Med. 5, 1413–1417 [DOI] [PubMed] [Google Scholar]
- 52. Bai F., Weis A., Takeda A. K., Chase P. B., Kawai M. (2011) Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophys. J. 100, 1014–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bing W., Knott A., Redwood C., Esposito G., Purcell I., Watkins H., Marston S. (2000) Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle α -tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. J. Mol. Cell. Cardiol. 32, 1489–1498 [DOI] [PubMed] [Google Scholar]
- 54. Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C., Warshaw D. M. (2007) Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291 [DOI] [PubMed] [Google Scholar]
- 55. Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K., Spudich J. A. (2013) Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. U.S.A. 110, 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yar S., Chowdhury S. A., Davis R. T., 3rd, Kobayashi M., Monasky M. M., Rajan S., Wolska B. M., Gaponenko V., Kobayashi T., Wieczorek D. F., Solaro R. J. (2013) Conserved Asp-137 is important for both structure and regulatory functions of cardiac α-tropomyosin (α-TM) in a novel transgenic mouse model expressing α-TM-D137L. J. Biol. Chem. 288, 16235–16246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hodges R. S., Mills J., McReynolds S., Kirwan J. P., Tripet B., Osguthorpe D. (2009) Identification of a unique “stability control region” that controls protein stability of tropomyosin: a two-stranded α-helical coiled-coil. J. Mol. Biol. 392, 747–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yap K. L., Ames J. B., Swindells M. B., Ikura M. (1999) Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins 37, 499–507 [DOI] [PubMed] [Google Scholar]
- 59. Rao V. S., Clobes A. M., Guilford W. H. (2011) Force spectroscopy reveals multiple “closed states” of the muscle thin filament. J. Biol. Chem. 286, 24135–24141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smith L., Tainter C., Regnier M., Martyn D. A. (2009) Cooperative cross-bridge activation of thin filaments contributes to the Frank-Starling mechanism in cardiac muscle. Biophys. J. 96, 3692–3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rieck D. C., Li K. L., Ouyang Y., Solaro R. J., Dong W. J. (2013) Structural basis for the in situ Ca2+ sensitization of cardiac troponin C by positive feedback from force-generating myosin cross-bridges. Arch. Biochem. Biophys. 537, 198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Desai R. A., Geeves M. A., Kad N. M. (2014) Using fluorescent myosin to directly visualize cooperative activation of thin filaments. J. Biol. Chem. 290, 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lehrer S. S., Geeves M. A. (1998) The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 277, 1081–1089 [DOI] [PubMed] [Google Scholar]
- 64. Kintses B., Gyimesi M., Pearson D. S., Geeves M. A., Zeng W., Bagshaw C. R., Málnási-Csizmadia A. (2007) Reversible movement of switch 1 loop of myosin determines actin interaction. EMBO J. 26, 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Behrmann E., Müller M., Penczek P. A., Mannherz H. G., Manstein D. J., Raunser S. (2012) Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jarraya M., Quijano-Roy S., Monnier N., Béhin A., Avila-Smirnov D., Romero N. B., Allamand V., Richard P., Barois A., May A., Estournet B., Mercuri E., Carlier P. G., Carlier R. Y. (2012) Whole-body muscle MRI in a series of patients with congenital myopathy related to TPM2 gene mutations. Neuromuscul. Disord. 22, S137–S147 [DOI] [PubMed] [Google Scholar]
- 67. Ochala J., Gokhin D. S., Pénisson-Besnier I., Quijano-Roy S., Monnier N., Lunardi J., Romero N. B., Fowler V. M. (2012) Congenital myopathy-causing tropomyosin mutations induce thin filament dysfunction via distinct physiological mechanisms. Hum. Mol. Genet. 21, 4473–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marston S., Memo M., Messer A., Papadaki M., Nowak K., McNamara E., Ong R., El-Mezgueldi M., Li X., Lehman W. (2013) Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 22, 4978–4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. van de Meerakker J. B., Christiaans I., Barnett P., Lekanne Deprez R. H., Ilgun A., Mook O. R., Mannens M. M., Lam J., Wilde A. A., Moorman A. F., Postma A. V. (2013) A novel α-tropomyosin mutation associates with dilated and non-compaction cardiomyopathy and diminishes actin binding. Biochim. Biophys. Acta 1833, 833–839 [DOI] [PubMed] [Google Scholar]
- 70. Morita H., Rehm H. L., Menesses A., McDonough B., Roberts A. E., Kucherlapati R., Towbin J. A., Seidman J. G., Seidman C. E. (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 358, 1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]