

Background: Retinoic acid regulates energy balance and induces phosphoenolpyruvate carboxykinase gene expression.
Results: Refeeding, glucose, and insulin decrease retinoic acid in vivo. Insulin suppresses retinol dehydrogenase gene expression through suppressing FoxO1.
Conclusion: Insulin inhibits retinoic acid biosynthesis through inhibition of FoxO1-induced Rdh10 gene expression.
Significance: Insulin and retinoic acid exert counter balancing effects in regulating energy status.
Keywords: Akt PKB, Dehydrogenase, Energy Metabolism, FOXO, Gene Regulation, Gluconeogenesis, Insulin, Retinoic Acid, Vitamin A
Abstract
All-trans-retinoic acid (atRA), an autacoid derived from retinol (vitamin A), regulates energy balance and reduces adiposity. We show that energy status regulates atRA biosynthesis at the rate-limiting step, catalyzed by retinol dehydrogenases (RDH). Six h after re-feeding, Rdh1 expression decreased 80–90% in liver and brown adipose tissue and Rdh10 expression was decreased 45–63% in liver, pancreas, and kidney, all relative to mice fasted 16 h. atRA in the liver was decreased 44% 3 h after reduced Rdh expression. Oral gavage with glucose or injection with insulin decreased Rdh1 and Rdh10 mRNA 50% or greater in mouse liver. Removing serum from the medium of the human hepatoma cell line HepG2 increased Rdh10 and Rdh16 (human Rdh1 ortholog) mRNA expression 2–3-fold by 4 h, by increasing transcription and stabilizing mRNA. Insulin decreased Rdh10 and Rdh16 mRNA in HepG2 cells incubated in serum-free medium by inhibiting transcription and destabilizing mRNA. Insulin action required PI3K and Akt, which suppress FoxO1. Serum removal increased atRA biosynthesis 4-fold from retinol in HepG2 cells, whereas dominant-negative FoxO1 prevented the increase. Thus, energy status via insulin and FoxO1 regulate Rdh expression and atRA biosynthesis. These results reveal mechanisms for regulating atRA biosynthesis and the opposing effects of atRA and insulin on gluconeogenesis, and also suggest an interaction between atRA and insulin signaling related diseases, such as type II diabetes and cancer.
Introduction
A wide scope of biological processes, including embryonic development, cell differentiation and proliferation, immune function, neurogenesis, and energy metabolism, depend on the vitamin A (retinol) metabolite all-trans-retinoic acid (atRA)3 (1–5). atRA controls energy balance by inhibiting differentiation of pre-adipocytes into mature white adipose, regulating the function of white adipose cells, and by regulating whole body lipid and carbohydrate metabolism (6–9). Dosing atRA to mice fed a chow diet, which contains ample vitamin A, affords resistance to diet-induced obesity (10, 11). Impairing atRA homeostasis causes abnormalities in intermediary metabolism. Mice with ablated cellular retinol-binding protein 1 (encoded by Rbp1), which regulates retinol homeostasis, experience glucose intolerance from enhanced gluconeogenesis, resulting from hyperglucagonemia, and also undergo increased adipocyte differentiation (12, 13). atRA functions through nuclear hormone receptors RARα, -β, and -γ and peroxisome proliferator-activated receptor δ, which affect transcription and translation (14, 15). Genes regulated by atRA through nuclear receptors include: Pck1, which expresses phosphoenolpyruvate carboxykinase, the enzyme that catalyzes the committed step in gluconeogenesis (16); Ucp1, which expresses uncoupling protein 1, a mediator of adaptive thermogenesis (17); and inhibitors of adipogenesis, including Pref1, Klf2, and Sox9 (18). Despite the impact of atRA on energy balance, little is known about whether energy balance might regulate atRA homeostasis.
Two successive dehydrogenations produce atRA from retinol (19). During limited or normal vitamin A nutriture, retinol dehydrogenases (RDH) of the short-chain dehydrogenase/reductase gene family catalyze the first and rate-limiting reaction to produce all-trans-retinal. Retinal dehydrogenases, of the aldehyde dehydrogenase gene family, catalyze the second reaction to produce atRA. Multiple isoforms of retinoid-metabolizing enzymes occur in both families, often in the same cell types, but these enzymes differ in subcellular expression loci, and can show differential cell expression patterns. RDH1 and RDH10 are the most characterized RDH in the path of atRA biosynthesis: both are expressed early in embryogenesis and throughout life in multiple tissues, and both contribute to atRA biosynthesis from retinol in intact cells in the presence of retinal dehydrogenases (20–22). Dehydrogenase reductase 3 (DHRS3) functions as a retinal reductase, which interacts with RDH10 to control retinoid homeostasis (23). Rdh1-null mice are born in Mendelian frequency, but experience increased weight and adiposity, which is prevented by feeding copious vitamin A (24). In contrast, most Rdh10-null mice die by embryonic day 12.5, but can be rescued by maternal administration of retinoids (25). Notably, like Rdh1-null mice, Rdh10-null mice do not exhibit total atRA deficiency, suggesting complementary actions of the two and/or occurrence of additional RDH.
Liver exerts a central function in maintaining whole body metabolic homeostasis (26). During feeding, liver catalyzes increased energy storage by generating glycogen and triacylglycerol. During fasting, liver generates glucose and ketone bodies from gluconeogenesis, glycogenolysis, and fatty acid oxidation. Glucose, insulin, and glucagon control the balance between energy storage during feeding and production of fuels during fasting.
A combination of portal vein glucose, insulin, and neuronal signal(s) causes liver to transition from catabolic to anabolic metabolism (27). Insulin binding to its cell surface receptors activates two canonical pathways in hepatocytes: the mitogen-activated protein kinase and phosphoinositide 3-kinase (PI3K). The mitogen-activated protein kinase pathway includes extracellular-signal regulated kinase (ERK) signaling and regulates transcription involved in differentiation, growth, and survival (28). The PI3K pathway activates Akt (PKB) to mediate metabolic transitions, including promotion of glycogen and lipid synthesis, and suppression of gluconeogenesis. Effectors downstream of PI3K/Akt include: glycogen synthase kinase-3 (GSK-3), which regulates glycogen synthesis; mammalian target of rapamycin complex 1 (mTORC1), which regulates protein synthesis; BCL2-associated agonist of cell death (BAD), which regulates cell survival; and forkhead box other (FoxO), which regulates transcription of genes required for gluconeogenesis, including glucose-6-phosphatase (Glc-6-P) (29, 30).
This report compares Rdh1 and Rdh10 expression in tissues of fasted versus re-fed mice, and focuses on atRA in liver. Liver served as the focus of this study because of its central contributions to both energy and retinoid homeostasis. We found that insulin, via inactivating FoxO1, represses Rdh1 and Rdh10 expression, with an associated decrease in atRA biosynthesis. These observations are consistent with the recent analysis that FoxO1 may link gluconeogenesis to retinoid homeostasis (31). Our results provide direct evidence showing that energy status regulates atRA biosynthesis, and provides new insight into the relationship between atRA biosynthesis and its regulation of energy balance.
EXPERIMENTAL PROCEDURES
Animals
C57Bl/6J male mice, age 2–3 months, bred in-house were housed up to 5 per cage with littermates, and fed an AIN93G semi-purified diet with 4 IU of vitamin A/g (Dyets, catalog number 110700), unless noted otherwise. The exceptions were: mice in the 12-h re-fed experiment were both male and female (no significant difference in atRA values was detected by sex); mice used for fasting and insulin or exendin-4 treatments were purchased from Jackson Laboratories (catalog number 000664) and were fed laboratory chow. Experimental groups were normalized to controls consisting of fasted mice of the same age, diet, and sex. Tissues were harvested immediately after euthanasia, frozen in liquid nitrogen, and stored at −80 until atRA quantification. Fasted groups were deprived of food for 16 h, beginning ∼2 h prior to initiation of the dark cycle. Re-fed groups were provided diet ad libitum. An oral gavage glucose dose of 2 g/kg body weight was delivered as 10 μl/g body weight of a 20% glucose solution in PBS. Intraperitoneal glucose was delivered in two injections of 2 g/kg body weight each at 0 and 45 min following a 16-h fast. Insulin (0.375 and 0.5 IU/kg, Humulin, Eli Lily number 8715) and exendin-4 (10 μg/kg, Sigma E7144) were delivered in two intraperitoneal injections of 1.8 and 2 μl/g of body weight, respectively. Animal experiments were approved by Office of Laboratory and Animal Care at University of California Berkeley.
Cell Culture
HepG2 cells (ATCC, HB-8065) were maintained in Eagle's minimal essential medium (ATCC 30-2003) supplemented with 10% fetal bovine serum (Life Technologies 10082), and incubated at 37 °C with 5% CO2. Medium containing serum is referred to as growth, and without serum as serum-free. Cells were treated with the following, delivered in medium or dimethyl sulfoxide: 25 μg/ml (20 μm) of actinomycin D (Life Technologies A7592), 10 μg/ml of cycloheximide (Sigma C4859), 10 nm human insulin (Sigma I9278), 100 nm glucagon (CalBiochem 05-23-2700), 50 μm LY294002 (Cell Signaling Technologies #9901), 1 μm triciribine (a.k.a. API-2, Cayman number 10010237), 200 nm rapamycin (Cayman number 13346), 20 μm PD98059 (CalBiochem number 513000). Adenovirus constructs dnFoxO1 (FoxO1Δ256) or lacZ were infected into 70% confluent HepG2 cells and collected for analysis after 48 h. Virus doses were tested empirically and selected based on the highest amount tolerated in HepG2 cells, according to published protocols (32).
Retinoid Quantification
Liver atRA was quantified in 6 to 9 mice per group. Tissues were collected under yellow light and frozen immediately in liquid nitrogen. On the day of analysis, tissues were weighed, thawed on ice, and hand homogenized in 0.9% saline. HepG2 cells were collected under yellow light in 1× reporter lysis buffer (Promega E3971), and stored at −80 °C. Cell protein was quantified with the Pierce BCA Protein Assay Kit (Thermo Scientific number 23227). For atRA biosynthesis in HepG2 cells, 50 nm (100 pmol) all-trans-retinol (Sigma R7632) was delivered in dimethyl sulfoxide 4 h before cell harvest. Cells were homogenized by pipetting and vortexing. Retinoids were recovered by a two-step acid and base extraction (33). All materials in contact with samples were glass or stainless steel. Internal standards were used to calculate extraction efficiency of retinoids. atRA was extracted and quantified by LC/MS/MS, with 4,4-dimethyl retinoic acid as an internal standard (34). Retinol was extracted and quantified by HPLC/UV, with 3,4-didehydroretinol as internal standard (35).
RNA Isolation and qPCR
Isolation of RNA from liver and cells was by the TRIzol reagent (Invitrogen) method. Liver was homogenized using a Qiagen Tissue Lyser II set at 30 Hz for 1 min. cDNA was prepared using iScript reagent kit (Bio-Rad 170-8891). Real-time qPCR was prepared with 2× TaqMan master mix (Life Technologies 4369016), and TaqMan Gene Expression Assays: mouse ACTB, Mm00607939_s1; Rdh10 Mm00467150_m1; G6pc, Mm00839363_m1; Rdh1, Mm00650636_m1; Dhrs3, Mm00488080_m1; human ACTB Hs01060665_g1; RDH10, Hs00416907_m1; Glc-6-P, Hs00609179_m1; Rdh16, Hs00559712_m1; Dhrs3, Hs00191073_m1 (Life Technologies), and run on an ABI 7900 thermocycler. Gene expression was analyzed by the ΔΔCt method, normalized to β-actin, and expressed as fold-change relative to expression in fasted liver or the reference condition described in cells.
Protein Expression
Western blots were done with the Mini PROTEAN system (Bio-Rad), 10% TGX gels (Bio-Rad number 456-1033), and semi-dry transfer (Bio-Rad Trans-blot SD) to nitrocellulose membranes (Whatman). Western blots were visualized with the Licor Odyssey system, and quantified by densitometry in reference to actin expression. Primary antibodies were actin (ProSci number 3779) and HA (12CA5) (Roche number 11583816001). Secondary antibodies were LI-COR IR Dye (680LT number 926-68020, 800CW number 926-32211).
Immunofluorescence
HepG2 cells were fixed in ice-cold acetone for 10 min, then stained with FoxO1 primary antibody (Santa Cruz number 11350) for 2 h at 37 °C, and incubated with Alexa Fluor 488 secondary antibody (Life Technologies number A11034) for 1 h at 37 °C. Coverslips were mounted onto glass slides with Vectashield mounting medium containing DAPI (Vector Labs number H-1200) and imaged by fluorescence microscopy (Zeiss AxioImager).
Statistics
Data are presented as mean ± S.E. and analyzed using two-tailed, unpaired Student's t tests or linear regression analysis.
RESULTS
Re-feeding Decreases Rdh mRNA in Multiple Mouse Tissues and atRA in Liver Versus Fasted Levels
To determine whether changes in energy status regulate atRA biosynthesis, we measured expression of Rdh1 and Rdh10 in tissues of mice fasted 16 h or re-fed ad libitum 6 h after a 16-h fast. In liver, Rdh1 and Rdh10 mRNA were decreased in re-fed animals 86 and 57%, respectfully, relative to expression in fasted animals (Fig. 1, A and B). In brown adipose tissue of re-fed mice, Rdh1 was decreased 77%, whereas Rdh10 expression was unchanged. In pancreata of re-fed mice, Rdh10 mRNA was decreased 43%, and Rdh1 expression was below the limit of detection. In epididymal white adipose tissue of re-fed mice, Rdh10 expression was unchanged and Rdh1 expression was below the limit of detection. In kidney of re-fed mice, Rdh10 expression was decreased 62%. Focusing on the liver, the atRA concentration was reduced 9 and 12 h after re-feeding, but not after 6 h (Fig. 1C). Thus, reduced Rdh mRNA preceded reduced atRA concentrations in liver of re-fed animals.
FIGURE 1.
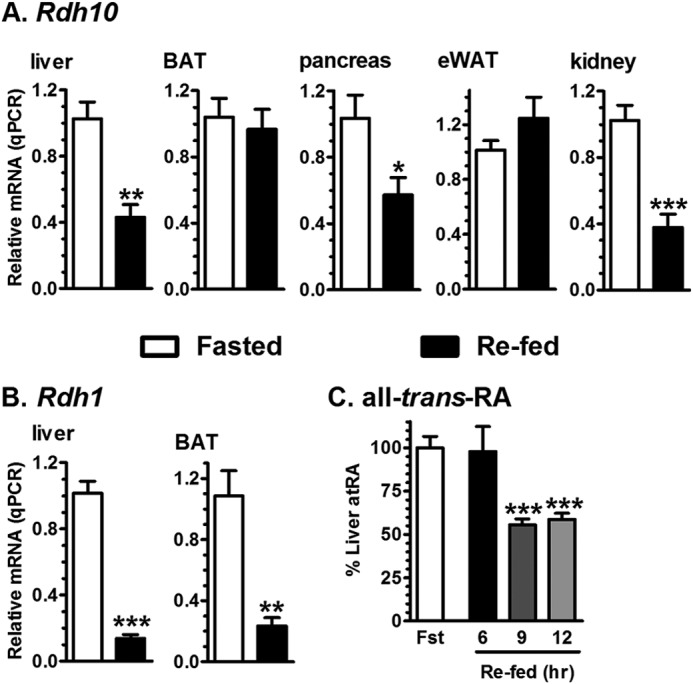
Regulation of Rdh expression and atRA concentrations by fasting and re-feeding. mRNA expression of Rdh in tissues of mice fasted 16 h, or fasted and re-fed ad libitum 6 h. Data show a representative experiment of 2–5 experiments, each with 4–8 mice per group/experiment. A, Rdh10. B, Rdh1. C, atRA in livers of mice fasted 16 h, or fasted and re-fed ad libitum. Each re-fed group was normalized to its fasted group. atRA data show a representative experiment of 3 experiments, each with 6–9 mice per group/experiment: *, p < 0.05; **, p < 0.005; ***, p < 0.0005, relative to fasted values.
Insulin and Oral Glucose Decrease Liver Rdh10 mRNA
We next measured Rdh10, Rdh1, and G6pc (as a positive control for FoxO1-induced hepatic gluconeogenic gene regulation) expression in liver of fasted mice versus mice fasted and dosed with glucose by oral gavage (Fig. 2A). Oral glucose repressed Rdh10, Rdh1, and G6pc expression similar to re-feeding. We elected to focus on Rdh10 because its relative expression levels exceed those of Rdh1 by ∼100-fold (Fig. 2B). An independent experiment verified that re-feeding reduced Rdh10 and G6pc expression at 6 h. Dhrs3 mRNA was decreased 50% in the livers of mice re-fed 12 h, relative to expression in fasted mice, with partial recovery of Rdh10 at 12 h (Fig. 2C). We tested Dhrs3 expression because during these studies a report concluded that Rdh10 and Dhrs3 form a heterodimer, which modulates the activities of both enzymes (23). As expected from the standard protocol of fasting and re-feeding, blood glucose from mice fasted 16 h and refed for 6 h increased significantly versus mice fasted 16 h (Fig. 2D).
FIGURE 2.
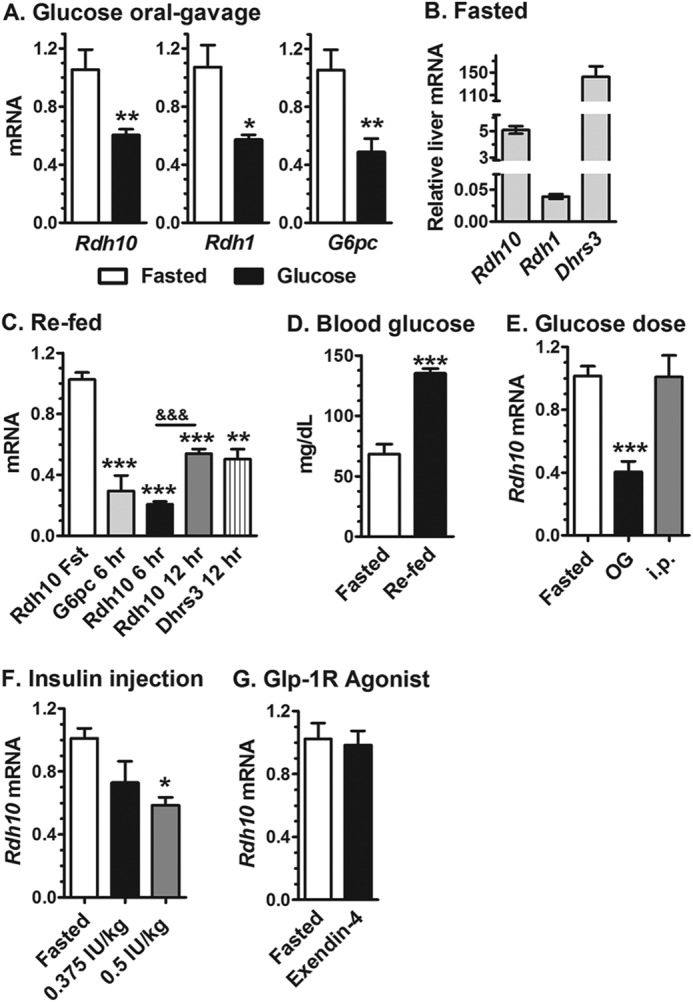
Regulation of liver Rdh expression by energy status. A, Rdh10, Rdh1, and Glc-6-P (G6pc) mRNA are relatively high in the livers of fasted mice and decreased in mice dosed with glucose by oral gavage. Samples were collected after a 16-h fast or 6 h after glucose dosing following a 16-h fast. B, relative expression levels of Rdh and Dhrs mRNA in liver of fasted mice, normalized to β-actin. C, effects of re-feeding on liver G6pc, Rdh10, and Dhrs3 mRNA in mice fasted 16 h and re-fed for the times indicated. D, blood glucose in mice after a 16-h fast or 6 h after re-feeding after a 16-h fast. E, glucose dosed by oral gavage (OG), but not intraperitoneal injection, decreased Rdh10 mRNA in liver of mice fasted 16 h relative to fasted mice. F, insulin dosing repressed Rdh10 mRNA in liver of mice fasted 16 h. G, the Glp-1 receptor agonist, exendin-4, did not repress Rdh10 mRNA expression. Treated groups were collected after 6 h with the exception of the 12-h re-fed group. qPCR data were normalized to their fasted groups: *, p < 0.05; **, p < 0.005; ***, p < 0.001; &&&, p < 0.001 between 6 and 12 h.
To determine whether the route of glucose dosing affected Rdh10 expression, we compared the impact of oral gavage and intraperitoneal injection. Glucose injected intraperitoneally had no effect on Rdh10 expression (Fig. 2D). Insulin, however, caused a dose-dependent decrease in liver Rdh10 expression that reached statistical significance at 0.5 IU/kg (Fig. 2F), a dose recommended for the insulin tolerance test (36). Because nutrients trigger incretin secretion, we tested the impact of glucagon-like peptide 1. Glucagon-like peptide 1 is short lived in circulation from inactivation by dipeptidyl peptidase-4 (37). Therefore, we used the stable glucagon-like peptide 1 receptor agonist, exendin-4 (38). Treatment with exendin-4 had no impact on Rdh10 expression (Fig. 2G). These data demonstrate that re-feeding, oral gavage with glucose, and intraperitoneal insulin each are sufficient to reduce liver Rdh10 expression from the higher fasted levels.
RDH10 Transcription Increases with Serum Removal and Is Attenuated by Insulin in HepG2 Cells
We used the human hepatoma cell line, HepG2, to study mechanisms of RDH10 regulation. The presence or absence of serum in the medium can model the impact of growth factors, including insulin (39, 40). Therefore, serum was removed from the medium, which prompted a 2–3-fold increase in RDH10 mRNA 4 h after the medium change (Fig. 3A). RDH10 mRNA remained elevated at least 16 h. The increase in RDH10 mRNA correlated with that of glucose-6-phosphatase (Glc-6-P), which is induced transcriptionally in liver during fasting (41). DHRS3 expression increased 1.5-fold after 4 h of serum-free medium, and insulin prevented the increase (Fig. 3B). Although insulin prevented the increase in RDH10 mRNA prompted by serum-free medium, neither high nor low glucose affected RDH10 expression (Fig. 3C).
FIGURE 3.
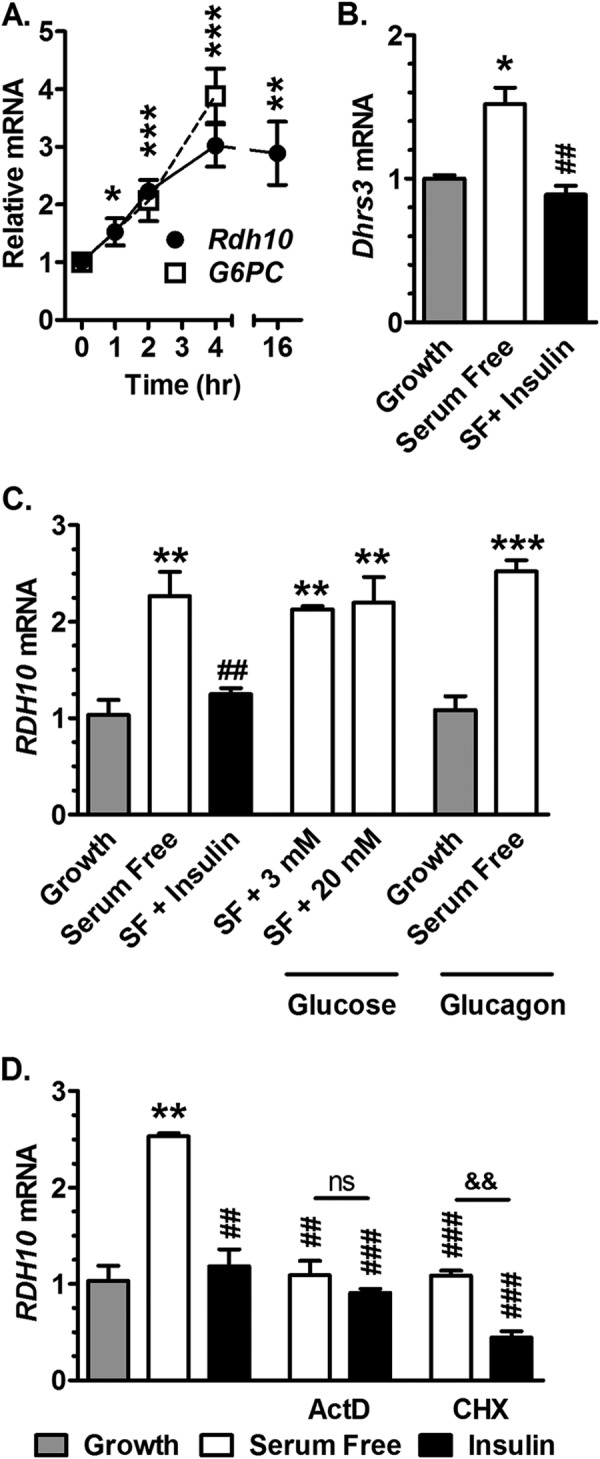
Insulin inhibits RDH10 and DHRS3 transcription. Cells were continued in growth medium or transitioned to serum-free medium. Additions of hormones, glucose, or inhibitors were made at the time of the transition to serum-free medium. A, HepG2 cells were transitioned from serum-containing (growth) to serum-free (SF) medium at 0 h and mRNA of RDH10 and glucose-6-phosphatase (G6PC) were measured. B, cells were maintained in growth medium or transitioned to serum-free medium and treated 4 h ± 10 nm insulin. C, cells were maintained in growth medium or transitioned to serum-free medium and treated 6 h with 10 nm insulin, or 3 or 20 mm glucose, or 100 nm glucagon. D, cells were maintained in growth or transitioned to serum-free medium and treated 4 h with 10 nm insulin, ± 25 μg/ml of ActD, or ± 10 μg/ml of CHX: *, p < 0.05; **, p < 0.005; ***, p < 0.0005 relative to growth values; ##, p < 0.005; ###, p < 0.001 relative to serum-free values; &&, p < 0.005. ns, not significantly different from growth medium.
Glucagon had no effect on RDH10 expression in either growth (10% serum) or serum-free medium. These data validate HepG2 cells as a model for insulin regulation of RDH10 expression in liver.
The transcription inhibitor actinomycin D (ActD) prevented the increase in RDH10 mRNA by removing serum (Fig. 3D). ActD effects were neither additive nor synergistic with those of insulin, indicating that both serum and insulin repress RDH10 transcription. The translation initiation inhibitor cycloheximide (CHX) prevented the increase in RDH10 expression upon serum exclusion, consistent with a requirement for protein synthesis for an increase in, or stabilization of, the mRNA. The actions of CHX and insulin were additive, suggesting insulin regulates RDH10 independently of translation.
Insulin Inhibits RDH10 Transcription via PI3K, Akt, and Suppression of FoxO1
PI3K or Akt inhibitors had no impact on RDH10 expression in serum-free medium, but prevented repression by insulin (Fig. 4A). Inhibition of mTORC1 or MEK/ERK did not prevent the increase in RDH10 expression induced by serum-free medium and did not ameliorate insulin inhibition. These data demonstrate that PI3K and Akt are required for suppression of RDH10 transcription by insulin and suggest that FoxO1, which insulin suppresses, induces RDH10 transcription. To test the need for FoxO1, we infected HepG2 cells with an adenovirus expressing a dominant-negative FoxO1 construct (dnFoxO1), which produces a truncated mutant that binds DNA, but lacks the transcription activation domain (32). Cells infected with dnFoxO1 did not increase RDH10 expression when exposed to serum-free medium. This is consistent with induction of FoxO1 activity by serum-free medium and prevention of FoxO1 activity by dnFoxO1 (32, 42). Insulin had no further effect on RDH10 expression in these cells. Expression of dnFoxO1 did not affect RDH10 expression in cells when exposed to growth medium. In contrast, adenovirus expressing lacZ responded to serum-free medium with an increase in RDH10 mRNA that was inhibited by insulin. G6pc expression followed the same pattern as RDH10 in the adenovirus experimental conditions (Fig. 4B). Western blot analysis confirmed the expression of HA-tagged dnFoxO1 (Fig. 4C). Insulin signaling induces phosphorylation and nuclear export of FoxO1 (43). Cells in serum-free medium had FoxO1 located in puncta within the nucleus. Cells treated with insulin had FoxO1 expressed in a diffuse pattern within the cytoplasm, consistent with nuclear export (Fig. 4D).
FIGURE 4.
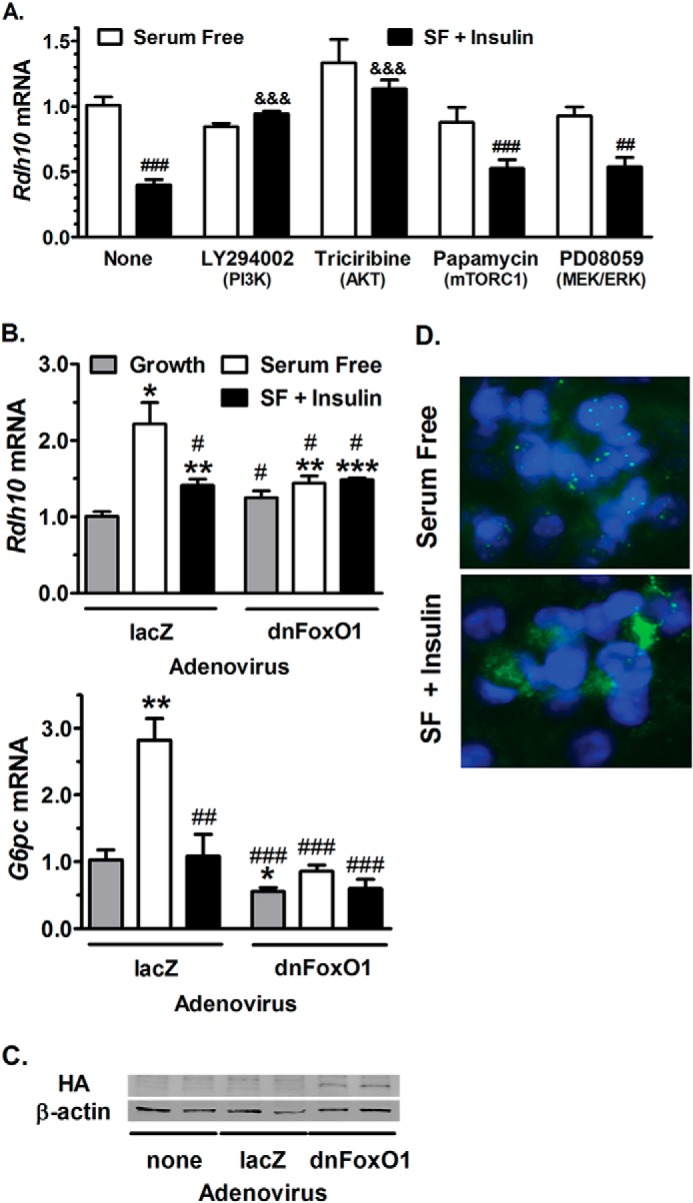
Transcriptional regulation of RDH10 expression by insulin requires FoxO1. A, RDH10 mRNA expression in HepG2 cells pre-treated with inhibitors or dimethyl sulfoxide vehicle for 1 h, followed by 4 h with fresh inhibitors in serum-free (SF) medium, with or without 10 nm insulin. PI3K was inhibited with 50 μm LY294002; Akt was inhibited with 1 μm triciribine; mTORC1 was inhibited with 200 nm rapamycin; MEK/ERK was inhibited with 20 μm PD98059. B, RDH10 and Glc-6-P (G6PC) mRNA expression in cells infected with adenovirus expressing dominant-negative FoxO1 (dnFoxO1) or lacZ for 48 h, and treated with growth or serum-free medium ± 10 nm insulin during the final 4 h. qPCR data were combined from two independent experiments with 3–4 replicates each. C, Western blotting verified expression of HA-tagged dnFoxO1 in cells infected with adenovirus dnFoxO1, and not in untreated or adenovirus lacZ cells. D, immunohistochemistry detection of FoxO1 (green) and DAPI (blue) in HepG2 cells treated 4 h with serum-free medium ± 10 nm insulin. Images represent one of two experiments. qPCR: #, p < 0.05; ##, p < 0.005; ###, p < 0.001 versus serum-free control; &&&, p < 0.0005 versus insulin-treated control (no inhibitor); *, p < 0.05; **, p < 0.005; ***, p < 0.001 versus growth medium control.
Serum-free Medium Increases and Insulin Decreases RDH10 mRNA Stability
We determined the elimination half-life (t½) of RDH10 mRNA in cells cultured 16 h in serum-free medium, or maintained in growth medium, and then exposed to ActD for 24 h (Fig. 5A). RDH10 mRNA had a t½ of 35 h in the absence of serum and insulin. In the absence of serum, but in the presence of insulin, the t½ decreased to 19 h. In the presence of serum and absence of insulin (growth medium), RDH10 mRNA had a biphasic t½. The mRNA was decreased 50% by 8 h; after 8 h the mRNA was stabilized with a t½ of 118 h. Adding serum for 8 h to cells exposed to serum-free medium for 16 h also decreased the amount of RDH10 mRNA by 50%. Because RDH10 expression is elevated 2–3-fold in serum-free medium, the amounts of message in cells with serum-free medium, with or without insulin, continued to exceed those of cells maintained in growth medium. Destabilization of RDH10 mRNA by insulin was attenuated by inhibition of PI3K or expression of dnFoxO1, and completely prevented by inhibition of Akt (Fig. 5B). In the absence of ActD, RDH10 expression in cells exposed to serum-free medium 16 h, then treated 4 h with insulin, was not changed significantly (Fig. 5C). In contrast, treatment with serum or serum and insulin reduced RDH10 mRNA to the amount in growth medium. These data show that mRNA induced by long-term exposure to serum-free medium is more sensitive to serum than insulin. In cells exposed to serum-free medium for 16 h, then treated 8 h with CHX and ActD versus ActD alone, maximum RDH10 mRNA stability required translation (Fig. 5D).
FIGURE 5.
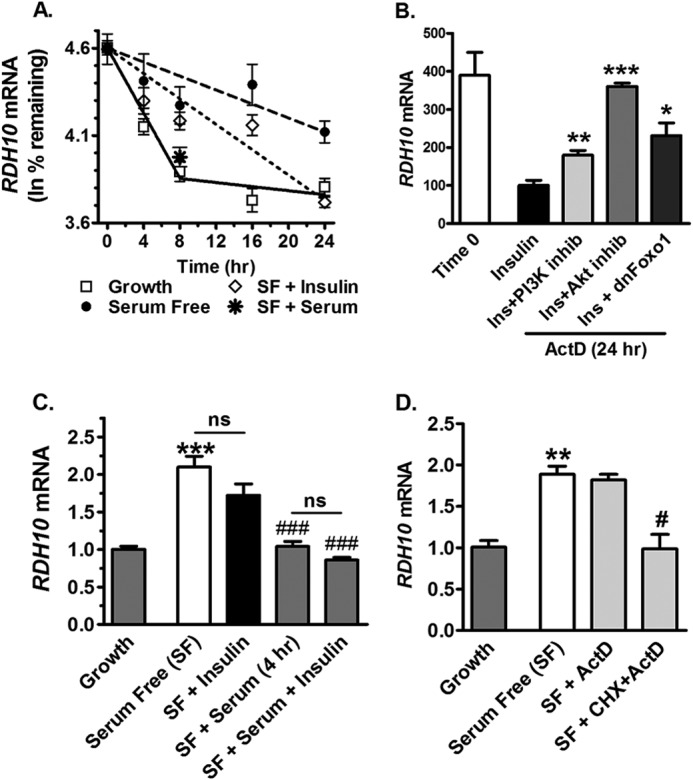
Insulin and serum decrease the elimination half-life of RDH10 mRNA. A, HepG2 cells were maintained in growth medium, or incubated in serum-free (SF) medium 16 h before time 0. At time 0, cells were treated with 25 μg/ml of ActD in growth medium (solid line) or in serum-free medium with (short dashed line) or without (long dashed line) 10 nm insulin or 10% serum. qPCR data were normalized to 100% relative expression at time 0 for each condition and plotted as ln % remaining. Half-lives (t½) were calculated as 0.693/slope, with the slope determined by linear regression analysis. B, cells were maintained in growth medium or incubated in serum-free medium 16 h before time 0. At time 0, cells that were transitioned to serum-free medium were treated a further 24 h in serum-free medium with the addition of 25 μg/ml of ActD and 10 nm insulin, or insulin and 50 μm PI3K inhibitor (LY294002), or insulin and 1 μm Akt inhibitor (triciribine), or insulin and adenovirus expressing dominant-negative FoxO1 (dnFoxO1). Adenovirus treatment began 48 h prior to collection. qPCR data were normalized to 100% expression relative to the insulin-treated group. C, cells were maintained in growth medium, or incubated in serum-free medium 16 h, then treated 4 h with 10 nm insulin and/or 10% serum. D, cells were maintained in growth medium or preincubated in serum-free medium 16 h, then treated 8 h with dimethyl sulfoxide (vehicle control), 25 μg/ml of ActD or ActD, and 10 μg/ml of cycloheximide: *, p < 0.05; **, p < 0.005; ***, p < 0.0001 versus insulin values; **, p < 0.005; ***, p < 0.001 versus growth values; #, p < 0.05; ###, p < 0.001 versus serum-free values. ns, not significantly different from growth medium.
RDH16 Is Regulated Similarly to RDH10
Because mouse Rdh1 expression was also reduced in liver of re-fed versus fasted mice, we examined its human ortholog RDH16 in HepG2 cells. Removing serum from the medium increased RDH16 expression within 2 h, rising to 4-fold by 16 h (Fig. 6A). Insulin prevented the increase in RDH16 mRNA that occurred in serum-free medium, but neither glucagon nor glucose had an effect (Fig. 6B). The increase in RDH16 mRNA that occurs in serum-free medium depended on transcription, as shown by ActD treatment (Fig. 6C). Interestingly, ActD treatment reduced RDH16 expression more than that of growth medium or insulin. Insulin had no effect in the presence of dnFoxO1, which reduced RDH16 mRNA to a greater extent than insulin treatment in serum-free medium (Fig. 6D).
FIGURE 6.
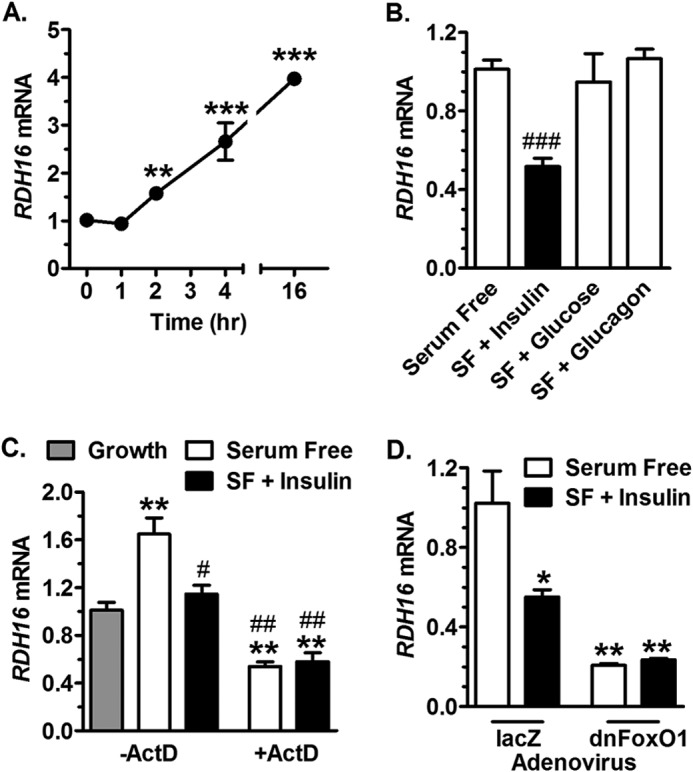
Serum and insulin repress RDH16 expression in HepG2 cells. A, cells were transitioned from growth (serum-containing) medium to serum-free (SF) medium at time 0 and assayed by qPCR for RDH16 mRNA. B, cells were exposed to serum-free medium and treated with 10 nm insulin, 20 mm glucose, or 100 nm glucagon for 6 h. C, cells were exposed to growth or serum-free medium and treated 4 h with 10 nm insulin and/or 25 μg/ml of ActD. D, cells were infected 48 h with adenovirus expressing dnFoxO1 or lacZ, and treated with serum free-medium ± 10 nm insulin during the final 4 h: *, p < 0.05; **, p < 0.005; ***, p < 0.0005 versus growth values; #, p < 0.05; ##, p < 0.005; ###, p < 0.0005 versus serum-free values.
FoxO1 Is Required for Elevated atRA Biosynthesis in Serum-free Medium
To test the impact of reduced RDH mRNA expression on atRA biosynthesis from retinol, HepG2 cells infected with dnFoxO1 or lacZ control were maintained 16 h in growth or serum-free medium, and then incubated 4 h with all-trans-retinol. atRA was increased ∼4-fold in cells incubated in serum-free relative to growth medium (Fig. 7). The increase was prevented by dnFoxO1. Quantification of retinol recovered in cells revealed greater uptake in cells maintained in serum-free relative to growth medium. Thus, despite the increased intracellular retinol concentration, atRA biosynthesis was decreased by dnFoxO1, relative to the rate observed in serum-free medium.
FIGURE 7.
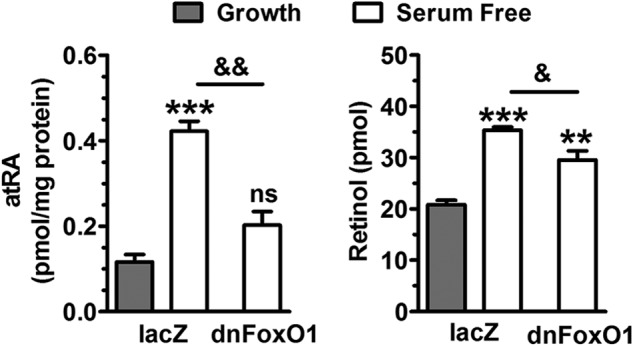
FoxO1 promotes atRA biosynthesis. HepG2 cells were infected with adenovirus 48 h prior to harvesting, incubated in growth or serum-free medium for 16 h, then treated with 50 nm (100 pmol) retinol in the final 4 h. Left, atRA synthesis is elevated in cells in serum-free medium. Dominant-negative FoxO1 (dnFoxO1) prevents the increase. Right, the amount of retinol recovered is elevated in cells incubated in serum-free medium: **, p < 0.005; ***, p < 0.0001 versus growth values; &, p < 0.05; &&, p < 0.005 as labeled; ns, not significantly different from growth medium.
DISCUSSION
A vitamin A-deficient diet results in glycogen deficiency because of impaired gluconeogenesis, caused by low Pck1 expression (44–46). Stimulation of gluconeogenesis is mediated directly by the vitamin A metabolite atRA through RA receptor response elements in the key gluconeogenic gene Pck1 (44, 45). Here we demonstrate reciprocal regulation by energy status of Rdh mRNA and atRA concentrations via insulin effects on FoxO1. Fasting-induced coordinate induction of both Rdh and Pck1 by FoxO1 stimulates gluconeogenesis (Fig. 8). In the fed state, insulin repression of FoxO1 suppresses gluconeogenesis via repressing both Rdh and Pck1. Although serum factor(s) other than insulin control Rdh expression and mRNA stability, we focused on insulin because of its critical role in regulating gluconeogenesis (9, 47).
FIGURE 8.
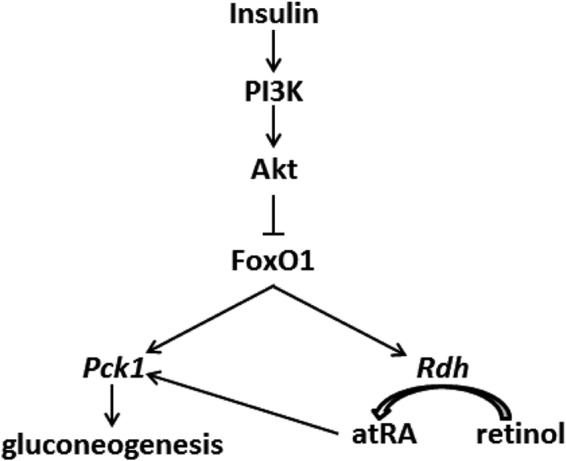
Regulation of Rdh expression by insulin signaling. Both atRA and FoxO1 induce Pck1 transcription to increase gluconeogenesis. FoxO1 induces atRA biosynthesis by inducing Rdh mRNA. Insulin suppresses FoxO1 activity, thereby suppressing gluconeogenesis and atRA biosynthesis through decreasing Rdh transcription and mRNA stability. Activation of PI3K/Akt represents a molecular indicator of cancer, implicating decreased atRA.
Tissue-specific changes in Rdh10 and Rdh1 expression during fasting and re-feeding are consistent with a complex contribution of atRA to regulating intermediary metabolism. Epididymal white adipose tissue and pancreata do not express Rdh1 (data not shown), but express Rdh10. The non-responsiveness of Rdh10 in epididymal white adipose tissue to changes in energy balance allows several interpretations, including the presence of another Rdh that responds to changes in energy status, or feeding versus fasting does not modify atRA epididymal white adipose tissue concentrations in adult mice. These possibilities are the subjects of ongoing studies. The decrease in Rdh10 expression in pancreas during re-feeding is consistent with the requirement for retinoids in pancreatic development (48, 49) and function (50, 51), and also a subject of ongoing studies. Suppression of Rdh1 in liver by re-feeding, and Rdh10 in liver and kidney, relate insulin action with atRA signaling in two gluconeogenic tissues. Decreased Dhrs3 expression in liver of re-fed versus fasted mice is consistent with a mutually activating interaction of Rdh10 and Dhrs3 (23), and the reduction in atRA biosynthesis. Evidently, energy metabolism affects Rdh expression in multiple tissues to regulate retinoid signaling effects as feedback to regulation of energy disposition by atRA.
Oral dosing has a singular effect on glucose uptake and insulin action in liver (27). Glucose delivery by the portal vein combined with insulin, and an as yet unidentified neuronal signal, increases net hepatic glucose uptake compared with peripheral delivery, with a concomitant impact on insulin-regulated processes (52, 53). A comparison of oral glucose dosing to infusion via the portal vein revealed similar enhancement in hepatic glucose uptake relative to peripheral presentation, which excludes a contribution from a gut-secreted factor, such as Glp-1 (54). Yet, secretion of Glp-1 upon feeding contributes to reduced glucose production in liver by augmenting glucose-stimulated insulin secretion from pancreas (55). Our data of oral but not peripheral glucose dosing reducing Rdh mRNA are consistent with these observations and support the physiological significance of the observation.
Reduced Rdh expression preceded the reduction in liver atRA, and was accompanied by a lower rate of atRA biosynthesis in HepG2 cells, consistent with a cause and effect relationship. atRA homeostasis autoregulates via induction of both catabolic cytochromes P-450 and the retinyl ester-forming lecithin:retinol acyltransferase (56–58). The former decreases atRA itself, whereas the latter decreases the amount of retinol available to support atRA biosynthesis. Changes in lecithin:retinol acyltransferase and cytochrome P-450 may have buffered atRA differences in the re-fed relative to the fasted liver. The 2-fold decrease in liver atRA resulting from insulin action seems remarkable in the context of this impetus to sustain atRA homeostasis.
FoxO (forkhead box “Other”) proteins constitute a subgroup of a family of evolutionarily conserved transcription factors that mediate insulin signaling in mammals, but also mediate metabolism and longevity in primitive organisms such as Caenorhabditis elegans and Drosophila (59, 60). FoxO proteins contribute to metabolic regulation through effects in liver, muscle, adipose tissue, and pancreas. Of these, FoxO1 stimulates a committed step in hepatic gluconeogenesis, catalyzed by phosphoenolpyruvate carboxykinase (PCK1), and also the last step catalyzed by glucose-6-phosphatase (Glc-6-P) (30, 32, 61, 63). Haploinsufficiency of FoxO1 restores insulin sensitivity in insulin-resistant mice (64). A constitutively active gain-of-function FoxO1 mutant targeted to liver and pancreas manifests a diabetic phenotype (64). Liver-specific inactivation results in decreased serum glucose at birth, and upon fasting in adult mice from impaired glycogenolysis and gluconeogenesis (63). A liver-specific FoxO1-null mouse crossed with an insulin receptor-null mouse has reduced glucose production and lacks the neonatal diabetes and hepatosteatosis that occur in insulin receptor-null mice (63). Liver-specific FoxO1-null mice treated with the β-cell toxin streptozotocin, have elevated VLDL secretion, cholesterol, and plasma-free fatty acids (65).
Insulin-stimulated phosphorylation of FoxO1 by Akt causes nuclear export, resulting in ubiquitination-mediated proteasomal degradation (66), and down-regulation of target gene transcription (43). Indeed, we confirmed the ability of insulin to cause nuclear export of FoxO1 in HepG2 cells (Fig. 4D). FoxO1 activity is also stimulated by deacetylation, catalyzed by sirtuin1 (67). Inhibition of sirtuin1 in HepG2 cells with Ex-527 incubated in serum-free medium did not affect RDH10 expression (data not shown), indicating that acetylation is not involved in regulation of RDH10, at least under the conditions tested.
Recent insight into the functions of insulin receptor substrates IRS1 and IRS2 indicate that both coordinate responses to insulin via FoxO1 (68, 69). During the re-fed state, insulin activates IRS1 to suppress FoxO1 and allow an increase in glucokinase and sterol regulatory element-binding transcription factor 1 (SREBF1, a.k.a. SREBP1) expression that promotes glycolysis and lipid biosynthesis, respectfully. In contrast, insulin inhibits IRS2. Because IRS2 stimulates gluconeogenesis by inducing PCK1 and G6PC in liver through FoxO1, the relatively low levels of insulin in the fasted state allow IRS2 to induce gluconeogenesis. Based on these insights, the higher insulin levels in the re-fed state would function through IRS1 to suppress RDH expression, whereas the lower insulin levels in the fasted state would allow IRS2 to induce RDH expression.
The actions of insulin and serum both prevented transcription and destabilized RDH mRNA. The effects differed in degree, and resulted in different levels of mRNA, but did not totally eliminate RDH mRNA. In both serum-free medium and insulin-containing serum-free medium, the amounts of RDH mRNA after 24 h remained 2–3-fold higher compared with serum-containing medium, because the initial concentrations were 2–3-fold higher. Destabilization of RDH mRNA by insulin depended on active PI3K, Akt, and inhibition of FoxO1. The reduction but not elimination of RDH mRNA by insulin and serum is compatible with the continued presence and biosynthesis of atRA, and a need for RDH to modulate basal gluconeogenesis and/or its other multiple functions, whether related or unrelated to energy balance, such as regulating proliferation (70).
FoxO1 binding sites have been identified in promoters of genes associated with retinoid metabolism, including dehydrogenase/reductase SDR family member 9 (Dhrs9), cellular retinol-binding protein type 1 (encoded by Rbp1), and Rdh8, but not in Rdh1 or Rdh10 (31). To address the impact of FoxO1-regulated expression of retinoid genes, atRA target genes Pck1 and Pdk4 were measured in cells with FoxO1 knocked down. Induction of both genes in response to atRA or to its precursor, retinol, were blunted. The dose (20 μm) of atRA used, however, exceeded those found in tissues by 400–4000-fold, and those used in vitro by 20-fold. Nevertheless, these results are also consistent with FoxO1 linking retinoid metabolism and hepatic gluconeogenesis. Multiple putative FoxO/A binding sites were found in the 5′-untranslated region of Rdh10 (Encyclopedia of DNA Elements Consortium, ENCODE; University of California Southern California genome browser). Future work will determine whether these or other sites are true response elements.
Regulation by insulin signaling prompts the question whether abnormal RDH expression associates with human disease. For example, impaired insulin secretion and insulin resistance during diabetes predicts elevated RDH expression and atRA synthesis (71). Conversely, enhancement of insulin signaling through PI3K/Akt from loss of phosphatase and tensin homolog deleted on chromosome 10 in cancer implicates decreased RDH (72). Reduced RDH expression would decrease atRA biosynthesis, which could affect tumor differentiation and/or aggressiveness. Various cancers show alterations in the atRA signaling pathway, such as loss of atRA receptors (73, 74) and down-regulation of atRA chaperons (75, 76). atRA biosynthesis is impaired in breast cancer cell lines relative to normal cells (77). Reduced atRA synthesis in the Rbp1-null mouse is consistent with increased mammary tumors, and suggests consequences of the epigenetic silencing of Rbp1 in 25% of human breast cancers (62). In contrast, overexpression of RDH10 in HepG2 cells reduces proliferation (70).
In summary, despite the importance of atRA in vertebrate physiology, little is known about regulation of the rate-limiting enzymes (Rdh) that catalyze its biosynthesis. This report reveals a reciprocal interaction in which energy status regulates atRA biosynthesis through insulin that would attenuate energy balance regulation by atRA. These data predict altered atRA concentrations in diseases characterized by dysfunctional insulin signaling, including diabetes and cancer.
Acknowledgment
We are grateful to Domenico Accili for a gift of the dominant-negative adenovirus construct (Δ256).
This work was supported, in whole or in part, by National Institutes of Health Grants DK090522 and AA017927 (to J. L. N.).
- atRA
- all-trans-retinoic acid
- ActD
- actinomycin D
- CHX
- cycloheximide
- dnFoxO1
- dominant-negative FoxO
- FoxO1
- forkhead box other 1
- RDH
- retinol dehydrogenase
- DHRS3
- dehydrogenase reductase 3
- mTORC1
- mammalian target of rapamycin complex 1
- Glc-6-P
- glucose-6-phosphatase
- G6pc
- glucose-6-phosphatase
- qPCR
- quantitative PCR.
REFERENCES
- 1. Ross A. C. (2007) Vitamin A supplementation and retinoic acid treatment in the regulation of antibody responses in vivo. Vitam. Horm. 75, 197–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maden M. (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755–765 [DOI] [PubMed] [Google Scholar]
- 3. Pino-Lagos K., Guo Y., Noelle R. J. (2010) Retinoic acid: a key player in immunity. BioFactors 36, 430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gudas L. J. (2012) Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 1821, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rhinn M., Dollé P. (2012) Retinoic acid signalling during development. Development 139, 843–858 [DOI] [PubMed] [Google Scholar]
- 6. Frey S. K., Vogel S. (2011) Vitamin A metabolism and adipose tissue biology. Nutrients 3, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berry D. C., Noy N. (2012) Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim. Biophys. Acta 1821, 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonet M. L., Ribot J., Palou A. (2012) Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 1821, 177–189 [DOI] [PubMed] [Google Scholar]
- 9. Shin D.-J., Odom D. P., Scribner K. B., Ghoshal S., McGrane M. M. (2002) Retinoid regulation of the phosphoenolpyruvate carboxykinase gene in liver. Mol. Cell. Endocrinol. 195, 39–54 [DOI] [PubMed] [Google Scholar]
- 10. Mercader J., Ribot J., Murano I., Felipe F., Cinti S., Bonet M. L., Palou A. (2006) Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology 147, 5325–5332 [DOI] [PubMed] [Google Scholar]
- 11. Berry D. C., Noy N. (2009) All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 29, 3286–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zizola C. F., Frey S. K., Jitngarmkusol S., Kadereit B., Yan N., Vogel S. (2010) Cellular retinol-binding protein type I (CRBP-I) regulates adipogenesis. Mol. Cell. Biol. 30, 3412–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kane M. A., Folias A. E., Pingitore A., Perri M., Krois C. R., Ryu J. Y., Cione E., Napoli J. L. (2011) CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol. Cell. Biol. 31, 3277–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Germain P., Chambon P., Eichele G., Evans R. M., Lazar M. A., Leid M., De Lera A. R., Lotan R., Mangelsdorf D. J., Gronemeyer H. (2006) International Union of Pharmacology: LX. retinoic acid receptors. Pharmacol. Rev. 58, 712–725 [DOI] [PubMed] [Google Scholar]
- 15. Shaw N., Elholm M., Noy N. (2003) Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem. 278, 41589–41592 [DOI] [PubMed] [Google Scholar]
- 16. Sugiyama T., Scott D. K., Wang J. C., Granner D. K. (1998) Structural requirements of the glucocorticoid and retinoic acid response units in the phosphoenolpyruvate carboxykinase gene promoter. Mol. Endocrinol. 12, 1487–1498 [DOI] [PubMed] [Google Scholar]
- 17. Mercader J., Palou A., Bonet M. L. (2010) Induction of uncoupling protein-1 in mouse embryonic fibroblast-derived adipocytes by retinoic acid. Obesity 18, 655–662 [DOI] [PubMed] [Google Scholar]
- 18. Noy N. (2013) The one-two punch: retinoic acid suppresses obesity both by promoting energy expenditure and by inhibiting adipogenesis. Adipocyte 2, 184–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Napoli J. L. (2012) Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 1821, 152–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang M., Chen W., Smith S. M., Napoli J. L. (2001) Molecular characterization of a mouse short chain dehydrogenase/reductase active with all-trans-retinol in intact cells, mRDH1. J. Biol. Chem. 276, 44083–44090 [DOI] [PubMed] [Google Scholar]
- 21. Belyaeva O. V., Johnson M. P., Kedishvili N. Y. (2008) Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J. Biol. Chem. 283, 20299–20308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu B. X., Chen Y., Chen Y., Fan J., Rohrer B., Crouch R. K., Ma J.-X. (2002) Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Invest. Ophthalmol. Vis. Sci. 43, 3365–3372 [PubMed] [Google Scholar]
- 23. Adams M. K., Belyaeva O. V., Wu L., Kedishvili N. Y. (2014) The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 289, 14868–14880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang M., Hu P., Krois C. R., Kane M. A., Napoli J. L. (2007) Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. FASEB J. 21, 2886–2896 [DOI] [PubMed] [Google Scholar]
- 25. Rhinn M., Schuhbaur B., Niederreither K., Dollé P. (2011) Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. U.S.A. 108, 16687–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van den Berghe G. (1991) The role of the liver in metabolic homeostasis: implications for inborn errors of metabolism. J. Inherit. Metab. Dis. 14, 407–420 [DOI] [PubMed] [Google Scholar]
- 27. Moore M. C., Coate K. C., Winnick J. J., An Z., Cherrington A. D. (2012) Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. 3, 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tobe K., Kadowaki T., Hara K., Gotoh Y., Kosako H., Matsuda S., Tamemoto H., Ueki K., Akanuma Y., Nishida E. (1992) Sequential activation of MAP kinase activator, MAP kinases, and S6 peptide kinase in intact rat liver following insulin injection. J. Biol. Chem. 267, 21089–21097 [PubMed] [Google Scholar]
- 29. Siddle K. (2011) Signalling by insulin and IGF receptors: supporting acts and new players. J. Mol. Endocrinol. 47, R1–R10 [DOI] [PubMed] [Google Scholar]
- 30. Vander Kooi B. T., Streeper R. S., Svitek C. A., Oeser J. K., Powell D. R., O'Brien R. M. (2003) The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J. Biol. Chem. 278, 11782–11793 [DOI] [PubMed] [Google Scholar]
- 31. Shin D.-J., Joshi P., Hong S.-H., Mosure K., Shin D.-G., Osborne T. F. (2012) Genome-wide analysis of FoxO1 binding in hepatic chromatin: potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Res. 40, 11499–11509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakae J., Kitamura T., Silver D. L., Accili D. (2001) The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 108, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kane M. A., Napoli J. L. (2010) Quantification of endogenous retinoids. Methods Mol. Biol. 652, 1–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kane M. A., Folias A. E., Wang C., Napoli J. L. (2008) Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem. 80, 1702–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kane M. A., Folias A. E., Napoli J. L. (2008) HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal. Biochem. 378, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heikkinen S., Argmann C. A., Champy M.-F., Auwerx J. (2007) Evaluation of glucose homeostasis. Curr. Protoc. Mol. Biol. Chapter 29, Unit 29B.3 [DOI] [PubMed] [Google Scholar]
- 37. Zarrinpar A., Loomba R. (2012) Review article: the emerging interplay among the gastrointestinal tract, bile acids and incretins in the pathogenesis of diabetes and non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 36, 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eng J., Kleinman W. A., Singh L., Singh G., Raufman J. P. (1992) Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 267, 7402–7405 [PubMed] [Google Scholar]
- 39. Pirkmajer S., Chibalin A. V. (2011) Serum starvation: caveat emptor. Am. J. Physiol. Cell Physiol. 301, C272–C279 [DOI] [PubMed] [Google Scholar]
- 40. Sancho P., Fabregat I. (2010) NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 285, 24815–24824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mithieux G., Vidal H., Zitoun C., Bruni N., Daniele N., Minassian C. (1996) Glucose-6-phosphatase mRNA and activity are increased to the same extent in kidney and liver of diabetic rats. Diabetes 45, 891–896 [DOI] [PubMed] [Google Scholar]
- 42. Frescas D., Valenti L., Accili D. (2005) Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 280, 20589–20595 [DOI] [PubMed] [Google Scholar]
- 43. Nakae J., Park B. C., Accili D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J. Biol. Chem. 274, 15982–15985 [DOI] [PubMed] [Google Scholar]
- 44. Wolf G., Wagle S. R., Lane M. D., Johnson B. C. (1958) Studies on the function of vitamin A in metabolism by the use of radioactive metabolic intermediates. Prog. Nucl. Energy 6 Biol. Sci. 2, 457–468 [PubMed] [Google Scholar]
- 45. Lucas P. C., O'Brien R. M., Mitchell J. A., Davis C. M., Imai E., Forman B. M., Samuels H. H., Granner D. K. (1991) A retinoic acid response element is part of a pleiotropic domain in the phosphoenolpyruvate carboxykinase gene. Proc. Natl. Acad. Sci. U.S.A. 88, 2184–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scott D. K., Mitchell J. A., Granner D. K. (1996) Identification and charaterization of the second retinoic acid response element in the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 271, 6260–6264 [DOI] [PubMed] [Google Scholar]
- 47. Kido Y., Nakae J., Accili D. (2001) Clinical review 125: the insulin receptor and its cellular targets. J. Clin. Endocrinol. Metab. 86, 972–979 [DOI] [PubMed] [Google Scholar]
- 48. Martín M., Gallego-Llamas J., Ribes V., Kedinger M., Niederreither K., Chambon P., Dollé P., Gradwohl G. (2005) Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev. Biol. 284, 399–411 [DOI] [PubMed] [Google Scholar]
- 49. Pérez R. J., Benoit Y. D., Gudas L. J. (2013) Deletion of retinoic acid receptor β (RARβ) impairs pancreatic endocrine differentiation. Exp. Cell Res. 319, 2196–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chertow B. S., Blaner W. S., Baranetsky N. G., Sivitz W. I., Cordle M. B., Thompson D., Meda P. (1987) Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J. Clin. Invest. 79, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao S., Li R., Li Y., Chen W., Zhang Y., Chen G. (2012) Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 90, 142–152 [DOI] [PubMed] [Google Scholar]
- 52. Ishida T., Chap Z., Chou J., Lewis R., Hartley C., Entman M., Field J. B. (1983) Differential effects of oral, peripheral intravenous, and intraportal glucose on hepatic glucose uptake and insulin and glucagon extraction in conscious dogs. J. Clin. Invest. 72, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Myers S. R., Biggers D. W., Neal D. W., Cherrington A. D. (1991) Intraportal glucose delivery enhances the effects of hepatic glucose load on net hepatic glucose uptake in vivo. J. Clin. Invest. 88, 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bergman R. N., Beir J. R., Hourigan P. M. (1982) Intraportal glucose infusion matched to oral glucose absorption: lack of evidence for “gut factor” involvement in hepatic glucose storage. Diabetes 31, 27–35 [DOI] [PubMed] [Google Scholar]
- 55. Ohlsson L., Kohan A. B., Tso P., Ahrén B. (2014) GLP-1 released to the mesenteric lymph duct in mice: effects of glucose and fat. Regul. Pept. 189, 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamamoto Y., Zolfaghari R., Ross A. C. (2000) Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. FASEB J. 14, 2119–2127 [DOI] [PubMed] [Google Scholar]
- 57. Ross A. C., Cifelli C. J., Zolfaghari R., Li N.-Q. (2011) Multiple cytochrome P-450 genes are concomitantly regulated by vitamin A under steady-state conditions and by retinoic acid during hepatic first-pass metabolism. Physiol. Genomics 43, 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuura T., Ross A. C. (1993) Regulation of hepatic lecithin:retinol acyltransferase activity by retinoic acid. Arch. Biochem. Biophys. 301, 221–227 [DOI] [PubMed] [Google Scholar]
- 59. Kousteni S. (2012) FoxO1, the transcriptional chief of staff of energy metabolism. Bone 50, 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barthel A., Schmoll D., Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 61. Hall R. K., Yamasaki T., Kucera T., Waltner-Law M., O'Brien R., Granner D. K. (2000) Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin: the role of winged helix/forkhead proteins. J. Biol. Chem. 275, 30169–30175 [DOI] [PubMed] [Google Scholar]
- 62. Pierzchalski K., Yu J., Norman V., Kane M. A. (2013) CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB J. 27, 1904–1916 [DOI] [PubMed] [Google Scholar]
- 63. Matsumoto M., Pocai A., Rossetti L., Depinho R. A., Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 64. Nakae J., Biggs W. H., 3rd, Kitamura T., Cavenee W. K., Wright C. V., Arden K. C., Accili D. (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 32, 245–253 [DOI] [PubMed] [Google Scholar]
- 65. Haeusler R. A., Han S., Accili D. (2010) Hepatic FoxO1 ablation exacerbates lipid abnormalities during hyperglycemia. J. Biol. Chem. 285, 26861–26868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matsuzaki H., Daitoku H., Hatta M., Tanaka K., Fukamizu A. (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 100, 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schwer B., Verdin E. (2008) Conserved metabolic regulatory functions of sirtuins. Cell Metab. 7, 104–112 [DOI] [PubMed] [Google Scholar]
- 68. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kubota N., Kubota T., Itoh S., Kumagai H., Kozono H., Takamoto I., Mineyama T., Ogata H., Tokuyama K., Ohsugi M., Sasako T., Moroi M., Sugi K., Kakuta S., Iwakura Y., Noda T., Ohnishi S., Nagai R., Tobe K., Terauchi Y., Ueki K., Kadowaki T. (2008) Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 8, 49–64 [DOI] [PubMed] [Google Scholar]
- 70. Rossi E., Picozzi P., Bodega B., Lavazza C., Carlo-Stella C., Marozzi A., Ginelli E. (2007) Forced expression of RDH10 gene retards growth of HepG2 cells. Cancer Biol. Ther. 6, 238–244 [DOI] [PubMed] [Google Scholar]
- 71. Lin H. V., Accili D. (2011) Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 14, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A., Jr., Kinzler K. W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chakravarti N., Lotan R., Diwan A. H., Warneke C. L., Johnson M. M., Prieto V. G. (2007) Decreased expression of retinoid receptors in melanoma: entailment in tumorigenesis and prognosis. Clin. Cancer Res. 13, 4817–4824 [DOI] [PubMed] [Google Scholar]
- 74. Tanabe K., Utsunomiya H., Tamura M., Niikura H., Takano T., Yoshinaga K., Nagase S., Suzuki T., Ito K., Matsumoto M., Hayashi S., Yaegashi N. (2008) Expression of retinoic acid receptors in human endometrial carcinoma. Cancer Sci. 99, 267–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Calmon M. F., Rodrigues R. V., Kaneto C. M., Moura R. P., Silva S. D., Mota L. D., Pinheiro D. G., Torres C., de Carvalho A. F., Cury P. M., Nunes F. D., Nishimoto I. N., Soares F. A., da Silva A. M., Kowalski L. P., Brentani H., Zanelli C. F., Silva W. A., Jr., Rahal P., Tajara E. H., Carraro D. M., Camargo A. A., Valentini S. R. (2009) Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia 11, 1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Campos B., Warta R., Chaisaingmongkol J., Geiselhart L., Popanda O., Hartmann C., von Deimling A., Unterberg A., Plass C., Schmezer P., Herold-Mende C. (2012) Epigenetically mediated downregulation of the differentiation-promoting chaperon protein CRABP2 in astrocytic gliomas. Int. J. Cancer 131, 1963–1968 [DOI] [PubMed] [Google Scholar]
- 77. Mira-Y-Lopez R., Zheng W. L., Kuppumbatti Y. S., Rexer B., Jing Y., Ong D. E. (2000) Retinol conversion to retinoic acid is impaired in breast cancer cell lines relative to normal cells. J. Cell. Physiol. 185, 302–309 [DOI] [PubMed] [Google Scholar]