

Background: LIP5 and ESCRT-III are regulators of VPS4 in biological processes that require the ESCRT function.
Results: Structural and functional analyses of human VPS4, LIP5, and ESCRT-III interactions are presented.
Conclusion: ESCRT-III protein CHMP5 inhibits LIP5-mediated VPS4 activation by inducing a moderate conformational change within LIP5.
Significance: This study reveals important mechanistic differences in VPS4 regulation between fungi and metazoans.
Keywords: ATPase, Crystal Structure, Endosomal Sorting Complexes Required for Transport (ESCRT), Membrane Trafficking, Protein-Protein Interaction, CHMP5, LIP5, MIT-MIM, VPS4, Multivesicular Body
Abstract
Disassembly of the endosomal sorting complex required for transport (ESCRT) machinery from biological membranes is a critical final step in cellular processes that require the ESCRT function. This reaction is catalyzed by VPS4, an AAA-ATPase whose activity is tightly regulated by a host of proteins, including LIP5 and the ESCRT-III proteins. Here, we present structural and functional analyses of molecular interactions between human VPS4, LIP5, and the ESCRT-III proteins. The N-terminal domain of LIP5 (LIP5NTD) is required for LIP5-mediated stimulation of VPS4, and the ESCRT-III protein CHMP5 strongly inhibits the stimulation. Both of these observations are distinct from what was previously described for homologous yeast proteins. The crystal structure of LIP5NTD in complex with the MIT (microtubule-interacting and transport)-interacting motifs of CHMP5 and a second ESCRT-III protein, CHMP1B, was determined at 1 Å resolution. It reveals an ESCRT-III binding induced moderate conformational change in LIP5NTD, which results from insertion of a conserved CHMP5 tyrosine residue (Tyr182) at the core of LIP5NTD structure. Mutation of Tyr182 partially relieves the inhibition displayed by CHMP5. Together, these results suggest a novel mechanism of VPS4 regulation in metazoans, where CHMP5 functions as a negative allosteric switch to control LIP5-mediated stimulation of VPS4.
Introduction
The endosomal sorting complex required for transport (ESCRT)3 machinery is responsible for catalyzing membrane fission reactions away from the cytoplasm (1–4). ESCRT was initially identified in the yeast multivesicular body biogenesis pathway (5) and was later shown to be involved in other biological processes in higher eukaryotic cells, including enveloped virus budding from the plasma membrane (6), resolution of the mid-body structure during cytokinesis (7), and, more recently, repair of small plasma membrane wounds (8). The core components of the ESCRT machinery have shown remarkable conservation in eukaryotic cells. Among them, Vps4 and the ESCRT-III complex are required for all known ESCRT-dependent cellular processes and are thought to constitute a minimal membrane fission machine.
The ESCRT-III complex consists of four core subunits: Vps20 (human ortholog: CHMP6), Snf7 (CHMP4), Vps24 (CHMP3), and Vps2 (CHMP2) (9, 10). These subunits normally exist as inactive, soluble proteins in the cytoplasm. They polymerize into membrane-bound helical filaments when activated by upstream factors (11–15). ESCRT-III polymerization plays a critical role in the membrane fission reaction (16–19). At the end of the reaction cycle, the ESCRT-III filaments are disassembled and extracted from the membrane and returned to their inactive states. Disassembly of the ESCRT-III complex is catalyzed by Vps4, an ATPase associated with diverse cellular activities (AAA-ATPase) (20–24). There is also evidence suggesting that Vps4 directly contributes to membrane fission in some cellular processes (8, 20, 25–28). Because Vps4 is the only energy-consuming enzyme in the ESCRT machinery, it has long been viewed as the master regulator of the system. Understanding the structure, function, and regulation of Vps4 is an important goal in understanding ESCRT biology.
The structure of Vps4 contains an N-terminal domain, a linker, and a C-terminal canonical AAA-ATPase cassette (29–31). The N-terminal domain is also a microtubule-interacting and transport (MIT) domain, which the protein uses to bind the MIT-interacting motifs (MIMs) located within the C terminus of the ESCRT-III proteins (32, 33). Unique to the Vps4 structure is a small domain of three anti-parallel β-strands called the “β-domain,” inserted within the AAA cassette. Vps4 can exist in two quaternary structures, an inactive monomer/dimer and an active high-order oligomer (34). Vps4 oligomerization and its membrane recruitment represent major steps in Vps4 regulation (35), which are mediated by a host of proteins, including its cofactor Vta1 (LIP5) and its substrates the ESCRT-III proteins (36–39).
The structure of Vta1 contains two domains that are connected by a long, non-conserved flexible linker (36, 41). The N-terminal domain consists of two tandem MIT domains (41). The C-terminal domain is known as the Vta1-SBP1-LIP5 (VSL) domain that directly binds to the β-domain of Vps4 (36). In yeast, Vta1 can stimulate the ATPase activity of Vps4 about 3-fold, and this stimulation is largely attributable to the fact that interaction between the VSL domain and Vps4 stabilizes the active Vps4 oligomeric structure (41). Recent work has also identified a region immediately N-terminal to the VSL domain in yeast Vta1 that can further stimulate Vps4 activity (named VSE) by increasing the catalytic efficiency of the ATPase (42).
The ESCRT-III proteins also have regulatory activity toward Vps4. Binding of the Vps4 MIT domain to the MIM sequences displayed by the membrane-bound ESCRT-III filaments can serve to recruit, assemble, and activate the ATPase (21, 25). Beyond the four core ESCRT-III subunits, there exist several proteins whose structural folds are similar to ESCRT-III, including Did2 (CHMP1), Vps60 (CHMP5), and Ist1 (IST1) (17, 43). They have also been implicated in Vps4 regulation and in some cases are thought to aid in the recruitment of Vps4 to the membrane (44). The regulatory role of Did2, Vps60, and Ist1, in particular, overlaps with that of Vta1. Biochemical analysis of protein-protein interactions has shown that Did2, Vps60, and Ist1 can bind to the N-terminal domain of Vta1 (41, 45). Studies in yeast revealed that binding of Did2 and Vps60 further stimulates Vps4 activity in a Vta1-dependent manner (37, 42). These results suggest that Did2 and Vps60 function as allosteric ligands of Vps4.
Most of our current understanding with regard to the mechanism of Vps4 regulation by Vta1 and the ESCRT-III proteins has been derived from studies in yeast. Because the metazoan ESCRT machinery performs additional functions not described in yeast, it is important that parallel mechanistic studies are carried out in the native context. To this end, we performed structural and functional analyses of molecular interactions between human VPS4, LIP5, and regulatory ESCRT-III proteins CHMP1B and CHMP5 to gain insights into regulation of the ESCRT machinery in metazoans. Although LIP5 strongly stimulates VPS4 ATPase activity, it requires both the N- and C-terminal domains of the protein for full stimulation. Furthermore, binding of CHMP5 to the N-terminal domain of LIP5 strongly inhibits LIP5-mediated VPS4 stimulation, which is diametrically opposed to what was observed in yeast (37). High-resolution crystal structure of human LIP5-CHMP1B-CHMP5 complex revealed that CHMP1B and CHMP5 utilize distinct binding mechanism to engage LIP5. In particular, binding of CHMP5 leads a moderate conformational change that alters the MIT domain interface in the N-terminal domain of LIP5. We propose a novel mechanism where induction of this conformational change precludes some critical interactions between LIP5 and VPS4, hence the inhibition of LIP5-mediated VPS4 stimulation by CHMP5 in metazoans.
EXPERIMENTAL PROCEDURES
Cloning and Expression
All proteins and protein fragments used in the study were expressed as N-terminal His6-SUMO-tagged fusion proteins by cloning encoding cDNAs into a modified pET-28b vector. Point mutations were generated using a standard PCR mutagenesis protocol (Stratagene). Native proteins were expressed in Escherichia coli Rosetta (DE3) cells. Briefly, bacterial cells were grown to mid-log phase in LB medium at 37 °C and induced with 0.2 mm isopropyl β-d-1-thiogalactopyranoside for an additional 16–20 h at 16 °C. Selenomethionyl derivative proteins were expressed in E. coli B834 (DE3) cells using a minimal medium where methionine was replaced with selenomethionine.
Protein Purification
All proteins were purified using a similar protocol with adjustments made when necessary. Bacterial cells were lysed by sonication in buffer A (25 mm Tris, pH 8.0, 300 mm NaCl, 5 mm 2-mercaptoethanol, and 10 μg/ml (w/v) phenylmethylsulfonyl fluoride). Cell lysate was cleared by centrifugation, and supernatant was loaded onto a Ni2+-nitrilotriacetic acid affinity column. Bound protein was washed with buffer A and eluted with buffer A supplemented with 250 mm imidazole. Fractions were pooled and digested with Ulp1 protease to cleave off the His6-SUMO tag while being dialyzed against 50 mm Tris, pH 8.0, 25 mm NaCl overnight. Dialyzed protein sample was passed through a second Ni2+-nitrilotriacetic acid column to remove the tag and the undigested protein.
Proteins used in crystallization were further purified. LIP51–162 was loaded on a Source-S (GE Healthcare) cation exchange column equilibrated in buffer C (50 mm HEPES, pH 7.0, 50 mm NaCl, 1 mm EDTA, 1 mm DTT) and eluted using a NaCl gradient in buffer C. Fractions containing the protein were pooled, concentrated, and loaded onto a SuperdexTM 75 (GE Healthcare) column equilibrated in buffer C. For the two protein complexes, CHMP1B176–199 and/or CHMP5151–190 were mixed with purified LIP51–162 in an ∼2:1 ratio. Protein mixtures were concentrated and loaded onto a SuperdexTM 200 column equilibrated in buffer C. Complex formation was confirmed by elution position change on the gel filtration column.
Crystallization and Data Collection
Crystals of the LIP5 N-terminal domain (LIP5NTD) were grown using the hanging drop vapor diffusion method. LIP51–162 (15 mg/ml) was mixed in a 1:1 ratio with a reservoir solution of 7.5–9% (w/v) polyethylene glycol 8000 (PEG 8000), 3% (v/v) 1,4-butanediol, 0.1 m cacodylate, pH 7.0, 0.2 m sodium acetate to a final volume of 4 μl, and the mixture was equilibrated against 500 μl of the reservoir solution at 4 °C. Small hexagonal crystals appeared after 2 weeks. They were transferred into a solution containing 20% (w/v) PEG 8000, 20% (v/v) glycerol, 8% (v/v) butanediol, 0.1 m cacodylate, pH 7.0, 0.2 m sodium acetate and immediately stored under liquid nitrogen. The selenomethionyl derivative LIP5NTD crystals were produced and harvested in a similar manner.
Crystals of the LIP5NTD-CHMP1B complex were grown using the sitting drop vapor diffusion method. LIP51–162-CHMP1B176–199 (12.5 mg/ml) was mixed in a 1:1 ratio with 16% (v/v) 2-methyl-2,4-pentanediol, 0.1 m Tris, pH 9.0, to a final volume of 4 μl, and the mixture was equilibrated against 500 μl of 8% (v/v) 2-methyl-2,4-pentanediol, 0.1 m Tris, pH 9.0, at 4 °C. Two crystals with different morphology appeared. Long needle-like crystals would appear immediately and then dissolve over time. Diamond-shaped crystals would appear later and were used for data collection. Crystals were dialyzed against 30% (v/v) 2-methyl-2,4-pentanediol, 0.1 m Tris, pH 9.0, for 16–24 h before being stored under liquid nitrogen.
Crystals of the LIP5NTD-CHMP1B-CHMP5 complex were also grown using the sitting drop vapor diffusion method. LIP51–162-CHMP1B176–199-CHMP5151–190 (13.7 mg/ml) was mixed in a 1:1 ratio with a reservoir solution of 19% (w/v) PEG 4000, 25 mm sodium acetate, pH 5.5, to a final volume of 4 μl, and the mixture was equilibrated against 500 μl of the reservoir solution at 4 °C. Large diamond-shaped crystals would appear after a few days. They were dialyzed against 25% (w/v) PEG 4000, 20% (v/v) ethylene glycol, 25 mm sodium acetate, pH 5.5, before being stored under liquid nitrogen.
All data were collected under liquid nitrogen temperature at Advanced Photon Source beamline 21-ID and processed and scaled using either XDS (46) or HKL2000 (HKL Research Inc.). Data collection statistics are shown in Table 1.
TABLE 1.
Crystallographic data statistics
| LIP5NTD |
LIP5NTD-CHMP1B (native) | LIP5NTD-CHMP1B-CHMP5 (native) | ||
|---|---|---|---|---|
| Native | SeMet | |||
| Data collection | ||||
| Space group | P6522 | P6522 | P212121 | P21 |
| Unit cell parameters | ||||
| a, b, c (Å) | 76.4, 76.4, 355.9 | 76.1, 76.1, 355.1 | 52.8, 55.2, 194.9 | 34.2, 60.5, 52.0 |
| α, β, γ (degrees) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 | 90.0, 90.5, 90.0 |
| Molecules/asymmetric unit | 3 | 3 | 2 | 1 |
| Wavelength (Å) | 0.9792 | 0.9792 | 0.9787 | 0.9998 |
| Resolution (Å) | 3.0 | 3.3 | 2.2 | 1.0 |
| Unique reflections | 13124 | 9907 | 29702 | 99846 |
| Redundancya | 6.9 (7.1) | 18.3 (19.0) | 7.3 (7.4) | 4.5 (3.5) |
| Completeness (%) | 98.8 (99.8) | 98.2 (99.1) | 99.9 (100) | 87.4 (46.1) |
| Average I/σ (I) | 18.8 (3.6) | 22.4 (5.0) | 19.4 (3.2) | 50.6 (5.4) |
| Rmerge | 0.10 (0.60) | 0.16 (0.56) | 0.11 (0.55) | 0.05 (0.27) |
| Refinement | ||||
| Residues in the structure | ||||
| LIP5 | 3–158 | 1–162 | 4–162 | |
| CHMP1B | 183–199 | 185–199 | ||
| CHMP5 | 151–190 | |||
| Resolution range (Å) | 50.0–3.0 | 50.0–2.2 | 50.0–1.0 | |
| Rwork (%) | 25.0 | 19.6 | 16.5 | |
| Rfree (%) | 28.7 | 22.7 | 18.1 | |
| Root mean square deviations | ||||
| Bond lengths (Å) | 0.010 | 0.009 | 0.008 | |
| Bond angles (degrees) | 1.06 | 1.22 | 1.28 | |
| B-Factor average (A2) | 71.1 | 37.6 | 8.5 | |
| Ramachandran plot | ||||
| Most favored (%) | 97.7 | 98.0 | 98.8 | |
| Allowed (%) | 1.2 | 1.7 | 1.2 | |
| Outliers (%) | 1.2 | 0.3 | 0.0 | |
| Protein Data Bank accession code | 4TXP | 4TXQ | 4TXR | |
a Values in parentheses are for the specified high-resolution bin.
Structure Determination and Refinement
The crystal structure of LIP5NTD was determined using the single-wavelength anomalous diffraction method. Twelve of 15 expected selenium sites (5 sites/LIP5 molecule) were located using Phenix (47). Phases were calculated and refined. After several rounds of density modification, the resulting electron density map would allow the building of a model for one of the three molecules in the asymmetric unit under the guidance of the yeast Vta1 structure (41). This model was partially refined and used to search for the other two molecules against the native data set by molecular replacement. BUSTER was used for further structure refinement with strict non-crystallographic symmetry applied throughout (48). The crystal structures of the LIP5NTD-CHMP1B complex and the LIP5NTD-CHMP1B-CHMP5 complex were solved by molecular replacement using the refined LIP5NTD structure as the initial search model. Both structures were refined using Phenix. Non-crystallographic symmetry restrictions were applied in the early rounds of refinement for the LIP5NTD-CHMP1B complex structure. COOT was used for model building and adjustment (49). Structure refinement statistics are shown in Table 1.
GST Pull-down Analysis
GST pull-down analysis was performed following standard protocols in phosphate-buffered saline (PBS) solution supplemented with 1 mm DTT and 0.1% (v/v) Tween 20 (30). Specific samples were also supplemented with 2 mm ATP. Briefly, 1 ml of bacterial cell lysate containing GST- or GST-SUMO-tagged “bait” proteins was incubated with 20 μl of glutathione-agarose bead slurry for 1 h at 4 °C. The protein-immobilized beads were then washed three times with the buffer and incubated with 1 ml of “prey” proteins for 1 h at 4 °C. Finally, the beads were washed three times with the buffer again before bound proteins were analyzed on SDS-PAGE and visualized by Coomassie Blue staining.
Malachite Green ATPase Assay
The Malachite Green assay was used to measure the ATPase activity of VPS4B as described previously with modifications (50). Proteins used in the assay were dialyzed in the ATPase buffer (100 mm Tris-HCl, pH 7.4, 20 mm KCl, 6 mm MgCl2) and stored at −80 °C. Briefly, VPS4B (or VPS4B79–444) was incubated with buffer or LIP5 and with additional ESCRT-III (all final concentrations) where indicated in a 15-μl protein mixture. The reaction was started with the addition of 2 mm ATP (final concentration) to a final reaction volume of 25 μl and was allowed to proceed for 1 h or another specified time. Malachite Green reagent (80 μl) was then added, and the reaction was quenched with the addition of 10 μl of 32% (w/v) sodium citrate. Samples were mixed and incubated at 37 °C for 10 min before A620 was measured on a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). To account for background ATP hydrolysis, signal from identically treated sample lacking ATPase was subtracted. Likewise, background activities of LIP5 or ESCRT-III were subtracted from samples with an identical composition but lacking ATPase. Each experiment was repeated three times in triplicate, where error bars represent S.E. Significances in the data were assessed using one-way analysis of variance in Prism5 (GraphPad).
RESULTS
The N-terminal Domain of LIP5 Is Required for LIP5-mediated VPS4 Stimulation
Previous studies in yeast have shown that Vta1 stimulated the ATPase activity of Vps4, and this stimulatory effect was attributed to the binding of its C-terminal domain (VSL or Vta1CTD) to Vps4 (37, 42). The N-terminal domain of Vta1 (Vta1NTD) was not required for the basal stimulatory activity, but binding of the ESCRT-III proteins Did2 and Vps60 to Vta1NTD provided an additional level of Vps4 stimulation. Based on these results, a model was proposed where the unstructured linker of Vta1 acts to inhibit the function of VSL and this inhibition is relieved upon binding of ESCRT-III to Vta1NTD (51). To determine whether a similar mechanism is utilized in metazoans, we probed LIP5-mediated regulation of VPS4 ATPase activity and its modulation by the ESCRT-III proteins. Earlier studies on human Vps4 orthologs (VPS4A and VPS4B) indicated that they have much weaker ATPase activity than yeast Vps4 (39). We observed similar results, but our study also indicated that VPS4B was slightly more active than VPS4A and more stable under experimental conditions. Therefore, VPS4B was used for the rest of our experiments.
We used the Malachite Green assay to measure the phosphate produced from ATP hydrolysis by VPS4B. At 2 mm ATP and submicromolar concentration of VPS4B, the rate of ATP hydrolysis was linear over 60 min with different ATPase and activator combinations used in our experiments (Fig. 1A). The ATPase activity of VPS4B was concentration-dependent and low at 125 nm protein concentration (3.6 ± 1.1 Pi/VPS4B/min). With the addition of 375 nm LIP5, we observed a dramatic increase in activity. Titration of LIP5 indicated concentration-dependent stimulation of VPS4B ATPase activity with a maximum activity of 39.2 ± 4.3 Pi/VPS4B/min (11-fold stimulation) (Fig. 1B).
FIGURE 1.

LIP5 stimulates the ATPase activity of VPS4. A, time course of ATP hydrolysis by VPS4B. Reactions with different combinations of VPS4B and LIP5 were allowed to proceed for 1 h. Production of inorganic phosphate was monitored at 10-min intervals. B, concentration-dependent stimulation of VPS4B by LIP5. The ATPase activity of VPS4B (125 nm) was measured in the presence of increasing concentrations of either LIP5 or LIP5253–307. Reactions were allowed to proceed for 1 h. C, schematic diagram showing the C-terminal fragments of LIP5 used in the assays and their corresponding Vta1 fragments. D, the C-terminal domain of LIP5 alone does not stimulate VPS4B. The ATPase activity of VPS4B (125 nm) was measured in the presence of various C-terminal fragments of LIP5 (375 nm). Reactions were allowed to proceed for 1 h. E, the C-terminal domain of LIP5 is responsible for binding VPS4B. Various C-terminal fragments of LIP5 were assayed for their binding affinity for VPS4B. The GST-tagged or GST-SUMO-tagged protein-immobilized glutathione beads were incubated with 1 ml of VPS4BE233Q (1 μm) for 1 h at 4 °C in the presence or absence of ATP. GST-SUMO was used as a tag for LIP5163–307 and LIP51–162/215–307, because the GST-tagged proteins run at a position similar to that of VPS4BE233Q. E233Q is an ATPase-deficient mutant of VPS4B. LIP51–162 lacks the VPS4B binding C-terminal domain and was intended as a negative control for binding. The arrow indicates the running position of VPS4BE233Q. Error bars, S.E.
To probe whether the C-terminal domain of LIP5 (LIP5CTD) alone was sufficient for this stimulatory activity, we generated a number of LIP5 truncation mutants that encompass the putative LIP5CTD plus different lengths of the linker region immediately N-terminal to LIP5CTD (Fig. 1C). The design of these mutants was based on the recent observation that whereas the VSL domain of Vta1 provided the basal Vps4 stimulatory activity, residues in the upstream VSE region could substantially potentiate the stimulation (42). Sequence alignment of LIP5/Vta1 homologs suggested that LIP5253–307 corresponds to Vta1275–330, which was the most potent stimulator of all Vta1 fragments. To our surprise, titration of LIP5253–307 to 125 nm VPS4B showed very low stimulatory activity toward the ATPase (Fig. 1B). Similarly, Vta1165–330, Vta1267–330, and Vta1290–330 were all stimulators of yeast Vps4, yet, correspondingly, LIP5 fragments (LIP5163–307, LIP5245–307, and LIP5265–307) were ineffective at a 375 nm protein concentration, where full-length LIP5 displayed maximum stimulatory activity (Fig. 1D) (42). Interestingly, although these LIP5 fragments were inactive in stimulation, they nevertheless retained binding to VPS4B to a similar extent as the full-length protein based on a GST pull-down analysis (Fig. 1E).
These observations suggested that residues beyond LIP5CTD and the linker were required for LIP5-mediated VPS4 stimulation. To further map the location of these residues, we generated additional LIP5 truncation mutants and examined their stimulatory activities toward VPS4B (Fig. 2A). As expected, a LIP5 fragment that contains LIP5CTD plus part of the linker (LIP5215–307) was not able to stimulate VPS4B. Neither was the N-terminal domain (LIP51–162) alone (Fig. 2B). However, we were able to partially restore the stimulatory activity of the full-length LIP5 by linking the above two fragments together (LIP51–162/215–307). Titration of LIP51–162/215–307 to 125 nm VPS4B showed concentration-dependent stimulation with a maximum activity of 11.8 ± 1.8 Pi/VPS4B/min (3-fold stimulation) (Fig. 2C). Hence, it appeared that both the N- and C-terminal domains of LIP5 were required for LIP5-mediated VPS4B stimulation. Interestingly, a physical connection between the two was important as adding them in trans could not reproduce the same effect (Fig. 2B).
FIGURE 2.
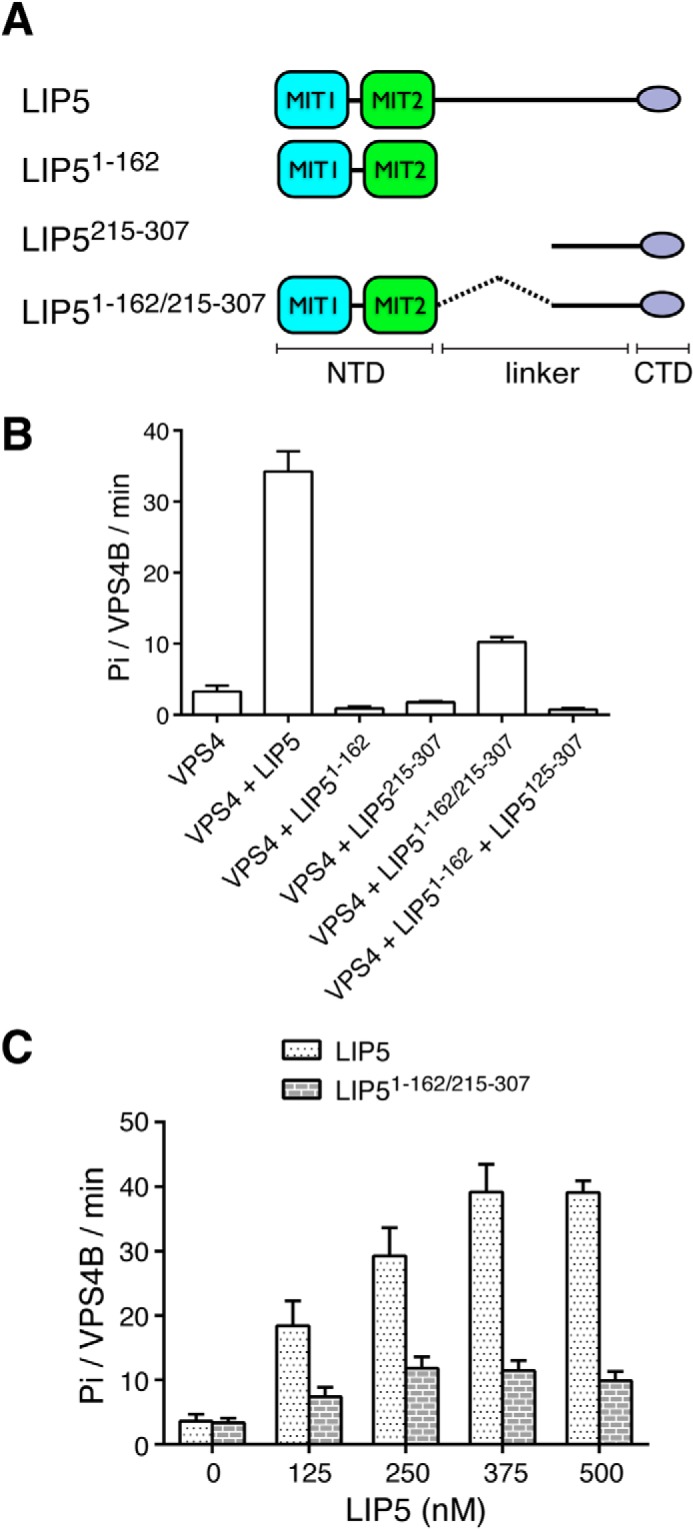
Both the N- and C-terminal domains of LIP5 are required for its VPS4 stimulatory activity. A, schematic diagram showing the LIP5 fragments used in the assays. B, linking the N- and C-terminal domains together (LIP51–162/215–307) provides partial VPS4B stimulation. The ATPase activity of VPS4B (125 nm) was measured in the presence of different LIP5 fragments (375 nm). Reactions were allowed to proceed for 1 h. Note that adding the N- and C-terminal domains in trans does not provide VPS4B stimulation. C, concentration-dependent stimulation of VPS4B by LIP51–162/215–307. The ATPase activity of VPS4B (125 nm) was measured in the presence of increasing concentrations of either LIP5 or LIP51–162/215–307. Reactions were allowed to proceed for 1 h. Error bars, S.E.
Binding of CHMP5 Inhibits LIP5-mediated VPS4 Stimulation
We next asked what effect metazoan ESCRT-III orthologs CHMP1B and CHMP5 would have on LIP5-mediated VPS4B stimulation. The minimal LIP5 binding fragments of CHMP1B and CHMP5 have been previously identified to be CHMP1B176–199 and CHMP5151–190 (37). These fragments were used in the following studies due to difficulties in purifying full-length proteins.
When CHMP5151–190 was included with 125 nm VPS4B and 375 nm LIP5 in the ATPase assay, a strong inhibition on the LIP5-mediated VPS4B stimulation was observed. The addition of 1.5 μm CHMP5 reduced the VPS4B ATPase activity to 21.5 ± 3.6 Pi/VPS4B/min, which represented a 44% inhibition (Fig. 3A). To determine whether this inhibitory effect was dependent on CHMP5 binding to LIP5, we used a previously described LIP5 binding-deficient mutant of CHMP5 (L163D/L167D/L170D/L174D or CHMP5L4D) (52). We saw that inclusion of 1.5 μm quadruple leucine mutant in the assay failed to reproduce a similar inhibitory effect (Fig. 3A). These data showed that although human CHMP5 regulated the VPS4B ATPase activity in a LIP5-dependent manner, its effect was diametrically opposed to what was observed in yeast, suggesting a different mechanism of regulation (37). Interestingly, the addition of 1.5 μm CHMP1B176–199 to the assay did not alter LIP5-mediated activation of the VPS4B ATPase activity (Fig. 3A). This suggested that CHMP1B and CHMP5 may play different roles in regulating LIP5 function.
FIGURE 3.

The ESCRT-III proteins regulate LIP5-mediated VPS4 stimulation. A, effect of CHMP5 or CHMP1B on LIP5-mediated VPS4B stimulation. The ATPase activity of VPS4B (125 nm) was measured in the presence of 375 nm LIP5 and increasing concentrations of CHMP5151–190, CHMP1B176–199, CHMP5151–190,L4D, or BSA. Reactions were allowed to proceed for 1 h. B, concentration-dependent stimulation of VPS4B79–444 by LIP5 or LIP51–162/215–307. The ATPase activity of VPS4B79–444 (375 nm) was measured in the presence of increasing concentrations of either LIP5 or LIP51–162/215–307. Reactions were allowed to proceed for 1 h (LIP5) or 15 min (LIP51–162/215–307). C, effect of CHMP5 or CHMP1B on LIP5-mediated VPS4B79–444 stimulation. The ATPase activity of VPS4B79–444 (375 nm) was measured in the presence of 375 nm LIP5 and increasing concentrations of CHMP5151–190, CHMP1B176–199, CHMP5151–190,L4D, or BSA. Reactions were allowed to proceed for 1 h. D, the inhibitory activity of CHMP5 does not depend on the linker region of LIP5. LIP5, LIP51–162/215–307, and LIP5165–307 (375 nm) were assayed for their abilities to stimulate VPS4B79–444 (375 nm) ATPase activity in the presence of increasing concentrations of CHMP5. Reactions were allowed to proceed for 1 h. Error bars, S.E.
The N terminus of VPS4B contains an MIT domain, which binds to the ESCRT-III proteins. This interaction has been shown to regulate the ATPase activity of VPS4B (39). To ascertain that the regulatory effects of CHMP5 seen above were solely due to its interaction with LIP5 but not with VPS4B, we repeated the above experiments using an MIT domain truncation mutant of VPS4B (VPS4B79–444). VPS4B79–444 had a weaker basal ATPase activity than the full-length protein at comparable protein concentrations (Fig. 1A) but displayed a similar concentration-dependent LIP5 stimulation profile (Fig. 3B). At 375 nm VPS4B79–444, maximum activity was reached at 750 nm LIP5 with more than 40-fold stimulation. The addition of CHMP5 to an ATPase assay containing 375 nm VPS4B79–444 and 375 nm LIP5 showed that CHMP5 had a pronounced inhibitory effect (70% inhibition at 1.5 μm) (Fig. 3C). This result suggested that the inhibitory effect of CHMP5 on VPS4B activity is due to its interaction with LIP5, probably LIP5NTD (see below). Interestingly, the addition of CHMP5L4D or CHMP1B to the same assay also produced a modest inhibitory effect (Fig. 3C).
We have seen that LIP5-mediated VPS4B stimulation did not depend upon much of the linker sequence of LIP5 (although a physical connection is required). In fact, LIP51–162/215–307 displayed an even higher activity toward VPS4B79–444 as compared with LIP5 (Fig. 3B). We asked whether the missing linker sequence would be required for the inhibitory effect of CHMP5 on LIP5-mediated VPS4B stimulation. Strong inhibition was observed for LIP51–162/215–307 when CHMP5 was added to the assay that contained 375 nm VPS4B79–444 and 375 nm LIP51–162/215–307 (Fig. 3D). As a control, there was no reduction in the moderate ATPase activity generated by VPS4B79–444 and LIP5163–307 when CHMP5 was added. Therefore, regulation of LIP5-mediated VPS4 stimulation by CHMP5 binding to LIP5NTD is unlikely to be mediated through the linker region, as was suggested for yeast Vta1 (42). Instead, CHMP5 could exert its function by modulating the interaction between LIP5 and VPS4 through a mechanism that involves conformational change in LIP5NTD.
The Atomic Resolution Crystal Structure of the LIP5NTD-CHMP1B-CHMP5 Complex
Although the NMR structures of LIP5NTD alone and LIP5NTD-CHMP5 binary complex were known, comparison between the two structures did not lead us to detect any obvious conformational change in LIP5NTD upon CHMP5 binding (52). We reasoned that CHMP5-binding induced LIP5 conformational change might be small and transient and thus not easily observable in the NMR structures. To that end, we embarked on determining the crystal structures of the LIP5NTD complexes.
Crystals that diffracted to an atomic resolution of 1.0 Å were obtained for the LIP5NTD-CHMP1B-CHMP5 complex. The structure was determined and refined with an R-factor of 16.5% and a free R-factor of 18.1%. The final model consists of LIP5 residues 4–162, CHMP1B residues 185–199 and CHMP5 residues 151–190. To identify conformational change in LIP5NTD associated with CHMP5 and CHMP1B binding, we also determined the crystal structures of LIP5NTD alone and LIP5NTD in complex with CHMP1B. The LIP5NTD alone structure was determined and refined to a 3.0 Å resolution with an R-factor of 24.8% and a free R-factor of 28.6%. The LIP5NTD-CHMP1B complex structure was determined and refined to 2.2 Å with an R-factor of 19.3% and a free R-factor of 22.7%.
CHMP1B Binds to the MIT1 Domain of LIP5
The Did2 binding site for yeast Vta1 has been previously mapped to the second MIT domain (MIT2) within Vta1NTD (37). However, crystal structures of both LIP5NTD-CHMP1B and LIP5NTD-CHMP1B-CHMP5 clearly showed that the ESCRT-III protein binds to the first MIT domain (MIT1) of human LIP5. This is also consistent with previous results from site-directed mutagenesis and NMR chemical shift mapping studies (52). The LIP5-binding sequence of CHMP1B forms a three-turn α-helix and sits on the surface groove formed by helices α2 and α3 of MIT1 (Fig. 4A). The binding mode of CHMP1B to LIP5NTD is similar to that observed in the VPS4A-CHMP1A complex structure (33), which is also more generally known as the MIT-MIM1 binding mode. As seen in other MIT-MIM1 structures, the cognate MIT domain presents an overall hydrophobic surface, which is necessary for tight binding (Fig. 4B). The binding between the two molecules buries ∼624 Å2 of surface area.
FIGURE 4.

The LIP5NTD-CHMP1B complex structure. A, an overview of the LIP5NTD-CHMP1B complex structure. Shown is a schematic representation of LIP5NTD in complex with CHMP1B176–199 in two orthogonal views. CHMP1B is colored in yellow. MIT1, MIT2, and the short linker between the two MIT domains of LIP5NTD are colored cyan, green, and white, respectively. Helices of LIP5NTD are labeled α1–α7. B, the CHMP1B-binding surface of LIP5NTD is hydrophobic. LIP5NTD is shown as a surface representation and colored based on the underlying atoms: hydrophobic side chain atoms (orange), polar and charged side chain atoms (blue), and main chain atoms (white). CHMP1B is shown as a yellow coil. C and D, detailed interactions at the LIP5NTD-CHMP1B interface. Shown is an enlarged schematic representation showing residues involved in hydrophobic (C) and polar (D) interactions between LIP5NTD and CHMP1B. Residues in contact are shown as stick models. Hydrogen bonds are denoted as dashed lines. Color schemes are the same as in A except for the following atoms: oxygen (red), nitrogen (blue), and sulfur (orange).
The bound peptide in the canonical MIT-MIM1 structure often utilizes a series of conserved aliphatic residues, in many cases leucines, for binding (32, 33). Here, Leu188, Leu192, and Leu195 of CHMP1B make intimate contacts with LIP5NTD, whose binding surface consists of Tyr36, Leu40, and Met47 from helix α2 and Leu60, Met64, and Leu67, from helix α3 (Fig. 4C). In addition to the leucines, two polar residues of CHMP1B, Arg191 and Arg196, are also present at the binding interface, with their aliphatic portion of the side chains contributing to hydrophobic binding and terminal groups contributing to polar interactions (Fig. 4D). Arg191 forms two salt bridges with Asp65 and Glu68 of LIP5, and Arg196 makes one hydrogen bond interaction with Gln44. Interestingly, Arg196 also makes a lone interaction with the MIT2 domain. Here, a hydrogen bond is formed between its side chain and the main chain carbonyl oxygen atom of Val130. A tabulated list of all interactions is shown in Table 2.
TABLE 2.
Detailed interactions between LIP5 and the ESCRT-III proteins CHMP5 and CHMP1B
| CHMP5 | LIP5 | CHMP1B |
|---|---|---|
| Hydrogen bond interactions | ||
| Arg57 NH1 | Asp186 OD1 | |
| Arg57 NH2 | Asp186 OD2 | |
| Glu68 OE2 | Arg191 NE | |
| Glu68 OE1 | Arg191 NH2 | |
| Asp65 OD1 | Arg191 NH2 | |
| Lys71 NZ | Leu195 O | |
| Glu83 OE2 | Arg196 NE | |
| Glu83 OE1 | Arg196 NH1 | |
| Gln44 OE1 | Arg196 NH2 | |
| Val130 O | Arg196 NH2 | |
| Glu83 N | Gln-198 OE1 | |
| Arg151 NH1 | Asp102 OD2 | |
| Arg151 NH1 | Asn103 OD1 | |
| Ser152 O | Arg106 NH2 | |
| Tyr153 OH | Asp102 OD2 | |
| Glu166 OE1 | Asn154 ND2 | |
| Asp177 OD2 | Lys116 NZ | |
| Asp177 O | Arg22 NH2 | |
| Asp179 O | Lys116 NZ | |
| Tyr182 OH | Glu26 OE2 | |
| Tyr182 OH | Thr120 OG | |
| Asp184 OD2 | Arg30 NE | |
| Ala186 O | Lys96 NZ | |
| Ala189 O | Lys96 NZ | |
| Van der Waals interactions | ||
| Met47, Arg57, Leu60, Ser61, Met64 | Leu188 | |
| Met64 | Arg191 | |
| Leu40, Met43, Gln44, Met47, Met64 | Leu192 | |
| Tyr36, Leu40, Met64, Leu67, Lys71 | Leu195 | |
| Leu40 | Arg196 | |
| Tyr153 | Phe98, Leu99, Arg106, His141, Tyr144 | |
| Thr155 | Lys143, Trp147 | |
| Pro156 | Trp147 | |
| Leu158 | Trp147 | |
| Leu163 | Arg146, Thr150 | |
| Glu166 | Thr150, Asn154 | |
| Leu167 | Tyr119, Arg146, Thr150 | |
| Leu170 | Ile115, Tyr119, Ala149, His153 | |
| Gly171 | Tyr119, His19 | |
| Asp172 | His19 | |
| Leu174 | Arg22, Tyr119, Thr120 | |
| Leu175 | Asn18, His19, Arg22 | |
| Tyr182 | Asn113, Lys116, Ser117 | |
| Leu183 | His27, Arg30, Met97, Tyr93 | |
| Glu185 | Tyr100, Asn113 | |
| Ala186 | Tyr100 | |
To evaluate the contribution of individual residues to the interaction between LIP5NTD and CHMP1B, site-directed mutagenesis and glutathione S-transferase (GST)-pull down analysis were performed. We used GST-tagged CHMP1B176–199 to test its binding to LIP5 and various LIP5 mutants, including LIP5L40A, LIP5Q44A, LIP5M64A, LIP5E68A, and LIP5E68A/K71A/K72A. Among the mutants, M64A has been previously shown to disrupt CHMP1B binding (52). We observed that both L40A and E68A disrupted CHMP1B binding to a similar extent as M64A, whereas Q44A had little effect on CHMP1B binding (Fig. 5A). However, to completely abolish the binding of CHMP1B, multiple mutations (LIP5E68A/K71A/K72A) on the LIP5NTD surface were required (Fig. 5A). We also mutated two CHMP1B residues, Leu195 and Arg196, and examined the ability of GST-tagged CHMP1B176–199, CHMP1B176–199/L195D, or CHMP1B176–199/R196A to bind LIP5. L195D did not bind LIP5 (Fig. 5B), demonstrating the importance of hydrophobic interaction to the overall binding. In contrast, R196A bound LIP5 to the same extent as the wild-type protein (Fig. 5B), suggesting that the peripheral polar interaction contributes to a lesser degree to the overall stability of the complex.
FIGURE 5.
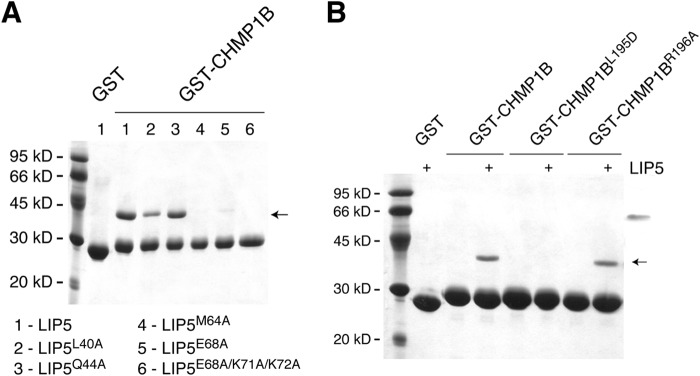
Critical residues at the LIP5NTD-CHMP1B interface. A, critical residues on LIP5NTD. GST-CHMP1B176–199 was used to analyze its interaction with LIP5 and various LIP5 mutants. B, critical residues on CHMP1B. GST-CHMP1B176–199, GST-CHMP1B176–199,L195D, or GST-CHMP1B176–199,R196A was used to analyze its interaction with LIP5. The GST-tagged protein-immobilized glutathione beads were incubated with 1 ml of LIP5 or LIP5 mutant (375 nm) for 1 h at 4 °C. The arrow indicates the running position of LIP5 or LIP5 mutants.
Comparison between the three crystal structures showed that binding of CHMP1B176–199 does not cause significant conformational change in the overall structure of LIP5NTD. Alignment of the LIP5NTD molecules in the apo- and CHMP1B complex structures showed a root mean square deviation of 0.34 Å between the Cα atom positions. Similarly, the presence of CHMP5151–190 also has minimal effect on the structure of the binding interface between CHMP1B and LIP5NTD.
CHMP5 Binds at the MIT1-MIT2 Domain Interface of LIP5
The mode of CHMP5 binding to LIP5NTD in the ternary complex structure largely mirrors that observed in the binary complex structure (52). CHMP5151–190 forms a loop-helix-turn-helix structure that wraps around the MIT2 domain of LIP5NTD but also makes significant contacts with MIT1 (Fig. 6A). The interface between LIP5 and CHMP5 is extensive, with a buried surface area of 1505 Å2. The CHMP5 conformation in the complex structure can be divided into three major parts (Fig. 6B). Residues 151–159 adopt an extended loop conformation and bind across the surface of helices α5 and α7 of MIT2. Residues 160–176 form a long helix that binds across helices α6 and α7 of MIT2 as well as making extensive contacts with helix α1 of MIT1. Following a three-residue turn, residues 181–189 form a short helix that binds across the surface of helices α5 and α6 of MIT2. When binding to the helices of the MIT2 domain, these CHMP5 structural elements all bind at a nearly perpendicular angle.
FIGURE 6.

The LIP5NTD-CHMP1B-CHMP5 complex structure. A, overview of the LIP5NTD-CHMP1B-CHMP5 complex structure. Shown is a schematic representation of LIP5NTD in complex with CHMP1B176–199 and CHMP5151–190 in front (left) and back (right) views. Color schemes and labeling schemes are the same as in Fig. 4A except that CHMP5 is colored in magenta. B, CHMP5 makes extensive contacts with LIP5NTD. The interface is divided and shown in three separate panels based on three sequence segments of CHMP5: 151–159 (left), 160–176 (center) and 176–190 (right). CHMP5 is shown as a magenta coil. LIP5NTD is shown as a semitransparent surface along with a stick representation of side chains that contribute to van der Waals interactions. Surface and sticks are colored using the following scheme: carbon atoms of MIT1 (cyan), carbon atoms of MIT2 (green), oxygen (red), and nitrogen (blue). C, Trp147 of LIP5 flips upon binding CHMP5. An enlarged schematic representation shows the conformation of Trp147 before (pale green) and after (green) binding CHMP5 (magenta).
Binding between CHMP5 and LIP5NTD is largely mediated by van der Waals interactions (Table 2). As noted previously, CHMP5 contains a “leucine collar” that consists of six leucine residues, Leu158, Leu163, Leu167, Leu170, Leu174, and Leu183 (52). These residues are highly conserved in metazoan CHMP5. They bind to a conserved hydrophobic LIP5NTD surface that is formed by aromatic side chains and aliphatic portions of basic residues (Fig. 5B). For example, the highly conserved Trp147 of LIP5 has been previously shown to be essential for high affinity binding (52). Comparison between the LIP5NTD-CHMP1B and LIP5NTD-CHMP1B-CHMP5 structures showed that the imidazole ring of the Trp147 side chain adopts a different rotamer conformation upon CHMP5 binding. It swings around the Cβ-Cγ bond by 180° to make room for CHMP5 binding (Fig. 5C). There are only a few specific hydrogen bond interactions between CHMP5 and LIP5NTD. CHMP5 residues involved in these interactions are clustered either at the N-terminal loop (Arg151 and Tyr153) or at the short linker between the two helices (Asp177, Asp179, Tyr182, and Asp184). These residues are invariant among metazoan CHMP5. There has been speculation that charge-charge interaction might contribute to the high-affinity interaction between the two molecules given that CHMP5 is acidic and LIP5NTD is basic (52). However, except for the few residues mentioned above, most of the acidic residues of CHMP5 point away from the interface and hence are unlikely to contribute directly to binding.
Conformational Change in LIP5 Associated with CHMP5 Binding
Comparison between the three crystal structures revealed moderate conformational adjustment in LIP5NTD associated with CHMP5 binding. Most notably, the MIT2 domain moves relative to the MIT1 domain due to the presence of the ESCRT-III protein at the MIT1-MIT2 interface (Fig. 7A). Helices α5 and α7 of MIT2 are pushed outward by about 2 Å. In the absence of CHMP5 binding, there are a number of hydrogen bonds between the two MIT domains. In particular, Glu26 of the MIT1 domain interacts with Lys116 of the MIT2 domain. This salt bridge is observed in each of the three molecules within the asymmetric unit of the LIP5NTD apo-structure and in each of the two molecules within the asymmetric unit of the LIP5NTD-CHMP1B complex structure as well (Fig. 7B); thus, it appears to be a conserved structural feature in the absence of CHMP5 binding. However, upon CHMP5 binding, the salt bridge is broken (Fig. 7B). Instead, these two residues now form new interactions with the bound CHMP5 molecule. Glu26 makes a hydrogen bond with the side chain of Tyr182. Lys116 makes hydrogen bonds with the side chain of Asp177 and with the main chain carbonyl oxygen of Asp179 (Fig. 7C). There are also additional interactions in the vicinity between CHMP5 and LIP5NTD, including His19, Arg22, and Arg30, that stabilize the new conformation of LIP5NTD (Table 2).
FIGURE 7.

Conformational change at the LIP5 MIT1-MIT2 domain interface. A, superimposition of the LIP5NTD-CHMP1B complex structure with the LIP5NTD-CHMP1B-CHMP5 complex structure. The color scheme used is the same as in Figs. 3A and 5A (the binary complex has a pale color tone). The view in the right panel is nearly orthogonal to that of the left panel and highlights the movement of the MIT domains. Helices are shown as cylinders, and loops in the right panel are omitted for clarity. B, Glu26-Lys116 salt bridge is broken in the ternary complex structure. Side chains of Glu26 and Lys116 in each of the three crystal structures as indicated are shown in stick representations. Carbon atoms are colored using the same scheme as in A. C, Glu26 and Lys116 are engaged in new interactions in the ternary complex structure. The left panel shows an enlarged view of interactions involving Glu26 and Lys116 before and after CHMP5 binding. Residues involved are shown as sticks and labeled. New hydrogen bonds are denoted as dashed lines with distances indicated. The right panel omits the binary complex structure but shows 2Fo − Fc electron density (2.0σ) associated with interacting residues in the ternary complex structure. D, insertion of Tyr182 at the MIT domain interface. Left, superimposition of the LIP5NTD-CHMP1B-CHMP5 structure with the LIP5NTD-CHMP5 structure (only one of the 10 NMR structure assemblies is shown) (52). Right, conformation of Tyr182 in the two structures. Tyr182 in the ternary complex structure adopts a “flipped in” conformation. Tyr182 in the binary complex structure can adopt either a “flipped in” or “flipped out” conformation.
Of particular interest is Tyr182 of CHMP5, which stands out as the potential culprit of the conformational change. In the ternary complex structure, Tyr182 drives itself between the MIT1 and MIT2 domains of LIP5NTD, where it makes both hydrogen bond and van der Waals interactions with LIP5NTD. In the NMR structure of the LIP5NTD-CHMP5 binary complex, five of the 10 structure ensembles have the Tyr182 pointed into the MIT domain interface, and the other five are oriented toward the solvent (Fig. 7B) (52). These data suggested that Tyr182 could exist in equilibrium between a “flipped in” and a “flipped out” conformation. Binding of CHMP1B appears to have stabilized Tyr182 in the “flipped in” conformation, as evidenced by the well defined electron density associated with the residue (Fig. 7C). Interestingly, Tyr182 of CHMP5 and Glu26 of LIP5 are strictly conserved in the metazoan protein sequences, suggesting a potentially conserved mechanism in higher eukaryotes.
We looked to see whether insertion of Tyr182 into the MIT domain interface might be structurally related to the inhibitory function of CHMP5 on LIP5-mediated VPS4 stimulation. A partial loss of inhibition was observed in CHMP5 when Tyr182 was mutated to alanine (Fig. 8A). Interestingly, this mutation had little impact on CHMP5 binding to LIP5 because GST-CHMP5Y182A could still bind a similar amount of LIP5 as the wild-type protein (Fig. 8B). These data suggested that the mere binding of CHMP5 to LIP5 might not be sufficient for its inhibitory effect on LIP5 function. Instead, the inhibitory effect is at least partially predicated on the conformational change in LIP5 associated with insertion of Tyr182 at the MIT domain interface.
FIGURE 8.
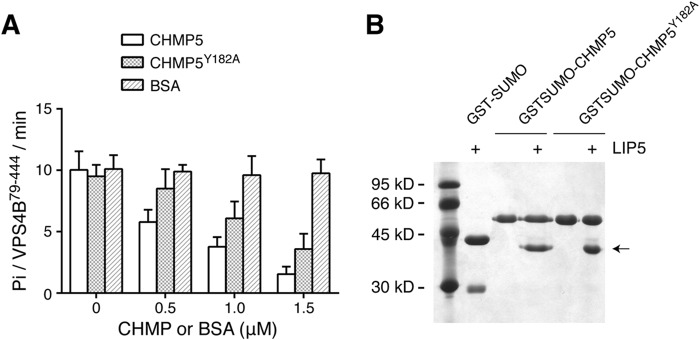
Tyr182 of CHMP5 is important for its inhibitory activity. A, Y182A partially reverses the inhibitory activity of CHMP5 toward LIP5-mediated VPS4B stimulation. The ATPase activity of VPS4B79–444 (375 nm) was measured in the presence of 375 nm LIP5 and increasing concentrations of CHMP5, CHMP5Y182A, or BSA. Reactions were allowed to proceed for 1 h. B, CHMP5Y182A binds to LIP5. GST-SUMO, GST-SUMO-CHMP5151–190, or GST-SUMO-CHMP5151–190,Y182A was used to analyze its interaction with LIP5. The GST-SUMO-tagged protein-immobilized glutathione beads were incubated with 1 ml of LIP5 (375 nm) for 1 h at 4 °C. GST-SUMO was used as a tag because GST-CHMP5151–190 runs at a position similar to that of LIP5. Arrow, running position of LIP5. Error bars, S.E.
DISCUSSION
Given the structural and functional complexity of the ESCRT machinery, it is not surprising that ESCRT is under tight spatial and temporal control at multiple steps. As the only energy-consuming enzyme within the ESCRT machinery, Vps4 plays a central role in ESCRT regulation. It engages in at least three types of protein-protein interactions: with itself, with ESCRT-III, and with Vta1. These interactions represent three general mechanisms that are used to control the activity of Vps4 in the cell: (a) ATP-dependent switching between “inactive” (unassembled) and “active” (assembled) states; (b) recruitment to the site of action through specific protein-protein interactions; and (c) protein cofactors that promote or inhibit its activity.
Vta1 is a protein cofactor in yeast that functions as a Vps4 activator (41, 50). The basal activity of Vta1 has been attributed to the structural features near the C terminus, whereas binding of the ESCRT-III proteins Did2 and Vps60 to the N terminus provides an additional level of stimulation (42). The human ortholog of Vta1, LIP5, also stimulates the activity of VPS4, but the mechanism of action appears to be different in the two systems. Although LIP5CTD is both necessary and sufficient for binding VPS4, the C-terminal domain alone cannot reproduce the stimulatory activity of LIP5. Instead, the full-scale activity requires the presence of both the N- and C-terminal domains. Furthermore, a physical connection between the two domains is necessary because domains added in trans are not active. The length as well as the sequence of the linker appears to be less critical. This is consistent with the fact that the sequence conservation at the linker region appears to be very low. We hypothesize that LIP5CTD makes an initial contact with VPS4, probably through the conserved β-domain (Fig. 9). This interaction brings LIP5NTD close to the ATPase because it is connected to LIP5CTD through the linker. This enables an additional interaction between LIP5NTD and VPS4, which otherwise would not occur due to its weak inherent binding affinity. Thus, the two domains of LIP5 play different roles in LIP5-mediated VPS4 stimulation. LIP5CTD is responsible for the high-affinity VPS4 binding, whereas the actual stimulation is probably the result of the combined actions of the two domains.
FIGURE 9.
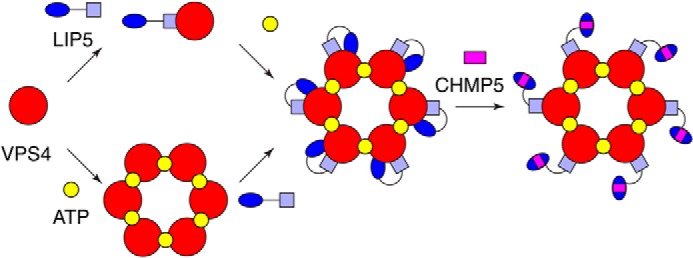
A model of VPS4 regulation by the LIP5-CHMP5 complex. The C-terminal domain of LIP5 makes an initial contact with VPS4. This interaction brings its N-terminal domain close to VPS4, which enables an additional interaction between the N-terminal domain and VPS4. The combined actions of the two domains lead to full activity of VPS4. Binding of CHMP5 to LIP5 weakens the interaction between the N-terminal domain and VPS4 and leads to an inhibition of LIP5-mediated VPS4 stimulation.
A number of MIM1-containing ESCRT-III proteins, including CHMP1, CHMP2, CHMP3, and IST1, have been previously shown to bind to LIP5 (38, 45). Our crystal structure of the LIP5NTD-CHMP1B complex confirmed that CHMP1B indeed binds to MIT1 of LIP5NTD as a MIM1. This predicts that similar MIMs in other ESCRT-III proteins will bind to LIP5 in a similar manner. Furthermore, our data have shown that binding of CHMP1B MIM1 to LIP5 does not induce significant conformational change in LIP5 or significantly alter its ability to stimulate VPS4 ATPase. This suggests that interaction between LIP5 and other MIM1-containing ESCRT-III proteins probably serves only to recruit the VPS4-LIP5 complex to the ESCRT-III-bound membrane.
In contrast to CHMP1B, binding of CHMP5 to LIP5 potently inhibits the stimulatory activity that LIP5 displays toward the VPS4 ATPase. This suggests that binding of CHMP5 could potentially serve as an allosteric power switch to spatially and temporally control the activity of VPS4. Sequence comparison has shown that MIT2 of LIP5 has diverged significantly from other known MIT domain sequences, suggesting that it might function as a specific CHMP5-binding domain. Although most of the CHMP5 binding occurs on the MIT2 domain, there is also significant binding to the MIT1 domain, particularly near the MIT1-MIT2 interface. The latter is significant because it leads to the movement of MIT2 relative to MIT1, in part due to the insertion of a highly conserved Tyr182 of CHMP5 at the domain interface. We suspect that these changes form the structural basis of the inhibitory effect of CHMP5 on LIP5 function. As we have seen, interaction between LIP5NTD and VPS4 is required for the full stimulatory activity of LIP5. Movement of MIT2, induced by CHMP5 binding, could conceivably alter the interaction between LIP5NTD and VPS4 and hence the inhibition (Fig. 9).
Although most ESCRT-III proteins promote the activity of Vps4, it is not unprecedented for one of them to exhibit an inhibitory function. Early work studying the role of ESCRT in HIV-1 release found that siRNA silencing of CHMP5 in co-transfected human cells caused an increase in the release of infectious virus-like particles, suggesting a possible negative regulatory role for CHMP5 in vivo (53). Additionally, Ist1 inhibits Vps4 oligomerization and ATP hydrolysis (44). Whereas Ist1 utilizes its C-terminal MIM for initial targeting to the MIT domain of Vps4, the subsequent association between the N-terminal domain of Ist1 and Vps4 is responsible for its inhibition. LIP5-CHMP5 inhibition of VPS4 mirrors this mechanism, with the LIP5CTD acting like the Ist1 MIM and the LIP5NTD-CHMP5 complex acting like the Ist1 N-terminal domain.
In summary, we have performed structural and functional analyses of molecular interactions between VPS4 and its various associated regulators to gain mechanistic insights into regulation of the ESCRT machinery in higher eukaryotes. The results showed an important mechanistic difference in yeast versus metazoans. Although Vta1 and LIP5 can both stimulate the ATPase activity of their cognate Vps4, the structural requirement appears to be different. The C-terminal domain of Vta1 is sufficient for full activity in yeast (42), whereas both the N- and C-terminal domains of LIP5 are required in metazoans. Furthermore, the effect of the ESCRT-III proteins is also dramatically different in terms of their ability to up- or down-regulate the stimulatory activity of Vta1/LIP5 toward the ATPase. Structural results from the current study, together with previous NMR structures of human LIP5-CHMP5 and yeast Vta1-Vps60 complexes (40, 52), suggest that they are probably the results of different molecular interactions. Interestingly, residues at the interface of LIP5 and CHMP5 are highly conserved throughout metazoans but much less so in fungi, suggesting that the allosteric regulatory mechanism of LIP5 by CHMP5 has diverged during evolution.
Acknowledgment
We thank the staff at the Advanced Photon Source LS-CAT Sector 21 (21-ID-D) for access and help with data collection.
This work was supported, in whole or in part, by National Institutes of Health Grant GM095769 (to Z. X.). Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy Office of Science by Argonne National Laboratory, was supported by the United States Department of Energy under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
The atomic coordinates and structure factors (codes 4TXP, 4TXQ, and 4TXR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ESCRT
- endosomal sorting complex required for transport
- AAA-ATPase
- ATPase associated with diverse cellular activities
- MIT
- microtubule-interacting and transport
- MIM
- MIT-interacting motif
- VSL
- Vta1-SBP1-LIP5
- SUMO
- small ubiquitin-like modifier.
REFERENCES
- 1. Hurley J. H. (2010) The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45, 463–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Henne W. M., Buchkovich N. J., Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 3. McCullough J., Colf L. A., Sundquist W. I. (2013) Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82, 663–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanson P. I., Cashikar A. (2012) Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28, 337–362 [DOI] [PubMed] [Google Scholar]
- 5. Katzmann D. J., Babst M., Emr S. D. (2001) Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106, 145–155 [DOI] [PubMed] [Google Scholar]
- 6. Sundquist W. I., Kräusslich H. G. (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2, a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carlton J. G., Martin-Serrano J. (2007) Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science 316, 1908–1912 [DOI] [PubMed] [Google Scholar]
- 8. Jimenez A. J., Maiuri P., Lafaurie-Janvore J., Divoux S., Piel M., Perez F. (2014) ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- 9. Schuh A. L., Audhya A. (2014) The ESCRT machinery: from the plasma membrane to endosomes and back again. Crit. Rev. Biochem. Mol. Biol. 49, 242–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adell M. A., Teis D. (2011) Assembly and disassembly of the ESCRT-III membrane scission complex. FEBS Lett. 585, 3191–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bajorek M., Schubert H. L., McCullough J., Langelier C., Eckert D. M., Stubblefield W. M., Uter N. T., Myszka D. G., Hill C. P., Sundquist W. I. (2009) Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muzioł T., Pineda-Molina E., Ravelli R. B., Zamborlini A., Usami Y., Göttlinger H., Weissenhorn W. (2006) Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell 10, 821–830 [DOI] [PubMed] [Google Scholar]
- 13. Lata S., Schoehn G., Solomons J., Pires R., Göttlinger H. G., Weissenhorn W. (2009) Structure and function of ESCRT-III. Biochem. Soc. Trans. 37, 156–160 [DOI] [PubMed] [Google Scholar]
- 14. Fabrikant G., Lata S., Riches J. D., Briggs J. A., Weissenhorn W., Kozlov M. M. (2009) Computational model of membrane fission catalyzed by ESCRT-III. PLoS Comput. Biol. 5, e1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elia N., Fabrikant G., Kozlov M. M., Lippincott-Schwartz J. (2012) Computational model of cytokinetic abscission driven by ESCRT-III polymerization and remodeling. Biophys. J. 102, 2309–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Henne W. M., Stenmark H., Emr S. D. (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 5, 10.1101/cshperspect.a016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nickerson D. P., West M., Henry R., Odorizzi G. (2010) Regulators of Vps4 ATPase activity at endosomes differentially influence the size and rate of formation of intralumenal vesicles. Mol. Biol. Cell 21, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baumgärtel V., Ivanchenko S., Dupont A., Sergeev M., Wiseman P. W., Kräusslich H. G., Bräuchle C., Müller B., Lamb D. C. (2011) Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 13, 469–474 [DOI] [PubMed] [Google Scholar]
- 19. Jouvenet N., Zhadina M., Bieniasz P. D., Simon S. M. (2011) Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 13, 394–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanson P. I., Roth R., Lin Y., Heuser J. E. (2008) Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 180, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saksena S., Wahlman J., Teis D., Johnson A. E., Emr S. D. (2009) Functional reconstitution of ESCRT-III assembly and disassembly. Cell 136, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies B. A., Azmi I. F., Payne J., Shestakova A., Horazdovsky B. F., Babst M., Katzmann D. J. (2010) Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21, 3396–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wollert T., Hurley J. H. (2010) Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 464, 864–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wollert T., Wunder C., Lippincott-Schwartz J., Hurley J. H. (2009) Membrane scission by the ESCRT-III complex. Nature 458, 172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adell M. A., Vogel G. F., Pakdel M., Müller M., Lindner H., Hess M. W., Teis D. (2014) Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J. Cell Biol. 205, 33–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morita E., Colf L. A., Karren M. A., Sandrin V., Rodesch C. K., Sundquist W. I. (2010) Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc. Natl. Acad. Sci. U.S.A. 107, 12889–12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morita E., Sandrin V., McCullough J., Katsuyama A., Baci Hamilton I., Sundquist W. I. (2011) ESCRT-III protein requirements for HIV-1 budding. Cell Host. Microbe 9, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wemmer M., Azmi I., West M., Davies B., Katzmann D., Odorizzi G. (2011) Bro1 binding to Snf7 regulates ESCRT-III membrane scission activity in yeast. J. Cell Biol. 192, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scott A., Chung H. Y., Gonciarz-Swiatek M., Hill G. C., Whitby F. G., Gaspar J., Holton J. M., Viswanathan R., Ghaffarian S., Hill C. P., Sundquist W. I. (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao J., Xia H., Yoshino-Koh K., Zhou J., Xu Z. (2007) Structural characterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J. Mol. Biol. 374, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gonciarz M. D., Whitby F. G., Eckert D. M., Kieffer C., Heroux A., Sundquist W. I., Hill C. P. (2008) Biochemical and structural studies of yeast Vps4 oligomerization. J. Mol. Biol. 384, 878–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Obita T., Saksena S., Ghazi-Tabatabai S., Gill D. J., Perisic O., Emr S. D., Williams R. L. (2007) Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 [DOI] [PubMed] [Google Scholar]
- 33. Stuchell-Brereton M. D., Skalicky J. J., Kieffer C., Karren M. A., Ghaffarian S., Sundquist W. I. (2007) ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744 [DOI] [PubMed] [Google Scholar]
- 34. Babst M., Wendland B., Estepa E. J., Emr S. D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monroe N., Han H., Gonciarz M. D., Eckert D. M., Karren M. A., Whitby F. G., Sundquist W. I., Hill C. P. (2014) The oligomeric state of the active Vps4 AAA ATPase. J. Mol. Biol. 426, 510–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Azmi I., Davies B., Dimaano C., Payne J., Eckert D., Babst M., Katzmann D. J. (2006) Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J. Cell Biol. 172, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Azmi I. F., Davies B. A., Xiao J., Babst M., Xu Z., Katzmann D. J. (2008) ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev. Cell 14, 50–61 [DOI] [PubMed] [Google Scholar]
- 38. Shim S., Merrill S. A., Hanson P. I. (2008) Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol. Biol. Cell 19, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merrill S. A., Hanson P. I. (2010) Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J. Biol. Chem. 285, 35428–35438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang Z., Vild C., Ju J., Zhang X., Liu J., Shen J., Zhao B., Lan W., Gong F., Liu M., Cao C., Xu Z. (2012) Structural basis of molecular recognition between ESCRT-III-like protein Vps60 and AAA-ATPase regulator Vta1 in the multivesicular body pathway. J. Biol. Chem. 287, 43899–43908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao J., Xia H., Zhou J., Azmi I. F., Davies B. A., Katzmann D. J., Xu Z. (2008) Structural basis of Vta1 function in the multivesicular body sorting pathway. Dev. Cell 14, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Norgan A. P., Davies B. A., Azmi I. F., Schroeder A. S., Payne J. A., Lynch G. M., Xu Z., Katzmann D. J. (2013) Relief of autoinhibition enhances Vta1 activation of Vps4 via the Vps4 stimulatory element. J. Biol. Chem. 288, 26147–26156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nickerson D. P., West M., Odorizzi G. (2006) Did2 coordinates Vps4-mediated dissociation of ESCRT-III from endosomes. J. Cell Biol. 175, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dimaano C., Jones C. B., Hanono A., Curtiss M., Babst M. (2008) Ist1 regulates Vps4 localization and assembly. Mol. Biol. Cell 19, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bajorek M., Morita E., Skalicky J. J., Morham S. G., Babst M., Sundquist W. I. (2009) Biochemical analyses of human IST1 and its function in cytokinesis. Mol. Biol. Cell 20, 1360–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER version 2.10, Global Phasing Ltd., Cambridge, United Kingdom [Google Scholar]
- 49. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 50. Vild C. J., Xu Z. (2014) Vfa1 binds to the N-terminal microtubule-interacting and trafficking (MIT) domain of Vps4 and stimulates its ATPase activity. J. Biol. Chem. 289, 10378–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Davies B. A., Norgan A. P., Payne J. A., Schulz M. E., Nichols M. D., Tan J. A., Xu Z., Katzmann D. J. (2014) Vps4 stimulatory element (VSE) of the cofactor Vta1 contacts the ATPase Vps4 α7 and α9 to stimulate ATP hydrolysis. J. Biol. Chem. 289, 28707–28718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skalicky J. J., Arii J., Wenzel D. M., Stubblefield W. M., Katsuyama A., Uter N. T., Bajorek M., Myszka D. G., Sundquist W. I. (2012) Interactions of the human LIP5 regulatory protein with endosomal sorting complexes required for transport. J. Biol. Chem. 287, 43910–43926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ward D. M., Vaughn M. B., Shiflett S. L., White P. L., Pollock A. L., Hill J., Schnegelberger R., Sundquist W. I., Kaplan J. (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 280, 10548–10555 [DOI] [PubMed] [Google Scholar]