

Background: Equol, a daidzein metabolite, is a potent regulator of the cancer-promoting effects of dietary daidzein.
Results: Silencing of eIF4G or c-Myc demonstrated that the equol-mediated c-Myc up-regulation is central to the pro-cancer effects of equol.
Conclusion: Equol may promote cancer via c-Myc.
Significance: Caution must be exercised regarding soyfood consumption for patients with ER (−) breast cancer.
Keywords: breast cancer, cancer stem cells, cell growth, eukaryotic translation initiation factor 4G (eIF4G), Myc (c-Myc), protein synthesis, equol, soy
Abstract
Epidemiological studies implicate dietary soy isoflavones as breast cancer preventives, especially due to their anti-estrogenic properties. However, soy isoflavones may also have a role in promoting breast cancer, which has yet to be clarified. We previously reported that equol, a metabolite of the soy isoflavone daidzein, may advance breast cancer potential via up-regulation of the eukaryotic initiation factor 4GI (eIF4GI). In estrogen receptor negative (ER−) metastatic breast cancer cells, equol induced elevated levels of eIF4G, which were associated with increased cell viability and the selective translation of mRNAs that use non-canonical means of initiation, including internal ribosome entry site (IRES), ribosome shunting, and eIF4G enhancers. These mRNAs typically code for oncogenic, survival, and cell stress molecules. Among those mRNAs translationally increased by equol was the oncogene and eIF4G enhancer, c-Myc. Here we report that siRNA-mediated knockdown of c-Myc abrogates the increase in cancer cell viability and mammosphere formation by equol, and results in a significant down-regulation of eIF4GI (the major eIF4G isoform), as well as reduces levels of some, but not all, proteins encoded by mRNAs that are translationally stimulated by equol treatment. Knockdown of eIF4GI also markedly reduces an equol-mediated increase in IRES-dependent mRNA translation and the expression of specific oncogenic proteins. However, eIF4GI knockdown did not reciprocally affect c-Myc levels or cell viability. This study therefore implicates c-Myc as a potential regulator of the cancer-promoting effects of equol via up-regulation of eIF4GI and selective initiation of translation on mRNAs that utilize non-canonical initiation, including certain oncogenes.
Introduction
Isoflavones are found in nutritionally relevant amounts in soybeans comprising ∼3.5 mg/gm soy protein in traditional soy foods. Studies have reported a range of 1–25 μm soy isoflavones in the human circulation following consumption of soy products, which is sufficient for physiological activity (1–4). Because of the structural similarity to 17β estradiol, these phytoestrogens have been extensively studied for their potential estrogenic or antiestrogenic effects in breast cancer (5, 6). Moreover, soy isoflavones may have additional estrogen-independent effects in aggressive estrogen receptor negative (ER−)2 breast cancers (5, 7).
Daidzein is the second most prominent isoflavone in soy and ∼70% of daidzein can be metabolized by the intestinal microflora to the metabolite equol (8). Only ∼30–50% of humans have the gut microflora necessary to convert daidzein to equol (8, 9); therefore, not all humans are affected by equol. Equol is also chemically similar to estrogen and has 80 times more ERβ affinity than daidzein (10–12). The effects of equol, specifically in ER− breast cancers, or established aggressive breast cancers, remains unclear and requires additional study (7, 8, 13, 14).
At high concentrations (50–350 μm), equol has been implicated in inhibition of cancer cell growth, invasion, tumor progression, and cancer risk (15–19), while low physiological concentrations of equol reportedly increases cancer cell proliferation (14, 20). In ER+ human breast cancer cells, equol increases estrogenic activity and cell proliferation, but does not affect tumor growth in mice (20–23). Dietary daidzein also failed to reduce mammary tumor growth in rats when metabolized to ∼1 μm equol in serum (24), although others have shown inhibitory effects of daidzein and equol on ER+ breast cancer cells and tumors (25, 26). Thus, whether daidzein and equol prevents or promotes breast cancer in experimental models is not established, and may be concentration and ER status dependent. Accordingly, the molecular mechanisms by which equol acts on breast cancer cells need to be better understood, enabling recommendations for soy consumption in breast cancer patients.
To this end, we previously tested the effect of individual and combined soy isoflavones on immune impaired (nude) mice with mammary tumors, established from ER− cancer cells. We reported that dietary daidzein increases mammary tumor growth and metastasis (27). These findings support our recent in vitro studies, which demonstrated that equol is the active metabolite of daidzein and increases breast cancer cell malignancy, primarily via up-regulation of the eukaryotic initiation factor eIF4GI and its translation of mRNA-encoding oncogenic proteins (28).
Translational control has received increased attention in recent years due to its emerging significance in cancer development and progression (29). Translation initiation is typically the rate-limiting step and therefore, a primary site for regulation. Accordingly, the levels of two eukaryotic initiation factors that are members of the cap-initiation eIF4F complex, consisting of eIF4E (Cap-binding protein) and eIF4G (initiation complex molecular scaffold), are frequently elevated in human cancers, and have been associated with poor prognosis and outcome (29–31). Overexpression of eIF4GI is critical for the modes of translation initiation in eukaryotic cells that bypass or have a reduced requirement for eIF4E, including IRES-dependent mechanisms that allow the 40 S ribosome to be directly recruited to the mRNA (32, 33). It is thought that under the physiological stress conditions that exist in large tumors (i.e. growth arrest, amino acid starvation, hypoxia), cancer cells rely on non-canonical, eIF4E-deficient, IRES-dependent translation of a subset of mRNAs encoding pro-growth, pro-angiogenic and pro-survival proteins, such as, BCL2, Bcl -Xl, c-Myc, p120 catenin, and vascular endothelial growth factor A (VEGF) A, among others (29, 34–36).
In our recent study using MDA-MB 435 ER− metastatic breast cancer cells treated with equol, we showed that the transcription factor c-Myc was also elevated in addition to up-regulated levels of eIF4GI and the increased translation of IRES-containing mRNAs that control cell survival and cell proliferation (28). This result is relevant because c-Myc is overexpressed in a variety of human cancers and plays an important role in multiple signaling pathways including cell growth, cell proliferation, metabolism, ribosome biogenesis, microRNA regulation, cell death, and cell survival (37–39).
With the objective of determining whether the equol-mediated up-regulation of eIF4G promotes preferential synthesis of c-Myc, which has an IRES element (40), or whether c-Myc up-regulation by equol leads to eIF4GI transcription, as shown in (41), we investigated the effects of silencing eIF4GI or c-Myc in equol-treated metastatic breast cancer cells. Here, we show that reducing eIF4GI levels results in a marked reduction in IRES-dependent mRNA translation, decreased polysomal association of those mRNAs, and corresponding oncogenic protein levels of specific IRES-containing mRNAs. The increase in cell viability and mammosphere formation (an indicator of tumorigenic potential (42, 43)) in response to equol was not affected until c-Myc was also targeted by siRNA, which in addition, significantly down-regulated eIF4GI levels.
Taken together, these data implicate c-Myc, and the consequent increase in eIF4GI followed by selective translation of oncogene mRNAs, in the pro-transforming effects of equol on breast cancer progression.
EXPERIMENTAL PROCEDURES
Cell Culture
Metastatic variant of MDA-MB-435 (ER−) (gift of Dr. Danny Welch, The University of Kansas Cancer Center) and Hs578t (ER-) metastatic human breast cancer cells (American Type Culture Collection (ATCCC), Manassas, VA) were maintained in complete culture medium: Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, Houston, TX) supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C in 5% CO2. The MCF-10A mammary epithelial cells (ATCCC) were maintained in DMEM supplemented with 10% horse serum, EGF, hydrocortisone, cholera toxin, and insulin, as described previously (44). Cell lines were authenticated and found to be mycoplasma free.
Cell Treatment
Quiescent mammary epithelial cells or metastatic cancer cells were treated with 0 (vehicle, 0.1% DMSO) or 25 μm of (R,S) Equol (LC Laboratories, Woburn, MA) in DMEM and 5% FBS medium for 24 h.
Western Blotting
Cells were lysed and Western blotted, as described in Ref. 27. Primary antibodies to eIF4E, eIF4GI, c-Myc, p120 catenin, Bcl-Xl, Cyclin-D1, GAPDH, JunB, and β-actin proteins (Epitomics, Burlingame, CA, Cell Signaling, Danvers, MA, Sigma-Aldrich) were used. The integrated density of positive bands was quantified using Image J software, as described in Ref. 27.
Cell Viability Assay
Cell viability was determined by the CellTiter 96 Non-Radioactive Cell Proliferation kit according to the manufacturer's instructions (Promega, Madison, WI). Briefly, quiescent 1 × 105 MDA-MB-435 cells were added to each well of a 96-well plate and treated for 24 h with vehicle or 25 μm equol. Following equilibration, 15 μl/well of MTT (3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide) reagent was added, and the plates incubated at 37 °C for 4 h. Stop solution (100 μl) was added to each well, and the plates incubated to facilitate solubilization of newly formed formazan salts. The absorbance at 570 nm was measured using an ELISA plate reader.
Real-time Reverse Transcriptase Polymerase Reaction (RT-PCR) Analysis
Real-time quantitative RT-PCR analysis was performed as described (28). Briefly, total RNA was extracted using the Qiagen RNeasy Kit (Qiagen). RNA concentration was detected using NanoDrop (NanoDrop Technologies, Wilmington, DE). RNA (0.5 μg) was used to synthesize cDNA using the iScript cDNA synthesis kit (Bio-Rad). RT-PCR primers were as follows. MYC: forward, 5′-TTCTCAGAGGCTTGGCGGGAAA-3′, reverse, 5′-TGCCTCTCGCTGGAATTACTACA-3′. B- 2-microglobulin (B2M): forward, 5′-GGCTATCCAGCGTACTCCAAA-3′, reverse, 5′-CGGCAGGCATACTCATCTTTTT-3′. GAPDH: forward, 5′-TTGCCATCAATGACCCCTTCA-3′, reverse, 5′-CGCCCCACTTGATTTTGGA-3′. CCND1: forward, 5′-TGGTGAACAAGCTCAAGTGGA-3′. reverse, 5′-TGATCTGTTTGTTCTCCTCCGCCT-3′. eIF4GI: forward, 5′-TTGTGGATGATGGTGGCT-3′ reverse, 5′-TTATCTGTGCTTTCTGTGGGT-3′. CTNND1: forward, 5′-TCCAGCAAACGATACAGTGG-3′, reverse, 5′-GAACCACCTCTGGCTGAAAT-3′. Real-time reactions were performed using iQ-SYBR-Green PCR Master Mix (Bio-Rad). The amplification reaction was performed for 40 cycles (10 s at 95 °C, 30 s at 59 °C, and 30 s at 72 °C). B2M mRNA was used as an internal control. The fold change was determined by the 2 ΔΔCT method as described (27, 28).
Polysome Fractionation
MDA-MB-435 cells (non-silencing control or eIFGI silenced), treated with vehicle or equol, were used for polysome profiling, as described in Ref. 36. Cells were pre-treated with 100 μg/ml cycloheximide (Calbiochem), washed twice in PBS with 100 μg/ml cycloheximide, pelleted, and resuspended in 700 μl of polysome isolation buffer (200 mm Tris, pH 7.5, 100 mm NaCl, and 30 mm MgCl2). After 5 min of incubation, 250 μl of detergent buffer (1.2% Triton, 0.2 m sucrose in polysome isolation buffer) was added, and cells were lysed. Clarified lysates were layered onto 10–50% sucrose gradients (Sigma-Aldrich) and sedimented at 36,000 rpm for 2 h in a SW40 rotor (Beckman Coulter) at 4 °C. Gradients were collected as 15 × 750 μl fractions, by pumping 60% sucrose into the bottom of the gradient and collecting from the top using an ISCO fraction collector while simultaneously monitoring absorbance at 254 nm. RNA was isolated by extraction with phenol/chloroform. Fractions 4–12, representing polysomes, were pooled and classified as light polysome fractions (2–3 ribosomes) and heavy polysome fractions (≥ 4 ribosomes). RNA preparations from each fraction were subjected to qRT-PCR for c-MYC, CTNND1 (p120-catenin), CCND1 (cyclin D1), eIF4G, GAPDH, and B2M, as described above.
shRNA/siRNA Transfection
eIF4GI knockdown using shRNA adenovirus (Ad) vectors were conducted as described (36). MDA-MB-435 and Hs578t cells were infected with Ad vectors twice over 4 days with non-silencing (NS control) or eIF4GI-silencing (Ad) shRNA vectors at a multiplicity of infection (MOI) of 100, then treated with vehicle or 25 μm equol for 24 h. Control or c-Myc siRNAs (Santa Cruz, CA) were transfected into MDA-MB-435 and Hs578t cells using Lipofectamine (Invitrogen) according to the manufacturer's instructions, followed by treatment with vehicle or equol for 24 h.
IRES-dependent Protein Synthesis
MDA-MB-435 cells with non-silencing control or eIF4GI siRNA were transfected with a bicistronic reporter or control plasmid containing the luciferase constructs without an IRES, using Lipofectamine 2000 (Invitrogen), as per the manufacturer's directions. As described (36), this plasmid contains a cap-dependent Renilla luciferase (RLuc) followed by a 5′-UTR containing the p120 catenin IRES driving a firefly luciferase (FLuc). 24 h following transfection, cells were treated with equol for an additional 24 h. Relative IRES activity was analyzed as 570 nm FLuc/480 nm RLuc in a luminometer using a dual luciferase assay kit (Promega), according to the manufacturer's instructions.
Mammosphere Formation Assay
Mammosphere assays were performed, as described in Ref. 42. MDA-MB-435 cells were seeded in ultra-low attachment plates (Corning) at a density of 500–5000 cells/well in serum-free mammary epithelium basal medium (Lonza) supplemented with 1% penicillin/streptomycin (Lonza), B27 supplement minus vitamin A (50×, Invitrogen), 5 μg/ml insulin (Invitrogen), 1 μg/ml hydrocortisone (Sigma), 20 ng/ml EGF, and 20 ng/ml fibroblast growth factor (Sigma). Mammospheres were counted using an inverted microscope after 3 days of incubation in 37°C, 5% CO2. Mammosphere-forming efficiency (MFE) was calculated as the number of mammospheres divided by the number of cells seeded per well and is expressed relative to cells expressing control siRNA treated with vehicle.
Statistical Analysis
Data were analyzed and reported as mean ± S.E. of at least three independent experiments. Statistical analyses were conducted using Microsoft Excel. Differences between means were determined using the Student's t-test, and values p ≤ 0.05 or p ≤ 0.01 were considered significant.
RESULTS
eIF4GI Silencing Partially Rescues the Pro-cancer Effects of Equol
We recently reported that the daidzein metabolite equol, through the up-regulation of eukaryotic protein synthesis initiation factor eIF4GI, may specifically direct the synthesis of IRES-containing mRNAs that induce cell survival and proliferation, and increase cancer progression. In the previous study, we used 0, 1, 5, 25, or 50 μm equol and reported that the effects of equol on cell viability and up-regulation of oncogenic protein expression saturated at 25 μm equol (28). Therefore, the present study was conducted with 25 μm equol, using two metastatic cancer cell lines.
To investigate whether equol regulates breast cancer cell malignancy via eIF4GI, we determined the effects of equol on MDA-MB-435 and Hs578t cells with eIF4GI knockdowns. Using eIF4GI shRNA vectors, eIF4GI protein expression was silenced by ∼60%, compared with cells expressing non-silenced vector controls, in both vehicle and equol-treated MDA-MB-435 and Hs578t cells (Fig. 1A). Notably, the ∼1.8-fold increase in eIF4GI mRNA levels in response to equol treatment was effectively abolished by the eIF4GI targeting, resulting in a 3-fold reduction in eIF4GI mRNA in the presence and absence of equol (Fig. 1C).
FIGURE 1.

Protein and mRNA levels in response to equol treatment of transformed MDA-MB435 and Hs578t cells expressing control or eIF4GI shRNAs. MDA-MB-435 and Hs578t cells were infected with non-silencing (control) or eIF4GI silencing (eIF4GI) adenovirus (Ad) shRNA vectors, then treated with vehicle or 25 μm equol for 24 h. Equal amounts of lysates were immunoblotted for the indicated proteins or mRNAs quantified by qRT-PCR. A, representative immunoblots. B, fold changes of protein expression compared with vehicle control, calculated from the integrated density of bands normalized for actin expression. C, fold changes in EIF4G, MYC, CTNND, and CCND1 mRNAs. B2M was invariant and used as an internal control. Results are shown as fold-changes in equol-treated cells relative to vehicle controls (n = 3). An asterisk indicates statistical significance at p ≤ 0.05. Two asterisks indicate statistical significance of p ≤ 0.01.
Because equol up-regulates expression of IRES containing oncogenic proteins (28), cells expressing control or eIF4GI knockdown were analyzed for protein expression levels of members of the eIF4F complex, as well as proteins from IRES containing mRNAs (Fig. 1, A and B). At 25 μm, equol protein expression in Hs578t and MB-435 cells, respectively increased levels of eIF4GI (∼1.4, 1.6-fold), c-Myc (∼1.5, 1.8-fold), Cyclin D (∼1.4, 1.5-fold), Bcl-Xl (∼1.3, 1.5-fold), and p120 catenin (∼1.3, 1.6-fold) in a statistically significant manner, compared with control shRNA-expressing cells. Cells expressing eIF4GI shRNA demonstrated a ∼50% reduction in eIF4GI in vehicle treatments and an 80∼120% reduction in equol-treated cells, while the eIF4E levels remained unchanged. Knockdown of eIF4GI also caused a marked ∼40–60% reduction in expression levels of proteins encoded by mRNAs with IRESs: Cyclin D, Bcl-Xl, and p120 catenin, from both vehicle and equol-treated cells. Protein expression of GAPDH, JunB, and Actin from mRNAs with short 5′-UTRs lacking an IRES were not affected by equol treatment or eIF4GI silencing. These results suggest that elevated eIF4GI levels are essential for the significant and selectively increased level of translation of IRES-containing cancer promoting mRNAs, in response to equol treatment. Interestingly, the equol-mediated ∼1.7-fold increase in c-Myc, a reported IRES containing mRNA, remained elevated regardless of the eIF4GI knockdown (Fig. 1). The reason for sustained c-Myc expression is unknown but may reflect an ability to use other eIF4GI homologs such as DAP5 (45), or contribution from eIF4E-mediated cap-dependent protein synthesis.
To determine whether silencing eIF4GI alters transcriptional activity of the mRNAs examined in Fig. 1, we quantified the mRNA levels of eIF4GI, C-MYC, CTNND1 (p120), and CCND1 (Cyclin D) in MDA-MB-435 cancer cells expressing control or eIF4GI shRNAs, treated with vehicle or 25 μm equol. Fig. 1C demonstrates that as previously reported by us from MDA-MB-435 mammary tumors and cells (27, 28), equol increased eIF4GI transcription by ∼1.8-fold. This increase was abolished in cells expressing eIF4GI shRNA, where eIF4GI expression was reduced by ∼3-fold in both vehicle and equol-treated cells. The mRNA levels of CTNND1 (p120) and CCND1 (CyclinD1) were not affected by equol treatment or eIF4GI knockdown, indicating that cyclin D1 and p120 catenin expression were regulated entirely at the translational level. However, c-Myc mRNA levels increased almost 2-fold in response to equol in both control and eIF4GI-silenced cells, confirming that the equol-mediated up-regulation in c-Myc protein levels is independent of eIF4GI (Fig. 1).
As previously reported, in metastatic cancer cells, equol at 1, 5, 10, 25, or 50 μm increased c-Myc protein and mRNA expression by 2-fold (28) (Fig. 2A). However, the immortalized but non-transformed mammary epithelial cell line MCF-10A did not demonstrate statistically significant changes in c-Myc protein expression in response to equol treatment at 5 or 25 μm (Fig. 2, B and C). Therefore, the equol-mediated up-regulation of c-Myc appears to be limited to transformed cancer cells.
FIGURE 2.
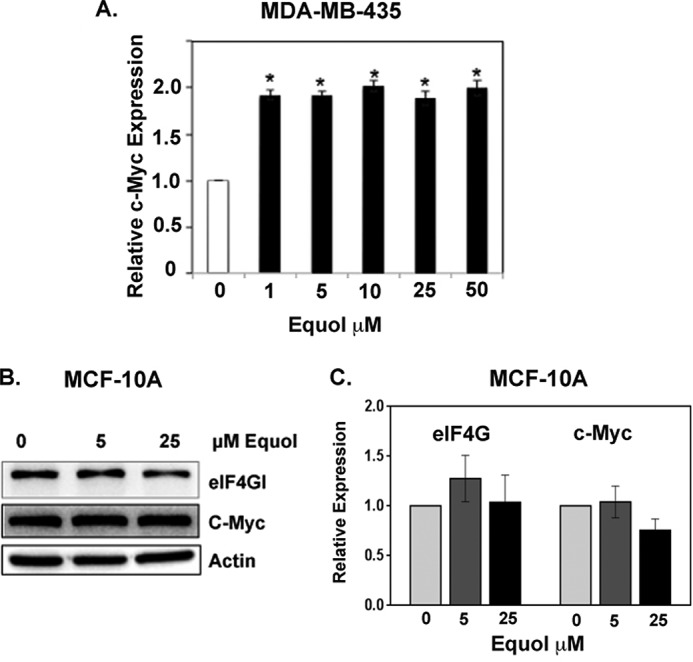
Effect of equol on eIF4G and c-Myc expression in cancer and non-cancer cells. Quiescent cells were treated with vehicle or equol (0–50 μm) for 24 h and equal amounts of lysates were immunoblotted for the indicated proteins. A, c-Myc expression in MDA-MB-435 human metatsatic cancer cells. B and C, eIF4G and c-Myc expression in MCF-10 mammary epithelial cells. Representative immunoblots (B) fold changes of protein expression compared with vehicle control, calculated from the integrated density of bands normalized for actin expression. Results are shown as fold-changes in equol-treated cells relative to vehicle controls (n = 3). An asterisk indicates statistical significance at p ≤ 0.05.
Next, we determined whether the elevated levels of eIF4GI, as found in highly transformed cells in response to equol treatment, might be necessary for efficient translation of IRES-containing mRNAs. Dual luciferase assays were performed to measure both cap-dependent and p120 catenin IRES-dependent protein synthesis in MDA-MB-435 cells expressing control or eIF4GI shRNA, treated with vehicle or equol. Data shown in Fig. 3 demonstrates that equol increased IRES-dependent mRNA translation in cells expressing control shRNA by ∼1.7-fold, while this increase was reduced by half when eIF4GI was silenced, effectively abolishing the effect of equol. Vehicle treatment of cells with eIF4GI knockdown did not statistically decrease IRES activity (data not shown). Thus, the elevated protein synthesis of mRNAs with IRESs can be attributed to the increased levels of eIF4GI in response to equol.
FIGURE 3.
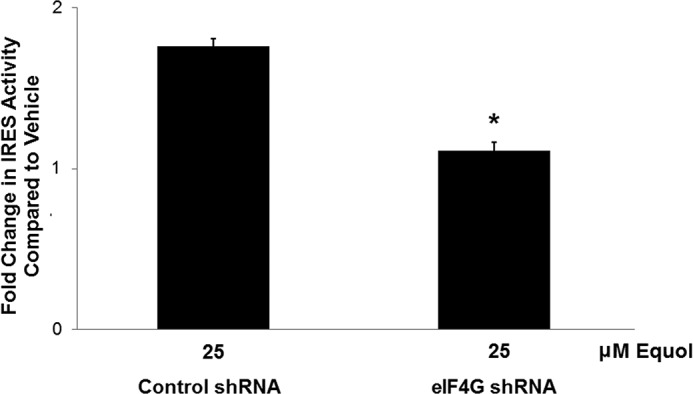
Effect of eIF4GI knockdown on IRES-dependent mRNA translation in response to equol treatment. MDA-MB-435 cells with control or eIF4GI silencing expressing a bicistronic reporter mRNA containing a cap-dependent Renilla luciferase (RLuc) followed by a 5′-UTR containing the p120 catenin IRES driving the firefly luciferase (FLuc), or a control without an IRES. Cells were treated with vehicle or equol for 24 h, lysed, and the relative IRES activity analyzed as 570 nm FLuc/480 nm RLuc from equal amounts of lysate. IRES activity was quantified relative to control activity for vehicle or equol treated cells. Results show fold change in IRES activity compared with vehicle for n = 3 ± S.E. An asterisk indicates statistical significance of p ≤ 0.05.
To investigate whether translation initiation is sensitive to the reduction in eIF4GI levels, we performed polysome profiling for control and eIF4GI-silenced MDA-MB-435 cells treated with vehicle or equol. The ribosome content on mRNA is an established surrogate for translation activity. As previously reported (28), Fig. 4A shows that equol increased the total mRNA associated with the polysomal fractions compared with vehicle alone, a manifestation of increased eIF4GI levels. In equol-treated MDA-MB-435 cells with eIF4GI knockdown, total RNA associated with polysomes was only very slightly decreased, again indicative of a selective reduction in translation of specific mRNAs rather than a global decrease. As expected in vehicle-treated cells, in the absence of equol, eIF4GI knockdown resulted in a general decreased association of RNA with both light and heavy polysomal fractions.
FIGURE 4.

Analysis of polysome profiles in response to vehicle or equol treatment of MDA-MB-435 cells expressing control or eIF4GI shRNA. Equal amounts of cytoplasmic cell lysates from vehicle control or 25 μm equol-treated MDA-MB-435 cells expressing control or eIF4GI shRNA were loaded onto 10–50% sucrose gradients and light (fractions 4–7, 2–4 ribosomes) and heavy polysome fractions (fractions 8–12, >4 ribosomes) collected. A, polysome profiles. B, C-MYC, EIF4G, CNND1, CTNND, and GAPDH mRNAs associated with polysome fractions, as detected by qRT-PCR. B2M was used as an internal control. Results are shown as fold-changes in equol-treated cells relative to vehicle controls (n = 3). An asterisk indicates statistical significance of p ≤ 0.05.
To identify specific mRNAs differentially associated with polysome fractions in control or eIF4GI silenced cells, real-time quantitative (q) RT-PCR was performed to measure IRES-containing mRNAs: CTNND1, CCND1, EIF4G, C-MYC, and the non-IRES containing housekeeping genes GAPDH and B2M. Fig. 4B demonstrates that in MDA-MB-435 cells expressing control shRNA, equol treatment increased the association of eIF4GI with both light and heavy polysomal fractions (∼1.7-fold), while the association of C-MYC, CTNND1, and CCND1 with the heavy polysomal fraction was increased by ∼1.5–1.8-fold. Equol therefore promoted a greater increase in the translation of these mRNAs by increasing their polysome content, a hallmark of increased initiation. In the MDA-MB-435 cells expressing eIF4GI shRNA, a ∼2.5-fold reduction in eIF4GI levels was reflected in decreased polysome association of CTNND1 and CCND1, by ∼1.2-fold in the light fractions and by ∼1.4-fold in the heavy fractions. However, the equol-stimulated C-MYC mRNA levels remained increased by 1.4- and 1.7-fold in the light and heavy fractions compared with vehicle controls, even in the cells with eIF4GI knockdown. The IRES-negative GAPDH mRNA was also not affected by eIF4GI silencing or equol treatment. These data are consisted with the protein abundance levels shown earlier under identical conditions. The observed down-regulation of equol-mediated lD and p120 catenin protein expression (Fig. 1, A and B) and decreased polysome association of their mRNAs in cells silenced for eIF4GI (Fig. 4B), indicates a dependence on increased eIF4GI abundance to enable preferential translation of some IRES-containing mRNAs in equol-treated cells.
c-Myc Knockdown Reduces eIF4GI Levels and Abrogates the Effects of Equol on Cell Viability and Mammosphere Formation
Our results show that equol up-regulates mRNA and protein levels and the polysomal association of the key transcription factor and oncogene c-Myc, independent of eIF4G (Figs. 1 and 4). Therefore, we tested the effect of reducing c-Myc expression in Hs578t and MDA-MB-435 cells to investigate the importance of its elevated expression in equol action. To achieve this, cellular c-Myc levels were only partially reduced by carefully titrated c-Myc siRNA. This strategy enabled us to specifically determine the physiological relevance of elevated c-Myc in response to equol. Cells transfected with c-Myc siRNA demonstrated a ∼40% reduction in c-Myc protein expression compared with control siRNA cells. The 1.5-fold increase in equol-mediated c-Myc protein expression in cells expressing the control siRNA was completely abolished in both Hs578t and MDA-MB-435 cells expressing c-Myc siRNA. This result indicates that the siRNA targeting of c-Myc is sufficient to inhibit the de novo protein synthesis of c-Myc in response to equol, without affecting its basal levels (Fig. 5A). Protein expression of eIF4GI and cyclin D were also significantly down-regulated in c-Myc-silenced cells, indicating that increased c-Myc abundance promotes increased eIF4GI mRNA expression by c-Myc. Interestingly, the levels of Bcl-Xl and p120 catenin proteins were not altered by c-Myc silencing. Rather, a statistically significant increase in Bcl-Xl and p120 catenin was demonstrated in response to equol in cells with control or c-Myc siRNA in the MDA-MB-435 cells. In the Hs578t cell line, protein expression of eIF4GI, cyclin D, and Bcl-Xl were down-regulated following c-Myc knockdown, while p120 catenin expression remained unchanged.
FIGURE 5.
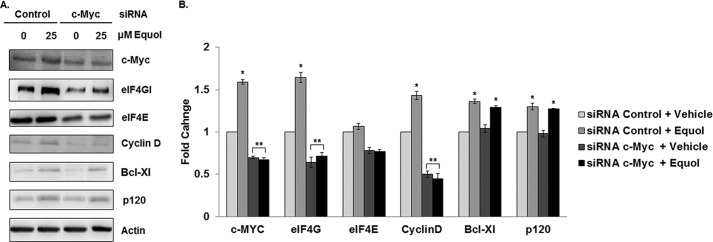
Expression of oncogenic proteins following equol treatment of MDA-MB 435 cells with c-Myc silencing. Quiescent MDA-MB-435 cells expressing control or c-Myc siRNA were treated with vehicle or equol for 24 h. Equal amounts of lysates were immunoblotted for the indicated proteins. A, representative Western blots. B, fold changes of protein expression compared with vehicle as calculated from the integrated density of positive bands normalized for actin expression (n = 3). An asterisk indicates statistical significance of p ≤ 0.05.
When eIF4GI was silenced, we observed a statistically significant decrease in p120 catenin and cyclin D1 expression while the other oncogenes tested, including c-Myc, remained unchanged. Therefore, to investigate the relative roles of eIF4GI and c-Myc on cell viability, Hs578t and MDA-MB-435 cancer cells expressing control, eIF4GI shRNA, or c-Myc siRNA were treated with vehicle or 25 μm equol, and subjected to cell viability analysis by MTT assay. Although the increase in eIF4GI levels in response to equol was effectively abolished by eIF4GI siRNA targeting (Fig. 1), the 1.4–1.7-fold increase in cell viability in response to 25 μm equol remained constant in MDA-MB-435 and Hs578t cells, despite eIF4GI silencing (Fig. 6A). These results indicate that the equol effect on cell viability is not eIF4G dependent. In contrast, Fig. 6B demonstrates that knockdown of c-Myc reduced the viability of MDA-MB-435 cells by 50% in control-treated cells and 50–80% in equol-treated cells.
FIGURE 6.
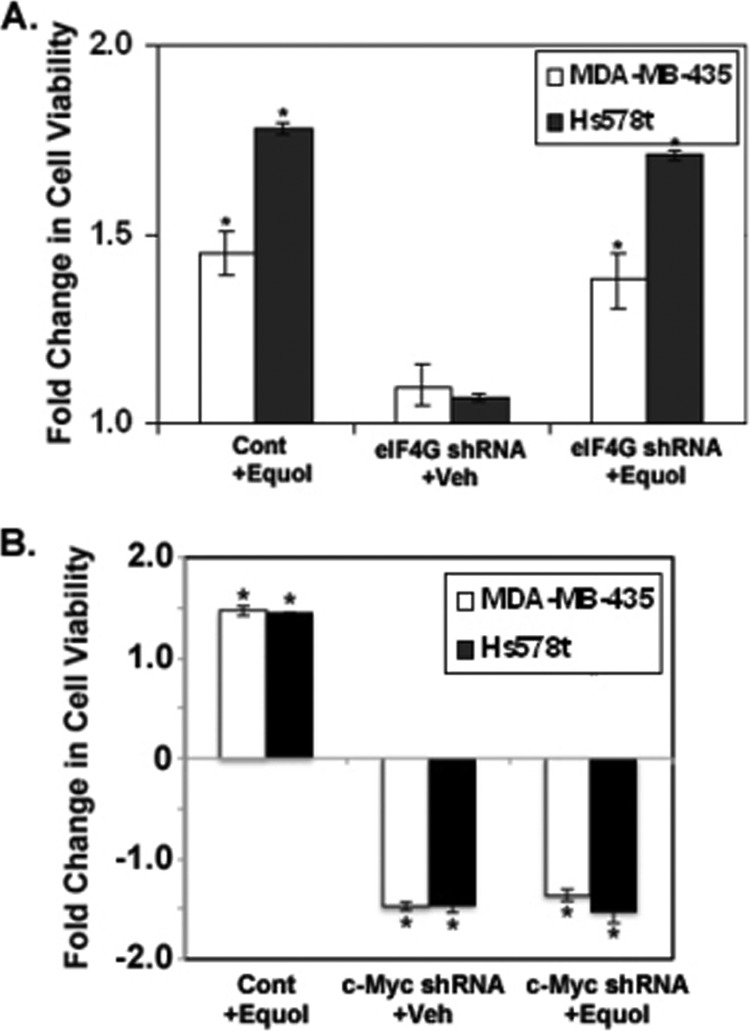
Effect of eIF4GI or c-Myc knockdown on MDA-MB-435 and Hs578t cell viability in response to equol treatment. A, viability of cells expressing control or eIF4G shRNA. Quiescent MDA-MB-435 cells or Hs578t cells were infected with non-silencing control or eIF4GI-silencing (eIF4G) adenovirus shRNA vectors, then treated with vehicle or 25 μm equol for 24 h. B, viability of cells expressing control or c-Myc siRNA. Following 48 h of control or c-Myc siRNA transfection, cells were treated with vehicle or 25 μm equol for 24 h. For both A and B, fold change in cell viability, from an MTT assay, is compared with vehicle treatments of cells expressing control vector (1.0). Mean ± S.E. (n = 3) is shown. An asterisk indicates statistical significance at p ≤ 0.05. Two asterisks indicate statistical significance of p ≤ 0.01.
To further investigate a role for c-Myc in regulating the effects of equol on cancer malignancy, non-adherent seeding of spheroids (mammosphere assays), which measure tumor initiating cell capacity, were performed in response to vehicle or equol using MDA-MB-435 cells expressing control or c-Myc siRNA. Our results show that equol increases the size and number of mammospheres formed by the MDA-MB-435 cell line by 1.5-fold compared with controls. Knockdown of c-Myc expression (as determined by Western blotting, data not shown) abolished the equol-mediated increase in mammosphere-forming efficiency (Fig. 7). Therefore, the breast cancer-promoting effects of equol appear to be dependent on its ability to increase c-Myc expression.
FIGURE 7.
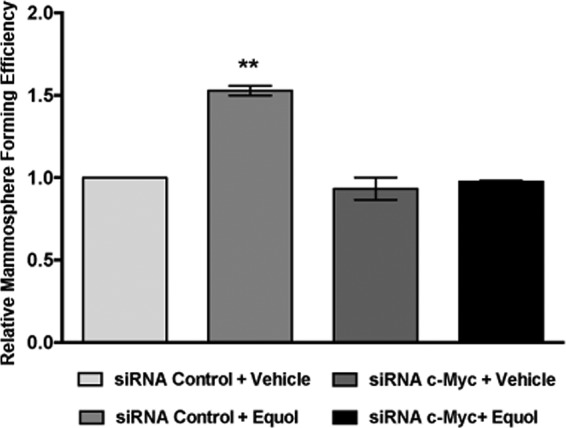
Mammosphere-forming efficiency (MFE) in MDA-MB-435 following equol treatment and c-Myc silencing. Quiescent MDA-MB-435 cells expressing control or c-Myc siRNA were treated with vehicle or equol for 24 h. Cells, seeded in duplicates in ultra-low attachments plates with 500–5000 cells/well, were treated with vehicle or equol, and incubated for 3 days to determine MFE. MFE was calculated by dividing the number of mammospheres formed by the number of cells seeded per well. The data are presented relative to vehicle-treated control siRNA-expressing cells. Mean ± S.E. (n = 3) is shown. Two asterisks indicate statistical significance of p < 0.01.
DISCUSSION
Dysregulation of translational control can have oncogenic consequences by altering global control of protein synthesis as well as selective translation of a subset of mRNAs important for cell growth, survival, metastasis, and proliferation (29, 46). Accordingly, eIF4F complex members eIF4E and eIF4GI are overexpressed in advanced cancers. Up-regulation of eIF4GI in aggressive breast cancers may preferentially enhance cap-independent and non-canonical means of initiation such as IRES-mediated translation; especially, when the cap-dependent scanning mechanism of translation initiation is compromised by stress conditions (47–49).
Therefore, our finding that eIF4GI is up-regulated by the dietary soy isoflavone equol in MDA-MB-435 human metastatic cancer cell tumors in immune-impaired mice, as well as in human metastatic breast cancer cells in vitro, has implicated the equol-stimulated eIF4GI in selective translational control (27, 28). We also showed that equol treatment of breast cancer cells results in increased IRES-dependent mRNA translation, protein expression, and polysome association of a number of mRNAs with IRES elements coding for proteins that regulate cancer cell survival, proliferation, and invasion (27). In this study, we tested the hypothesis that elevated levels of eIF4GI in response to equol contribute to breast cancer malignancy.
As shown by our results comparing metastatic cancer cells and non-cancer cells, the elevated c-Myc and eIF4G expression in response to equol is a cancer cell-specific effect. This may be due to the complex deregulation of c-Myc expression in cancer that would be more responsive to the effects of equol (50).
Our strategy in this study was to achieve only a partial silencing of eIF4GI levels in equol treated cells; thus, reducing it to levels found in untreated cells. In so doing, we found that the increased viability of metastatic cancer cells in response to equol was insensitive to eIF4GI knockdown. This result corroborates previous reports of eIF4GI depletion, which found only a small reduction in overall protein synthesis, cell viability, and only slightly impaired cell proliferation (35, 36). Nevertheless, similarly partial eIF4GI silencing has been shown previously to decrease the translation of the IRES-containing mRNAs such as p120 catenin, which has been implicated in cancer cell invasion (36).
Confirming this report, we found eIF4GI knockdown in metastatic breast cancer cells to result in a significant reduction in the equol-mediated increase in protein expression of p120 catenin, Cyclin D1, and Bcl-Xl, without affecting their mRNA levels. These mRNAs all contain an IRES in their 5′-UTR and have been shown to be under eIF4GI regulation during cancer progression (36, 51–53). Their decreased IRES-dependent translation and the associated polysome association also validates the mechanism that equol increases expression of eIF4GI, which in turn favors the increased translation of certain IRES-containing, cancer promoting mRNAs. When eIF4GI was silenced by 50–60%, we observed a parallel 50–60% decrease in the translation of these IRES-containing mRNAs. The eIF4GI knockdown was sufficient to reduce 100% of the equol-mediated increases in Cyclin D1, Bcl-Xl, and p120 catenin, proteins that were specifically dependent on elevated eIF4GI levels.
A central finding from our research is that equol increases mRNA and protein levels of eIF4GI and c-Myc, selectively in cancer cells. Intriguingly, c-Myc and eIF4E protein levels were not affected by silencing eIF4GI. The oncogenic transcription factor c-Myc regulates a large variety of cellular functions including cell cycle progression, protein synthesis, metabolism, apoptosis, and genomic stability through several signaling pathways. Accordingly, c-Myc is increased in many human cancers, including breast cancer, and is regulated via direct and indirect mechanisms (38, 54–57). We found that reducing the levels of eIF4GI was not sufficient to down-regulate the equol increased levels of c-Myc nor to abolish the increase in cell viability in response to equol. Therefore, equol may activate c-Myc transcription independently of eIF4GI up-regulation.
Overall, c-Myc appears to be a major player in the cancer promoting effects of equol in MDA-MB-435 and Hs578t metastatic cancer cells. Both eIF4E and eIF4GI are reported to be transcriptionally regulated by c-Myc (41, 58). Accordingly, we found a significant reduction in eIF4GI and a trend toward eIF4E reduction in c-Myc-silenced cells. C-Myc knockdown resulted in a significant decrease in Cyclin D1 levels, which were also reduced in cells with eIF4GI knockdown. c-Myc is an established transcriptional regulator of Cyclin D1, which likely explains the decreased Cyclin D1 expression and cell viability we observed in breast cancer cells expressing c-Myc siRNA. However, c-Myc knockdown did not affect Bcl-Xl and p120 catenin proteins. Since elevated Bcl-Xl and p120-catenin levels in equol treated cells were inhibited by partial eIF4GI silencing, as well as with c-Myc knockdown, this result indicates that equol mediated c-Myc up-regulation leads to the transcriptional activation of eIF4GI. The resultant elevated eIF4GI protein levels are predicted to stimulate preferential cap-independent synthesis of IRES-containing mRNAs such as Cyclin D1, Bcl-Xl, and p120 catenin. However, when c-Myc is silenced and the equol-elevated eIF4GI is abolished, cap-dependent protein synthesis may account for the increased Bcl-Xl and p120 expression.
Validating the role of c-Myc in control of cell viability in multiple cancers (59, 60), knockdown of c-Myc to basal levels of expression as in non-equol treated (control) Hs578t and MDA-MB-435 cells, completely abolished the increase in cell viability in response to equol. Furthermore, knockdown of c-Myc expression abolished the effect of equol on enhanced mammosphere formation. Mammosphere assays have been used to identify stem cell-like properties of self-renewal and tumorigenesis in aggressive cancer cell populations (42, 43, 61). Enhanced c-Myc expression and the associated increase in translation initiation have been implicated with mammary stem cell amplification and tumorigenic potential (62–64); especially in human growth factor receptor 2 (HER2)-amplified breast cancers, such as the MDA-MB-435 HER2++ cell line used in this study (65). Therefore, collectively, our data suggest that equol-mediated amplification of c-Myc and eIF4G may direct increased cancer stem cell-like and tumorigenic properties.
At present, the mechanism by which equol up-regulates c-Myc transcription, is not known. Equol is structurally similar to estrogen with 80 times more ERβ affinity than their precursor, daidzein (66). c-Myc is a recognized estrogen and estrogen mimetics regulated oncogene. However, unlike other estrogen stimulated genes, the mechanism by which c-Myc is regulated is not completely understood (56). c-Myc transcription is regulated by cell surface receptors that include Wnt/β-catenin and growth factor or signaling pathways that activate transcription factors such as TCF/LEF and AP-1, known to regulate c-Myc expression in breast cancer (37, 56, 67). Although the role of equol in Wnt signaling is not yet established, equol has been implicated in enhanced AP-1 activity (68). In our study, we used ER− breast cancer cell lines; however, these cells still expresses steroid receptors and may even express estrogen related receptors (66, 69). Moreover, MDA-MB-435 cells have been shown to express splice variants of the ER that may still be responsive to phytoestrogens (70). Therefore, equol may regulate c-Myc transcription via a variety of mechanisms, some of which may involve estrogen receptor-related pathways and crosstalk with growth factor receptor signaling.
c-Myc is highly amplified in human cancers (37). c-Myc regulates ∼15% of the human genome, and can also control multiple stages of ribosome biogenesis including the expression of translation initiation factors that are important for both cap-dependent and cap-independent translation (71, 37). Since equol did not significantly affect c-Myc or eIF4G levels in non-transformed cells, equol is not expected to affect the function of normal cells. However, in cancer cells that already have amplified c-Myc, the 2-fold up-regulation of c-Myc may have a profound effect on cancer potential. Increased c-Myc and eIF4G levels can promote the selective translation of mRNAs that use non-canonical eIF4E-independent modes of initiation; thus, enhancing the survival of cancer cells in the nutrient-deprived and hypoxic conditions that exist in solid tumors. Moreover, the equol-mediated c-Myc up-regulation may enhance the stem cell-like properties of a subset of cancer cells. This action is also expected to increase the potential for malignant properties such as therapy resistance and survival during nutrient deprivation.
Taken together, this study demonstrates that equol possesses pro-cancer properties and may influence cancer potential via up-regulation of c-Myc transcription leading to both c-Myc dependent and -independent eIF4G-mediated translation initiation of oncogenes and increased cancer cell survival. Thus, this study contributes to an understanding of the possible mechanisms by which soy isoflavones can affect breast cancer by demonstrating a key role for c-Myc up-regulation.
Acknowledgment
We thank Dr. Deborah Silvera (NYU School of Medicine) for providing the eIF4G shRNA constructs.
This research was supported by United States Army/BCRP Grant W81XWH-11-1-0199 (to C. D.), National Institutes of Health/NIGMS Grant SC3GM084824 (to S. D.), National Institutes of Health/NIMHHD Grants G12RR035051 and R25GM061838 (to L. D. B.) to UPR MSC, and the Breast Cancer Research Foundation and Avon Foundation (to R. J. S.).
- ER−
- estrogen receptor negative
- eIF
- eukaryotic protein synthesis initiation factors
- IRES
- internal ribosome entry site
- 4E-BP
- 4E-binding protein
- O-DMA
- O-desmethylangolensin
- RT-PCR
- Real-Time reverse transcriptase polymerase reaction
- CCND1
- Cyclin D1
- CTNNB1
- Catenin (cadherin-associated protein) β1
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HER2
- human epidermal growth factor receptor 2
- JUN
- Jun oncogene
- VEGF
- vascular endothelial growth factor
- RLuc
- Renilla luciferase
- FLuc
- firefly luciferase
- shRNA
- short hairpin RNA
- siRNA
- small interfering RNA
- UTR
- untranslated region.
REFERENCES
- 1. Xu X., Wang H. J., Murphy P. A., Cook L., Hendrich S. (1994) Daidzein is a more bioavailable soymilk isoflavone than is genistein in adult women. J. Nutr. 124, 825–832 [DOI] [PubMed] [Google Scholar]
- 2. Zava D. T., Duwe G. (1997) Estrogenic and antiproliferative properties of genistein and other flavonoids in human breast cancer cells in vitro. Nutr. Cancer 27, 31–40 [DOI] [PubMed] [Google Scholar]
- 3. Okabe Y., Shimazu T., Tanimoto H. (2011) Higher bioavailability of isoflavones after a single ingestion of aglycone-rich fermented soybeans compared with glucoside-rich non-fermented soybeans in Japanese postmenopausal women. J. Sci. Food Agric. 91, 658–663 [DOI] [PubMed] [Google Scholar]
- 4. Haron H., Ismail A., Shahar S., Azlan A., Peng L. S. (2011) Apparent bioavailability of isoflavones in urinary excretions of postmenopausal Malay women consuming tempeh compared with milk, Int. J. Food Sci. Nutr. 62, 642–650 [DOI] [PubMed] [Google Scholar]
- 5. Douglas C. C., Johnson S. A., Arjmandi B. H. (2013) Soy and its isoflavones: the truth behind the science in breast cancer. Anticancer Agents Med. Chem. 13, 1178–1187 [DOI] [PubMed] [Google Scholar]
- 6. Hilakivi-Clarke L., Andrade J. E., Helferich W. (2010) Is soy consumption good or bad for the breast? J. Nutr. 140, 2326S–2334S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bosviel R., Durif J., Déchelotte P., Bignon Y. J., Bernard-Gallon D. (2012) Epigenetic modulation of BRCA1 and BRCA2 gene expression by equol in breast cancer cell lines. Br. J. Nutr. 108, 1187–1193 [DOI] [PubMed] [Google Scholar]
- 8. Setchell K. D., Clerici C. (2010) Equol: pharmacokinetics and biological actions. J. Nutr. 140, 1363S–8S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolca S., Possemiers S., Herregat A., Huybrechts I., Heyerick A., De Vriese S., Verbruggen M., Depypere H., De Keukeleire D., Bracke M., De Henauw S., Verstraete W., Van de Wiele T. (2007) Microbial and dietary factors are associated with equol producer phenotype in healthy postmenopausal women. J. Nutr. 137, 2242–2246 [DOI] [PubMed] [Google Scholar]
- 10. Carreau C., Flouriot G., Bennetau-Pelissero C., Potier M. (2009) Respective contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor α induced transcriptional activity by isoflavones and equol: consequence on breast cancer cell proliferation. Mol. Nutr. Food Res. 53, 652–658 [DOI] [PubMed] [Google Scholar]
- 11. Setchell K. D., Clerici C. (2010) Equol: history, chemistry, and formation. J. Nutr. 140, 1355S–1362S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muthyala R. S., Ju Y. H., Sheng S., Williams L. D., Doerge D. R., Katzenellenbogen B. S., Helferich W. G., Katzenellenbogen J. A. (2004) Equol, a natural estrogenic metabolite from soy isoflavones: convenient preparation and resolution of R- and S-equols and their differing binding and biological activity through estrogen receptors α and β. Bioorg. Med. Chem. 12, 1559–1567 [DOI] [PubMed] [Google Scholar]
- 13. Setchell K. D., Brown N. M., Lydeking-Olsen E. (2002) The clinical importance of the metabolite equol-a clue to the effectiveness of soy and its isoflavones. J. Nutr. 132, 3577–3584 [DOI] [PubMed] [Google Scholar]
- 14. Liu H., Du J., Hu C., Qi H., Wang X., Wang S., Liu Q., Li Z. (2010) The clinical importance of the metabolite equol-a clue to the effectiveness of soy and its isoflavones. J. Nutr. Biochem. 21, 390–39619427779 [Google Scholar]
- 15. Taghizadeh B., Ghavami L., Nikoofar A., Goliaei B. (2013) Equol as a potent radiosensitizer in estrogen receptor-positive and -negative human breast cancer cell lines. Breast Cancer 10.1007/s12282-013-0492-0 [DOI] [PubMed] [Google Scholar]
- 16. Magee P. J., Raschke M., Steiner C., Duffin J. G., Pool-Zobel B. L., Jokela T., Wahala K., Rowland I. R. (2006) Equol: a comparison of the effects of the racemic compound with that of the purified S-enantiomer on the growth, invasion, and DNA integrity of breast and prostate cells in vitro. Nutr. Cancer 54, 232–242 [DOI] [PubMed] [Google Scholar]
- 17. Magee P. J., McGlynn H., Rowland I. R. (2004) Differential effects of isoflavones and lignans on invasiveness of MDA-MB-231 breast cancer cells in vitro. Cancer Lett. 208, 35–41 [DOI] [PubMed] [Google Scholar]
- 18. Choi E. J., Kim T. (2008) Equol induced apoptosis via cell cycle arrest in human breast cancer MDA-MB-453 but not MCF-7 cells. Mol. Med. Report. 1, 239–244 [PubMed] [Google Scholar]
- 19. Choi E. J., Ahn W. S., Bae S. M. (2009) Equol induces apoptosis through cytochrome c-mediated caspases cascade in human breast cancer MDA-MB-453 cells. Chem. Biol. Interact. 177, 7–11 [DOI] [PubMed] [Google Scholar]
- 20. Ju Y. H., Fultz J., Allred K. F., Doerge D. R., Helferich W. G. (2006) Effects of dietary daidzein and its metabolite, equol, at physiological concentrations on the growth of estrogen-dependent human breast cancer (MCF-7) tumors implanted in ovariectomized athymic mice. Carcinogenesis 27, 856–863 [DOI] [PubMed] [Google Scholar]
- 21. Welshons W. V., Murphy C. S., Koch R., Calaf G., Jordan V. C. (1987) Stimulation of breast cancer cells in vitro by the environmental estrogen enterolactone and the phytoestrogen equol. Breast Cancer Res. Treat. 10, 169–175 [DOI] [PubMed] [Google Scholar]
- 22. Tonetti D. A., Zhang Y., Zhao H., Lim S. B., Constantinou A. I. (2007) The effect of the phytoestrogens genistein, daidzein, and equol on the growth of tamoxifen-resistant T47D/PKCα. Nutr. Cancer 58, 222–229 [DOI] [PubMed] [Google Scholar]
- 23. Onoda A., Ueno T., Uchiyama S., Hayashi S., Kato K., Wake N. (2011) Effects of S-equol and natural S-equol supplement (SE5-OH) on the growth of MCF-7 in vitro and as tumors implanted into ovariectomized athymic mice. Food Chem. Toxicol. 49, 2279–2284 [DOI] [PubMed] [Google Scholar]
- 24. Lamartiniere C. A., Wang J., Smith-Johnson M., Eltoum I. E. (2002) Daidzein: bioavailability, potential for reproductive toxicity, and breast cancer chemoprevention in female rats. Toxicol Sci 65, 228–238 [DOI] [PubMed] [Google Scholar]
- 25. Charalambous C., Pitta C. A., Constantinou A. I. (2013) Equol enhances tamoxifen's anti-tumor activity by induction of caspase-mediated apoptosis in MCF-7 breast cancer cells. BMC. Cancer 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X., Suzuki N., Santosh Laxmi Y. R., Okamoto Y., Shibutani S. (2012) Anti-breast cancer potential of daidzein in rodents. Life Sci. 91, 415–419 [DOI] [PubMed] [Google Scholar]
- 27. Martínez-Montemayor M. M., Otero-Franqui E., Martinez J., De La Mota-Peynado A., Cubano L. A., Dharmawardhane S. (2010) Individual and combined soy isoflavones exert differential effects on metastatic cancer progression. Clin. Exp. Metastasis 27, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de la Parra C., Otero-Franqui E., Martinez-Montemayor M., Dharmawardhane S. (2012) The soy isoflavone equol may increase cancer malignancy via upregulation of eukaryotic protein synthesis initiation factor eIF4G. J. Biol. Chem. 287, 41640–41650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Silvera D., Formenti S. C., Schneider R. J. (2010) Translational control in cancer. Nat. Rev. Cancer 10, 254–266 [DOI] [PubMed] [Google Scholar]
- 30. Yi T., Papadopoulos E., Hagner P. R., Wagner G. (2013) Hypoxia-inducible factor-1α (HIF-1α) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. J. Biol. Chem. 288, 18732–18742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee T., Pelletier J. (2012) Eukaryotic initiation factor 4F: a vulnerability of tumor cells. Future. Med. Chem. 4, 19–31 [DOI] [PubMed] [Google Scholar]
- 32. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kong J., Lasko P. (2012) Translational control in cellular and developmental processes. Nat. Rev. Genet. 13, 383–394 [DOI] [PubMed] [Google Scholar]
- 34. Cuesta R., Gupta M., Schneider R. J. (2009) The regulation of protein synthesis in cancer. Prog. Mol. Biol. Transl. Sci. 90, 255–292 [DOI] [PubMed] [Google Scholar]
- 35. Ramírez-Valle F., Braunstein S., Zavadil J., Formenti S. C., Schneider R. J. (2008) eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 181, 293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Silvera D., Arju R., Darvishian F., Levine P. H., Zolfaghari L., Goldberg J., Hochman T., Formenti S. C., Schneider R. J. (2009) Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 11, 903–908 [DOI] [PubMed] [Google Scholar]
- 37. Dang C. V. (2012) MYC on the path to cancer. Cell 149, 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Riggelen J., Yetil A., Felsher D. W. (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 [DOI] [PubMed] [Google Scholar]
- 39. Eilers M., Eisenman R. N. (2008) Myc's broad reach. Genes Dev. 22, 2755–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stoneley M., Paulin F. E., Le Quesne J. P., Chappell S. A., Willis A. E. (1998) C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene 16, 423–428 [DOI] [PubMed] [Google Scholar]
- 41. Lin C. J., Cencic R., Mills J. R., Robert F., Pelletier J. (2008) c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 68, 5326–5334 [DOI] [PubMed] [Google Scholar]
- 42. Shaw F. L., Harrison H., Spence K., Ablett M. P., Simões B. M., Farnie G., Clarke R. B. (2012) A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary. Gland. Biol. Neoplasia. 17, 111–117 [DOI] [PubMed] [Google Scholar]
- 43. Grimshaw M. J., Cooper L., Papazisis K., Coleman J. A., Bohnenkamp H. R., Chiapero-Stanke L., Taylor-Papadimitriou J., Burchell J. M. (2008) Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 10, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Montalvo-Ortiz B. L., Castillo-Pichardo L., Hernández E., Humphries-Bickley T., De la Mota-Peynado A., Cubano L. A., Vlaar C. P., Dharmawardhane S. (2012) Characterization of EHop-016, small molecule inhibitor of Rac GTPase. J. Biol. Chem. 287, 13228–13238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lewis S. M., Cerquozzi S., Graber T. E., Ungureanu N. H., Andrews M., Holcik M. (2008) The eIF4G homolog DAP5/p97 supports the translation of select mRNAs during endoplasmic reticulum stress. Nucleic Acids Res. 36, 168–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willis A. E. (1999) Translational control of growth factor and proto-oncogene expression. Int. J. Biochem. Cell Biol. 31, 73–86 [DOI] [PubMed] [Google Scholar]
- 47. Braunstein S., Karpisheva K., Pola C., Goldberg J., Hochman T., Yee H., Cangiarella J., Arju R., Formenti S. C., Schneider R. J. (2007) A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 28, 501–512 [DOI] [PubMed] [Google Scholar]
- 48. Spriggs K. A., Cobbold L. C., Jopling C. L., Cooper R. E., Wilson L. A., Stoneley M., Coldwell M. J., Poncet D., Shen Y. C., Morley S. J., Bushell M., Willis A. E. (2009) Canonical initiation factor requirements of the Myc family of internal ribosome entry segments. Mol. Cell. Biol. 29, 1565–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bauer C., Brass N., Diesinger I., Kayser K., Grässer F. A., Meese E. (2002) Overexpression of the eukaryotic translation initiation factor 4G (eIF4G-1) in squamous cell lung carcinoma. Int. J. Cancer 98, 181–185 [DOI] [PubMed] [Google Scholar]
- 50. Wolf E., Lin C. Y., Eilers M., Levens D. L. (2014) Taming of the breast: shaping Myc-dependent amplification. Trends Cell Biol. S0962-8924(14)00193-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sherrill K. W., Byrd M. P., Van Eden M. E., Lloyd R. E. (2004) BCL-2 translation is mediated via internal ribosome entry during cell stress. J. Biol. Chem. 279, 29066–29074 [DOI] [PubMed] [Google Scholar]
- 52. Pieters T., van H. J., van R. F. (2012) Functions of p120ctn in development and disease. Front. Biosci. 17, 760–783 [DOI] [PubMed] [Google Scholar]
- 53. Silvera D., Schneider R. J. (2009) Inflammatory breast cancer cells are constitutively adapted to hypoxia. Cell Cycle 8, 3091–3096 [DOI] [PubMed] [Google Scholar]
- 54. Xu J., Chen Y., Olopade O. I. (2010) MYC and Breast Cancer. Genes Cancer 1, 629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meyer N., Penn L. Z. (2008) Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 [DOI] [PubMed] [Google Scholar]
- 56. Wang C., Mayer J. A., Mazumdar A., Fertuck K., Kim H., Brown M., Brown P. H. (2011) estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 25, 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frenzel A., Lovén J., Henriksson M. A. (2010) Targeting MYC-Regulated miRNAs to Combat Cancer. Genes Cancer 1, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones R. M., Branda J., Johnston K. A., Polymenis M., Gadd M., Rustgi A., Callanan L., Schmidt E. V. (1996) An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol. Cell. Biol. 16, 4754–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Y. H., Liu S., Zhang G., Zhou C. Q., Zhu H. X., Zhou X. B., Quan L. P., Bai J. F., Xu N. Z. (2005) Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res. 7, R220–R228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin C. Y., Lovén J., Rahl P. B., Paranal R. M., Burge C. B., Bradner J. E., Lee T. I., Young R. A. (2012) Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 151, 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Badve S., Nakshatri H. (2012) Breast cancer stem cells- beyond semantics. Lancet Oncol. 13, e43–e48 [DOI] [PubMed] [Google Scholar]
- 62. Moumen M., Chiche A., Decraene C., Petit V., Gandarillas A., Deugnier M. A., Glukhova M. A., Faraldo M. M. (2013) Myc is required for β-catenin-mediated mammary stem cell amplification and tumorigenesis. Mol. Cancer 12, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Avdulov S., Herrera J., Smith K., Peterson M., Gomez-Garcia J. R., Beadnell T. C., Schwertfeger K. L., Benyumov A. O., Manivel J. C., Li S., Bielinsky A., Yee D., Bitterman P. B., Polunovsky V. A. (2015) eIF4E threshold levels differ in governing normal and neoplastic expansion of mammary stem and luminal progenitor cells. Cancer Res. 10.1158/0008-5472.CAN-14-2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yi T., Kabha E., Papadopoulos E., Wagner G. (2014) 4EG1 targets breast cancer stem cells by selective inhibition of translation that persists in CSC maintenance, proliferation, and metastasis. Oncotarget. 5, 6028–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nair R., Roden D. L., Teo W. S., McFarland A., Junankar S., Ye S., Nguyen A., Yang J., Nikolic I., Hui M., Morey A., Shah J., Pfefferle A. D., Usary J., Selinger C., Baker L. A., Armstrong N., Cowley M. J., Naylor M. J., Ormandy C. J., Lakhani S. R., Herschkowitz J. I., Perou C. M., Kaplan W., O'Toole S. A., Swarbrick A. (2014) c-Myc and Her2 cooperate to drive a stem cell-like phenotype with poor prognosis in breast cancer. Oncogene 33, 3992–4002 [DOI] [PubMed] [Google Scholar]
- 66. Davison Z., de Blacquière G. E., Westley B. R., May F. E. (2011) nsulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: implications for therapy. Neoplasia. 13, 504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yochum G. S., Cleland R., Goodman R. H. (2008) A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 28, 7368–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park J., Kim S. H., Cho D., Kim T. S. (2005) Formononetin, a phyto-oestrogen, and its metabolites up-regulate interleukin-4 production in activated T cells via increased AP-1 DNA binding activity. Immunology 116, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Magklara A., Grass L., Diamandis E. P. (2000) Differential steroid hormone regulation of human glandular kallikrein (hK2) and prostate-specific antigen (PSA) in breast cancer cell lines. Breast Cancer Res. Treat. 59, 263–270 [DOI] [PubMed] [Google Scholar]
- 70. Poola I., Koduri S., Chatra S., Clarke R. (2000) Identification of twenty alternatively spliced estrogen receptor α mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J. Steroid Biochem. Mol. Biol. 72, 249–258 [DOI] [PubMed] [Google Scholar]
- 71. Mateyak M. K., Obaya A. J., Sedivy J. M. (1999) c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell. Biol. 19, 4672–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]