

Background: The regulatory mechanism of PHD2 function and expression under inflammatory conditions in the nucleus pulposus is unknown.
Results: PHD2 controls TNF-α effects by positively regulating NF-κB signaling, whereas NF-κB mediates cytokine-dependent PHD2 expression.
Conclusion: PHD2 and NF-κB forms a functional circuit that promotes the catabolic effects of TNF-α in the intervertebral disc.
Significance: PHD2 may play an important role in pathogenesis of disc disease.
Keywords: Cytokine Action, Hypoxia-inducible Factor (HIF), NF-kappa B (NF-KB), Nucleus Pulposus, Tumor Necrosis Factor (TNF), Intervertebral Disc, Prolyl Hydroxylase
Abstract
Prolyl-4-hydroxylase (PHD) proteins are key in sensing tissue hypoxia. In nucleus pulposus (NP) cells, our previous work demonstrated that PHD isoforms have a differential contribution in controlling hypoxia-inducible factor (HIF)-α degradation and activity. Recently we have shown that a regulatory relationship exists between PHD3 and inflammatory cytokines in NP cells. With respect to PHD2, the most abundant PHD isoform in NP cells, very little is known concerning its function and regulation under inflammatory conditions that characterize intervertebral disc degeneration. Here, we show that PHD2 is a potent regulator of the catabolic activities of TNF-α; silencing of PHD2 significantly decreased TNF-α-induced expression of catabolic markers including SDC4, MMP-3, MMP-13, and ADAMTS5, as well as several inflammatory cytokines and chemokines, while partially restoring aggrecan and collagen II expression. Use of NF-κB reporters with ShPHD2, SiHIF-1α, as well as p65−/−, PHD2−/−, and PHD3−/− cells, shows that PHD2 serves as a co-activator of NF-κB/p65 signaling in HIF-1-independent fashion. Immunoprecipitation of endogenous and exogenously expressed tagged proteins, as well as fluorescence microscopy, indicates that following TNF-α treatment, PHD2 interacts and co-localizes with p65. Conversely, loss of function experiments using lentivirally delivered Sh-p65, Sh-IKKβ, and NF-κB inhibitor confirmed that cytokine-dependent PHD2 expression in NP cells requires NF-κB signaling. These findings clearly demonstrate that PHD2 forms a regulatory circuit with TNF-α via NF-κB and thereby plays an important role in enhancing activity of this cytokine. We propose that during disc degeneration PHD2 may offer a therapeutic target to mitigate the deleterious actions of TNF-α, a key proinflammatory cytokine.
Introduction
The intervertebral disc is a complex tissue that allows a range of motions between adjacent vertebrae and accommodates high biomechanical forces applied to the spine. Commonly, the disc undergoes degeneration, a condition frequently linked to neck and back pain and requiring pharmacological or surgical intervention. Degenerative disc disease is characterized by elevated levels of proinflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-8, as well as down-regulation of expression of the important matrix molecules aggrecan and collagen II (1–4). TNF-α and IL-1β stimulate production of NGF, BDNF, and VEGF, molecules associated with nerve ingrowth and pain as well as angiogenesis (5). Recent work has shown that both TNF-α and IL-1β promote NF-κB signaling in the NP and control expression of syndecan-4 (SDC4),3 a heparan sulfate proteoglycan involved in ADAMTS5/aggrecanase-2 activation, as well as MMP-3 expression (6, 7).
One clue to the pathogenesis of disc disease is the limited vascular supply to the NP, the aggrecan-rich gelatinous tissue that occupies the central compartment of the disc. Thus, blood vessels arising in the vertebral body penetrate the superficial zone of the endplate cartilages; however, none infiltrate the NP. With respect to the ligamentous annulus fibrosus, this tissue is avascular except for small discrete capillary beds in the dorsal and ventral surfaces; in no case does the annular vasculature enter the NP (8–11). Thus, the NP is completely devoid of any vasculature and is hypoxic.
We have previously shown that members of the 2-oxoglutarate/Fe2+-dependent dioxygenase superfamily, PHD1–3, are expressed in the hypoxic NP in either hypoxia-inducible factor 1 (HIF-1)- or HIF-2-dependent fashion, with PHD2 being the most abundant isoform (12, 13). These enzymes hydroxylate-specific proline residues in the oxygen-dependent degradation domain of HIF-α subunits and target the protein for Von Hippel-Lindau-dependent proteasomal degradation. It is noteworthy that our work has shown that although PHD2 partially controls the oxygen-dependent degradation of HIF-1α, PHD3 plays no role in either HIF-1α or HIF-2α degradation; instead, in hypoxia it controls HIF-1α transcriptional activity (14). Very recently we found that a regulatory relationship existed between inflammatory cytokines TNF-α and IL-1β and PHD3 in NP cells, where PHD3 positively regulates the catabolic actions of these cytokines. It is also significant that the role and contribution of PHDs to the inflammatory response is somewhat controversial, playing both pro- and anti-inflammatory roles. Supporting a proinflammatory function, it has been shown that inhibition of PHDs through activation of HIF-1 blocked expression of cytokines, chemokines, and inflammation-associated molecules (15, 16). On the other hand, in macrophages, it was reported that independent of HIFs, the pan hydroxylase inhibitor dimethyloxalylglycine, by blocking NF-κB pathway, significantly suppressed LPS-induced expression of proinflammatory genes (17). In contrast to these reports, some studies indicate an anti-inflammatory role of individual PHD isoforms through mechanisms that involve blocking the activity of components of IKKα/β/γ complex in hydroxylase-dependent (18) or -independent fashion (19), implying that the contribution of PHDs and the mechanism of their action in modulating inflammatory response is cell type-specific (20). Relevant to NP cells, despite being the most abundant PHD isoform, it is not known whether there is cross-talk between PHD2 and components of the inflammatory pathways.
The major objective of the study was to examine the relationship between PHD2 and the inflammatory cytokines TNF-α and IL-1β in cells of the NP. We show for the first time that by positively controlling NF-κB signaling activity, PHD2 promotes the catabolic effects of the inflammatory cytokines on NP cells. Our findings also show that TNF-α and IL-1β maintain PHD2 expression in a NF-κB-dependent fashion, indicating the existence of a regulatory circuit between PHD2 and cytokines through NF-κB. These results suggest that PHD2 may play an important role in the pathogenesis of disc disease and may offer a therapeutic target to treat this debilitating and painful degenerative condition.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
Plasmids were kindly provided by Dr. Kenneth Thirstrup of Denmark (lentiviral ShPHD2 co-expressing enhanced GFP) (21), Dr. Andree Yeramian of the University of Lleida (lentiviral Shp65, ShIKKβ co-expressing YFP) (22), Mark B. Taubman of the University of Rochester (NF-κB luciferase reporter) (23), Dr. Nianli Sang of Drexel University (pcDNA3.1-PHD2 and -PHD3] (5), and Dr. Jitendra Gautam of the University of Virginia (SDC4 reporter, wild type, and NF-κB mutant) (25). psPAX2 (catalog no. 12260) and pMD2G (catalog no. 12259) developed by Dr. Didier Trono, p65 (catalog no. 20012) developed by Dr. Inder Verma, and HRE-Luc (catalog no. 26731) developed by Navdeep Chandel were obtained from Addgene. As an internal transfection control, vector pRL-TK (Promega) containing Renilla reniformis luciferase gene was used. HEK293T cells were provided by Dr. Aviva Symes. p65 null and wild type MEFs were a kind gift from Dr. Denis Guttridge of Ohio State University, Columbus (26). PHD2f/f; CreER(+) and PHD2f/f; Cre-ER(+); PHD3−/− LT K1 MEFs were a kind gift from Dr. William G. Kaelin of Harvard Medical School (27).
Isolation of NP Cells and Cell Treatments
NP cells were isolated from intervertebral discs of skeletally mature Wistar rats using a method reported earlier by Risbud et al. (28). NP cells were maintained in DMEM and 10% FBS supplemented with antibiotics. To investigate effect of cytokines, cells were treated with IL-1β (10 ng/ml) or TNF-α (50 ng/ml) (Peprotech) for 1–24 h. To delete PHD2 through activation of Cre-ER, 4-hydroxytamoxifen (Sigma-Aldrich) was added to the medium at a final concentration of 200 nm for 72 h.
Transfections and Dual Luciferase Assay
Cells were transferred to 48-well plates at a density of 2 × 104 cells/well 1 day before transfection. For each transfection, plasmids were premixed with the transfection reagent (Lipofectamine 2000; Invitrogen). To measure the effect of cytokine treatment on NRE and SDC4 reporter activity, 48 h after transfection, cells in some wells were treated with TNF-α or IL-1β for 24 h. The next day, the cells were harvested, and a Dual-LuciferaseTM reporter assay system (Promega) was used for sequential measurements of firefly and Renilla luciferase activities. Quantification of luciferase activities and calculation of relative ratios were carried out using a luminometer (TD-20/20; Turner Designs).
Real Time RT-PCR Analysis
Total RNA was extracted from rat NP cells using RNAeasy mini columns (Qiagen). Before elution from the column, RNA was treated with RNase-free DNase I (Qiagen). The purified, DNA-free RNA was converted to cDNA using EcoDryTM premix (Clontech). Template cDNA and gene-specific primers were added to the SYBR Green master mixture (Applied Biosystems), and mRNA expression was quantified using the StepOnePlus real time PCR System (Applied Biosystems). Hprt1 and β-actin were used to normalize gene expression for rat and human samples, respectively. Melting curves were analyzed to verify the specificity of the RT-PCR and the absence of primer dimer formation. Each sample was analyzed in duplicate and included a template-free control. All primers used were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA).
Protein Extraction, Immunoprecipitation, and Western Blotting
Cells were placed on ice immediately and washed with ice-cold Hanks' balanced salt solution. All the wash buffers and final resuspension buffer included 1× protease inhibitor mixture (Roche), NaF (5 mm), and Na3VO4 (200 μm). Nuclear and cytosolic proteins were prepared using the CellLytic NuCLEAR extraction kit (Sigma). Immunoprecipitation was performed using protein A/G PLUS-agarose beads (Santa Cruz) following standard protocol. Before protein extraction, the cells were treated with 2 mm dithiobis[succinimidylpropionate] (Pierce), an amine-reactive cross-linker, for 30 min at room temperature. Proteins were resolved on 8–12% SDS-polyacrylamide gels and transferred by electroblotting to PVDF membranes (Bio-Rad). The membranes were blocked with 5% nonfat dry milk in TBST (50 mm Tris, pH 7.6, 150 mm NaCl, 0.1% Tween 20) and incubated overnight at 4 °C in 5% nonfat dry milk in TBST with the with the anti-PHD2 (catalog no. 4835), anti-p65 (catalog no. 6956), anti-IKKβ (catalog no. 2678), anti-Cox2 (catalog no. 1228), anti-YFP/GFP (catalog no. 2956), anti-FLAG (catalog no. 8146), anti-Lamin A/C (catalog no. 2032) (1:1000, Cell Signaling), anti-PHD3 (catalog no. NB100–139A2, 1:1000) and anti-GAPDH (catalog no. NB300–221SS, 1:3000, Novus), anti-SDC4 (catalog no. AB24511, 1:750), and anti-MMP3 (catalog no. AB52915, 1:1000, Abcam) recognizing processed form of MMP-3. Immunolabeling was detected using the ECL reagent (Amersham Biosciences). Relative expression levels were determined by quantitative densitometric analysis (Image Quant TL, GE Bioscience).
Immunofluorescence Microscopy
Cells were plated in flat bottom 96-well plates (5 × 103/well) and treated with TNF-α or IL-1β for 1–4 h. After incubation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS for 10 min, blocked with PBS containing 5% FBS, and incubated with antibodies against PHD2 (1:200, Novus) and p65 (1:200, Cell Signaling) at 4 °C overnight. As a negative control, cells were reacted with isotype IgG under similar conditions. After washing, the cells were incubated with Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 594-conjugated anti-rabbit secondary antibodies (Invitrogen) at a dilution of 1:200 for 1 h at room temperature. Cells were imaged using a fluorescence microscope (Nikon Eclipse E600).
Lentiviral Production and Transduction
HEK 293T cells were seeded in 10-cm plates (3 × 106 cells/plate) in DMEM with 10% heat-inactivated FBS 1 day before transfection. Cells were transfected with 9 μg of Shp65 or ShIKKβ or ShPHD2 plasmids along with 6 μg of psPAX2 and 3 μg of pMD2G using calcium phosphate transfection kit (CalPhos, Clontech). After 16 h, transfection media were removed and replaced with DMEM with 10% heat-inactivated FBS. Lentiviral particles were harvested at 48 and 60 h post-transfection. NP cells in 10-cm plates were transduced with 8 ml of media containing viral particles along with 6 μg/ml Polybrene. After 24 h, viral medium was removed and replaced with DMEM with 10% FBS. 5 days following viral transduction, cells were cultured under serum-free conditions with or without TNF-α for 24 h prior to protein, RNA, and conditioned medium collection. Transduction efficiency of 80–90% was achieved as determined from the number of GFP- or YFP-positive cells.
Statistical Analysis
All measurements were performed in triplicate, and data are presented as the means ± S.D. Differences between groups were analyzed by the Student's t test and one-way analysis of variance (*, p < 0.05).
RESULTS
PHD2 Regulates Catabolic Effects of TNF-α on NP Cells
To determine whether PHD2 plays a role in controlling catabolic effects of TNF-α on NP cells, we first transduced rat disc cells with a lentivirus expressing ShPHD2. Assessment of GFP-positive cells confirmed that the transduction efficiency was high: ∼80% (Fig. 1A). Real time RT-PCR (Fig. 1B) and Western blot analysis (Fig. 1, I and J) indicate a robust suppression of PHD2 expression in ShPHD2 group compared with cells transduced with control ShRNA. Note that TNF-α-dependent induction in expression of PHD3 (Fig. 1C), SDC4 (Fig. 1D), MMP13 (Fig. 1E), and ADAMTS5 (Fig. 1F), catabolic marker genes that are hallmark of disc degeneration, is significantly reduced in PHD2 silenced cells. Furthermore, silencing of PHD2 significantly restored the TNF-α-mediated reduction in aggrecan (Fig. 1G) and collagen II (Fig. 1H), which are critical matrix genes in NP cells. Importantly, none of these genes showed a significant change in baseline expression in cells transduced with ShPHD2 in the absence of TNF-α (data not shown). Western blot analysis of cellular protein fraction confirmed that the TNF-α-mediated increase in levels of SDC4, COX2, and PHD3 is significantly lower in PHD2 silenced cells (Fig. 1, I and J). We also analyzed the conditioned medium to examine the levels of secreted proteases. Western blot analysis using an antibody that recognizes the processed/active form of MMP-3 clearly showed that the level of MMP3 protease is significantly down-regulated in the ShPHD2 group (Fig. 1, I and J). To summarize these findings, PHD2 controls the expression of many of the pro-catabolic and inflammatory genes that are known to be involved with the degenerative cascade and matrix breakdown.
FIGURE 1.

PHD2 positively regulates catabolic effects of TNF-α on NP cells. A, immunofluorescence analysis of NP cells transduced with lentivirus co-expressing Sh-PHD2 (Sh-PHD2) and GFP indicates high transduction efficiency. Original magnification, ×20. B, real time RT-PCR analysis confirmed that PHD2 mRNA expression is significantly silenced in TNF-α-treated NP cells transduced with Sh-PHD2. C–F, TNF-α-induced mRNA expression of PHD3 (C), SDC4 (D), MMP13 (E), and ADAMTS5 (F) is significantly suppressed in NP cells transduced with Sh-PHD2. G and H, TNF-α-dependent suppression of aggrecan (G) and collagen II (H) mRNA expression is almost completely restored in NP cells transduced with Sh-PHD2. I, Western blot analysis of NP cells transduced by Sh-control or Sh-PHD2 with or without TNF-α treatment. The induction of COX2, PHD3, and SDC4 in cellular fraction and secreted MMP3 in conditioned medium (C.M.) is abolished in PHD2-silenced cells. J, densitometric analysis of multiple blots from the experiment described in I above. The data are represented as means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.05.
Knockdown of PHD2 Inhibits TNF-α-mediated Inflammatory Cytokine and Chemokine Production by NP Cells
To delineate the broader effects of PHD2 silencing on TNF-α-dependent mediators of the inflammatory response, we analyzed the conditioned medium of rat NP cells transduced with lentivirus expressing either a control-ShRNA or ShPhD2 using a cytokine/chemokine protein array. As expected, following TNF-α treatment, there is marked increase in expression of inflammatory molecules especially IL-1α, IL-6, IFN-γ, MCP-1, MIP-3α/CCL20, and ICAM-1 (Fig. 2, A and B). Importantly, knockdown of PHD2 results in a significant down-regulation of 19 cytokines/chemokines and molecules associated with inflammatory cascade, including those that are elevated by TNF-α in control-ShRNA group (see Fig. 2B for detailed list). The results of this study further implicate PHD2 in the expression of most of the major proinflammatory cytokines, as well as chemokines implicated in the pathogenesis of disc disease.
FIGURE 2.

PHD2 controls TNF-α-mediated expression of inflammatory cytokines, chemokines, and inflammation-related molecules in NP cell. A, Conditioned medium was collected following TNF-α treatment of NP cells transduced with either lentivirus expressing Sh-PHD2 or Sh-control and analyzed by inflammation pathway-focused protein array. Although TNF-α treatment induces levels of several molecules including IFN-γ, IL-1α, MIP-3α, and MMP-8, silencing of PHD2 results in their suppression. B, densitometric analysis shows significant down-regulation in levels of molecules associated with inflammatory response following PHD2 silencing in presence of TNF-α. The data are represented as means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.1.
PHD2 Positively Controls Activity of Cytokine-dependent NF-κB Signaling in NP Cells Independent of HIF-1
Because PHD2 controlled expression of catabolic marker genes and inflammatory cytokines/chemokine, we determined whether regulation is NF-κB-dependent. Thus, we performed PHD2 loss and gain of function experiments employing a prototypic NF-κB-responsive reporter (NRE-luc), as well as SDC4 reporter (SDC4-WT) shown earlier to be regulated by TNF-α and IL-1β in a NF-κB-dependent manner (29) (Fig. 3A). As a control, a reporter harboring NF-κB binding site mutation was utilized (SDC4-MT). The basal activities of NRE (Fig. 3B) and WT-SDC4 (Fig. 3C) are significantly suppressed when co-transfected with increasing concentrations of Sh-PHD2. Silencing of PHD2 also suppressed the TNF-α and IL-1β-dependent increase in activities of both reporters (Fig. 3, D–G). However, the loss of PHD2 function failed to suppress the basal (Fig. 3C) and cytokine-induced activity of SDC4-MT reporter harboring NF-κB binding site mutation (Fig. 3, F and G), suggesting that PHD2 may exert its effects through NF-κB. Furthermore, because silencing of PHD2 is expected to increase HIF-1α levels, a known modulator of NF-κB signaling (30), we investigated whether PHD2-mediated NF-κB regulation is HIF-1-dependent. Notably, similar to inhibition of PHD2 alone, co-silencing of HIF-1α and PHD2 also suppressed TNF-α-dependent induction in NRE reporter activity (Fig. 3H). In contrast to the suppression of PHD2, overexpression of PHD2 did not change the basal activities of NRE and SDC4 reporter (Fig. 4A) nor synergize the inductive effect of TNF-α (Fig. 4B) or IL-1β (Fig. 4C). These findings implicate for the first time that in disc cells, PHD2 exerts its proinflammatory function through regulation of the NF-κB signaling pathway and that this regulation was independent of HIF-1.
FIGURE 3.

PHD2 positively controls TNF-α- and IL-1β-dependent activity of NF-κB-responsive reporters in NP cells. A, schematic of NRE and SDC4 luciferase reporters used for transfections. MT, mutant κB. B and C, the activity of NRE and WT-SDC4 is significantly suppressed when co-transfected with increasing concentration of Sh-PHD2. +, 50 ng; ++, 100 ng; +++, 200 ng. PHD2 silencing has no effect on the activity of MT-SDC4 reporter. D and E, TNF-α- and IL-β-dependent induction of NRE reporter activity is significantly suppressed when PHD2 is silenced. +, 75 ng; ++, 150 ng. F and G, TNF-α- and IL-β-dependent induction of SDC4-WT reporter activity is significantly suppressed when PHD2 was silenced. +, 75 ng; ++, 150 ng. The activity of SDC4-MT reporter did not respond to cytokine treatment and to PHD2 silencing. H, PHD2 effect on NF-κB activity is HIF-1-independent. TNF-α-dependent induction of NRE reporter activity is significantly suppressed when either ShPHD2 alone (100 ng) or SiHIF-1α and ShPHD2 (100 ng each) are co-transfected. The data represent means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.05. ns, not significant.
FIGURE 4.
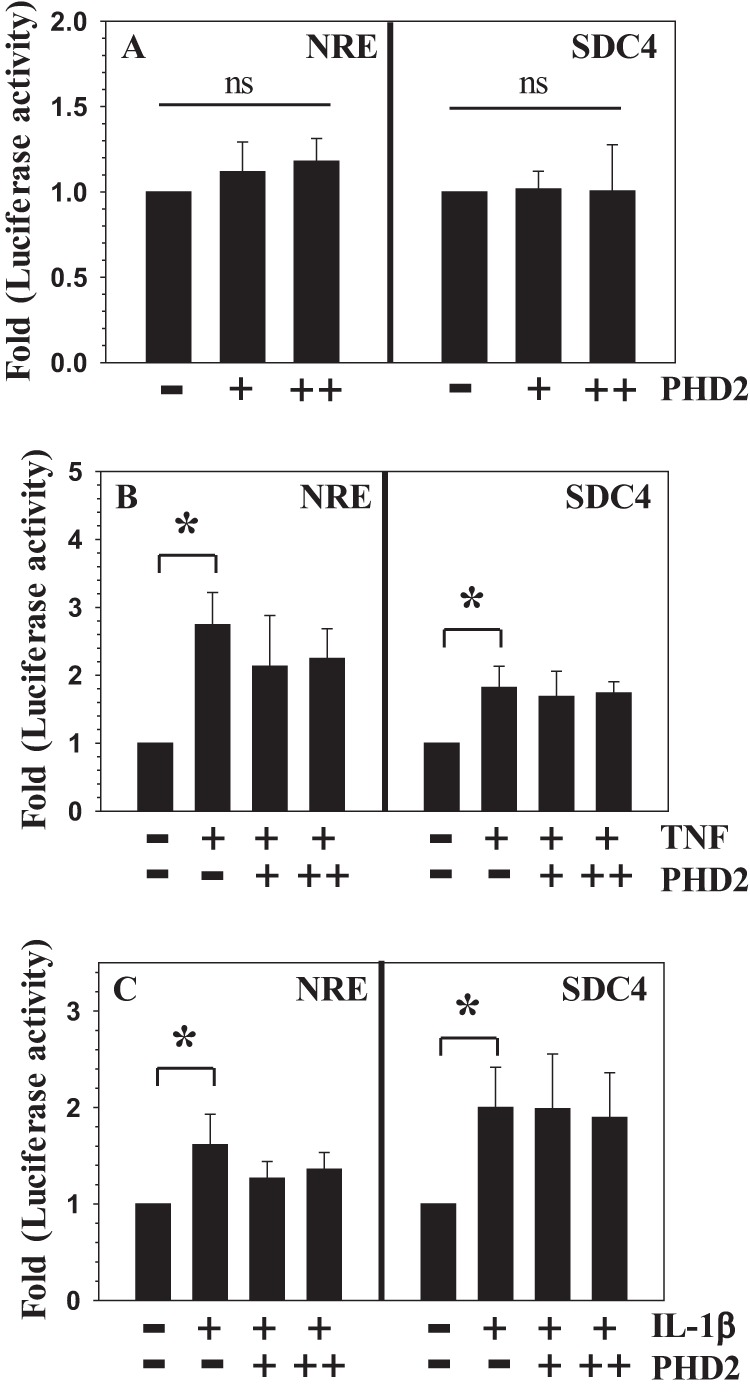
PHD2 overexpression does not up-regulate the activity of NF-κB-responsive reporters in NP cells. A, the basal activity of NRE and SDC4 reporters in NP cells is not significantly affected by transfection with an increasing dose of PHD2. +, 75 ng; ++, 150 ng. B and C, overexpression of PHD2 did not enhance the TNF-α-dependent (B) or IL-1β-dependent (C) activation of NRE and SDC4. The data represent means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.05. ns, not significant.
PHD2 Controls p65 Activity in NP Cells
We explored the mechanism by which PHD2 controlled the activity of the NF-κB signaling pathway. For this purpose, we investigated whether p65/RelA transcriptional activity is controlled by PHD2. Fig. 5 (A and B) shows that suppression of PHD2 in NP cells significantly decreases the inductive effect of exogenously added p65 on NRE and SDC4 reporter activities, respectively. However, co-transfection of PHD2 with p65 did not cause a synergistic increase in NRE reporter activity when compared with p65 alone (Fig. 5C). To determine whether high endogenous levels of PHD2 in NP cells caused p65 insensitivity toward exogenously expressed PHD2, we stably silenced PHD2 and performed a classical rescue experiment. Notably, in PHD2 silenced NP cells, p65 activity was further enhanced by co-addition of PHD2 (Fig. 5D). Our previous studies have shown that MEFs mimic many aspects of p65 transcriptional response in NP cells (29, 31). We therefore used MEFs derived from p65−/− and PHD2f/f CreER(+); PHD3−/− mice to further validate the role of PHD2 in p65 transactivation. In p65−/− (Fig. 5E) and PHD2−/−;PHD3−/− (Fig. 5F) cells, both PHD2 and PHD3 enhance the inductive effect of p65 on NRE-luc activity. Importantly, compared with PHD2 or PHD3 alone, co-transfection of PHD2 and PHD3 results in synergistic activation of NRE reporter (Fig. 5, E and F). Again, PHD2 or PHD3 overexpression fails to enhance the basal activity of NRE in PHD2−/−, PHD3−/−, and PHD2−/−;PHD3−/−, as well as p65−/− cells (Fig. 5G). To summarize these findings, results of this study so far point to the importance of the p65 component of the NF-κB signaling pathway in mediating the proinflammatory effects of PHD2.
FIGURE 5.

PHD2 controls p65 transcriptional activity in NP cells. A and B, silencing of PHD2 using increasing dose of Sh-PHD2 (50–150 ng) resulted in suppression of p65-dependent induction in both NRE (A) and SDC4 (B) reporter activities. C, co-transfection of p65 (15 ng) with PHD2 (+; 75 ng, ++; 150 ng) showed no additive effect on NRE reporter activity when compared with p65 alone. D, in PHD2 stably silenced NP cells, PHD2 addition resulted in synergistic increase in p65 dependent increase in NRE reporter activity when compared with p65 alone. +, 75 ng; ++, 150 ng. E and F, in p65 null (E) and PHD2−/−PHD3−/− MEFs (F), overexpression of either PHD2 or PHD3 (50 ng each) increases p65-dependent induction in NRE reporter activity. Co-transfection of PHD2 and PHD3 results in synergistic activation of NRE reporter. G, overexpression of PHD2 or PHD3 (50 ng each) does not affect baseline NRE reporter activity in PHD2−/− or PHD3−/− or PHD2−/−;PHD3−/− or p65−/− cells. The data are represented as means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.05. ns, not significant.
PHD2 Interacts with and Co-localizes with p65 in NP Cells
We used immunoprecipitation-Western blot to determine whether PHD2 interacts with p65 in NP cells. Pulldown of endogenously expressed PHD2 in rat NP cells results in co-precipitation of p65; the amount of p65 associated with PHD2 remained unaffected by TNF-α treatment. Note, when an isotype IgG is used in place of the PHD2 antibody, p65 is not co-precipitated (Fig. 6A). Notably, although our previous study has shown an association of PHD3 with p65 in NP cells (31), we were unable to pull down PHD3 using anti-PHD2 antibody in NP cells, implying that both PHDs may form distinct complexes with p65. To further confirm interaction of PHDs with p65, we transfected HEK293T cells with constructs expressing FLAG-p65 along with YFP-PHD2 and untagged PHD3. Immunoprecipitation results clearly show that pulldown using anti-FLAG antibody results in co-precipitation of PHD2-YFP. Similarly, Western blot using anti-PHD3 showed association of PHD3 with FLAG-p65 (Fig. 6B). These results suggest that both PHD2 and PHD3 can interact with p65.
FIGURE 6.

PHD2 interacts with p65 in NP cells. A, immunoprecipitation (IP) of PHD2 from both TNF-α-treated and untreated (CTR) NP cells followed by Western blot analysis using p65 antibody showed association between PHD2 and p65. When an isotype IgG was used in place of the PHD2 antibody, p65 was not co-precipitated. Input blot (10% input) was also performed to ensure expression of protein in cell lysate used for immunoprecipitation. H.C., heavy chain. B, total cell lysates from HEK293T transfected with the indicated plasmids were immunoprecipitated with anti-FLAG or mouse IgG. FLAG-p65 interacted with both YFP-PHD2 and PHD3 (endogenous and exogenously expressed) in HEK2943T cells. C, fluorescence microscopy was used to examine localization of PHD2 and p65 following 1 and 4 h of TNF-α treatment of NP cells. Cytokine treatment promotes nuclear localization of p65. The merged image (yellow signal) indicates co-localization of PHD2 and p65 in treated cells. Scale bars, 50 μm. D, Western blot analysis of cellular localization of PHD2 and p65 in NP cells treated with TNF-α for 4–24 h. Unlike p65, which showed increased localization into nuclear fraction, PHD2 expression remained unaffected in both nuclear and cytoplasmic compartments by cytokine treatment. Lamin A/C and GAPDH was used as loading control for nuclear and cytoplasmic fractions, respectively.
To delineate the cellular localization of the PHD-p65 complex, fluorescence microscopy was performed. Fig. 6C shows that PHD2 is present in both the nuclear and the cytoplasm compartment of NP cells under basal conditions. Following TNF-α treatment, there is an increase in p65 translocation to cell nucleus at both 1 and 4 h; importantly, there is co-localization of PHD2 with p65 under these conditions (Fig. 6C). Using Western blot analysis, we evaluated the expression of PHD2 and p65 in both the nuclear and cytoplasmic fractions of NP cells treated with TNF-α for up to 24 h. In agreement with the microscopic studies, PHD2 is present in both cellular compartments. Moreover, whereas p65 levels in the nuclear fraction showed increases at all the time points, expression levels of PHD2 remained more or less unaffected by TNF-α treatment (Fig. 6D). Based on these findings, it can be deduced that the mechanism by which PHD2 promotes the expression of proinflammatory cytokines like TNF-α is by interacting with the p65 protein component of NF-κB signaling pathway.
Cytokine-dependent PHD2 Expression in NP Cells Requires NF-κB Signaling
Although it is clear that PHD2 promoted the proinflammatory function of cytokines, it was not known whether cytokines concerned with disc degeneration regulated PHD2 expression in NP cells. To investigate this relationship, NP cells were treated with TNF-α and IL-1β and PHD2 expression analyzed. We found that PHD2 protein level remain unaffected by cytokine treatment (Fig. 7, A and B). To investigate whether maintenance of PHD2 expression involved components of the NF-κB signaling pathway, we first treated NP cells with SM7368, an inhibitor of NF-κB signaling. Western blot analysis showed that inhibitor treatment results in a decrease in PHD2 protein levels (Fig. 7C). To further explore this relationship, we transduced human NP cells with lentivirus co-expressing YFP and p65 shRNA or IKKβ shRNA or control-shRNA. For both the lentiviruses, infection efficiency is high (80–90%) as assessed from YFP signal (Fig. 7D). Real time PCR (Fig. 7E) and Western blot and corresponding densitometric analysis (Fig. 7, F–H) following cytokine treatment showed that there is a significant decrease in expression of p65 and IKKβ in cells transduced with the respective specific ShRNAs compared with cells receiving control-ShRNA. Moreover, p65 and IKKβ silenced cells exhibit a decrease in PHD2 mRNA (Fig. 7E) and protein (Fig. 7, F–H) levels in the presence of TNF-α. In addition, when NP cells were treated with increasing doses of SM7368 (10–20 μm) in the absence of TNF-α, basal PHD2 mRNA expression is blocked (Fig. 8A). Western blot (Fig. 8B) and corresponding densitometric analysis (Fig. 8C) confirmed that inhibitor treatment significantly suppresses PHD2 protein levels. To further clarify the role of NF-κB in PHD2 regulation and to determine whether this mechanism is cell type-specific, we measured the baseline expression of PHD2 in p65 null and wild type MEFs. Western blot analysis shows that, compared with wild type cells, levels of PHD2 protein are much lower in p65 null cells (Fig. 8, D and E). Taken together, these results suggest that in NP cells, TNF-α and PHD2 form a functional circuit via p65/RelA (Fig. 8F). In other words, aside from the feed forward effects of PHD2 on cytokine expression, via p65/RelA, which serves to promote a proinflammatory state, NF-κB/p65 regulation of PHD2 expression may indirectly control the activities of other PHD-dependent pathways in NP cells.
FIGURE 7.

Involvement of NF-κB in TNF-α-dependent PHD2 expression in NP cells. A and B, Western blot (A) and corresponding densitometric analysis (B) of PHD2 expression in NP cells treated with TNF-α or IL-1β for 24 h. There is no significant change of PHD2 expression following cytokine treatment. CTR, control. C, PHD2 expression is suppressed by SM7368 (10 μm), an NF-κB signaling inhibitor in presence of cytokines. D, immunofluorescence analysis of NP cells transduced with lentivirus co-expressing shp65 and YFP or shIKKβ and YFP shows 80–90% transduction efficiency. Original magnification, ×20. E, real time RT-PCR analysis confirmed that PHD2 mRNA expression is significantly down-regulated when either IKKβ or p65 is silenced. F–H, Western blot (F) and corresponding densitometric analysis (G and H) of cells transduced with lentivirus expressing sh-control (shCTR) and shIKKβ (F and G) or shCTR and shp65 (F and H) following treatment with TNF-α. Expression level of IKKβ and p65 are suppressed by respective gene targeting shRNA when compared with shCTR cells. Note that the PHD2 expression showed a significant decrease in IKKβ or p65 silenced cells. The data are represented as means ± S.D. of three independent experiments performed in triplicate (n = 3). *, p < 0.05.
FIGURE 8.

NF-κB controls basal PHD2 expression in NP cells. A, real time RT-PCR analysis shows that basal PHD2 mRNA expression is significantly suppressed by NF-κB signaling inhibitor SM7368 (10–20 μm). CTR, control. B and C, Western blot (B) and corresponding densitometric analysis (C) of PHD2 expression in NP cells treated with increasing dose of SM7368 (10–20 μm). PHD2 expression is significantly suppressed by NF-κB inhibition. D and E, Western blot analysis shows that PHD2 protein level is significantly lower in p65−/− compared with p65+/+ MEFs. E, densitometric analysis of multiple blots from the experiment described in D. The data are represented as means ± S.D. of three independent experiments (n = 3). *, p < 0.05. F, schematic showing the role of PHD2 in controlling NF-κB/p65 signaling. PHD2 forms functional circuit with TNF-α through p65 and controls its catabolic actions on NP cells.
DISCUSSION
The experiments in this investigation demonstrated for the first time that PHD2 has a proinflammatory effect in NP cells and promoted cytokine-induced NF-κB/p65 signaling activity. Loss of function studies using a lentiviral knockdown of PHD2 down-regulated TNF-α-dependent expression of catabolic markers such as MMP-3, MMP-13, SDC4, ADAMTS-5, and COX2, as well as inhibiting production of a number of critical cytokines/chemokines and inflammation-associated molecules. Expression of each of these molecules is closely linked to degeneration of the intervertebral disc. Paradoxically, we noted that when NF-κB/p65 signaling was blocked or silenced the baseline expression of PHD2 in NP cells was inhibited. Taken together, PHD2 forms one arm of a feed-forward regulatory circuit with TNF-α via NF-κB; the second arm of this circuit is mediated by NF-κB/p65 signaling feeding backwards to maintains the basal expression of PHD2. Because PHD2 is the nexus of these two signals, it provides a new molecular target for treating or preventing degenerative disc disease.
Intervertebral disc degeneration is characterized by expression of inflammatory molecules that elicit catabolic changes in proteins and proteoglycans of the NP extracellular matrix (32–40). That PHD2 mediates this response was evident because silencing significantly down-regulated TNF-α-induced expression of MMP-3, MMP-13, SDC4, COX2, PHD3, and ADAMTS5; these catabolic factors promote degradation of the disc extracellular matrix, and their expression is closely tied to the pathogenesis of disc disease (29, 35, 41, 42). Moreover, silencing of PHD2 partially restored TNF-α-mediated suppression of aggrecan and collagen II, matrix genes critical for maintenance of the NP tissue structure and biomechanical function (38, 43). The cytokine array study that showed a positive relationship between PHD2 and a number of inflammatory cytokines in NP cells, including IL-1α, IL-6, INF-γ, suggested that PHD2 may play a broader role in NP tissue inflammation. It is likely that PHD2 may promote TNF-α-dependent recruitment of immune cells to inflamed and herniated discal tissues (44), because the array study indicated the expression of ICAM-1, a molecule critical in leukocyte adhesion through LFA-1 and CD11b/CD18, and chemokines such as MCP-1, CCL-20, and thymus chemokine-1/CXCL7 is PHD-dependent. Relevant to this discussion is the observation that by regulating the expression of β-NGF and CNTF, PHD2 may possibly modulate sprouting of sensory axons and control innervation of herniated disc by nociceptive sensory neurons (45). It is noteworthy but not unexpected that the role of PHD2 reported here and that of PHD3 earlier (31) is markedly different from that of other cell types (15–17). Our studies of NP cells have shown that PHDs positively control cytokine-mediated gene expression independent of their hydroxylase activity (31) and thus possibly independent of HIF-1α accumulation. Other workers have attributed an anti-inflammatory role to enzymatic function of PHDs with (15, 16) or without HIF-1α (17) involvement, indicating a strong cell- and tissue-type dependence.
The changes seen in TNF-dependent expression of several catabolic and inflammation-associated molecules in PHD2 silenced NP cells raised an important question: does PHD2 target the NF-κB signaling pathway, and more importantly did this provide a mechanism for transducing cytokine activity? Use of NRE and SDC4 luciferase reporter assays coupled with loss of function studies suggested that PHD2 controlled TNF-α/IL-1β-induced NF-κB activity. Importantly, the preserved ability of ShPHD2 to block cytokine-mediated NF-κB transactivation in HIF-1α suppressed cells suggested that the regulation was HIF-1-independent. This observation supports our previous finding that cytokine-mediated catabolic gene expression was insensitive to dimethyloxalylglycine treatment (31). The decrease in the ability of p65/RelA to induce NRE and SDC4 reporters following suppression of PHD2 offered further insights into the regulatory mechanism. Pulldown of endogenous PHD2 in NP cells with co-precipitation of p65 suggested that a transcriptional complex was formed consisting of both these proteins. In further support of complex formation was the observation that co-expressed FLAG-p65 was bound to YFP-PHD2. Note that, in agreement with our previous studies, the observed interaction of FLAG-p65 with PHD3 confirmed the specificity of the binding studies. That a distinct transcriptional complex was formed between PHD2 and p65 was supported by the observation that although p65 can interact with both PHD isoforms, there was no interaction between PHD2 and PHD3 in NP cells. Fluorescence microscopy and cell fractionation experiments confirmed that p65-PHD2 were present in same cellular compartment in cytokine-treated cells. Based on these findings, we conclude that PHD2 promotes NF-κB transcriptional activity through interaction with p65.
Relevant to this discussion, it is important to comment that the mechanism of PHD control of NF-κB signaling is unique in NP cells when compared with other cell types (18–20, 24). For example, in several tumor cell lines, PHD1 and PHD2 hydroxylate IKKβ in a conserved LXXLAP motif, resulting in decreased NF-κB activity (18). In contrast, Fu and Taubman (19) have shown that hydroxylase-independent suppression of NF-κB is mediated through blocking activating K63-linked ubiquitination of IKKγ by PHD3 but not PHD1 and PHD2. Whereas, Chan et al. (24) observed that PHD2, independent of both its hydroxylase function and HIF signaling, inhibits NF-κB signaling in tumor cells. These studies clearly indicate that regulation of NF-κB signaling by individual PHD isoforms is cell type-specific.
The observation that PHD2 overexpression did not increase basal or cytokine-dependent reporter activities was of some interest. However, because the inductive effect of PHD2 was seen in a rescue experiment using PHD2 suppressed NP cells, one plausible explanation for this finding was that in NP cells there is a higher endogenous level of PHD2 compared with the levels of p65 in the NF-κB pool. Accordingly, a further increase in PHD2 levels would not be expected to enhance NF-κB activity. Importantly, the ability of exogenously added PHD2 and PHD3 to enhance the transcriptional activity of FLAG-p65 in PHD2−/−;PHD3−/−, as well as p65−/− cells that have low levels of endogenous PHD3 (31) and PHD2, supports the notion of saturating levels of PHD2-p65 complexes in NP cells. Finally, lending strong support to our earlier report showing that PHD3 controls NF-κB activity in NP cells (31), studies clearly indicate that an active PHD2-p65 complex exists in NP cells under basal conditions and that a cytokine stimulus is not necessary for its formation.
Although it is clear that PHD2 has a feed forward role in controlling NF-κB activity, the question arose regarding whether NF-κB could in turn control PHD2 expression in NP cells. Studies using pharmacological inhibitor as well as silencing of individual NF-κB signaling components clearly showed that this is the case; NF-κB controlled basal as well as cytokine-dependent levels of PHD2. When compared with PHD3, although cytokines increase the level of this isoform through NF-κB pathway in NP cells (31), NF-κB maintained only baseline expression of PHD2. The observation that MEFs from p65 null mice displayed diminished PHD2 expression also indicates that this mechanism may be broadly shared by different cell types. Importantly, these studies suggest that NF-κB and PHD2 constitute a functional circuit, each regulating the activity of each other. Consequently, by regulating the expression of PHD2, NF-κB/p65 signaling may indirectly control the function of other PHD-dependent pathways in NP cells. Together, our findings demonstrate for the first time that PHD2 is an important mediator of inflammatory response in NP cells and thus provides new insights into the understanding of intervertebral disc degeneration. It is not unreasonable to assume that PHD2 may offer an attractive therapeutic target to treat catabolic events in the pathogenesis of intervertebral disc disease.
This work was supported, in whole or in part, by National Institutes of Health Grants AR055655, AR064733, and AR050087.
- SDC
- syndecan
- PHD
- prolyl-4-hydroxylase domain containing protein
- NRE
- NF-κB response element
- NP
- nucleus pulposus
- HIF
- hypoxia-inducible factor.
REFERENCES
- 1. Appelhoff R. J., Tian Y. M., Raval R. R., Turley H., Harris A. L., Pugh C. W., Ratcliffe P. J., Gleadle J. M. (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 279, 38458–38465 [DOI] [PubMed] [Google Scholar]
- 2. Stiehl D. P., Wirthner R., Köditz J., Spielmann P., Camenisch G., Wenger R. H. (2006) Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 281, 23482–23491 [DOI] [PubMed] [Google Scholar]
- 3. Ivan M., Kaelin W. G., Jr. (2001) The von Hippel-Lindau tumor suppressor protein. Curr. Opin. Genet. Dev. 11, 27–34 [DOI] [PubMed] [Google Scholar]
- 4. Pientka F. K., Hu J., Schindler S. G., Brix B., Thiel A., Jöhren O., Fandrey J., Berchner-Pfannschmidt U., Depping R. (2012) Oxygen sensing by the prolyl-4-hydroxylase PHD2 within the nuclear compartment and the influence of compartmentalisation on HIF-1 signalling. J. Cell Sci. 125, 5168–5176 [DOI] [PubMed] [Google Scholar]
- 5. Huang J., Zhao Q., Mooney S. M., Lee F. S. (2002) Sequence determinants in hypoxia-inducible factor-1α for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J. Biol. Chem. 277, 39792–39800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaelin W. G., Jr., Ratcliffe P. J. (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 7. Wang X., Wang H., Yang H., Li J., Cai Q., Shapiro I. M., Risbud M. V. (2014) Tumor necrosis factor-α- and Interleukin-1β-dependent matrix metalloproteinase-3 expression in nucleus pulposus cells requires cooperative signaling via syndecan 4 and mitogen-activated protein kinase-NF-κB axis: implications in inflammatory disc disease. Am. J. Pathol. 184, 2560–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts S., Evans H., Trivedi J., Menage J. (2006) Histology and pathology of the human intervertebral disc. J. Bone Joint Surg. Am. 88, 10–14 [DOI] [PubMed] [Google Scholar]
- 9. Urban J. P., Smith S., Fairbank J. C. (2004) Nutrition of the intervertebral disc. Spine (Phila Pa 1976) 29, 2700–2709 [DOI] [PubMed] [Google Scholar]
- 10. Podichetty V. K. (2007) The aging spine: the role of inflammatory mediators in intervertebral disc degeneration. Cell Mol. Biol. (Noisy-le-grand) 53, 4–18 [PubMed] [Google Scholar]
- 11. Bartels E. M., Fairbank J. C., Winlove C. P., Urban J. P. (1998) Oxygen and lactate concentrations measured in vivo in the intervertebral discs of patients with scoliosis and back pain. Spine (Phila Pa 1976) 23, 1–7 [DOI] [PubMed] [Google Scholar]
- 12. Fujita N., Chiba K., Shapiro I. M., Risbud M. V. (2012) HIF-1α and HIF-2α degradation is differentially regulated in nucleus pulposus cells of the intervertebral disc. J. Bone Miner. Res. 27, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujita N., Markova D., Anderson D. G., Chiba K., Toyama Y., Shapiro I. M., Risbud M. V. (2012) Expression of prolyl hydroxylases (PHDs) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: distinct roles of PHD2 and PHD3 proteins in controlling HIF-1α activity in hypoxia. J. Biol. Chem. 287, 16975–16986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gu Y. Z., Moran S. M., Hogenesch J. B., Wartman L., Bradfield C. A. (1998) Molecular characterization and chromosomal localization of a third α-class hypoxia inducible factor subunit, HIF3α. Gene Expr. 7, 205–213 [PMC free article] [PubMed] [Google Scholar]
- 15. Keely S., Campbell E. L., Baird A. W., Hansbro P. M., Shalwitz R. A., Kotsakis A., McNamee E. N., Eltzschig H. K., Kominsky D. J., Colgan S. P. (2014) Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 7, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Natarajan R., Salloum F. N., Fisher B. J., Ownby E. D., Kukreja R. C., Fowler A. A., 3rd. (2007) Activation of hypoxia-inducible factor-1 via prolyl-4 hydoxylase-2 gene silencing attenuates acute inflammatory responses in postischemic myocardium. Am. J. Physiol. Heart Circ. Physiol. 293, H1571–H1580 [DOI] [PubMed] [Google Scholar]
- 17. Takeda K., Ichiki T., Narabayashi E., Inanaga K., Miyazaki R., Hashimoto T., Matsuura H., Ikeda J., Miyata T., Sunagawa K. (2009) Inhibition of prolyl hydroxylase domain-containing protein suppressed lipopolysaccharide-induced TNF-α expression. Arterioscler. Thromb. Vasc. Biol. 29, 2132–2137 [DOI] [PubMed] [Google Scholar]
- 18. Cummins E. P., Berra E., Comerford K. M., Ginouves A., Fitzgerald K. T., Seeballuck F., Godson C., Nielsen J. E., Moynagh P., Pouyssegur J., Taylor C. T. (2006) Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. U.S.A. 103, 18154–18159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu J., Taubman M. B. (2013) EGLN3 inhibition of NF-κB is mediated by prolyl hydroxylase-independent inhibition of IκB kinase gamma ubiquitination. Mol. Cell. Biol. 33, 3050–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hams E., Saunders S. P., Cummins E. P., O'Connor A., Tambuwala M. T., Gallagher W. M., Byrne A., Campos-Torres A., Moynagh P. M., Jobin C., Taylor C. T., Fallon P. G. (2011) The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock 36, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johansen J. L., Sager T. N., Lotharius J., Witten L., Mørk A., Egebjerg J., Thirstrup K. (2010) HIF prolyl hydroxylase inhibition increases cell viability and potentiates dopamine release in dopaminergic cells. J. Neurochem. 115, 209–219 [DOI] [PubMed] [Google Scholar]
- 22. Yeramian A., Santacana M., Sorolla A., Llobet D., Encinas M., Velasco A., Bahi N., Eritja N., Domingo M., Oliva E., Dolcet X., Matias-Guiu X. (2011) Nuclear factor-κB2/p100 promotes endometrial carcinoma cell survival under hypoxia in a HIF-1α independent manner. Lab. Invest. 91, 859–871 [DOI] [PubMed] [Google Scholar]
- 23. Fu J., Taubman M. B. (2010) Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-κB-dependent pathway. J. Biol. Chem. 285, 8927–8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan D. A., Kawahara T. L., Sutphin P. D., Chang H. Y., Chi J. T., Giaccia A. J. (2009) Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 15, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith M. F., Jr., Novotny J., Carl V. S., Comeau L. D. (2006) Helicobacter pylori and toll-like receptor agonists induce syndecan-4 expression in an NF-κB-dependent manner. Glycobiology 16, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hertlein E., Wang J., Ladner K. J., Bakkar N., Guttridge D. C. (2005) RelA/p65 regulation of IκBβ. Mol. Cell. Biol. 25, 4956–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minamishima Y. A., Moslehi J., Padera R. F., Bronson R. T., Liao R., Kaelin W. G., Jr. (2009) A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol. Cell. Biol. 29, 5729–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Risbud M. V., Guttapalli A., Stokes D. G., Hawkins D., Danielson K. G., Schaer T. P., Albert T. J., Shapiro I. M. (2006) Nucleus pulposus cells express HIF-1α under normoxic culture conditions: a metabolic adaptation to the intervertebral disc microenvironment. J. Cell. Biochem. 98, 152–159 [DOI] [PubMed] [Google Scholar]
- 29. Wang J., Markova D., Anderson D. G., Zheng Z., Shapiro I. M., Risbud M. V. (2011) TNF-α and IL-1β promote a disintegrin-like and metalloprotease with thrombospondin type I motif-5-mediated aggrecan degradation through syndecan-4 in intervertebral disc. J. Biol. Chem. 286, 39738–39749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walmsley S. R., Print C., Farahi N., Peyssonnaux C., Johnson R. S., Cramer T., Sobolewski A., Condliffe A. M., Cowburn A. S., Johnson N., Chilvers E. R. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J. Exp. Med. 201, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujita N., Gogate S. S., Chiba K., Toyama Y., Shapiro I. M., Risbud M. V. (2012) Prolyl hydroxylase 3 (PHD3) modulates catabolic effects of tumor necrosis factor-α (TNF-α) on cells of the nucleus pulposus through co-activation of nuclear factor κB (NF-κB)/p65 signaling. J. Biol. Chem. 287, 39942–39953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mern D. S., Fontana J., Beierfuss A., Thomé C., Hegewald A. A. (2013) A combinatorial relative mass value evaluation of endogenous bioactive proteins in three-dimensional cultured nucleus pulposus cells of herniated intervertebral discs: identification of potential target proteins for gene therapeutic approaches. PLoS One 8, e81467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mern D. S., Beierfubeta A., Fontana J., Thomé C., Hegewald A. A. (2014) Imbalanced protein expression patterns of anabolic, catabolic, anti-catabolic and inflammatory cytokines in degenerative cervical disc cells: new indications for gene therapeutic treatments of cervical disc diseases. PLoS One 9, e96870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Le Maitre C. L., Freemont A. J., Hoyland J. A. (2005) The role of interleukin-1 in the pathogenesis of human intervertebral disc degeneration. Arthritis Res. Ther. 7, R732–R745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seguin C. A., Pilliar R. M., Roughley P. J., Kandel R. A. (2005) Tumor necrosis factor-α modulates matrix production and catabolism in nucleus pulposus tissue. Spine (Phila Pa 1976) 30, 1940–1948 [DOI] [PubMed] [Google Scholar]
- 36. Weiler C., Nerlich A. G., Bachmeier B. E., Boos N. (2005) Expression and distribution of tumor necrosis factor α in human lumbar intervertebral discs: a study in surgical specimen and autopsy controls. Spine (Phila Pa 1976) 30, 44–53 [DOI] [PubMed] [Google Scholar]
- 37. Yoshida M., Nakamura T., Sei A., Kikuchi T., Takagi K., Matsukawa A. (2005) Intervertebral disc cells produce tumor necrosis factor α, interleukin-1β, and monocyte chemoattractant protein-1 immediately after herniation: an experimental study using a new hernia model. Spine (Phila Pa 1976) 30, 55–61 [DOI] [PubMed] [Google Scholar]
- 38. Le Maitre C. L., Pockert A., Buttle D. J., Freemont A. J., Hoyland J. A. (2007) Matrix synthesis and degradation in human intervertebral disc degeneration. Biochem. Soc. Trans. 35, 652–655 [DOI] [PubMed] [Google Scholar]
- 39. Millward-Sadler S. J., Costello P. W., Freemont A. J., Hoyland J. A. (2009) Regulation of catabolic gene expression in normal and degenerate human intervertebral disc cells: implications for the pathogenesis of intervertebral disc degeneration. Arthritis Res. Ther. 11, R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shamji M. F., Setton L. A., Jarvis W., So S., Chen J., Jing L., Bullock R., Isaacs R. E., Brown C., Richardson W. J. (2010) Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis Rheum. 62, 1974–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao C. Q., Zhang Y. H., Jiang S. D., Li H., Jiang L. S., Dai L. Y. (2011) ADAMTS-5 and intervertebral disc degeneration: the results of tissue immunohistochemistry and in vitro cell culture. J. Orthop. Res. 29, 718–725 [DOI] [PubMed] [Google Scholar]
- 42. Roberts S., Caterson B., Menage J., Evans E. H., Jaffray D. C., Eisenstein S. M. (2000) Matrix metalloproteinases and aggrecanase: their role in disorders of the human intervertebral disc. Spine (Phila Pa 1976) 25, 3005–3013 [DOI] [PubMed] [Google Scholar]
- 43. Yu Z. G., Xu N., Wang W. B., Pan S. H., Li K. S., Liu J. K. (2009) Interleukin-1 inhibits Sox9 and collagen type II expression via nuclear factor-κB in the cultured human intervertebral disc cells. Chin. Med. J. 122, 2483–2488 [PubMed] [Google Scholar]
- 44. Haro H., Komori H., Okawa A., Murakami S., Muneta T., Shinomiya K. (1997) Sequential dynamics of monocyte chemotactic protein-1 expression in herniated nucleus pulposus resorption. J. Orthop. Res. 15, 734–741 [DOI] [PubMed] [Google Scholar]
- 45. Abe Y., Akeda K., An H. S., Aoki Y., Pichika R., Muehleman C., Kimura T., Masuda K. (2007) Proinflammatory cytokines stimulate the expression of nerve growth factor by human intervertebral disc cells. Spine (Phila Pa 1976) 32, 635–642 [DOI] [PubMed] [Google Scholar]