

Background: The function of Ndfip1 in DNA repair is unknown.
Results: Ndfip1 is required during stress for ubiquitinating and trafficking BRAT1 into the nucleus.
Conclusion: Ndfip1 is required for activating the ATM (ataxia telangiectasia mutated) pathway for DNA repair during stress injury.
Significance: We describe a new mechanism for ubiquitinating and trafficking of BRAT1 into the nucleus for DNA repair.
Keywords: DNA Damage Response, Intracellular Trafficking, Neuroprotection, Nuclear Transport, Ubiquitylation (ubiquitination)
Abstract
During injury, cells are vulnerable to apoptosis from a variety of stress conditions including DNA damage causing double-stranded breaks. Without repair, these breaks lead to aberrations in DNA replication and transcription, leading to apoptosis. A major response to DNA damage is provided by the protein kinase ATM (ataxia telangiectasia mutated) that is capable of commanding a plethora of signaling networks for DNA repair, cell cycle arrest, and even apoptosis. A key element in the DNA damage response is the mobilization of activating proteins into the cell nucleus to repair damaged DNA. BRAT1 is one of these proteins, and it functions as an activator of ATM by maintaining its phosphorylated status while also keeping other phosphatases at bay. However, it is unknown how BRAT1 is trafficked into the cell nucleus to maintain ATM phosphorylation. Here we demonstrate that Ndfip1-mediated ubiquitination of BRAT1 leads to BRAT1 trafficking into the cell nucleus. Without Ndfip1, BRAT1 failed to translocate to the nucleus. Under genotoxic stress, cells showed increased expression of both Ndfip1 and phosphorylated ATM. Following brain injury, neurons show increased expression of Ndfip1 and nuclear translocation of BRAT1. These results point to Ndfip1 as a sensor protein during cell injury and Ndfip1 up-regulation as a cue for BRAT1 ubiquitination by Nedd4 E3 ligases, followed by nuclear translocation of BRAT1.
Introduction
Following traumatic injury or stroke, neurons in affected brain tissues undergo apoptosis, whereas others adopt defense strategies to stay alive (1). Neuronal apoptosis occurs mostly in tissues surrounding the damaged core and can continue for days during the secondary phase of injury. During this phase, cytotoxic factors are released in response to glutamate excitotoxicity, inflammation, and hypoxia. The injury process also dramatically increases DNA damage and production of reactive oxygen species, resulting in excess redox activity and oxidative stress (2). The resulting free radicals inflict damage to cell membranes and produce detrimental reactions with proteins, lipids, and DNA, resulting in cell death (3).
A central line of defense to sudden DNA damage and oxidative stress is activation of the ATM (ataxia telangiectasia mutated) protein for DNA repair. This kinase is a member of the phosphatidylinositol 3-kinase-like kinase family of Ser/Thr-protein kinases and a key player in redox homeostasis and the DNA damage response (4). Following double-strand DNA breaks and oxidation, ATM is recruited and activated by cell sensor proteins, including the MRE11-RAD50-NBS1 complex for DNA repair by homologous end joining (5). Aside from promoting genome stability to assist cell survival, ATM also activates cell survival pathways linked to PI3K and mammalian target of rapamycin complex, as well as checkpoint proteins p53 and checkpoint kinase 2 to modulate the cell cycle (6).
ATM mobilization and activation at sites of broken DNA requires the participation of multiple sensor proteins. Aside from the MRE11-RAD50-NBS1 complex, a number of other sensor proteins have been identified including TP53BP1 (p53-binding protein 1), BRCA1 (breast cancer type 1), and MDC1 (mediator of DNA damage checkpoint protein 1) (6). In addition, BRAT1 (BRCA1-associated ATM activator 1) has been identified to be important for ATM phosphorylation at Ser1981 and complex assembly (7).
Previous work from our laboratory has identified Ndfip1 as a regulator of neuroprotective mechanisms to improve neuron survival following brain injury or cerebral ischemia (8–11). Ndfip1 is an adaptor and activator for Nedd4 family of ubiquitin ligases, and in the adult brain, Ndfip1 is normally present in cortical neurons at low levels within vesicular membranes (12). Following injury or stress, Ndfip1 is selectively up-regulated in certain neurons, resulting in their survival (8). The precise circumstances favoring up-regulation of Ndfip1 in some neurons but not in others are poorly understood. However, recent work from our group has defined a number of mechanisms through which Ndfip1 can combat apoptosis. These include degradation of DMT1 (divalent metal transporter 1) to prevent metal poisoning of neurons (13) and transient shuttling of the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) into the nucleus to promote pAkt-mediated cell survival (9). Given the importance of the DNA damage response following injury, we set out to investigate whether or not Ndfip1 is a participant in the ATM-mediated DNA repair response.
EXPERIMENTAL PROCEDURES
Animals and Traumatic Brain Injury (TBI)
All procedures were approved by the Florey Institute of Neuroscience and Mental Health Animal Ethics Committee and in accordance with the ARRIVE guidelines for reporting in vivo experiments. C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were obtained from the Animals Resource Center of Australia. All animals were housed under a 12-h light/dark cycle in temperature-controlled rooms (22 °C) and allowed free access to standard chow and water. The method for TBI induction has been previously described (11).
Cell Culture and Transfection
HEK293T cells and mouse embryonic fibroblasts (MEFs)3 were cultured in DMEM (Invitrogen) supplemented with 10% FCS, 2 mm l-glutamate, and 50 g/ml penicillin/streptomycin. SH-SY5Y cells were cultured in RPMI media (Invitrogen) supplemented with 15% FCS, 2 mm l-glutamate, and 50 g/ml penicillin/streptomycin. Cultures were maintained in 37 °C in a 5% CO2 atmosphere. HEK293T cells and MEFs were transfected with the appropriate constructs using Effectene transfection reagent kit (Qiagen) according to the manufacturer's instruction. The procedures for the production of lentiviral particles containing Ndfip1-Flag and inducible cell lines (HEK293T and SH-SY5Y) sensitive to 4-hydroxytamoxifen for Ndfip1-Flag expression has been described (9).
For production of HEK293T cells capable of responding to doxycyline for expressing Flag-Ndfip1, we used the pF TRE3G PGK puro vector (gift of David Vaux, Walter and Eliza Hall Institute of Medical Research, Parkville, Australia). Human Ndfip1 cDNA flanked with BamHI and NheI restriction sites was generated by PCR with the forward primer 5′-CG CGG ATC CAT GGC GTT GGC GTT GGC-3′ and reverse primer 5′-TCT AGC TAG CTT AAT AAA TAA AGA GAA CTC TGG TC-3′. The purified Ndfip1 PCR fragment was digested with BamHI and NheI restriction enzyme and cloned into the pF TRE3G PGK puro vector and sequence-verified. To generate lentiviral particles, packaging constructs pCMV ðR8.2 and VSVg and the relevant lentiviral plasmids were co-transfected into HEK293T cells using Effectene reagent (Qiagen). Supernatants were harvested at 100,000 × g. HEK293T cells were infected with viral supernatant for 24 h. Infected cells were selected using puromycin (2 μg/ml; Sigma-Aldrich), and tested for Flag-Ndfip1 expression following exposure to doxycycline (2 ng/ml).
To induce DNA damage, the topoisomerase II inhibitor etoposide (Sigma-Aldrich) was dissolved in DMSO and added to the medium to final concentrations of 2 and 20 μm for 2 h. Control experiments showed that DMSO alone had no effect on any of the parameters measured.
Cell Survival Measured by MTT Assay
MEFs were grown in 24-well plates at a density of 5 × 104 cells-well in DMEM. Etoposide (2 μm) was added to the medium for 24 h followed by 100 μl of MTT solution (5 mg/ml in PBS) (Sigma-Aldrich) and incubated for 3 h at 37 °C. After aspirating off the medium, 200 μl of DMSO was added and incubated for 10 min at 37 °C before colorimetry. The absorbance of the dye was measured at 550 nm. Three separate assays were conducted for each experimental condition.
Plasmid Constructs
To visualize protein binding, plasmids were created for bimolecular fluorescent complementation (BiFC) of Venus using N-terminal (VN) and C-terminal (VC) reporter fragments as previously described (14). To create a BRAT1-VC155–238 construct, human BRAT1 cDNA flanked with EcoRI and BglII restriction sites was generated by PCR with the forward primer 5′-CCC GAA TTC ATG GAC CCA GAA TGC GCC CA-3′ and reverse primer 5′-TAG AGA TCT GTA GCA GTC GGC CTC GTC CC-3′. The purified BRAT1 PCR fragment was digested with EcoRI and BglII restriction enzymes and cloned into pBiFC-VC155 (addgene Plasmid 22011). To create a Strep-Flag-BRAT1 construct, human BRAT1 cDNA flanked with NheI restriction sites was generated by PCR with the forward primers 5′-TCT AGC TAG CAT GGA CCC AGA ATG CGC CCA-3′ and reverse primer 5′-TCC GCT AGC GCT CAG TAG CAG TCG GC-3′. The purified BRAT1 PCR fragment was digested with NheI restriction enzymes and cloned into Strep-Flag pcDNA3 vector (gift of Christian Gloeckner (Institute of Human Genetics, Munich-Neuherberg, Germany). To map binding of BRAT1 with Ndfip1, BRAT1 deletion mutant constructs were generated using Gibson assembly (New England Biolabs) with the following primers: BRAT1 Δ1–100 forward, 5′-AGT TCG AGA AAG GAG CTA GCG GGC TCT TTG GGG AGC CAG G-3′; BRAT1 Δ1–200 forward, 5′-AGT TCG AGA AAG GAG CTA GCC ACG TTG AAG AGT CCT TGT G-3′; and BRAT1 reverse, 5′-CGC CAA CGC CAT GCT AGC GCT CAG TAG CAG TCG GCC TCG T-3′.
Other constructs used were SF-Ndfip1 (Strep-Flag-Ndfip1 in pcDNA3), Ndfip1 in pBiFC-VN173, His-Ubiquitin in pcDNA3, Flag-Nedd4-1 in pcDNA3, Flag-Nedd4-2 in pcDNA3, Flag-Itch in pcDNA3, Ub in pBiFC-VN173, F-BimL in pBiFC-VN173, Flag-BRAT1 and HA-BRAT1 in pcDNA3 (14, 15), and BRAT1 shRNA plasmid (16).
Protein Lysate Preparation and Immunoprecipitation Assay
HEK293T and SH-SY5Y cells were lysed in ice-cold radioimmunoprecipitation assay buffer (50 mm, pH 7.2, 0.15 m NaCl, 1% Nonidet P-40, and 0.1% SDS) with protease inhibitor mixture (Complete Mini; Roche) for 20 min at 4 °C. Cell lysates were cleared of insoluble debris by centrifugation at 21,500 × g for 15 min at 4 °C. Protein concentration of lysate was measured using detergent-compatible protein assay according to the manufacturer's instructions (Bio-Rad). For immunoprecipitation experiments, Strep-Tactin (Qiagen) beads were used for precipitate SF-Ndfip1 and SF-BRAT1. For each immunoprecipitation assay, beads were washed three times with radioimmunoprecipitation assay buffer before elution using the manufacturer's instructions. For the ubiquitination assay, HEK293T cells were transfected with His-Ubiquitin, SF-Ndfip1, HA-BRAT1, Flag-Nedd4-1, Flag-Nedd4-2, and Flag-Itch alone or in combination. Twenty-four hours after transfection, lysates were immunoprecipitated with HisLink beads (Promega) under denaturing condition using 6 m guanidine HCl and N-ethylamaleimide to inhibit deubiquitinase enzymes. Beads were washed three times using radioimmunoprecipitation assay buffer before elution using 300 mm imidazole. Eluted fractions were suspended in Laemmli buffer for SDS-PAGE and analyzed by Western blotting.
Western Blot Analysis
Lysates or immunoprecipitates were resolved on SDS-PAGE gels followed by transfer onto Hybond-C nitrocellulose membrane (GE Healthcare). Membranes were blocked for 1 h at room temperature in 5% nonfat milk in TBS and 0.05% Tween 20. Blots were incubated overnight with primary antibodies at 4 °C followed by appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. Blots were exposed using ECL according to the manufacturer's instructions (GE Healthcare). Alternatively, gel imaging system ChemiDoc MP (Bio-Rad) was used to visualize the blot. Primary antibodies used were rabbit polyclonal anti-Ndfip1 (1:2000), rabbit polyclonal anti-BRAT1 (1:500, Abcam), purified rabbit polyclonal anti-Nedd4-2 (1:1000, Abcam), mouse monoclonal anti-Nedd4-1 (1:1,000; BD), mouse monoclonal anti-Itch (1:1,000; BD), mouse monoclonal anti-ATM (1:1,000; Abcam), rabbit monoclonal anti-pATM (1:1,000; Abcam), mouse monoclonal anti-pH2AX (1:1,000), Millipore, mouse monoclonal anti–β-actin (1:5,000; Sigma-Aldrich), and mouse monoclonal anti-Flag M2 (1:1,000; Sigma-Aldrich). The HRP-conjugated secondary antibodies used were goat polyclonal anti-rabbit (1:10,000; Millipore) and goat polyclonal anti-mouse (1:15,000; Millipore).
Immunohistochemistry
Mice were euthanized (sodium pentobarbitone, 80 mg/kg, intraperitoneal) by transcardial perfusion of PBS (pH 7.4) followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB). The brains were cryoprotected in 20% sucrose in 0.1 m PB at 4 °C overnight and followed by embedding in Tissue-Tek OCT compound (Sakura Finetek, Tokyo, Japan) before coronal sectioning with a cryostat (12 μm). Perfusion-fixed sections were washed with 0.1 m PB, antigens were retrieved with sodium citrate buffer (10 mm sodium citrate), permeabilized with 0.3% Triton X-100 in 0.1 m PB, and then blocked with 10% FCS in 0.1% Triton X-100 with 0.1 m PB. The sections were incubated with primary antibodies overnight followed by appropriate secondary antibodies for 1 h at room temperature. Coverslips were then mounted on slides using ProLong® Gold antifade reagent with DAPI (Invitrogen).
Cells on coverslips were washed with PBS (pH 7.4) followed by fixation with 4% paraformaldehyde in 0.1 m PB for 15 min. Permeabilization was carried out with 0.3% Triton X-100 in 0.1 m PB and then blocked with 10% FCS in 0.1% Triton X-100 with 0.1 m PB. The cells were incubated with primary antibodies overnight followed by appropriate secondary antibodies for 1 h at room temperature. Coverslips were then mounted on slides using ProLong® Gold antifade reagent with DAPI (Invitrogen).
Primary antibodies used were: rat monoclonal anti-Ndfip1 (1:1000), rabbit polyclonal anti-BRAT1, (1:500; Abcam), and mouse monoclonal anti-Flag M2 (1:500; Sigma-Aldrich). Secondary antibodies were: Alexa Fluor 594-conjugated goat anti-rat IgG (1:500; Molecular Probes), Alexa Fluor 488-conjugated goat anti-mouse IgG (1:500, Molecular Probes), and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500; Molecular Probes). Imaging was performed on a Zeiss 780 confocal microscope.
Bimolecular Fluorescent Complementation Experiments
MEFs on coverslips were transfected with the appropriate plasmids. At 24 h post-transfection, MEFs were washed once with 0.1 m PB and then fixed in 4% paraformaldehyde in PBS for 15 min. After fixation, the coverslips were incubated for 5 min with 0.1 m PB phalloidin marker (50 μg/ml) before a final wash in 0.1 m PB. Coverslips were then mounted on slides using ProLong® Gold antifade reagent with DAPI (Invitrogen). Imaging was performed on a Zeiss 780 confocal microscope. The fluorescence intensity of the nuclear BiFC signal for each cell was measured using ImageJ. A minimum of 15 cells from two independent experiments were conducted.
Statistical Analysis
All data were analyzed using Prism software and presented as means ± S.E. unless otherwise indicated. For measure of statistical significance, a minimal p < 0.05 cutoff was employed.
RESULTS
BRAT1 Is a Novel Binding Partner of Ndfip1
BRAT1 was initially identified in a screen of interacting proteins for Ndfip1 in HEK293T cells using tandem affinity purification of proteins bound to Strep-Flag-Ndfip1 (SF-Ndfip1), followed by mass spectrometry analysis.4 To confirm the interaction between BRAT1 and Ndfip1, co-immunoprecipitation (co-IP) was performed following overexpression of HA-BRAT1 and SF-Ndfip1 in HEK293T cells. The results showed that co-IP with Strep-Tactin-coated beads produced the correctly sized band for HA-BRAT1 (Fig. 1A), whereas the reverse pulldown with Strep-Flag-BRAT1 (SF-BRAT1) also revealed a specific band for Ndfip1-mCherry (Fig. 1B).
FIGURE 1.

Ndfip1 interaction with BRAT1. A, HA-BRAT1 co-precipitated with Strep-Flag-Ndfip1 (SF-Ndfip1) after co-expression in HEK293T cells. IB, immunoblotting. B, Ndfip1-mCherry co-precipitated with Strep-Flag-BRAT1 (SF-BRAT1) after co-expression in HEK293T cells. C, schematic diagram of the BiFC system. Ndfip1 is conjugated to the N-terminal fragment of Venus (VN-Ndfip1), and BRAT1 is conjugated to the C-terminal fragment of Venus (BRAT1-VC). In the event of Ndfip1 and BRAT1 interaction, complementation of Venus fragments can be detected by a fluorescent signal. D, BiFC experiments demonstrate binding between BRAT1 and Ndfip1 in the cytoplasm (upper panels). Nuclear staining with DAPI (blue) and F-actin staining (red) using phalloidin provided cellular contrast. A control BiFC experiment (lower panels) showed no interaction between BimL-VN and BRAT1-VC. Scale bar, 10 μm.
Previous reports had shown that Ndfip1 is a cytoplasmic protein (8, 17), whereas BRAT1 can shuttle between the cytoplasm and nucleus (7, 18). Thus, we investigated the whereabouts of the interaction between Ndfip1 and BRAT1 using BiFC with the Venus reporter protein.
Ndfip1 was fused with the N-terminal fragment of Venus protein (Ndfip1-VN1–173) and BRAT1 fused with a complementary C-terminal fragment of Venus (BRAT1-VC155–238) (Fig. 1C). Following co-expression of these plasmids in MEFs, the BiFC fluorescent signal was observed in the cytoplasm, confirming their interaction in this cellular location (Fig. 1D, top panels). A control experiment using BimL, a protein that is not known to interact with BRAT1, did not yield a BiFC signal (BimL-VN1–173 and BRAT1-VC155–238) (Fig. 1D, bottom panels). In aggregate, these experiments suggest that Ndfip1 and BRAT1 interact in the cytoplasm.
Mapping the Interaction of BRAT1 with Ndfip1
To further identify the responsible domain in BRAT1, we performed a series of co-IP experiments using deletion mutants. Initially, we performed a pulldown assay with a mutant form of BRAT1 (c.638_639insA) (BRAT1 IMut) that previously was reported to be associated with lethal neonatal rigidity and seizure syndrome (18). The protein sequence of BRAT1 c.638_639insA produced a frameshift from amino acid 231 resulting in premature termination at amino acid 401 and a reduced molecular mass of 42 kDa (Fig. 2A). The co-IP was performed by overexpression of SF-BRAT1 IMut and Ndfip1-mCherry in HEK293T cells. As controls, wild-type SF-BRAT1 and an empty vector were used. The results showed binding of Ndfip1-mCherry with both wild-type SF-BRAT1 and SF-BRAT1 IMut (Fig. 2B). This would suggest that the binding site for BRAT1 might lie upstream of amino acid 231.
FIGURE 2.

Mapping the site of Ndfip1-BRAT1 interaction. A, a schematic diagram showing the various constructs used for binding assays with Ndfip1-mCherry. In addition to wild-type BRAT1, three deletion constructs were employed: IMut, BRAT1 c.638_639insA; BRAT1 Δ1–100 (B1); and BRAT1 Δ1–200 (B2). B, Ndfip1-mCherry co-precipitated with both wild-type Strep-Flag-BRAT1 and SF-BRAT1 IMut, suggesting that the binding site might lie upstream of amino acid 213. The empty vector served as a negative control. IB, immunoblotting. C, immunoprecipitation experiments revealed binding of BRAT1 Δ1–100 but not BRAT1 Δ1–200 with Ndfip1-mCherry, suggesting that the binding site lies between amino acids 100 and 200.
To identify this, deletion mutants of BRAT1 with varying lengths of N-terminal truncations were produced, and co-IP experiments with Ndfip1-mCherry were performed as before (Fig. 2C). The results showed that SF-BRAT1 Δ1–100 (B1) was able to bind to Ndfip1-mCherry, but SF-BRAT1 Δ1–200 (B2) was not, indicating that the putative binding site was likely to be contained between amino acids 100–200.
Ndfip1 Enhanced Ubiquitination of BRAT1 by Nedd4 Family Ubiquitin Ligases
BRAT1 has been shown to be an important mediator of the DNA damage response (7, 19). However, it is unclear whether BRAT1 was post-translationally modified by addition of ubiquitin in this pathological process. Given the role of Ndfip1 as an adaptor and activator for Nedd4 family E3 ligases during injury in brain cells (9, 10, 13), we explored a role for Ndfip1/Nedd4 family proteins in the ubiquitination of BRAT1. A ubiquitination assay was performed by overexpression of His-ubiquitin and HA-BRAT1 in HEK293T cells, with and without Ndfip1, and Nedd4 family E3 ligases. Ubiquitinated proteins were precipitated from lysates under denaturing conditions to prevent nonspecific binding. The immunoprecipitate of ubiquitinated proteins was probed using anti-BRAT1 antibodies to assess BRAT1 ubiquitination (Fig. 3). The results showed that expression of BRAT1 alone was not sufficient for ubiquitination of the protein (Fig. 3, lane 1). When co-expressed with E3 ubiquitin ligases, but in the absence of Ndfip1, BRAT1 showed only a small degree of ubiquitination by members of the Nedd4 E3 ligases (Nedd4-1, Nedd4-2, and Itch; lanes 2, 3, and 4, respectively). Interestingly, Itch was more effective (compared with Nedd4-1 and Nedd4-2) in promoting the ubiquitination of BRAT1 (Fig. 3, ∼96-kDa bands in lane 4). With the addition of Ndfip1 in the assay (lanes 5–8), the ubiquitination of BRAT1 was dramatically enhanced by all three E3 ligases, particularly more so by Itch (lane 8). Indeed, Ndfip1 alone (lane 5) demonstrated increased ubiquitination of BRAT1, presumably by recruiting endogenous E3 ligases present in HEK293T cells (9). The pattern of ubiquitination suggests both mono- and polyubiquitination of BRAT1, with the monoubiquitinated band showing strong intensity at ∼96 kDa. These experiments indicate that BRAT1 is subject to post-translational modification by the Ndfip1/Nedd4 ubiquitination system.
FIGURE 3.
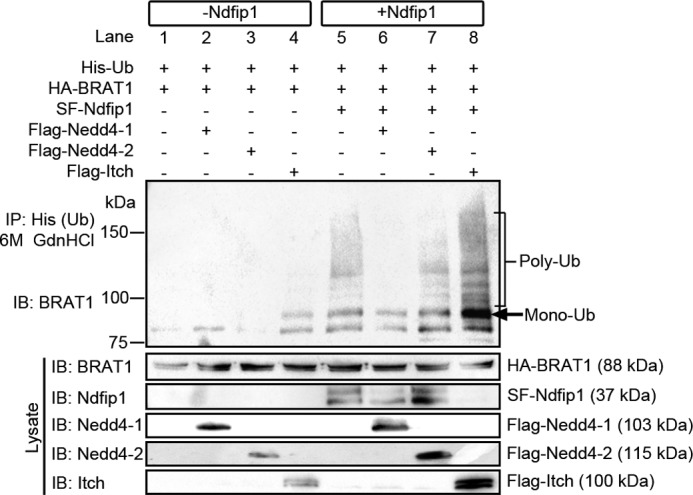
Ndfip1 enhances BRAT1 ubiquitination by Nedd4 E3 ligases. BRAT1 ubiquitin (Ub) assay with Ndfip1 and E3 ligases Nedd4-1, Nedd4-2 and Itch in HEK293T cells. IB, immunoblotting. Lanes without Ndfip1 co-expression (lanes 1–4) show decreased BRAT1 ubiquitination compared with lanes expressing Ndfip1 (lanes 5–8). In particular, the co-transfection of Ndfip1 with Itch (lane 8) produced the characteristic monoubiquitinated ladder together with higher molecular mass polyubiquitinated smear. Ndfip1 band is not visible on lane 8, suggesting Ndfip1 degradation by Itch.
Ndfip1 Does Not Promote Degradation of BRAT1 but It Is Required for Its Nuclear Trafficking
Previous experiments from our laboratory have shown that, depending on the physiological context, protein targets that bind to Ndfip1 have different fates following ubiquitination. In the presence of excess transition metals such as Co2+ and Fe2+, Ndfip1 in neurons targets DMT1 for polyubiquitination and degradation to prevent metal poisoning (13). On the other hand, during brain ischemia, Ndfip1 mediates the monoubiquitination of the tumor suppressor PTEN, causing PTEN to traffic into the nucleus and allowing the up-regulation of PI3K activity to promote neuron survival (9). To examine whether overexpressed Ndfip1 can lead to BRAT1 degradation, an inducible system was employed whereby the addition of 4-hydroxytamoxifen can activate the mutant estrogen receptor to drive the expression of Ndfip1-Flag using the yeast Gal4/UAS transcription system (13). In HEK293T cells, 4-hydroxytamoxifen induction of Ndfip1 expression did not result in the reduction of endogenous BRAT1, compared with uninduced cells (Fig. 4, A and B). A similar result was obtained when using 4-hydroxytamoxifen on the neuroblastoma cell line, SH-SY5Y (Fig. 4, C and D). Conversely, the use of shRNA to knock down Ndfip1 did not change the abundance of BRAT1 (Fig. 4, E and F). These results suggest that the Ndfip1 (and by inference the ubiquitination of BRAT1) did not lead to degradation of BRAT1.
FIGURE 4.

BRAT1 abundance in cells is unaffected by overexpression of Ndfip1. A–D, overexpression of Ndfip1 by 4-hydroxytamoxifen-induction in two different cell lines (HEK293T and SH-SY5Y) showed no difference in BRAT1 abundance, suggesting that Ndfip1-mediated ubiquitination of BRAT1 does not result in degradation. E and F, treatment of SH-SY5Y with Ndfip1 shRNA did not result in changes to BRAT1 abundance. The values are means ± S.E., n = 3 experiments. IB, immunoblotting.
Because the overexpression of Ndfip1 did not lead to the degradation of BRAT1, we then tested for the possibility that Ndfip1 is required for nuclear trafficking BRAT1. To test this, BiFC constructs were used to examine the physical location of ubiquitinated BRAT1 in Ndfip1+/+ and Ndfip1−/− MEFs (Fig. 5A). In Ndfip1+/+ MEFs, co-expression of BRAT1-VC155–238 and VN1–173-Ubiquitin resulted in nuclear localization of ubiquitinated BRAT1 (Fig. 5B, top panels). In contrast, the same co-transfection experiments conducted in Ndfip1−/− MEFs demonstrated only BiFC fluorescence in the cytoplasm (Fig. 5B, bottom panels). A requirement for Ndfip1 was also demonstrated using transfected Flag-BRAT1 (rather than complementation by BiFC); this showed nuclear translocation of BRAT1 only in Ndfip1+/+ MEFs (Fig. 5C, top panels). Quantification of the BiFC signal in nuclei in both genotypes showed a significant increase of BiFC signal intensity in Ndfip1+/+ MEFs, when compared with Ndfip1−/− MEFs (p < 0.001) (Fig. 5D), suggesting that nuclear trafficking of ubiquitinated BRAT1 is enhanced by Ndfip1.
FIGURE 5.

Ndfip1 is required for nuclear localization of ubiquitinated BRAT1 in vitro. A, schematic diagram of the BiFC system employed to track the spatial localization of ubiquitinated BRAT1. B, in Ndfip1+/+ MEFs, ubiquitinated BRAT1 was observed in both the perinuclear and nuclear regions of the cell (upper panels). In contrast, Ndfip1−/− MEFs showed only perinuclear and cytoplasmic distribution of ubiquitinated BRAT1 (lower panels). C, confocal microscopy to study cellular localization of overexpressed Flag-BRAT1 in Ndfip1+/+ and Ndfip1−/− MEFs subjected to etoposide treatment. Flag-BRAT1 can be detected in both cytoplasm and nucleus of Ndfip1+/+ MEFs at all concentrations of etoposide treatment. In contrast, Flag-BRAT1 is excluded from the nucleus of Ndfip1−/− MEFs with or without etoposide treatment. Scale bar, 10 μm. D, quantification of nuclear versus non-nuclear distribution of ubiquitinated BRAT1. Nuclear volume was determined by DAPI staining. A significant decrease in the distribution of ubiquitinated BRAT1 was observed in Ndfip1−/− MEFs. Nuclei were stained with DAPI (blue). The values are the means ± S.E., unpaired t test, n = 20 each group. ***, p < 0.001. E, MTT cell survival assay shows reduced cell survival in Ndfip1−/− MEFs compared with wild-type MEFs following etoposide treatment. The values are the means ± S.E., two-way analysis of variance test, n = 24 each group. ****, p < 0.0001.
Ndfip1 Is Required for Nuclear Distribution of BRAT1 under Stress Conditions of Cells in Vitro and the Injured Brain in Vivo
The requirement for Ndfip1 to facilitate nuclear localization of ubiquitinated BRAT1 was also tested under stress conditions. In vitro, Flag-BRAT1 was transfected into MEFs, and its cellular distribution was examined following treatment with different concentrations of the chromosomal mutagen etoposide (Fig. 5C) (20). Immunostaining with anti-Flag antibodies revealed that the Flag-BRAT1 fusion protein was distributed in both nucleus and cytoplasm in wild-type MEFs regardless of etoposide concentration (Fig. 5C, Ndfip1+/+ panels). In contrast, the BRAT1 fusion protein was restricted only to the cytoplasm of Ndfip1−/− MEFs at all concentrations of etoposide used (Fig. 5C, Ndfip1−/− panels). Thus, during genotoxic stress, BRAT1 translocation to the nucleus appeared to require Ndfip1. Following on from this, we hypothesized that any reduction in nuclear translocation of BRAT1 during genotoxic stress would impact upon cell survival. To test this, we performed MTT cell survival assays following treatment of wild-type and Ndfip KO MEFs with 2 μm etoposide for 24 h. This showed a small but significant reduction (p < 0.0001) in the survival of Ndfip1 KO cells following etoposide treatment (Fig. 5E).
Ndfip1 was first discovered as a neuroprotective protein following traumatic brain injury (8). A number of mechanisms for Ndfip1-mediated neuroprotection have been proposed, including restricting cytoplasmic entry of toxic metals into neurons and promoting nuclear entry of PTEN, which is known for DNA repair (9, 13, 21). To test the hypothesis that Ndfip1-mediated ubiquitination of BRAT1 promotes its entry into the nucleus for initiating the DNA damage response, we examined the cellular distribution of BRAT1 in the cortex following brain injury (Fig. 6A). In the uninjured contralateral cortex, there was only basal level of Ndfip1 expression; this was coincident with mainly cytoplasmic BRAT1 (Fig. 6B, upper panels). In the injured ipsilateral cortex, there was marked Ndfip1 expression, which was accompanied by increased nuclear distribution of BRAT1 (Fig. 6B, bottom panels). Thus, there is a strong relationship between increased Ndfip1 expression in neurons following injury, together with increased nuclear localization of BRAT1.
FIGURE 6.
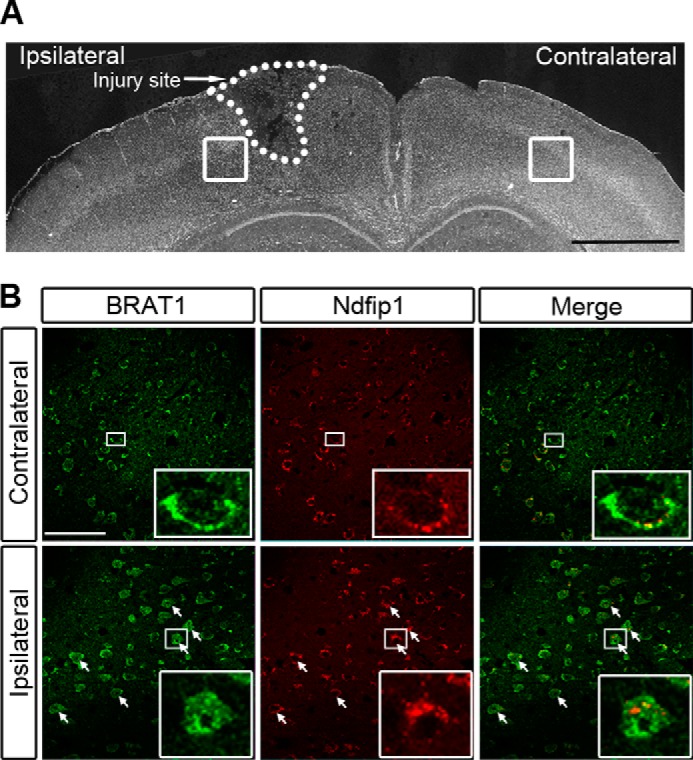
Ndfip1 is required for nuclear distribution of endogenous BRAT1 in brain neurons following traumatic injury. A, low power micrograph of a coronal section of a mouse brain showing TBI in the ipsilateral hemisphere and uninjured contralateral hemisphere. Boxed areas depict locations of higher power figures in B. B, confocal microscopy of cortex sections following immunocytochemical staining for Ndfip1 and BRAT1 24 h following TBI (ipsilateral hemisphere). The uninjured contralateral hemisphere provides an internal control, which shows cytoplasmic BRAT1 and a low abundance of Ndfip1 in neurons. High power views (insets) show cytoplasmic distribution of Ndfip1 and BRAT1. In the ipsilateral cortex, injured neurons (arrows) show increased Ndfip1 in the cytoplasm, and these neurons demonstrate increased abundance of BRAT1 in their nuclei (insets show high power views of one neuron). Scale bars, 1 mm (A) and 50 μm (B).
BRAT1 Is Required for Increasing the Abundance of the DNA Repair Protein pATM by Ndfip1
Previous studies have shown that a number of sensor proteins, including BRAT1, are involved in the activation the DNA repair protein ATM (6, 7). So far, we have demonstrated that driven by Ndfip1, ubiquitinated BRAT1 is trafficked into the cell nucleus. What is missing is a demonstrated link between BRAT1 and increasing abundance of Ndfip1 and pATM under DNA damage conditions. To pursue this, we studied the relative abundance of pATM and Ndfip1, by Western blot analysis, under different concentrations of etoposide treatment of the neuroblastoma cell line SH-SY5Y. The results showed a dose-related increase in the abundance of both Ndfip1 and pATM (Fig. 7A). In contrast, unphosphorylated ATM was unchanged, whereas the DNA damage marker protein pH2AX, which lies downstream of pATM (22), was also increased (Fig. 7A). Parallel analysis of this relationship was also performed in HEK293T cells engineered to express Flag-Ndfip1 by doxycycline (“Experimental Procedures”). Ndfip1 induction in HEK293T cells was performed for 18 h followed by etoposide treatment (0, 2, and 20 μm) for 2 h prior to Western blot analysis. In the absence of Ndfip1 induction, 2 and 20 μm of etoposide treatment elicited a mild increase in pATM (Fig. 7B, lanes 2 and 3). However, induction of Ndfip1 with etoposide treatment in these cells resulted in increased abundance of pATM (Fig. 7B, lanes 5 and 6). To test the requirement for BRAT1, RNA interference was performed by introducing BRAT1 shRNA in the presence of Ndfip1 and examining pATM. The results showed a reduction in pATM abundance (Fig. 7B, lanes 8 and 9), suggesting that BRAT1 is required for the co-up-regulation of pATM with Ndfip1 under DNA damage conditions. To view the above results in cultured cells, immunocytochemical analysis was performed using HEK293T cells with or without Ndfip1 induction (Fig. 7C). With Ndfip1 induction (with doxycycline), there was increased abundance of pATM with 2 μm etoposide treatment (5-fold increase, p < 0.001) (Fig. 7D), and this increase was accentuated with 20 μm etoposide treatment (4-fold difference, p < 0.001) (Fig. 7C, arrow). Importantly, knockdown of BRAT1 by RNA interference reversed the increase in pATM abundance in the presence of 20 μm etoposide (Fig. 7C, arrowhead). In summary, induction of DNA damage in two separate cell lines revealed that Ndfip1 increase was associated with elevation of the DNA repair protein, pATM, and this elevation required the presence of BRAT1.
FIGURE 7.

BRAT1 is required for increasing pATM by Ndfip1 under DNA damage conditions. A, treatment of SH-SY5Y neurons with increasing concentrations of etoposide (2 and 20 μm for 2 h) showed increased Ndfip1 and pATM in Western blots but unaltered levels of ATM and control proteins. IB, immunoblotting. B, induction of Flag-Ndfip1 using doxycycline shows increased Ndfip1 and pATM abundance in HEK293T cells treated with epotoside. DNA damage, revealed by the marker pH2AX (lanes 5 and 6), appears to be reduced. In contrast, without Flag-Ndfip1 induction, there is less pATM expression and more DNA damage as revealed by pH2AX (lanes 2 and 3). RNA interference of BRAT1 with BRAT1 shRNA showed reduced pATM and increased DNA damage marker pH2AX (lanes 8 and 9). C, immunocytochemical confirmation of results in B was obtained by confocal microscopy. This showed increasing levels of pATM in cells (arrow) treated with etoposide in the presence of Flag-Ndfip1 (+Dox) compared with cells without Ndfip1 (−Dox). However, pATM was reduced when induced cells were also treated with BRAT1 shRNA (arrowhead). D, quantitative analysis of results obtained in C show increased nuclear pATM at higher concentrations of etoposide following induction of Flag-Ndfip1. The pATM nuclear signal was determined as a percentage of nuclear volume (demarcated by DAPI staining). The values are means ± S.E., two-way analysis of variance test, n = 50 cells in each group. ***, p < 0.001.
DISCUSSION
Our previous studies have demonstrated a novel mechanism for increasing neuron survival in stress situations such as traumatic brain injury and cerebral ischemia (8, 9, 11). In those experiments, the principal pathway of neuroprotection was mediated by Ndfip1-dependent trafficking of the tumor suppressor PTEN into the nucleus. PTEN is the central inhibitor of the PI3K/pAkt signaling pathway (23), and PTEN loss leads to cancer by increasing cell proliferation and cell survival. During brain injury, nuclear sequestration of PTEN by Ndfip1 from its cytoplasmic location promotes pAkt generation by membrane phosphatidylinositol 3,4,5-triphosphate. Because postmitotic neurons do not proliferate, the response to increased pAkt does not lead to tumorigenesis but cell survival (9–11).
During cellular stress, a major cause of apoptosis is oxidative stress-induced DNA damage. Nuclear PTEN offers additional benefits against apoptosis by promoting DNA repair (24, 25) and protection against genotoxic stress (21). However, the present study offers further evidence that Ndfip1-mediated ubiquitination and nuclear trafficking of other proteins such as BRAT1 may also be involved in protection against DNA damage. BRAT1 has previously been shown to bind to and is required to maintain the phosphorylation state of ATM, a central participant in the DNA damage response (7). There is accumulating evidence that increasing BRAT1 is beneficial for cell survival, most likely through its regulation of ATM phosphorylation (7). Indeed, reduction of BRAT1 leads to reduced cell proliferation and tumorigenesity (16), mirroring the effects of reduced PI3K signaling and cell survival, although the extent of molecular cross-talk between BRAT1 and PI3K remains elusive. Nonetheless, there is gathering evidence that BRAT1 is a vital part of the armamentarium of the cell against apoptosis during the DNA damage response (19).
If the above hypothesis is correct, there should be a mechanism for BRAT1 protein trafficking into the cell nucleus during cell stress. Our results point to Ndfip1-mediated ubiquitination as the most likely driver of BRAT1 entry into the nucleus. Here, BRAT1 would most likely participate in activating cell survival mechanisms, possibly mediated by pATM and also BRCA1. BRAT1 is known to promote the longevity of pATM by hindering its dephosphorylation by the phosphatase PP2A (7). Thus, Ndfip1 up-regulation followed by ubiquitination of BRAT1 provides the mechanism for BRAT1 trafficking. Our loss of function experiments showed that in the absence of Ndfip1, BRAT1 failed to translocate to the cell nucleus. In summary, BRAT1 ubiquitination by the Ndfip1/Nedd4 system is a necessary prerequisite for combating DNA damage via pATM. Indeed, we showed a strong correlation in the up-regulation of Ndfip1 together with pATM following treatment with the genotoxic agent epotoside in two different cell lines, and in one of these lines, BRAT1 knockdown reversed pATM abundance. This suggests a direct association of Ndfip1 up-regulation via BRAT1 nuclear translocation to activate pATM-mediated DNA repair pathways.
Because we have previously demonstrated that Ndfip1 is pivotal for the nuclear transport of PTEN with survival benefits mediated by pAkt, it would suggest that Ndfip1-mediated ubiquitination of different proteins for nuclear transport is a multifaceted cell survival strategy. Ndfip1 acts both as adaptor and activator of Nedd4 family E3 ubiquitin ligases (26). However, not all E3 ligases are uniformly capable of participating in Ndfip1-mediated ubiquitination of target proteins. Instead, which Nedd4 family member is recruited appears to be target-dependent (10). In the case of PTEN during brain injury and stroke, we showed that Nedd4-2 is the preferred E3 ligase (11). To avoid metal poisoning, the E3 ligase for degrading the divalent metal transporter DMT1 has been shown to be Nedd4-2 (13). Under DNA damage conditions performed in the current study, the preferred E3 ligase for BRAT1 ubiquitination in vitro is Itch, although Nedd4-1 and Nedd4-2 can also participate to a certain extent. This diversification of ligase choice adds a further layer of molecular specificity during the recruitment of survival proteins under stress conditions. Additional studies would be required to determine the subcellular localization and cellular conditions that dictate ligase choice for BRAT1.
Finally, it is noteworthy that exome sequencing data from an Amish community have identified a homozygous truncating mutation (c.638_639insA) in BRAT1 as the cause of its failure to localize to the nucleus (18). This condition is associated with lethal neonatal rigidity and seizures, with pathological signs of neuron death and degenerative brain foci. Despite the mutation, we found that this protein was capable of binding to Ndfip1, although its failure to translocate to the nucleus is likely to arise from problems with ubiquitination of a truncated protein. More recent reports consistently associate BRAT1 mutations with neonatal epileptic encephalopathy (27), suggesting additional roles for BRAT1 during brain development.
In conclusion, the present study has identified Ndfip1/Nedd4 system as an important driver of BRAT1 ubiquitination and subsequent nuclear localization. Because the up-regulation of Ndfip1 is a feature of cells undergoing stress situations, including DNA damage, it is likely to function as an early sensor protein for mobilizing DNA repair during brain injury.
This work was supported by the National Health and Medical Research Council and the Victorian Government through the Operational Infrastructure Scheme.
L.-H. Low, J. Chow, Y, Li, C.-P. Goh, U. Putz, J. Silke, T. Ouchi, J. Howitt, and S.-S. Tan, unpublished data.
- MEF
- mouse embryonic fibroblast
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BiFC
- bimolecular fluorescent complementation
- VN
- plasmid created by BiFC of Venus using N-terminal reporter fragment
- VC
- plasmid created by BiFC of Venus using C-terminal reporter fragment
- PB
- phosphate buffer
- IP
- immunoprecipitation.
REFERENCES
- 1. Loane D. J., Faden A. I. (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewén A., Matz P., Chan P. H. (2000) Free radical pathways in CNS injury. J. Neurotrauma 17, 871–890 [DOI] [PubMed] [Google Scholar]
- 3. Hall E. D., Andrus P. K., Yonkers P. A. (1993) Brain hydroxyl radical generation in acute experimental head injury. J. Neurochem. 60, 588–594 [DOI] [PubMed] [Google Scholar]
- 4. Ditch S., Paull T. T. (2012) The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem. Sci. 37, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. You Z., Shi L. Z., Zhu Q., Wu P., Zhang Y. W., Basilio A., Tonnu N., Verma I. M., Berns M. W., Hunter T. (2009) CtIP links DNA double-strand break sensing to resection. Mol. Cell 36, 954–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shiloh Y., Ziv Y. (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 [PubMed] [Google Scholar]
- 7. Aglipay J. A., Martin S. A., Tawara H., Lee S. W., Ouchi T. (2006) ATM activation by ionizing radiation requires BRCA1-associated BAAT1. J. Biol. Chem. 281, 9710–9718 [DOI] [PubMed] [Google Scholar]
- 8. Sang Q., Kim M. H., Kumar S., Bye N., Morganti-Kossman M. C., Gunnersen J., Fuller S., Howitt J., Hyde L., Beissbarth T., Scott H. S., Silke J., Tan S. S. (2006) Nedd4-WW domain-binding protein 5 (Ndfip1) is associated with neuronal survival after acute cortical brain injury. J. Neurosci. 26, 7234–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howitt J., Lackovic J., Low L. H., Naguib A., Macintyre A., Goh C. P., Callaway J. K., Hammond V., Thomas T., Dixon M., Putz U., Silke J., Bartlett P., Yang B., Kumar S., Trotman L. C., Tan S. S. (2012) Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J. Cell Biol. 196, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lackovic J., Howitt J., Callaway J. K., Silke J., Bartlett P., Tan S. S. (2012) Differential regulation of Nedd4 ubiquitin ligases and their adaptor protein Ndfip1 in a rat model of ischemic stroke. Exp. Neurol. 235, 326–335 [DOI] [PubMed] [Google Scholar]
- 11. Goh C. P., Putz U., Howitt J., Low L. H., Gunnersen J., Bye N., Morganti-Kossmann C., Tan S. S. (2014) Nuclear trafficking of Pten after brain injury leads to neuron survival not death. Exp. Neurol. 252, 37–46 [DOI] [PubMed] [Google Scholar]
- 12. Putz U., Howitt J., Doan A., Goh C. P., Low L. H., Silke J., Tan S. S. (2012) The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 5, ra70. [DOI] [PubMed] [Google Scholar]
- 13. Howitt J., Putz U., Lackovic J., Doan A., Dorstyn L., Cheng H., Yang B., Chan-Ling T., Silke J., Kumar S., Tan S. S. (2009) Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 15489–15494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y., Low L. H., Putz U., Goh C. P., Tan S. S., Howitt J. (2014) Rab5 and Ndfip1 are involved in Pten ubiquitination and nuclear trafficking. Traffic 15, 749–761 [DOI] [PubMed] [Google Scholar]
- 15. So E. Y., Ouchi T. (2011) Functional interaction of BRCA1/ATM-associated BAAT1 with the DNA-PK catalytic subunit. Exp. Ther. Med. 2, 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. So E. Y., Ouchi T. (2014) BRAT1 deficiency causes increased glucose metabolism and mitochondrial malfunction. BMC Cancer 14, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Putz U., Howitt J., Lackovic J., Foot N., Kumar S., Silke J., Tan S. S. (2008) Nedd4 family-interacting protein 1 (Ndfip1) is required for the exosomal secretion of Nedd4 family proteins. J. Biol. Chem. 283, 32621–32627 [DOI] [PubMed] [Google Scholar]
- 18. Puffenberger E. G., Jinks R. N., Sougnez C., Cibulskis K., Willert R. A., Achilly N. P., Cassidy R. P., Fiorentini C. J., Heiken K. F., Lawrence J. J., Mahoney M. H., Miller C. J., Nair D. T., Politi K. A., Worcester K. N., Setton R. A., Dipiazza R., Sherman E. A., Eastman J. T., Francklyn C., Robey-Bond S., Rider N. L., Gabriel S., Morton D. H., Strauss K. A. (2012) Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One 7, e28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ouchi M., Ouchi T. (2010) Regulation of ATM/DNA-PKcs phosphorylation by BRCA1-associated BAAT1. Genes Cancer 1, 1211–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizumoto K., Rothman R. J., Farber J. L. (1994) Programmed cell death (apoptosis) of mouse fibroblasts is induced by the topoisomerase II inhibitor etoposide. Mol. Pharmacol. 46, 890–895 [PubMed] [Google Scholar]
- 21. Bassi C., Ho J., Srikumar T., Dowling R. J., Gorrini C., Miller S. J., Mak T. W., Neel B. G., Raught B., Stambolic V. (2013) Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuo L. J., Yang L. X. (2008) Gamma-H2AX: a novel biomarker for DNA double-strand breaks. In Vivo 22, 305–309 [PubMed] [Google Scholar]
- 23. Chalhoub N., Baker S. J. (2009) PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 4, 127–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang C. J., Mulholland D. J., Valamehr B., Mosessian S., Sellers W. R., Wu H. (2008) PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell Biol. 28, 3281–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song M. S., Carracedo A., Salmena L., Song S. J., Egia A., Malumbres M., Pandolfi P. P. (2011) Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mund T., Pelham H. R. (2009) Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 10, 501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saitsu H., Yamashita S., Tanaka Y., Tsurusaki Y., Nakashima M., Miyake N., Matsumoto N. (2014) Compound heterozygous BRAT1 mutations cause familial Ohtahara syndrome with hypertonia and microcephaly. J. Hum. Genet. 59, 687–690 [DOI] [PubMed] [Google Scholar]