

Background: The non-nuclear BRCA1 tumor suppressor can stimulate cell death, but the mechanisms are unknown.
Results: BRCA1 binds to the inositol 1,4,5-trisphophate receptor (IP3R) calcium channel at the endoplasmic reticulum to stimulate apoptotic calcium release.
Conclusion: BRCA1 tumor suppressor activity includes direct stimulation of apoptotic cell death via increased IP3R activity.
Significance: We identify a novel role for the tumor suppressor BRCA1.
Keywords: Apoptosis; BRCA1; Calcium; Cancer; Inositol 1,4,5-Trisphosphate Receptor (IP3R); Lipid; Lipid Binding
Abstract
The inositol 1,4,5-trisphosphate receptor (IP3R) is a ubiquitously expressed endoplasmic reticulum (ER)-resident calcium channel. Calcium release mediated by IP3Rs influences many signaling pathways, including those regulating apoptosis. IP3R activity is regulated by protein-protein interactions, including binding to proto-oncogenes and tumor suppressors to regulate cell death. Here we show that the IP3R binds to the tumor suppressor BRCA1. BRCA1 binding directly sensitizes the IP3R to its ligand, IP3. BRCA1 is recruited to the ER during apoptosis in an IP3R-dependent manner, and, in addition, a pool of BRCA1 protein is constitutively associated with the ER under non-apoptotic conditions. This is likely mediated by a novel lipid binding activity of the first BRCA1 C terminus domain of BRCA1. These findings provide a mechanistic explanation by which BRCA1 can act as a proapoptotic protein.
Introduction
The IP3R2 is a ligand-gated calcium channel localized primarily to ER membranes. The IP3R is a tetrameric protein, and each subunit consists of an N-terminal ligand binding domain, a C-terminal transmembrane pore domain, and an intervening modulatory domain. Calcium release through the IP3R regulates multiple cellular processes, including, but not limited to, gene expression, metabolism, and apoptosis (1, 2). Numerous IP3R protein-protein interactions allow for signal integration of diverse signaling pathways (1). Several proto-oncogenes and tumor suppressors interact with and regulate the IP3R, including Bcl-2, PTEN, and PML (3–5). These proteins regulate IP3R activity and apoptotic calcium release through multiple mechanisms. In general, proto-oncogenes cause reduced IP3R activity, whereas tumor suppressors cause increased IP3R activity to regulate cell death (4).
Breast and ovarian cancer susceptibility gene 1 (BRCA1) is a tumor suppressor well known for its function during homologous recombination and repair of DNA double strand breaks (6–8). The presence of nuclear export and import signals suggests a regulated transport of BRCA1 into and out of the nucleus (9, 10). Several studies have suggested a pro-apoptotic role for BRCA1 linked to its nuclear export and cytosolic localization, but the mechanisms by which BRCA1 stimulates cell death outside of the nucleus are unclear (11–13).
Here we show that BRCA1 stimulates apoptosis through a physical and functional interaction with the IP3R. BRCA1 is recruited to the IP3R during apoptosis and stimulates apoptotic calcium release and cell death. Furthermore, we identified a pool of BRCA1 that is constitutively localized to the ER via binding of the BRCA1 C terminus (BRCT) domain to phospholipids, which may facilitate the rapid recruitment of BRCA1 to the IP3R during cell death. Therefore, BRCA1 mediates its proapoptotic effects by binding to and modulating apoptotic calcium release through the IP3R.
EXPERIMENTAL PROCEDURES
Materials and Antibodies
Paclitaxel was purchased from Sigma-Aldrich and resuspended in dimethyl sulfoxide. The Cell Signaling Technology anti-BRCA1 antibody (catalog no. 9010) was used on HeLa cell lysates and in immunoprecipitations because of questions regarding the specificity of the widely used MS110 monoclonal BRCA1 antibody (14). We tested all other commercially available BRCA1 antibodies and determined the Cell Signaling rabbit anti-BRCA1 antibody to be the most specific. The specificity of this antibody for detecting endogenous human BRCA1 was confirmed in knockdown experiments (Fig. 3A). The Bethyl anti-BRCA1 antibody (catalog no. A301-378A) was used on DT40 lysates because this antibody reacts with the chicken BRCA1 isoform. The IP3R-1 antibody has been described previously (15). The actin antibody used for coimmunoprecipitation was purchased from Abcam (catalog no. ab6276). The anti-LRP130 antibody was purchased from Thermo Scientific (PA5-22034). The anti-α-fodrin clone AA6 was from EMD Millipore (catalog no. MAB1622). Cytochrome c oxidase, histone H1, and lactate dehydrogenase-HRP antibodies were purchased from Invitrogen (catalog no. 338500), Cell Signaling Technology (catalog no. 2576), and Chemicon (no longer sold), respectively. The Sigma-Aldrich anti-GST antibody (catalog no. G1160) was used to detect GST-tagged proteins on PIP strips. Fura-2/AM (catalog no. F1221), Mag-Fura-2/AM (catalog no. M1292), and Rhod-2/AM (catalog no. R1244) were purchased from Molecular Probes.
FIGURE 3.

Endogenous BRCA1 modulates IP3R function. A, Western blot analysis of HeLa cells with siRNA-mediated knockdown of endogenous BRCA1. Two siRNAs (s458 and s459) alone and in combination significantly reduced endogenous BRCA1 expression, whereas a control (Con) siRNA did not. Probing with an anti-IP3R-1 antibody revealed that siRNA s458 also increased IP3R levels. Blotting with anti-α-fodrin was used as a loading control. Because full-length α-fodrin is cleaved very efficiently during cell death (49), this result also indicates that the siRNAs had limited or no toxicity. Finally, this result also validates the specificity of the BRCA1 antibody used in this study. B, representative traces of histamine release in BRCA1 knockdown cells or cells transfected with control siRNA. The control siRNA is commercially available and is not homologous to any sequence in the vertebrate transcriptome (Life Technologies, catalog no. 12935-300). In this and all subsequent panels, control siRNA is shown in black, and BRCA1 siRNA is shown in red. C, peak release in control and BRCA1 knockdown cells in response to 100 nm histamine. D, percent responders in control and BRCA1 knockdown cells in response to 100 nm histamine. E, oscillation frequency in response to 1 and 10 μm histamine in control and BRCA1 knockdown cells. F, peak release in control and BRCA1 siRNA-expressing cells in response to 1 and 10 μm histamine. For C–F, n = 36 for control siRNA and 34 for BRCA1 siRNA-expressing cells. All data were derived from HeLa cells.
Cell Lines and Transfections
The HeLa (CCL-2), UWB1.289 (CRL-2945), and UWB1.289-BRCA1 (CRL-2946) cell lines were purchased from the ATCC and maintained in the medium suggested by the ATCC. DT40 and DT40 TKO cells were cultured as described previously (15). HeLa cells were transfected with Lipofectamine 2000 (Invitrogen). DT40 and DT40 TKO cells were transfected with the Invitrogen Neon transfection system using 1 pulse of 1375 V for 40 ms.
Protein Purification and in Vitro Binding
GST-tagged BRCA1 RING (amino acids 1–112), the BRCA1 BRCT (amino acids 1528–1863), the GST-IP3R modulatory domain (amino acids 922–1581), and the GST-IP3R tail domain (amino acids 2589–2749) were purified using glutathione-agarose beads (Pierce). GST was cleaved from GST-BRCA1 RING with thrombin protease (GE Health Sciences). BRCA1 RING (225 μg) free of cleaved GST was conjugated to 0.1 g of cyanogen bromide-activated agarose (Sigma-Aldrich) to make RING-agarose. In vitro binding assays were performed using RING-agarose as bait and 200 μg of the GST-IP3R modulatory domain and GST-IP3R tail domain as prey in PBS with 1% Triton-X100.
PIP Strip Binding
PIP strips were purchased from Echelon Biosciences (catalog no. P-6001). Binding was performed as suggested by the manufacturer using 5 μg/ml of GST-RING and GST-BRCT.
Subcellular Fractionation
Cell homogenization and purification of the 10,000 × g pellet (P2), the 100,000 × g pellet (P3), the 100,000 × g supernatant (S3), and the 100,000 × g pellet (P3) fractions were performed as described previously (15). The 1000 × g pellet (P1) was resuspended in 1 ml of buffer A (1.62 m sucrose, 10 mm HEPES (pH 7.5), and 2 mm MgCl2) and underlain with 326 μl of buffer B (2.3 m sucrose, 10 mm HEPES (pH 7.5), 2 mm MgCl2) and centrifuged for 1 h at 40,000 rpm. The supernatant was removed by aspiration. P1 pellets were resuspended in buffer A. Resuspended pellets were sonicated in a bath sonicator in ice water for 30 min before being passed through a 23-gauge needle 10 times to shear genomic DNA.
Cell Death Assays
Propidium iodide and caspase-3 measurements were performed as described previously (16).
Cytosolic Calcium Imaging
HeLa cells were transfected with full-length YFP-BRCA (9). After 48 h, cells were loaded with 2.5 μm Fura-2 in imaging buffer composed of 1% BSA, 107 mm NaCl, 20 mm HEPES, 2.5 mm MgCl2, 7.25 mm KCl, 11.5 mm glucose, 1 mm CaCl2 for 30 min at room temperature. The solution was replaced with imaging solution without Fura-2 for an additional 30 min prior to imaging. Coverslips were then imaged on a Nikon TiS inverted microscope with a ×40 oil objective as described previously (17). Responses to 100 nm, 1 μm, and 10 μm histamine were recorded in YFP-BRCA1 cells and adjacent YFP-negative control cells from four separate coverslips. A total of 25 individual YFP-BRCA1-positive and 24 YFP-negative cells were quantified and pooled from the four coverslips. Peak release was determined in the MetaFluor acquisition software. Oscillation frequency was determined manually, where an oscillation was defined as a rise and fall of the Fura-2 ratio of at least 0.01 units. This same threshold was used to determine the percentage of responding cells at 100 nm histamine. All cells responded at 1 and 10 μm histamine and, therefore, were not quantified. For the siRNA knockdown experiments, a similar approach was used. HeLa cells were transfected with two different siRNAs targeting human BRCA1 (Ambion/Life Technologies siRNAs s458 and s459). The total amount was 12.5 pmol/well of a 6-well dish. Transfected cells were identified by cotransfection with YFP. Cells were imaged after 48 h. We found that both siRNAs efficiently knocked down BRCA1 expression (Fig. 3A). However, siRNA s458 also up-regulated IP3R-1 expression (Fig. 3A). Therefore, only siRNA s459 was used for the calcium imaging experiments. The siRNA data were quantified exactly as above, with the exception that the control cells were transfected with control siRNA (control siRNA, medium GC content, Life Technologies, catalog no. 12935-300) on separate coverslips and identified by YFP expression exactly as described for the BRCA1-targeted siRNA.
Mitochondrial Calcium Imaging
Mitochondrial calcium was quantified by Rhod-2 imaging exactly as described previously (17). The response to 10 μm histamine was quantified and pooled from five coverslips comprising 18 YFP-BRCA1 and 18 YFP-negative cells.
ER Calcium Imaging
HeLa cells were loaded with 5 μm Mag-Fura-2/AM in imaging solution (1% BSA, 107 mm NaCl, 20 mm HEPES, 2.5 mm MgCl2, 7.25 mm KCl, 11.5 mm glucose, and 1 mm CaCl2) for 20 min at room temperature. To obtain cytosolic access and image ER calcium, cells were permeabilized with 120 μg/ml saponin in intracellular solution (125 mm KCl, 19 mm NaCl, 10 mm HEPES, 1 mm EGTA, and 0.4 mm CaCl2) until permeabilization was obvious via imaging (∼1 min). Images were taken every 3 s during continuous recording of the response to various additions. Measurement of steady-state changes in cytosolic calcium after 24 h of paclitaxel treatment (Fig. 4D) was performed using Fura-2 as described previously (15).
FIGURE 4.
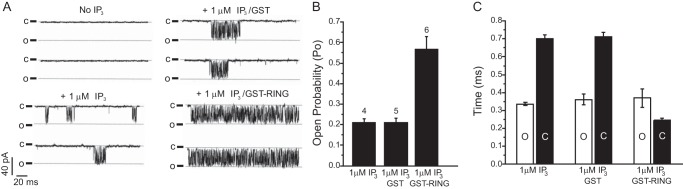
BRCA1 increases IP3R single-channel activity. A, representative traces of nuclear patches of triple IP3R knockout DT40 cells expressing recombinant rat IP3R-1. Traces were recorded with no IP3 in the pipette, 1 μm IP3, 1 μm IP3 with 30 nm GST, or 1 μm IP3 with 30 nm GST-BRCA1 RING domain. B, open probability (Po) of IP3R-1 channels recorded in the presence of IP3, IP3 and GST, or IP3 and the GST-BRCA1 RING domain. The number of channels analyzed is indicated above the bars (n = 4, 5, and 6, respectively). C, mean open (O, white) and closed (C, black) times for each condition.
IP3R Single Channel Recording
Recombinant rat IP3R-1 was stably expressed in triple IP3R knockout DT40 cells. Single channel recording by nuclear patch clamp technique was performed exactly as described previously (18). The recording solution contained 140 mm KCl, 10 mm HEPES, 5 mm NaATP, 100 μm BAPTA, 200 nm free calcium, and 1 μm IP3. Channels were recorded at −100 mV, sampled at 20 kHz, and filtered at 5 kHz. A minimum of 15 s of recording from patches containing a single channel was used for analysis. The total number of channels analyzed for each condition are indicated over the bar graph in Fig. 4B. Where indicated, either 30 nm BRCA1 GST-RING or 30 nm GST alone was included in the patch pipette.
FRET Imaging
HeLa cells were transfected with YFP-BRCA1 (9) and CFP-IP3R (15). After 48 h, cells were imaged on a Nikon TiS inverted microscope with a ×100 SuperFluor objective, and images were acquired every 30 s with a Photometrics Evolve EMCCD camera. Excitation and emission filters for acquiring donor and acceptor channels were from Chroma Technology (set 89002). Cells were maintained at 37 °C during imaging. Data were quantified by taking a region of interest from the cytosol and nucleus and taking the ratio of the acceptor fluorescence (excitation, 430 nm; emission, 535 nm) over the donor fluorescence (excitation, 430 nm; emission, 470 nm) for each region of interest. The data in Fig. 5B were pooled from 20 cells (CFP-IP3R/YFP-BRCA1) and 22 cells (CFP-IP3R/YFP) from at least three separate experiments.
FIGURE 5.

BRCA1 interaction with IP3R increases during apoptosis and mediates cell death. A, representative images of CFP-IP3R, YFP-BRCA1, and the FRET signal (CFP excitation, YFP emission; see “Experimental Procedures”). B, live cell imaging of FRET dynamics in HeLa cells expressing YFP-BRCA1 and CFP-IP3R. Cells were treated with 1 μm paclitaxel (PTX) at the indicated time. Data from FRET ratio of cytosolic and nuclear regions of interest (normalized acceptor/donor) are reported on the x axis. The data are pooled from 20 cells from five separate experiments and are presented as the mean ± S.E. C, live cell imaging of FRET dynamics in cells expressing YFP only and CFP-IP3R. Cells were treated as in B. The data are pooled from 22 cells from five separate experiments. D, cytosolic calcium measurements of cells treated with 100 nm paclitaxel for 0 and 24 h. The cytosolic calcium concentration was measured with the ratiometric calcium indicator Fura-2/AM. E, -fold increase in caspase-3 activity in cells treated with 100 nm paclitaxel for 24 h compared with cells treated with dimethyl sulfoxide. Caspase-3-like activity was assessed by measuring the linear rate of cleavage of a fluorescent substrate (Ac-DEVD-R110). F, cell death percentage as measured by propidium iodide staining of cells treated with 100 nm paclitaxel for 0 and 24 h. For D–F, the data are pooled from at least three separate experiments. *, p < 0.05. The data in all panels are presented as the mean ± S.E. The data in A–C were obtained from HeLa cells. The data in D–F are from UWB cells or UWB cells stably expressing BRCA1.
Fatty Acid Binding Site Prediction
To screen for a putative fatty acid binding region in BRCA1, a position-specific scoring matrix (PSSM)-based method, described previously in Ko et al. (19), was used. An initial PSSM library was defined from the experimentally determined fatty acid binding protein regions in 42 well characterized lipid binding crystal structures collected from the Protein Data Bank (20–23). This initial PSSM library was leveraged to search for more fatty acid binding protein regions using psi-blast and, therefore, expanded to 1185 fatty acid binding protein-specific PSSM libraries. Human BRCA1 was aligned with this expanded fatty acid binding protein-specific PSSM library. All positive alignments were recorded. From these positive PSSMs, a residue score was calculated that represents the occurrence of identical and similar residues from each query-PSSM alignment above the threshold. The Smith-Waterman algorithm was used to re-evaluate PSSM comparisons with the query sequence that resulted in a positive alignment, as in 24, 25. Then raw scores for each residue were calculated by scoring a value of 2 for identities and a value of 1 for positive substitutions from each alignment. These values were summed for all alignments at each position to obtain a total raw residue score.
RESULTS
BRCA1 Binds to the IP3R
In a yeast-two-hybrid experiment using the C-terminal tail domain of IP3R (amino acids 2589–2749) as bait, the RING domain of BRCA1 was isolated as an interacting clone (amino acids 1–112). To confirm this direct interaction, we used purified recombinant protein in an in vitro binding experiment. The purified RING domain was covalently conjugated to cyanogen bromide-activated agarose beads, and the ability of the purified GST-IP3R tail domain and modulatory domain (amino acids 923–1582) to bind to the conjugated beads was tested. The RING domain of BRCA1 was able to specifically pull down the IP3R tail domain, suggesting a direct protein-protein interaction between BRCA1 and the IP3R tail (Fig. 1A). To test for a physical interaction between full-length BRCA1 and IP3R in cells, we performed a coimmunoprecipitation experiment. Using an IP3R antibody, we were able to coimmunoprecipitate endogenous IP3R-1 with endogenous BRCA1 in HeLa cells (Fig. 1B). We could not perform a reciprocal coimmunoprecipitation of the IP3R with BRCA1 because we were unable to identify an antibody suitable for immunoprecipitation of BRCA1. This may be related to epitope masking in Triton X-100 lysates of BRCA1. Regardless, yeast two-hybrid screening, coimmunoprecipitation, and direct binding of purified components strongly suggest a direct physical interaction between BRCA1 and the IP3R mediated by the BRCA1 RING domain and the IP3R tail domain.
FIGURE 1.
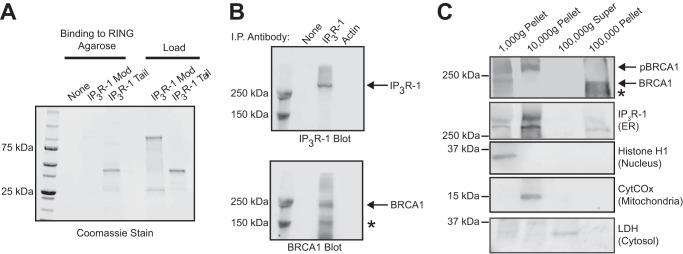
BRCA1 binds to the IP3R. A, in vitro binding of purified GST-tagged fragments of IP3R to RING-agarose beads. The first three lanes include RING-agarose beads, either alone (lane 1) or mixed with the indicated IP3R domains. Mod, modulatory domain; Tail, C-terminal tail domain. The load lanes include 5 μg of the indicated GST-tagged protein. B, immunoprecipitation (I.P.) of HeLa lysates with no antibody (lane 1), IP3R-1 antibody (lane 2), or actin antibody (lane 3) and Western blotted with either IP3R-1 (top panel) or BRCA1 (bottom panel) antibodies. The asterisk indicates a possible spliceoform of BRCA1 that is detected by the antibody. In our hands, commercial antibodies to BRCA1 were not suitable for reciprocal coimmunoprecipitation. C, subcellular fractionation by differential centrifugation of HeLa cells. BRCA1 expression was detected at the predicted molecular weight (BRCA1) and a slower migrating band that is likely hyperphosphorylated (pBRCA1). The asterisk indicates a possible spliceoform of BRCA1 that is detected by the antibody. Fraction purity was evaluated with the organelle controls lactate dehydrogenase (LDH, cytosol), IP3R-1 (endoplasmic reticulum), cytochrome c oxidase (CytCOx, mitochondria), and histone H1 (nucleus). The experiments in B and C utilized HeLa cell lysates.
The existence of BRCA1 nuclear import and export signals indicates the regulated transport of the protein into and out of the nucleus. However, the subcellular localization of BRCA1 has been a subject of debate (26). To determine the subcellular localization of BRCA1, we performed subcellular fractionation using differential centrifugation of HeLa cell homogenates. We used HeLa cells because IP3R function, subcellular localization, and protein interactions during apoptosis have been studied extensively and are well characterized in this cell line (15, 27). Furthermore, HeLa cells express endogenous wild-type BRCA1 (28). After differential centrifugation, the purity of the individual fractions was confirmed by stripping and reprobing the same blot with IP3R-1 (endoplasmic reticulum), histone H1 (nucleus), cytochrome c oxidase (mitochondria), and lactate dehydrogenase (cytosol). BRCA1 was expressed in all cell fractions except the 100,000 × g supernatant (representing cytosol), and, surprisingly, the majority of BRCA1 was localized to the 100,000 × g microsomal ER-enriched fraction (Fig. 1C). IP3Rs in this fraction also specifically bind to cytochrome c to regulate apoptotic calcium release (15). Interestingly, the phosphorylated form of BRCA1 was also found in the 10,000 × g pellet, which is enriched in IP3R and mitochondria, consistent with a previous report (28). Therefore, non-nuclear BRCA1 is abundant and localized to the same subcellular compartments as the IP3R and could potentially modulate its activity.
BRCA1 Modulates IP3R Function
We next chose to examine the effect of BRCA1 on IP3R function by expressing full-length BRCA1 fused to the C terminus of YFP in HeLa cells and examining the response to escalating doses of histamine. HeLa cells were used in these experiments because of their ease of transfection and well characterized calcium release characteristics after histamine challenge (29–31). The YFP-BRCA1 fusion protein has been characterized previously and is functionally comparable with the wild-type protein (12, 32). As shown in Fig. 2A, YFP-BRCA1 is localized to both nuclear and non-nuclear compartments. We measured calcium release in response to 100 nm, 1 μm, and 10 μm histamine in cells expressing YFP-BRCA1 and adjacent cells not expressing the fusion protein. As shown in Fig. 2, B–E, BRCA1 expression significantly sensitized HeLa cells to a 100 nm histamine challenge, increasing both peak release (Fig. 2, C and D) and the number of responding cells (Fig. 2E). Expression of BRCA1 did not appear to affect peak release of calcium in response to 1 μm histamine or a saturating dose of 10 μm histamine (Fig. 2, F and G). However, at both of these doses there is a significant increase in the oscillation frequency, which would be expected to have profound implications for downstream signaling events (Fig. 2H) (33, 34). Therefore, expression of BRCA1 has significant effects on calcium signaling through IP3R-coupled pathways. It has been shown that BRCA1, and the BRCA1 binding protein, and homologue BARD1 are targeted to mitochondria (28, 35), and our subcellular fractionation results indicate that a significant amount of BRCA1 is present in a fraction that also contains mitochondria (Fig. 1C). We hypothesized that BRCA1 may facilitate calcium transfer into mitochondria. To test this hypothesis, we stimulated HeLa cells with 10 μm histamine and measured calcium uptake into mitochondria using Rhod-2. As shown in Fig. 2I, BRCA1 expression has no effect on calcium uptake into mitochondria.
FIGURE 2.

Overexpressed BRCA1 modulates IP3R function. A, representative image of a YFP-BRCA1-expressing HeLa cell demonstrating both nuclear and non-nuclear localization. B, representative single cell traces of calcium release induced by 100 nm, 1 μm, and 10 μm histamine. In this and all subsequent panels, data from control cells are black, and data from BRCA1 expressing cells are red. C, peak release (change in ratio) induced by 100 nm histamine pooled from all cells from four coverslips (n = 24 control, n = 25 BRCA-expressing). D, release induced by 100 nm histamine normalized to the response to a saturating dose of histamine in the same cell. The results are comparable with C. E, percentage of cells responding to 100 nm histamine. F, peak release (change in ratio) induced by 1 and 10 μm histamine pooled from all cells as in C. G, release induced by 1 μm histamine normalized to the response to a saturating dose of histamine in the same cell. H, oscillation frequency in response to 1 and 10 μm histamine. No cells oscillated in response to 100 nm histamine. I, change in mitochondrial (Mito) calcium (ΔF/F0 of Rhod-2 fluorescence) in response to 10 μm histamine (n = 18 control and 18 BRCA1-expressing cells from five coverslips). J, representative MagFura-2 traces of ER calcium release in response to IP3 of two cells on one coverslip. One cell is transfected with YFP-BRCA1 (red). Medium was exchanged at the indicated points with the indicated solutions. The traces begin after permeabilization and ER calcium store loading. Data are reported as the ratio of 340/380 nm and are indicative of the relative calcium concentration. K, calcium released (ΔF/F0) from the ER after addition of a subsaturating (200 nm) and saturating (10 μm) dose of IP3. Control, n = 11 for 200 nm and 40 for 10 μm; BRCA1, n = 10 for 200 nm and 28 for 10 μm. The data in C–I and K are presented as the mean ± S.E. p values are indicated above each set of bars. All data were derived from HeLa cells.
The above results indicate that BRCA1 has significant effects on IP3R-coupled calcium release induced by histamine but does not determine whether BRCA1 has a direct effect on IP3R function. To directly measure IP3R activity, we used saponin-permeabilized cells loaded with MagFura-2 to measure ER calcium release induced by IP3 addition to the bath. MagFura-2 imaging of ER stores is a well established method for measuring ER calcium store depletion in response to IP3R activation (36, 37). Cell membrane permeabilization also allows for access to the cytosol and the ability to directly activate IP3R with addition of IP3 to the bath (38). We found that cells expressing YFP-BRCA1 released significantly more calcium after addition of both subsaturating (200 nm) and saturating (10 μm) doses of IP3 (Fig. 2, J and K). This indicates that expression of BRCA1 directly increases IP3R activity, which is consistent with the functional effects on histamine-induced calcium release.
The experiments in Fig. 2 examined the effects of overexpressed BRCA1 on IP3R function. We next wished to determine whether endogenous BRCA1, which we found to bind endogenous IP3R-1 (Fig. 1B), also modulates calcium release. We obtained two commercial siRNA duplexes targeting human BRCA1 and determined their efficiency in knocking down endogenous BRCA1 expression in HeLa cells. We found that both siRNA duplexes almost completely abrogated BRCA1 expression in HeLa cells (Fig. 3A). We found that one of the siRNA complexes (s458) also up-regulated IP3R-1 expression, which may reflect a compensatory mechanism. Regardless, further experiments were performed with the siRNA duplex that did not affect IP3R expression (s459). Knockdown of BRCA1 expression had the opposite effect of overexpression, including decreased peak release, decreased oscillation frequency, and a dramatic reduction in the number of percent responders to 100 nm histamine compared with cells expressing control siRNA (Fig. 3, B–E). In addition, BRCA1 knockdown also decreased peak release to 1 and 10 μm histamine (Fig. 3F). Therefore, endogenous BRCA1 has significant effects on the activity of endogenous IP3R channels in HeLa cells.
BRCA1 Increases IP3R Open Probability
To determine the direct effects of BRCA1 on IP3R activity, we recorded single-channel currents on isolated nuclei that express recombinant rat IP3R-1 (Ref. 18 and “Experimental Procedures”). As shown in Fig. 4, A and B, when the BRCA1 GST-RING domain was included in the patch pipette, the open probability of the IP3R-1 channel in the presence of a subsaturating concentration of IP3 (1 μm) was increased dramatically (control, 0.21 ± 0.02; GST-RING, 0.57 ± 0.06; GST only, 0.21 ± 0.02). This was due to a destabilization of the closed state of the channel (Fig. 4C). Therefore, the BRCA1 RING domain directly and potently increases IP3R activity by modulating channel gating.
BRCA1 Binds to IP3Rs during Cell Death to Stimulate Calcium-dependent Apoptosis
We hypothesize that BRCA1 binding to IP3R increases during cell death and that this is an essential component of its tumor suppressor activity. To measure changes in the BRCA1/IP3R interaction in living cells during apoptosis, we used the FRET pair CFP-IP3R and YFP-BRCA1. We used the clinically relevant chemotherapeutic paclitaxel to induce apoptosis and measured dynamic changes in the FRET ratio (and, therefore, binding) in HeLa cells transfected with CFP-IP3R-1 and YFP-BRCA1 or YFP alone (Fig. 5A). We deliberately avoided DNA-damaging chemotherapeutics such as cisplatin so that the DNA repair activity of BRCA1 would not confound the results. We found a significant increase in cytosolic FRET after treatment with paclitaxel (1 μm) in cells expressing both CFP-IP3R-1 and YFP-BRCA1 (Fig. 5B). The kinetics of association are consistent with the time course of activation of cell death proteins such as JNK1 in response to paclitaxel treatment (39). We saw no increase in FRET after treatment with paclitaxel in the nucleus of cells transfected with CFP-IP3R-1 and YFP-BRCA1 (Fig. 5B) or in cells transfected with CFP-IP3R-1 and YFP only (Fig. 5C). These results indicate that BRCA1 is recruited to IP3R channels on the ER during apoptosis and support the hypothesis that, under resting (i.e. non-apoptotic) conditions, only a subpopulation of IP3Rs are bound to BRCA1.
We next examined the effect of BRCA1 expression on paclitaxel-induced apoptosis. In these experiments, we used the patient-derived, BRCA1-mutated cell line UWB1.289 (UWB) isolated from an ovarian carcinoma, and this same cell line stably rescued wild-type BRCA1 (UWB-BRCA1). The BRCA1 mutations in the parental UWB line eliminate expression of the BRCA1 protein. Treatment of UWB cells with paclitaxel for 24 h did not cause an elevation of cytosolic calcium, whereas UWB cells with rescued BRCA1 expression had significantly elevated calcium consistent with apoptotic calcium release (Fig. 5D). This indicates that BRCA1 expression is required for paclitaxel-induced apoptotic calcium release via the IP3R. Measurement of apoptosis by both caspase-3 enzymatic activity and propidium iodide staining indicated that BRCA1 expression was required for efficient paclitaxel-induced cell death of ovarian carcinoma cells (Fig. 5, E and F). This suggests that BRCA1 expression restores paclitaxel sensitivity to the BRCA1-null cells. This is consistent with similar findings by other groups (40).
BRCA1 Is Recruited to the ER via IP3Rs and a Novel Lipid Binding Domain
We next examined whether IP3R expression is required for BRCA1 localization in non-nuclear compartments. We used DT40 cells as well as DT40 IP3R triple knockout (DT40 TKO) cells. This is the only cell line currently available that is deficient in all three IP3R isoforms (41). We transiently transfected DT40 and DT40 TKO cells with YFP-BRCA1 and nuclear localized DsRed (Fig. 6A, left panel) and scored cells with nuclear only or nuclear and cytosolic expression in a blinded manner. The number of cells with BRCA1 non-nuclear localization was significantly higher in DT40 cells expressing IP3R compared with DT40 TKO cells (Fig. 6A, right panel). This indicates that IP3R expression is a partial determinant of BRCA1 localization outside of the nucleus.
FIGURE 6.

BRCA1 is recruited to the ER during apoptosis and has lipid binding activity. A, distribution of YFP-BRCA1 in nucleus only versus nuclear and cytosol localization in wild-type DT40 cells or DT40 cells with triple IP3R knockout. Nuclear localization was established by colocalization with nuclearly (nuc) localized DsRed. Representative confocal images are shown in the left panel. Images of cells were scored in a blinded manner as being nuclear only or having both nuclear and cytosolic expression. DT40, n = 122; DT40 TKO, n = 143. Significance was determined by Fisher's exact test using a 2 × 2 contingency table (p = 0.0157). B, subcellular localization of BRCA1 in DT40 cells treated with or without paclitaxel. DT40 and DT40 TKO cells were treated with 500 nm paclitaxel for 48 h. Cells were harvested and fractionated using differential centrifugation as in Fig. 1C. 20 μg of each fraction was used for Western blotting with anti-BRCA1 antibody (top row). IP3R antibody was used to show the purity of the ER-enriched fraction (center row). LRP130, a mitochondrial marker, was used to show the absence of mitochondrial proteins in the microsomal ER-enriched fraction (bottom row). The loss of LRP130 from the mitochondrial fraction after paclitaxel treatment may be related to its putative role in Taxol-mediated cell death signaling (50, 51). For simplicity, only the mitochondrial and ER fractions are shown. The amount of BRCA1 protein present in the ER (100,000 × g) fraction was quantified and graphed from three separate experiments and is presented as the -fold increase over basal (no paclitaxel). p < 0.05. C, positional score of amino acid residues in BRCA1 that have homology to 1185 fatty acid binding protein-specific PSSM libraries (see “Experimental Procedures”). A high-scoring region is present in the first BRCT repeat encompassing amino acids 1664–1696 (solid line). The relative positions of domains in BRCA1 are shown above the graph. D, amino acids encompassing the putative lipid binding motif. Basic residues and adjacent threonine residues found in some lipid binding pockets (42, 43) are highlighted in purple. E, ribbon diagram (left panel) and space-fill diagram (right panel) of the tandem BRCT domains of BRCA1. Purple residues correspond to the highlighted residues labeled in D. F, lipid binding of GST-RING and GST-BRCT to PIP strips. Lipid spots present on the PIP strips are indicated in the left panel. LPA, Lysophosphatidic Acid; LPC, lysophosphatidylcholine; PtdIns, phosphotidylinositol; PI-3-P, phosphatidylinositol 3-phosphate; PI-4-P, phosphatidylinositol 4-phosphate; PI-5-P, phosphatidylinositol 5-phosphate; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine 1-phosphate; PI-3,4-P2, phosphatidylinositol 3,4-bisphosphate; PI-3,5-P2, phosphatidylinositol 3,5-bisphosphate; PI-4,5-P2, phosphatidylinositol 4,5-bisphosphate; PI-3,4,5-P3, phosphatidylinositol 3,4,5-trisphosphate; PA, phosphatidic acid; PS, phosphatidylserine. The data in A and B are from WT and TKO DT40 cells.
To determine whether BRCA1 is recruited to ER membranes during apoptosis, we treated DT40 and DT40 TKO cells with paclitaxel and purified ER-enriched 100,000 × g fractions as in Fig. 1C. BRCA1 ER localization increased with paclitaxel treatment, and this increase was dependent on IP3R expression (Fig. 6B). Interestingly, loss of IP3R expression did not completely prevent constitutive ER localization of BRCA1. This suggests that a subpopulation of BRCA1 is associated with the ER independent of both IP3R expression and apoptosis. We hypothesized that recruitment of BRCA1 to the ER may be mediated by an intrinsic lipid binding activity in the BRCA1 protein. We identified a potential lipid binding domain within the C-terminal tandem BRCT domain of BRCA1 using Adaptive-BLAST (24, 25). Specifically, we found that amino acid residues 1664–1696 within the first BRCT repeat have a strong potential for lipid binding on the basis of homology to fatty acid binding proteins (Fig. 6C and “Experimental Procedures”). Examination of high-scoring residues within this sequence identified a basic region with several threonine residues common to lipid binding pockets, including phosphatidic acid binding proteins ((Fig. 6D) (42, 43)). When mapped onto the structure of the tandem BRCT domains, these residues were solvent-accessible and, therefore, potentially able to participate in lipid binding (Fig. 6E).
To experimentally test this hypothesis, we purified recombinant GST-BRCT and performed a lipid strip overlay experiment. The GST-RING domain was used as a negative control. GST-BRCT was capable of binding several lipids, including phosphatidic acid, phosphatidyl inositol-3,5-P2 and phosphatidyl inositol-4,5-P2, whereas GST-RING was unable to bind to any lipids (Fig. 6F). Therefore, the BRCT domain of BRCA1 has a previously uncharacterized lipid binding activity that may mediate constitutive localization at ER membranes. Constitutive BRCA1 association with the ER may allow for rapid recruitment to the IP3R upon apoptotic induction. Of note, clinically relevant mutations of residues within the BRCT domain alter the subcellular distribution of BRCA1, which may be related to the lipid binding activity of the BRCT domain (44). It is also possible that the BRCA1 lipid binding domain may interact with other cellular membranes in addition to the ER. Although this may indeed be the case and may have functional relevance, with respect to this study we have shown that BRCA1 is recruited to the IP3R at ER membranes during apoptosis to mediate apoptotic calcium release (Fig. 5) and, further, that BRCA1 retention at the ER is reduced in IP3R null cells (Fig. 6, A and B). Therefore, at least some of the proapoptotic activities of BRCA1 are mediated by ER localization and binding to IP3R channels.
DISCUSSION
In this study, we established for the first time a possible mechanism through which BRCA1 exerts its proapoptotic function. Our data suggests a physical and functional interaction between BRCA1 and IP3R-1 that increases IP3R activity. We showed that BRCA1 is recruited to the ER during apoptosis and that loss of IP3R expression abolishes this recruitment. Finally, we identified a novel lipid binding activity in BRCA1 with significant implications for BRCA1 function. Future work will investigate the lipid binding activity of BRCA1 in more detail.
Although BRCA1 mutations and loss of expression are considered contributing causes of hereditary breast and ovarian cancer, studies have suggested that BRCA1 down-regulation may be present in a large number of breast tumors without mutations in BRCA1 (45–47). Furthermore, it has been shown that cytosolic localization of BRCA1 can be correlated with a better disease prognosis (48). We suggest that the proapoptotic function of BRCA1 is to increase IP3R-mediated apoptotic calcium release. This has significant implications for both hereditary and non-hereditary breast and ovarian cancers because BRCA1 expression mediates cellular responses to chemotherapeutics (40). The addition of yet another tumor suppressor, BRCA1, as a regulator of IP3Rs adds even more importance to the need to understand the role of calcium homeostasis in tumor progression.
This work was supported, in whole or in part, by National Institutes of Health Grant GM081685 (to D. B.) and by National Institutes of Health Grant DE014756 (to D. I. Y.). This work was also supported by seed funds provided by the Department of Obstetrics and Gynecology, University of Texas Medical Branch (Galveston, TX) and startup funds provided by the Department of Biochemistry and Molecular Biology, University of Texas Health Science Center at Houston (Houston, TX) (to D. B.); by Cancer Institute of New South Wales, Australia Grant 12/CDF/2-12 (to B. R. H.); and by a grant from the Pennsylvania Department of Health using tobacco settlement funds (to D. V. R.).
- IP3R
- inositol 1,4,5-trisphosphate receptor
- ER
- endoplasmic reticulum
- PTEN
- phosphatase and tensin homolog
- PML
- promyelocytic leukemia protein
- BRCT
- BRCA1 C terminus
- CFP
- cyan fluorescent protein
- PSSM
- position-specific scoring matrix
- UWB
- UWB1.289.
REFERENCES
- 1. Patterson R. L., Boehning D., Snyder S. H. (2004) Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73, 437–465 [DOI] [PubMed] [Google Scholar]
- 2. Decuypere J. P., Monaco G., Bultynck G., Missiaen L., De Smedt H., Parys J. B. (2011) The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 1813, 1003–1013 [DOI] [PubMed] [Google Scholar]
- 3. Rong Y.-P., Barr P., Yee V. C., Distelhorst C. W. (2009) Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta 1793, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akl H., Bultynck G. (2013) Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim. Biophys. Acta 1835, 180–193 [DOI] [PubMed] [Google Scholar]
- 5. Giorgi C., Ito K., Lin H.-K., Santangelo C., Wieckowski M. R., Lebiedzinska M., Bononi A., Bonora M., Duszynski J., Bernardi R., Rizzuto R., Tacchetti C., Pinton P., Pandolfi P. P. (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roy R., Chun J., Powell S. N. (2012) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 12, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoeijmakers J. H. (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374 [DOI] [PubMed] [Google Scholar]
- 8. Clark S. L., Rodriguez A. M., Snyder R. R., Hankins G. D. V., Boehning D. (2012) Structure-function of the tumor suppressor BRCA1. Comput. Struct. Biotechnol. J. 10.5936/csbj.201204005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodríguez J. A., Henderson B. R. (2000) Identification of a functional nuclear export sequence in BRCA1. J. Biol. Chem. 275, 38589–38596 [DOI] [PubMed] [Google Scholar]
- 10. Chen C. F., Li S., Chen Y., Chen P. L., Sharp Z. D., Lee W. H. (1996) The nuclear localization sequences of the BRCA1 protein interact with the importin-α subunit of the nuclear transport signal receptor. J. Biol. Chem. 271, 32863–32868 [DOI] [PubMed] [Google Scholar]
- 11. Shao N., Chai Y. L., Shyam E., Reddy P., Rao V. N. (1996) Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 13, 1–7 [PubMed] [Google Scholar]
- 12. Fabbro M., Schuechner S., Au W. W., Henderson B. R. (2004) BARD1 regulates BRCA1 apoptotic function by a mechanism involving nuclear retention. Exp. Cell Res. 298, 661–673 [DOI] [PubMed] [Google Scholar]
- 13. Henderson B. R. (2012) The BRCA1 breast cancer suppressor: regulation of transport, dynamics, and function at multiple subcellular locations. Scientifica 2012, 796808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milner R., Wombwell H., Eckersley S., Barnes D., Warwicker J., Van Dorp E., Rowlinson R., Dearden S., Hughes G., Harbron C., Wellings B., Hodgson D., Womack C., Gray N., Lau A., O'Connor M. J., Marsden C., Kvist A. J. (2013) Validation of the BRCA1 antibody MS110 and the utility of BRCA1 as a patient selection biomarker in immunohistochemical analysis of breast and ovarian tumours. Virchows Arch. 462, 269–279 [DOI] [PubMed] [Google Scholar]
- 15. Boehning D., Patterson R. L., Sedaghat L., Glebova N. O., Kurosaki T., Snyder S. H. (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 16. Wozniak A. L., Wang X., Stieren E. S., Scarbrough S. G., Elferink C. J., Boehning D. (2006) Requirement of biphasic calcium release from the endoplasmic reticulum for Fas-mediated apoptosis. J. Cell Biol. 175, 709–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borahay M. A., Kilic G. S., Yallampalli C., Snyder R. R., Hankins G. D., Al-Hendy A., Boehning D. (2014) Simvastatin potently induces calcium-dependent apoptosis of human leiomyoma cells. J. Biol. Chem. 289, 35075–35086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wagner L. E., 2nd, Yule D. I. (2012) Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J. Physiol. 590, 3245–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ko K. D., Liu C., Rwebangira M. R., Burge L., Southerland W. (2011) The development of a proteomic analyzing pipeline to identify proteins with multiple RRMs and predict their domain boundaries. IEEE Int. Conf. Bioinform. Biomed. Workshops 374–381 [Google Scholar]
- 20. Bernier I., Jollès P. (1987) A survey on cytosolic non-enzymic proteins involved in the metabolism of lipophilic compounds: from organic anion binders to new protein families. Biochimie 69, 1127–1152 [DOI] [PubMed] [Google Scholar]
- 21. Ockner R. K. (1990) Historic overview of studies on fatty acid-binding proteins. Mol. Cell. Biochem. 98, 3–9 [DOI] [PubMed] [Google Scholar]
- 22. Glatz J. F., van der Vusse G. J. (1990) Cellular fatty acid-binding proteins: current concepts and future directions. Mol. Cell. Biochem. 98, 237–251 [DOI] [PubMed] [Google Scholar]
- 23. Zanotti G., Scapin G., Spadon P., Veerkamp J. H., Sacchettini J. C. (1992) Three-dimensional structure of recombinant human muscle fatty acid-binding protein. J. Biol. Chem. 267, 18541–18550 [DOI] [PubMed] [Google Scholar]
- 24. Hong Y., Chalkia D., Ko K. D., Bhardwaj G., Chang G. S., van Rossum D. B., Patterson R. L. (2009) Phylogenetic profiles reveal structural and functional determinants of lipid-binding. J. Proteomics Bioinform. 2, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong Y., Chintapalli S. V., Bhardwaj G., Zhang Z., Patterson R. L., Van Rossum D. B. (2011) Adaptive-BLAST: a user-defined platform for the study of proteins. J. Integr. OMICS 1, 88–101 [Google Scholar]
- 26. Scully R., Ganesan S., Brown M., De Caprio J. A., Cannistra S. A., Feunteun J., Schnitt S., Livingston D. M. (1996) Location of BRCA1 in human breast and ovarian cancer cells. Science 272, 123–126 [DOI] [PubMed] [Google Scholar]
- 27. Boehning D., van Rossum D. B., Patterson R. L., Snyder S. H. (2005) A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coene E. D., Hollinshead M. S., Waeytens A. A., Schelfhout V. R., Eechaute W. P., Shaw M. K., Van Oostveldt P. M., Vaux D. J. (2005) Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 16, 997–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bootman M., Niggli E., Berridge M., Lipp P. (1997) Imaging the hierarchical Ca2+ signalling system in HeLa cells. J. Physiol. 499, 307–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sauvé R., Diarra A., Chahine M., Simoneau C., Morier N., Roy G. (1991) Ca2+ oscillations induced by histamine H1 receptor stimulation in HeLa cells: Fura-2 and patch clamp analysis. Cell Calcium 12, 165–176 [DOI] [PubMed] [Google Scholar]
- 31. Diarra A., Wang R., Garneau L., Gallo-Payet N., Sauvé R. (1994) Histamine-evoked Ca2+ oscillations in HeLa cells are sensitive to methylxanthines but insensitive to ryanodine. Pfluegers Arch. Eur. J. Physiol. 426, 129–138 [DOI] [PubMed] [Google Scholar]
- 32. Fabbro M., Rodriguez J. A., Baer R., Henderson B. R. (2002) BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 277, 21315–21324 [DOI] [PubMed] [Google Scholar]
- 33. Hajnóczky G., Robb-Gaspers L. D., Seitz M. B., Thomas A. P. (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 [DOI] [PubMed] [Google Scholar]
- 34. Tomida T., Hirose K., Takizawa A., Shibasaki F., Iino M. (2003) NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 22, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tembe V., Henderson B. R. (2007) BARD1 translocation to mitochondria correlates with Bax oligomerization, loss of mitochondrial membrane potential, and apoptosis. J. Biol. Chem. 282, 20513–20522 [DOI] [PubMed] [Google Scholar]
- 36. Churchill G. C., Louis C. F. (1999) Imaging of intracellular calcium stores in single permeabilized lens cells. Am. J. Physiol. 276, C426–34 [DOI] [PubMed] [Google Scholar]
- 37. Gerasimenko J. V., Sherwood M., Tepikin A. V., Petersen O. H., Gerasimenko O. V. (2006) NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J. Cell Sci. 119, 226–238 [DOI] [PubMed] [Google Scholar]
- 38. Betzenhauser M. J., Wagner L. E., 2nd, Won J. H., Yule D. I. (2008) Studying isoform-specific inositol 1,4,5-trisphosphate receptor function and regulation. Methods 46, 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee L.-F., Li G., Templeton D. J., Ting J. P. (1998) Paclitaxel (Taxol)-induced gene expression and cell death are both mediated by the activation of c-Jun NH2-terminal kinase (JNK/SAPK). J. Biol. Chem. 273, 28253–28260 [DOI] [PubMed] [Google Scholar]
- 40. Quinn J. E., Kennedy R. D., Mullan P. B., Gilmore P. M., Carty M., Johnston P. G., Harkin D. P. (2003) BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res. 63, 6221–6228 [PubMed] [Google Scholar]
- 41. Sugawara H., Kurosaki M., Takata M., Kurosaki T. (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones J. A., Rawles R., Hannun Y. A. (2005) Identification of a novel phosphatidic acid binding domain in protein phosphatase-1. Biochemistry 44, 13235–13245 [DOI] [PubMed] [Google Scholar]
- 43. Stace C. L., Ktistakis N. T. (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761, 913–926 [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez J. A., Au W. W., Henderson B. R. (2004) Cytoplasmic mislocalization of BRCA1 caused by cancer-associated mutations in the BRCT domain. Exp. Cell Res. 293, 14–21 [DOI] [PubMed] [Google Scholar]
- 45. Kennedy R. D., Quinn J. E., Mullan P. B., Johnston P. G., Harkin D. P. (2004) The role of BRCA1 in the cellular response to chemotherapy. J. Natl. Cancer Inst. 96, 1659–1668 [DOI] [PubMed] [Google Scholar]
- 46. Thompson M. E., Jensen R. A., Obermiller P. S., Page D. L., Holt J. T. (1995) Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat. Genet. 9, 444–450 [DOI] [PubMed] [Google Scholar]
- 47. Magdinier F., Ribieras S., Lenoir G. M., Frappart L., Dante R. (1998) Down-regulation of BRCA1 in human sporadic breast cancer: analysis of DNA methylation patterns of the putative promoter region. Oncogene 17, 3169–3176 [DOI] [PubMed] [Google Scholar]
- 48. Mylona E., Melissaris S., Nomikos A., Theohari I., Giannopoulou I., Tzelepis K., Nakopoulou L. (2014) Effect of BRCA1 immunohistochemical localizations on prognosis of patients with sporadic breast carcinomas. Pathol. Res. Pract. 210, 533–540 [DOI] [PubMed] [Google Scholar]
- 49. Akimzhanov A. M., Barral J. M., Boehning D. (2013) Caspase 3 cleavage of the inositol 1,4,5-trisphosphate receptor does not contribute to apoptotic calcium release. Cell Calcium 53, 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu L., Amy V., Liu G., McKeehan W. L. (2002) Novel complex integrating mitochondria and the microtubular cytoskeleton with chromosome remodeling and tumor suppressor RASSF1 deduced by in silico homology analysis, interaction cloning in yeast, and colocalization in cultured cells. In Vitro Cell. Dev. Biol. Anim. 38, 582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Michaud, Barakat, Magnard, Rigal, Baggetto (2010) Leucine-rich protein 130 contributes to apoptosis resistance of human hepatocarcinoma cells. Int. J. Oncol. 38, 169–178 [PubMed] [Google Scholar]