

Background: Polyamine metabolism is relevant in neuroblastoma progression.
Results: Polyamine synthesis is hampered by glycolysis inhibition in neuroblastoma cells.
Conclusion: Glycolysis inhibition triggers signaling events leading to decrease of N-Myc and ODC levels, resulting in decreased polyamine synthesis.
Significance: Combined therapies targeting glucose metabolism and polyamine synthesis could be effective in the treatment of n-myc-expressing tumors.
Keywords: Glycolysis, Myc (C-Myc), Neuroblastoma, Polyamine, Tumor Metabolism, N-Myc, Cancer Metabolism, Polyamines
Abstract
Polyamines are essential for cell proliferation, and their levels are elevated in many human tumors. The oncogene n-myc is known to potentiate polyamine metabolism. Neuroblastoma, the most frequent extracranial solid tumor in children, harbors the amplification of n-myc oncogene in 25% of the cases, and it is associated with treatment failure and poor prognosis. We evaluated several metabolic features of the human neuroblastoma cell lines Kelly, IMR-32, and SK-N-SH. We further investigated the effects of glycolysis impairment in polyamine metabolism in these cell lines. A previously unknown linkage between glycolysis impairment and polyamine reduction is unveiled. We show that glycolysis inhibition is able to trigger signaling events leading to the reduction of N-Myc protein levels and a subsequent decrease of both ornithine decarboxylase expression and polyamine levels, accompanied by cell cycle blockade preceding cell death. New anti-tumor strategies could take advantage of the direct relationship between glucose deprivation and polyamine metabolism impairment, leading to cell death, and its apparent dependence on n-myc. Combined therapies targeting glucose metabolism and polyamine synthesis could be effective in the treatment of n-myc-expressing tumors.
Introduction
Ornithine-derived polyamines (PAs)5 are putrescine (Put), spermidine (Spd), and spermine (Spm). They are able to interact with DNA, RNA, and proteins, which explains their ability to modulate cell growth, survival, and proliferation (1). PA metabolism (summarized in Fig. 1A) (2–4) is strictly controlled under physiological conditions. The two rate-limiting enzymes of polyamine biosynthesis are ornithine decarboxylase (ODC) and S-adenosylmethionine decarboxylase (SAMDC), whereas the rate-limiting enzyme of catabolism is spermidine/spermine N1-acetyltransferase (SSAT). A recent review has highlighted the mitogenic role of PAs in the stromal and immune cells in health and disease (5). Interestingly, PAs can be found at high levels in several tumors (6), and inhibition of PA synthesis has shown promising anti-tumor effects on prostate (7), colon (8), and skin cancers (9), among others.
FIGURE 1.

Polyamines and myc. A, polyamine metabolism. Synthesis reactions are depicted with dark gray arrows, whereas degradation reactions are represented with pale gray arrows. Reactions inside the circle take place in mitochondria. B, Myc oncoprotein is a common transcription factor controlling PA metabolism and glycolysis. Myc transactivates several PA synthetic genes and glycolytic enzymes and also the translation initiation factor eIF5A2, which has a unique posttranslational modification dependent on spermidine (2); in turn, spermidine stimulates myc transcription (3). ADC, arginine decarboxylase (controversial (4)); APAO, acetylpolyamine oxidase; MAT, methionine adenosyltransferase; SMO, spermine oxidase; SpmS, spermine synthase; SpdS, spermidine synthase.
C-Myc, N-Myc, and L-Myc transcriptional regulators are deregulated in many human cancers (10). The c-myc oncogene is closely involved in PA metabolism. It has been described to transactivate ODC (11), SAMDC (12), and Spd synthase (13). Furthermore, Myc is able to induce the expression of eIF5A2, a translation initiation factor showing a unique Spm-dependent post-translational modification (14). It has been suggested that the promotion of high levels of polyamines and production of active eIF5A2 could explain the oncogenic activity of Myc in certain cell and tissue types (15). In addition, c-myc transcription is stimulated by PAs (16), suggesting a positive feedback system. Amplified n-myc has been detected in several neuroendocrine tumors, like neuroblastoma (17). Neuroblastoma (ORPHA635) is the most frequent pediatric extracranial solid tumor, and it accounts for 10–15% of oncologic deaths in children (18). It has been known for a long time that N-Myc directly potentiates ODC expression (19) and that ODC levels positively correlate with neuroblastoma malignancy stage and indicate a poor prognosis of neuroblastoma even without N-Myc amplification (20). In addition, n-myc-amplified neuroblastoma tumors present elevated levels of every enzyme involved in PA synthesis, whereas some genes related to PA degradation are down-regulated in advanced stage neuroblastoma (20, 21). Thus, the overexpression of N-Myc potentiates neuroblastoma cell proliferation through several mechanisms (22), including the promotion of PA synthesis. Fig. 1B depicts the positive feedback loop established between PA metabolism and Myc oncoprotein levels.
Macromolecular synthesis is essential for cell growth and division. Therefore, rapidly proliferating tumor cells experience a metabolic reprogramming that includes high glycolysis and dependence on glutamine (23). N-Myc seems to be relevant for aerobic glycolysis in N-Myc-overexpressing tumors (24).
Current literature suggests that tumor remodeling might involve the coordination of all of the modules necessary for proliferation, including the metabolism of common energy fuels and other molecules relevant for cell cycle progression, such as PAs. As depicted in Fig. 1B, we hypothesize that PA metabolism could be linked to glycolysis through their mutual dependence on Myc functioning. Consequently, here we report for the first time an earlier unknown connection between glucose and PA metabolisms mediated by N-Myc, using neuroblastoma cell lines as model systems. Several metabolic features in these cells have been studied, and it has been shown that glycolysis inhibition with 2-deoxy-d-glucose (2-DG) is able to reduce the polyamine content in neuroblastoma cells as an early effect before cell death can be detected. This work represents an integrative advance in the knowledge about the metabolic reprogramming in neuroblastoma as a model of N-Myc-expressing malignancy.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
Cell lines were purchased from the European Collection of Cell Cultures. Kelly cells were cultured in RPMI 1640, and IMR-32 and SK-N-SH cells were cultured in Eagle's minimal essential medium. HEK293 cells were cultured in DMEM. Incubations were performed at 37 °C in a humidified atmosphere with 5% CO2. Cells between passages 8 and 23 (Kelly and SK-N-SH) or 68 and 80 (IMR-32) were used for experiments. Except as otherwise specified, cells were treated for 24 h with 3 mm 2-DG or 20 μm BPTES freshly diluted in cell culture medium/DMSO and sterilized by filtration.
Cell Transfection
SK-N-SH cells were transfected with empty pcMV6XL4 vector or pcMV6XL4 containing human n-myc cDNA. Plasmids were purchased from OriGene Technologies. Transfections were performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions, with a DNA/reagent ratio of 1:2. Cells were incubated in the presence of the transfection mix for 24 h, and then the medium was replaced, and 2-DG was added to the corresponding samples for another 24 h. After incubation with 2-DG, cell pellets were harvested and kept at −80 °C until analysis. Transfections were checked by Western blot against N-Myc.
Cell Growth Curves
Cell suspensions of 80,000 cells/ml were plated in 24-well plates with a final volume of 500 μl/well. After 24 h, control cells (time 0) were collected and counted, and 3 mm 2-DG was added to the corresponding wells. For the next 5 days, cells from four wells were detached with trypsin daily and counted using a Beckman Coulter Counter device, diluting cells at 1:20 with Isoton®.
Cell Cycle Analysis by Flow Cytometry
5 × 106 cells/ml were stained with propidium iodide as described previously (25). Then 10,000 cells/sample were analyzed with a MoFlo flow cytometer. The resulting data were analyzed with the free software WinMDI.
Glucose, Glutamine, and Lactate Determination in Protein-free Medium
Fresh culture media and culture media incubated with cells for 24 h were deproteinized with 10% (v/v) HClO4 (media/HClO4, 1:1), and neutralized with 20% (w/v) KOH. Deproteinized samples were immediately analyzed or kept at −20 °C until used. Glucose content in deproteinized culture media was determined by the glucose oxidase-peroxidase method (26) with modifications; a colorimetric reaction was performed with 0.2 mg/ml 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid). After 30 min of incubation at 37 °C in the dark, absorbance was measured at 725 nm. Lactate measurements in culture media were performed based on the method described elsewhere (27). Glutamine concentration in cell culture media was determined with the l-glutamine/ammonia (Rapid) kit (Megazyme) according to the manufacturer's instructions. In all cases, absorbances were determined with a Cary WinUV spectrophotometer (Varian); data were acquired with WinUV software.
ATP/ADP Ratio
ATP/ADP ratios were determined with the ADP/ATP ratio assay kit (Sigma) according to the manufacturer's instructions. Briefly, 5000 cells/well were seeded in 96-well plates. Cells were allowed to adhere for 24 h. 3 mm 2-DG was added to the corresponding wells, and the assay was performed after an incubation of 24 h. Measurements were performed in triplicate, and three independent replicates were assayed.
Polyamine Quantification by HPLC
Intracellular levels of Put, Spd, and Spm were simultaneously determined by fluorometry using a separation by HPLC after acid extraction from cell pellets. 1,8-diaminooctane was used as an internal standard added to the cell extract before derivatization with dansyl chloride, based on the method described previously (28). PA values were normalized to total protein contents estimated by a Bio-Rad protein assay.
Western Blots
Cell pellets of 2 × 106 cells were lysated with 100 μl of 1× denaturing loading buffer with added protease inhibitor (Roche Complete Mini) and phosphatase inhibitors (1 mm Na3VO4 and 30 mm NaF). Samples were heated, sonicated, and separated on 10% polyacrylamide gels. Proteins were transferred to nitrocellulose membranes and afterward blocked with 10% (w/v) semiskimmed dried milk. Blocked membranes were incubated overnight with primary antibodies, washed, and later incubated with the peroxidase-linked secondary anti-rabbit antibody for 1 h at room temperature. Membranes were washed and finally incubated with the Supersignal® West Pico chemiluminescent substrate system (Thermo Scientific). Image captions were taken with the ChemiDocTM XRS+ System (Bio-Rad), and densitometry analyses were made with Image LabTM software. Anti-GAPDH rabbit monoclonal antibody and anti-protein kinase B (AKT), anti-phospho-AKT (Ser-473), anti-GSK3β, anti-phospho-GSK3β (Ser-9), and anti-N-Myc rabbit polyclonal antibodies were from Cell Signaling Technology. Anti-phospho-Myc (Thr-58) rabbit polyclonal antibody was from Abcam. ECL anti-rabbit IgG horseradish peroxidase-linked antibody was from GE Healthcare. For the calculation of the phosphorylation ratios, the following formula was used (29).
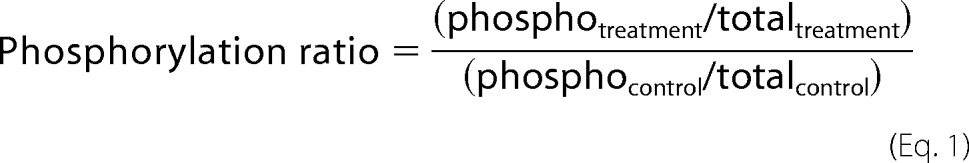 |
ODC Activity
Cell pellets of 20 × 106 cells were lysed with 800 μl of standard lysis buffer. Ornithine decarboxylase activity was determined in the supernatants by measuring the release of 14CO2 from [1-14C]ornithine based on the method described previously (30). Radioactivity was measured with a Microbeta scintillation counter. Protein quantification was performed in supernatants.
High Resolution Respirometry
Oroboros Oxygraph 2K high resolution respirometer and DatLab software were used for analyzing total cell respiration and respiratory rates. 106 to 2 × 106 cells were introduced in each chamber, and, after signal stabilization, 1 μl of 4 mg/ml oligomycin in ethanol was added per chamber for F0-ATP synthase inhibition. Then a titration with 10 mm carbonyl cyanide p-trifluoromethoxyphenylhydrazone uncoupler in ethanol was performed until the value of maximum oxygen consumption capacity was reached. 1 μl/chamber 0.2 mm rotenone and 5 mm antimycin A in ethanol was added for non-electron transport chain-related oxygen consumption measurement. Data were used for calculating the following respiratory flux control ratios, as described earlier (31).
Routine Control Ratio (R/E)
R/E represents routine oxygen consumption/uncoupled oxygen consumption. It indicates the percentage of electron transport capacity utilized in routine respiration in intact cells. The R/E control ratio is an expression of how close routine respiration operates to electron transport chain (ETC) capacity.
Leak Control Ratio (L/E)
L/E is the “leak” (remaining oxygen consumption after F0-ATP synthase inhibition)/uncoupled oxygen consumption. It represents the percentage of the electron transport capacity that is related to non-phosphorylating respiration (proton leak). The L/E ratio is an index of uncoupling or decoupling at constant ETC capacity. L/E increases with uncoupling from a theoretical minimum of 0.0 for a fully coupled system to 1.0 for a fully uncoupled system.
Net Routine Control Ratio (netR/E)
netR/E represents the leak-corrected routine oxygen consumption/uncoupled oxygen consumption. It shows the percentage of the total (uncoupled) electron transport capacity functionally activated for oxidative phosphorylation.
Real-time PCR
Cell pellets were frozen with liquid nitrogen and kept at −80 °C until used. Total mRNA was isolated from 5 × 106 cells with the GenElute mammalian total RNA purification kit plus on-column DNase I digestion set (Sigma). cDNA was synthesized from 1 μg of RNA using the Reverse transcription-PCR (RT-PCR) iScript cDNA synthesis kit (Bio-Rad). cDNA was kept at −20 °C until used. Semiquantitative real-time PCRs were run in triplicate with the Kapa SYBR Fast qPCR kit (Kapa Biosystems) in an Eco Ilumina device. All experiments included three independent replicates, each one assayed in triplicate. The thermal profile was the following: 95 °C for 10 min; 95 °C for 20 s, Tm for 30 s, and 72 °C for 20 s for 40 cycles; 95 °C for 15 s; 55 °C for 20 s; 95 °C for 15 s. Efficiencies were calculated with LinReg software. To determine the relative expression ratios of each gene, the gene expression's Ct difference method (32) was used, with β-actin as the reference gene. Genomic contamination was examined by quantitative PCR of RNA samples. Negative control included all reagents except for cDNA. Primers, amplicon sizes, and optimal Tm values are summarized in Table 1.
TABLE 1.
Primers used in qPCR experiments
| Target | 5′–3′ sequence | Amplicon size | Optimal Tm |
|---|---|---|---|
| bp | °C | ||
| Human SSAT | |||
| Forward | TGGCTAAATTCGTGATCC | 492 | 57 |
| Reverse | AGCAAGTACTCCTTGTCG | ||
| Human ODC | |||
| Forward | GATATCGATGCCTTCTATGTG | 595 | 57 |
| Reverse | AAGCTTCCGCCAATATCAAG | ||
| Human SAMDC | |||
| Forward | CGATATCCCAAGATCTGAGTG | 540 | 57 |
| Reverse | AAGCTTGGTATCAGGTCACG | ||
| Human β-actin | |||
| Forward | ACCTCATGAAGATCCTGAC | 521 | 57 |
| Reverse | ACTCCTGCTTGCTGATCC | ||
Statistics
Graphs were prepared using Prism version 5. Statistical analyses were made with GraphPad Prism QuickCalcs. In all graphs, except as otherwise indicated, means ± S.D. values are depicted, and statistical significance was measured with unpaired Student's t tests. Statistical significance is represented as follows: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
RESULTS
Characterization of Energy and Polyamine Metabolic Features in Cultured Human Neuroblastoma Cells
Three human neuroblastoma cell lines were used: n-myc-amplified Kelly and IMR-32 cells and n-myc non-amplified SK-N-SH cells (33). N-Myc protein levels were compared in the three cell lines by Western blot, showing that the protein amount is related to the previously described gene amplification: the N-Myc/GAPDH ratio was 7.4 ± 1.8 in Kelly cells, 0.12 ± 0.003 in IMR-32 cells, and undetectable in SK-N-SH cells. Several metabolic features were characterized in order to gain some understanding of the metabolic reprogramming of these cells. Glucose and glutamine consumption and lactate production were determined in Kelly, IMR-32, and SK-N-SH culture media after 24 h of incubation. The N-Myc-amplified cell lines showed significantly higher glucose (Fig. 2A) and glutamine (Fig. 2C) consumption rates after 24 h than SK-N-SH cells, whereas lactate production was quite similar in the three cell lines (Fig. 2B). The sum of glucose and glutamine consumed and not used for lactate production is around 60% of the total quantity in Kelly cells, 56% in IMR-32 cells, and only around 37% in SK-N-SH cells. No difference was observed in O2 consumption between Kelly and SK-N-SH cells (N-Myc-non-amplified), whereas IMR-32 cells had a lower O2 consumption than the other cell lines (Fig. 2D). When comparing respiratory flux control ratios, L/E is lower in the SK-N-SH cells than in the other ones, showing that in SK-N-SH cells, ETC capacity is less related to non-phosphorylating respiration (mainly proton leak) than in the case of Kelly and IMR-32 cells (Fig. 2E). In contrast, IMR-32 cells exhibited higher netR/E values than Kelly and SK-N-SH cells, indicating that they devote a greater percentage of their ETC capacity to oxidative phosphorylation (OXPHOS) than the other cell lines.
FIGURE 2.

Nutrient and oxygen usage in neuroblastoma cells. A, glucose consumption; B, lactate production; C, glutamine consumption; D, O2 consumption; E, respiratory flux control ratios. Measurements of the metabolites were made in cell culture media after 24 h of incubation and compared with initial metabolite content in culture media. Experiments were performed in triplicate. Total protein content was determined in the cell pellets. For O2 measurements, cells were collected and counted, and cell suspensions were used for high resolution respirometry measurements in an Oxygraph 2-k device. Experiments were performed in quadruplicate. Data are shown as means ± S.D. (error bars). Statistical analysis was with Student's unpaired t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
PA contents were compared in the three cell lines (Fig. 3A). In general, Kelly cells had higher PA levels than the other ones, especially in the case of Put and Spd in Kelly versus SK-N-SH cells. In the three cell lines, individual PA contents showed the same tendency: [Put] < [Spd] < [Spm]. When comparing the total amount of PAs in the three cell lines, Kelly cells have 28 ± 3 pmol/μg protein, IMR-32 cells have 22 ± 1 pmol/μg protein, and SK-N-SH cells have 20 ± 1 pmol/μg protein. The mRNA levels of the three rate-limiting enzymes of PA metabolism were compared in the two cell lines with greater differences of N-Myc content (Kelly and SK-N-SH) (Fig. 3, B–D), in all of the cases showing higher content in Kelly cells.
FIGURE 3.

PA metabolism in neuroblastoma cells. A, PA levels. Experiments were performed in triplicate. PAs were extracted from frozen cell pellets, derivatized with dansyl-chloride, and analyzed by HPLC. Kelly and IMR-32 cells exhibited higher Put levels than SK-N-SH cells. Put levels were not statistically different between IMR-32 and SK-N-SH cells. Spd levels in Kelly cells were higher than those of IMR-32 and SK-N-SH cells (which did not present statistically significant differences between them). Differences in Spm levels were only statistically significant between IMR-32 and SK-N-SH. B, ODC mRNA levels. C, SAMDC mRNA levels. D, SSAT mRNA levels. mRNA was isolated in triplicate and used for cDNA synthesis. Each triplicate measurement was used for qPCR in triplicate measurements. The mRNA levels of the three genes were higher in N-Myc-amplified than in N-Myc-non-amplified cells. Statistical analysis was with Student's unpaired t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
Characterization of the Effects of Glycolysis Inhibition on Energy and Polyamine Metabolism and Cell Cycle
Because increased glycolysis plays an essential role in tumor cell proliferation (34), we decided to analyze the effects of glycolysis inhibition in human neuroblastoma cell lines. We used the well characterized competitive and allosteric hexokinase inhibitor 2-DG (35).
First, the effects of glycolysis inhibition with 3 mm 2-DG for 24 h on lactate production and glutamine consumption were analyzed. As expected, lactate production was reduced by 2-DG treatment, more markedly in Kelly than in SK-N-SH cells (Fig. 4D). Glutamine consumption increased in 2-DG-treated Kelly cells after 24 h, whereas no statistically significant effect was observed on SK-N-SH cells under the same conditions (Fig. 4E). 2-DG treatment had the general effect of reducing O2 consumption after F0-ATP-synthase inhibition (Leak) with oligomycin in the three cell lines (Fig. 4, F–H). The treatment also reduced maximum O2 consumption capacity (ETS) in Kelly and SK-N-SH cells, but in IMR-32 cells, the effect was not statistically significant. No effects on basal (Routine) O2 consumption and in residual (Rox) O2 consumption were observed in any of the cell lines. Regarding the respiratory flux control ratios (Fig. 4, I–K), treatment with 2-DG affected Kelly, IMR-32, and SK-N-SH in a very similar manner, increasing R/E and netR/E ratios, indicating that the treatment enhanced the use of the ETC and the OXPHOS, respectively. In contrast, 2-DG differentially affected the L/E ratio in the three cell lines. It caused a slight drop of L/E in Kelly cells and a more significant reduction in IMR-32 and, to a minor extent, in SK-N-SH cells. This overall reduction of the L/E ratio indicates a minor proton leak when cells are treated with 2-DG.
FIGURE 4.

Effect of 3 mm 2-DG on cell proliferation and nutrient consumption in neuroblastoma cells. Growth curves of control and treated Kelly (A) and SK-N-H (B) cells. Cells seeded in 24-well plates were treated 36 h after plating. Cells were detached and counted in quadruplicate every 24 h (day 0 being the first day of treatment). Results are expressed as a percentage of cells on day 0. C, effect of treatment on cell cycle distribution of Kelly cells after 24 and 48 h. Cells were incubated for 24 or 48 h, harvested in PIPES buffer supplemented with propidium iodide, and analyzed by flow cytometry. Experiments were performed in triplicate. Results are expressed as a percentage of control cells. D, effect of a 24-h treatment on lactate production of Kelly and SK-N-SH cells. E, glutamine consumption of Kelly and SK-N-SH control and treated cells. Experiments were performed in triplicate. Metabolite measurements were made in cell culture media after 24 h of culture and compared with lactate/glutamine content in non-used culture media. Total protein content was determined in the cell pellets. Results are expressed as a percentage of controls. Shown are effects of 3 mm 2-DG on O2 consumption in Kelly (F), IMR-32 (G), and SK-N-SH (H) cells treated with 3 mm 2-DG for 24 h. Cells were collected and counted, and cell suspensions were used for high resolution respirometry measurements in an Oxygraph 2-k device. Experiments were performed in quadruplicate. Shown are flux control ratios in Kelly cells (I), IMR-32 cells (J), and SK-N-SH (K) cells. High resolution respirometry measurements were performed in an Oxygraph 2-k device. Routine, O2 consumption in non-perturbed cells; Leak, O2 consumption in cells after F0-ATP synthase inhibition; ETS, maximum O2 consumption reached after titration with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP); Rox, residual, non-ETC-related O2 consumption after F0-ATP synthase and complex I/III inhibition. Experiments were performed in quadruplicate. Results are expressed as a percentage of controls. Statistical analysis was with Student's unpaired t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
When we measured PA content in 2-DG-treated cells, we observed an effect not described previously; the treatment was able to lower the levels of all PAs in Kelly cells, and it reduced the levels of Put and Spd in IMR-32 cells, whereas only Put levels were lowered in SK-N-SH cells (Fig. 5, A–C).
FIGURE 5.

Effects of 3 mm 2-DG on PA metabolism of neuroblastoma cells. PA levels in Kelly (A), IMR-32 (B), and SK-N-SH cells (C). Experiments were performed in triplicate. PAs were extracted from frozen cell pellets, derivatized with dansyl-chloride, and analyzed by HPLC. Data are shown as percentages of the controls. D–F, 2-DG effects on PA levels in Kelly cells cultured in low serum and in both low serum and glucose (Glc)-free medium. Experiments were performed in triplicate. PAs were extracted from frozen cell pellets, derivatized with dansyl-chloride, and analyzed by HPLC. Data are shown as percentages of the controls. 0.5% FBS is compared with complete medium (control); 0.5% FBS, 3 mm 2-DG, glucose-free 0.5% FBS medium is compared with 0.5% FBS (control); and glucose-free 0.5% FBS, 3 mm 2-DG is compared with glucose-free 0.5% FBS (control). 2-DG treatment diminishes ODC activity in Kelly (G) and SK-N-SH cells (H). ODC activity was determined by measuring the release of 14CO2 from [1-14C]ornithine. Experiments were performed in triplicate (and each sample was measured in duplicate). I, effect of treatment on relative ODC mRNA levels in Kelly cells. mRNA was isolated in triplicate and used for cDNA synthesis. Each triplicate measurement was used for qPCR in triplicate measurements. Statistic analyses was with unpaired Student's t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
Cell growth curves were determined for untreated and 3 mm 2-DG-treated Kelly and SK-N-SH cells (Fig. 4, A and B). The treatment affected the proliferation of Kelly cells as early as 24 h after 2-DG addition, whereas SK-N-SH cells were not significantly affected until 3 days after the beginning of the treatment. We also analyzed the cell cycle of Kelly cells by flow cytometry after 24 and 48 h of treatment (Fig. 4C). We observed an accumulation of cells in G1 phase, with a concomitant reduction of cells in S/G2/M after 24 h, which was more prominent after 48 h. An increase of dead cells (sub-G1) was detected after 48 h. No changes were observed in cell cycle distribution of 3 mm 2-DG-treated SK-N-SH cells with respect to controls after 24 h (data not shown).
2-DG-mediated Reduction of PA Levels Is Caused by Glycolysis Inhibition
Beyond its role as a glycolytic inhibitor, 2-DG has been described to indirectly activate insulin-like growth factor receptor and downstream signaling pathways in a serum-dependent manner in several tumor cell lines (29). To test whether the decrease in PA levels caused by 2-DG is dependent on a similar signaling mechanism, we measured PAs in Kelly cells (whose PA levels were the most sensitive to 2-DG treatment) cultured in low serum (0.5% FBS) with and without 2-DG. As shown in Fig. 5D, serum deprivation (from 10 to 0.5%) for 24 h increased Spd and Spm but did not affect Put. The treatment with 3 mm 2-DG in low serum medium dropped Put to undetectable levels and decreased Spd, although Spm was not affected (Fig. 5E). To elucidate whether the effect on PA levels exerted by 2-DG is dependent on glucose depletion or involves other unknown mechanisms, we measured PAs in Kelly cells cultured in glucose-free (and low serum, because serum contains glucose) medium, with and without 2-DG. The absence of glucose was enough to reduce Put and Spd levels in these cells, and the addition of 2-DG further decreased the levels of Spd and Spm and dropped Put levels to undetectable values (Fig. 5F).
PA Reduction Is Not Due to Energy Depletion and It Is Not Induced by Glutaminolysis Inhibition
The synthesis of the polyamine precursor, S-adenosylmethionine, requires ATP. Also, the rate-limiting enzymes of PA metabolism (ODC, SAMDC, and SSAT) have a rapid turnover (36), and protein synthesis also requires ATP. For these reasons, we measured the ATP/ADP ratio in control versus 2-DG-treated cells. As shown in Fig. 6, A and B, 3 mm 2-DG during 24 h causes a slight reduction (around 15%) in the ATP/ADP ratio of Kelly cells, whereas SK-N-SH cells do not show a statistically significant difference. To test whether the reduction of PA levels is a general response to nutrient depletion, Kelly and SK-N-SH cells were incubated with or without 20 μm BPTES (an inhibitor of glutaminase) for 24 h. Fig. 6, C and D, shows that the treatment does not reproduce the effect of 2-DG on PA levels. An induction of Put levels is observed, whereas the levels of the other PAs remain unchanged.
FIGURE 6.

PA reduction is not associated to energy depletion, and it is not induced by glutaminolysis inhibition. ATP/ADP ratio in Kelly (A) and SK-N-SH cells (B) treated with or without 3 mm 2-DG for 24 h. Experiments were performed in triplicate according to the manufacturer's instructions. C and D, effects of 24-h 20 μm BPTES on PA levels in Kelly (C) and SK-N-SH (D) cells. Experiments were performed in triplicate. PAs were extracted from frozen cell pellets, derivatized with dansyl-chloride, and analyzed by HPLC. Statistical analyses were with Student's unpaired t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01. Error bars, S.D.
2-DG Treatment Diminishes ODC mRNA Levels and Enzyme Activity
N-Myc stimulates both glycolysis and ODC expression (19, 24). Because we observed a sensitivity of PA metabolism to glycolytic impairment, we hypothesized that ODC could be involved in the 2-DG-induced PA reduction. To test this hypothesis, ODC activity was measured in Kelly and SK-N-SH cells. 3 mm 2-DG treatment was able to reduce ODC activity as soon as after 6 h of treatment in both cell lines (Fig. 5, G and H). Kelly cells experience a reduction of almost 75% after 6 h of treatment and 96% after 24 h, whereas in the case of SK-N-SH cells, the reduction of ODC activity is 57% after 6 h and 67% after 24 h. A direct inhibitory effect of 2-DG on ODC enzyme was discarded by measuring enzyme activity in control Kelly cell lysates with 3 mm 2-DG added to the enzymatic assay mixture (43.5 ± 2.2 versus 46.3 ± 5.2 pmol·h−1·μg of protein−1). The mRNA levels of ODC were analyzed by real-time PCR in control and 2-DG-treated Kelly cells. The treatment resulted in a decrease of the relative expression level of the ODC mRNA compared with control cells (Fig. 5I).
2-DG Treatment Affects the AKT-signaling Pathway, Leading to a Reduction of N-Myc Protein Levels
The analysis of the activation status of cancer-related signaling pathways with a protein antibody microarray (results not shown) suggested that 2-DG treatment could affect AKT-dependent signaling pathways. Therefore, we investigated by Western blot the phosphorylation status of AKT, its downstream target glycogen synthase kinase 3β (GSK3β), and N-Myc, which is a phosphorylation target of the former. As shown in Fig. 7A, Kelly cells treated for 24 h with 3 mm 2-DG showed a reduction in the phosphorylation levels of AKT and GSK-3β accompanied by a great reduction of total and phospho-Thr-58 N-Myc. The phosphorylation ratios of Ser-473 in AKT and Ser-9 in GSK-3β were significantly reduced by the treatment, whereas the phosphorylation of Thr-58 in N-Myc was augmented. In the case of IMR-32 cells, phospho-AKT could not be detected, but a significant reduction of phospho-Ser-9 in GSK-3β and both total and phospho-N-Myc was observed in 2-DG-treated cells. However, the phosphorylation ratio was not significantly affected by the treatment. Although phospho-AKT and total or phosphorylated N-Myc were not detectable in SK-N-SH cells by Western blot, a reduction of phospho-GSK-3β was observed when cells were treated with 2-DG.
FIGURE 7.

2-DG treatment increases the phosphorylation ratio of N-Myc in Thr-58. A, Western blot results for total and phosphorylated AKT, GSK3β, and N-Myc. GAPDH (not shown) was used for normalization. Samples were obtained and assayed in triplicate. Densitometry analyses were made with Image LabTM software. Phosphorylation ratios were calculated as the ratios phosphotreatment/totaltreatment and phosphocontrol/totalcontrol. Resulting data were analyzed with a one-sample t test (hypothetical mean = 1, 95% confidence interval). B, overexpression of n-myc in SK-N-SH cells. Cells were transfected with empty or n-myc-containing pcMV6XL4 vector and incubated for 24 h. Then medium was changed, and 3 mm 2-DG was added for a further 24 h. Western blot results for total and phosphorylated N-Myc are shown. GAPDH (not shown) was used for normalization. Samples were obtained and assayed in triplicate. Densitometry analyses were made with Image LabTM software. Phosphorylation ratios were analyzed with a one-sample t test (hypothetical mean = 1, 95% confidence interval). C, overexpression of n-myc increases the sensitivity of PA metabolism to 2-DG in SK-N-SH cells. Experiments were performed in triplicate. PAs were extracted from frozen cell pellets, derivatized with dansyl-chloride, and analyzed by HPLC. Statistic analyses were with Student's unpaired t test: 95% confidence interval; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
N-Myc Overexpression Increases the Sensitivity of PA Metabolism to 2-DG Treatment in SK-N-SH Cells
To reinforce evidence about the role of N-Myc protein in the sensitivity of PA metabolism to glycolysis inhibition, we overexpressed the human n-myc gene in SK-N-SH cells. After 24 h in the presence of transfection mix, medium was changed, and then cells were treated with 2-DG for 24 h. Western blot analysis showed that the levels of both total and phospho-N-Myc were extremely augmented in the cells transfected with the human n-myc construction (Fig. 7B). Furthermore, the phosphorylation ratio of N-Myc at Thr-58 was also augmented in n-myc-transfected cells treated with 3 mm 2-DG relative to untreated cells. PA measurement showed that cells overexpressing n-myc experience a significant rise of Spd levels. When treated with 2-DG, n-myc-overexpressing cells suffer a reduction of about 72% of Put, whereas cells transfected with the empty vector experience a reduction of around 50% in Put contents (Fig. 7C). Spd levels experience a reduction of around 37% in N-Myc-overexpressing cells treated with 2-DG, whereas cells transfected with the empty vector do not show changes in Spd in the same conditions. Spm levels were not affected under any condition.
DISCUSSION
Here we studied some general metabolic properties of three neuroblastoma cell lines and the connection existing between glucose and polyamine metabolism. By measuring glucose and lactate production and oxygen consumption, it has been shown that the three cell lines are competent for both glycolysis and OXPHOS (Fig. 2). Our results show that Kelly cells, which, in accordance with their status as N-Myc-amplified cells, are more proliferative than SK-N-SH cells (compare control curves in Fig. 4, A and B), as well as IMR-32 cells perform a more conservative use of glucose than SK-N-SH cells (Fig. 2; see “Results”). This cell line, less proliferative and without N-Myc amplification, consumes less glucose than the other ones (Fig. 2A), and its lactate production is less affected by 2-DG (Fig. 4D), probably indicating that SK-N-SH cells are less dependent on aerobic glycolysis for survival. The Warburg effect or, more widely, “metabolic reprogramming” supports the production of essential intermediaries for the synthesis of the building blocks necessary for cell growth and proliferation (23). Thus, an exacerbated waste of carbon can only be positively selected in a population of rapidly growing cells as long as it does not exceed some limits. In that context, it makes sense that a more “conservative” use of carbon is associated with a more malignant phenotype, as is the case of Kelly compared with SK-N-SH cells.
Our results do not show a homogeneous pattern for respiratory properties among the three cell lines studied (Fig. 2, D and E). It is interesting to note that N-Myc seems to be more relevant for the uptake of cell fuels than for their degradation (Fig. 2). On the other hand, 2-DG has the general ability to reduce proton leak and the maximum capacity of oxygen consumption while increasing net ETC and OXPHOS efficiencies (Fig. 4, F–K), possibly indicating that cells compensate for the reduction of energy obtained from glucose by increasing the use of other oxidizable substrates. This could be related to the increase of glutamine consumption in 2-DG-treated Kelly cells (Fig. 4E), which could balance their carbon requirement. This is an interesting finding that should be further characterized to understand metabolic remodeling in human neuroblastoma.
As far as we know, no previous relationship has been described between glucose deprivation/glycolysis inhibition and altered PA metabolism in any experimental model. We have observed that 2-DG reduces the levels of Put in the three neuroblastoma cell lines, Spd in Kelly and IMR-32 and Spm only in Kelly cells (Fig. 5, A–C). It is interesting that the sensitivity of PA metabolism to glycolysis inhibition seems to be related to the level of N-Myc (Kelly, around 100 n-myc copies; IMR-32, 25 n-myc copies; SK-N-SH does not show n-myc amplification (33)), although the number of cell lines tested is insufficient to state a correlation. Spd and Spm are fundamental for cell proliferation (37) and mediate the expression of the immediate early gene after mitogen stimuli (38), and ODC has been suggested to be a key regulator of growth-related signal transduction in tumors (39). Thus, the effects of 2-DG on cell cycle could result not only from the inhibition of glycolysis but from the consequent indirect down-regulation of ODC. Put levels show a higher sensitivity to glycolysis inhibition when compared with the other PAs. This is probably due, in the first place, to the fact that Put is the direct product of ODC, which is down-regulated by the treatment, whereas Spd and Spm synthesis requires further reactions. But it is also important to note that PA metabolism is a robust system very resistant to perturbations (40), and several previous works have shown that even treatments directly targeting PA biosynthesis cause rapid decreases of putrescine levels after 24 h, whereas higher PAs (Spd and Spm) start to show clear changes after 48 h (41, 42).
2-DG has been reported to indirectly enhance the activation of signaling pathways downstream insulin-like growth factor receptor in a serum-dependent manner in several cancer cells (29). We have discarded the possibility that a similar mechanism is involved in the sensitivity of PA metabolism to 2-DG in Kelly cells, given that 2-DG reduces PA levels even when serum concentration is reduced from 10 to 0.5% (Fig. 5E). Moreover, the absence of glucose itself is able to reduce PA levels, although the effect is enhanced in the presence of 2-DG (Fig. 5F), possibly due to the inhibition of the use of the endogenous glucose pool from intracellular sources like gluconeogenesis or glycogenolysis (that neuroblastoma cells are able to perform (43, 44)). These results indicate that the 2-DG effect on PA metabolism is mediated, at least partially, by its ability to inhibit glucose utilization and not by some other 2-DG-dependent signaling mechanisms. Furthermore, an energy depletion caused by the treatment does not seem to be the cause of PA reduction, because Kelly cells experience a reduction of the ATP/ADP ratio of only 15% after 24 h, and in SK-N-SH cells, the effect is not significant (Fig. 6, A and B). This is much lower than the reduction of ATP levels caused by 5 mm 2-DG on endothelial cells at shorter times (45). We have also discarded the possibility that the reduction of PA levels could be a general response to carbon deprivation because the inhibition of glutaminase using 20 μm BPTES for 24 h did not reproduce the effect of 2-DG on PA levels. The reason why Put levels are increased in both Kelly and SK-N-SH cells when treated with BPTES remains to be studied. However, because BPTES prevents glutamine from being used as an anaplerotic source for the tricarboxylic acid cycle through glutaminase, free glutamine could be the cause of this increment of Put due to the fact that amino acids, including glutamine, have been described to activate ODC through the mTOR-dependent regulation of antizyme synthesis (46).
When investigating the mechanisms behind the effect of 2-DG treatment on PA levels, we observed a great decrease of ODC activity in Kelly and SK-N-SH cells (Fig. 5, G and H). Because a direct inhibitory effect of 2-DG on ODC activity has been discarded, our data suggest that 2-DG treatment, through its ability to reduce glucose utilization, triggers some mechanisms that are capable of regulating ODC and PA levels. Those mechanisms, as shown in Fig. 7A, seem to be related to the AKT-signaling pathway, as a general effect of the inhibitor in these neuroblastoma cell lines. Fig. 7 shows that 2-DG treatment causes a reduction of AKT activation and of both phosphorylated GSK3β and total N-Myc as well as an increase of the phosphorylation level of N-Myc Thr-58 (unfortunately, all of the elements of the pathway could not be detected in the three cell lines). Newly synthesized Myc protein is unstable and quickly degraded in a ubiquitin-dependent manner (47). The phosphorylation of Myc at Ser-62 by ERK proteins greatly increases its protein stability (48). GSK3β is able to subsequently phosphorylate N-Myc in Thr-58, thus making it less stable and targeting it for degradation. Active AKT phosphorylates and inactivates GSK3β, thus enhancing N-Myc stability (48) (Fig. 8). Thus, our results indicate that 2-DG triggers N-Myc degradation through the inactivation of AKT and subsequent activation of GSK3β. Because ODC expression is under control of N-Myc in neuroblastoma, reduced N-Myc levels are probably responsible for the reduction of ODC mRNA levels and activity (Fig. 5, G–I) and for the reduction of PA levels in glucose-deprived cells and 2-DG-treated cells (Fig. 5, A–F).
FIGURE 8.

Effect of 2-DG treatment on PA metabolism of human neuroblastoma cells. The inhibition of hexokinase by 2-DG or glucose deprivation triggers the inactivation of AKT (confirmed in this work). Decreased AKT activity reduces the stability and levels of N-Myc (confirmed in this work) through the reduction of phosphorylation and activation of GSK3β (confirmed in this work). Diminished N-Myc leads to a reduction of ODC (and probably other PA biosynthetic protein) expression and protein levels (confirmed in this work), which affects actual PA pool (confirmed). The extent to which 2-DG treatment affects PAs seems to be related to specific kinetic properties of the PA bi-cycle, depending on cell type, as a result of the complex regulatory mechanisms controlling PA metabolism. The reduction of both glucose consumption and PA levels could be coordinately responsible for the blockade of cell cycle and increase of cell death.
Given that “futile” cycles, such as the PA bi-cycle, are more sensitive to changes in metabolism when subjected to an accelerated operation (49), we hypothesize that the sensitivity of PA metabolism to 2-DG treatment in neuroblastoma cells could depend on the velocity of the operation of the PA bi-cycle, as suggested in Kelly cells by the higher levels of expression of PA-related genes, higher levels of PAs, and higher ODC activity when compared with SK-N-SH cells (Figs. 3 and 5, G and H). This is in agreement with the previous observations that myc oncogenes control the expression of ODC (11) and SAMDC (12). Considering these results together, Kelly cells seem to maintain an accelerated flux through the PA metabolic bi-cycle. In fact, although ODC activity is reduced in SK-N-SH cells after 6 h of treatment, their PA levels do not suffer great changes in the next 24 h. Taking into account metabolic flux theory (49), this would mean a higher capacity of Kelly cells to amplify the response to regulatory factors of the pathway (50).
Finally, after the overexpression of n-myc in SK-N-SH cells, we have observed that 2-DG promotes an increase of the phosphorylation ratio of N-myc protein, in accordance with the effect exerted by the compound on naturally myc-amplified neuroblastoma cells (Fig. 7B). However, the total levels of N-Myc and P-Myc remain unchanged, possibly due to the artificially forced high expression of the protein in the transfected cells. A reduction was also observed in Put and Spd that was greater in the n-myc-overexpressing cells than in the cells transfected with the empty vector (Fig. 7C) in response to 2-DG treatment. The lack of effect on Spm could be due to the fact that the Spm pool is relatively resistant to disruptions in PA metabolism because of its association with macromolecules (51). The effect on Put and Spd indicates that N-Myc expression significantly influences the extent to which glycolysis inhibition affects PA metabolism. Similar results were observed when n-myc was overexpressed in the non-tumor human cell line HEK293 (results not shown). Fig. 8 includes our proposed model explaining the main changes taking place in neuroblastoma cells in response to 2-DG treatment.
In summary, it has been described here that impairment of glucose metabolism is able to affect the AKT-dependent signaling pathway, leading to a strong reduction of N-Myc protein levels and a consequent decrease of PA synthesis. As far as we know, it has never been stated before that impairment in the usage of a primary nutrient such as glucose could affect PA metabolism. Metabolism-targeted therapies are emerging as new approaches for cancer treatment (52–54). In fact, 2-DG, as well as several PA pathway-targeted drugs, have been tested in clinical trials (6, 55). New anti-tumor strategies could take advantage of the direct relationship between glucose deprivation and PA metabolism impairment, leading to cell death as described above and its apparent dependence on n-myc in the case of neuroblastoma. Combined therapies targeting glucose metabolism and PA synthesis could be effective in the treatment of N-Myc-expressing tumors, especially if elevated levels of elements related to PA metabolism are detected. The discovery of new cross-talks between metabolic pathways, signaling elements, and transcriptional networks in tumor cells would allow the identification of “Achilles heel” points that could selectively impair cancer growth when perturbed in a coordinated manner. Starting the search of those points by the analysis of pillar elements of tumors, like the main metabolic pathways or the controlling elements related to multifunctional molecules such as PAs, could accelerate the development of novel and more effective anticancer strategies for personalized oncology.
Acknowledgments
We thank Dr. Esther Melgarejo-Páez for assistance with real-time PCR and the technician Anne Karppinen for assisting M. V. R. P. during ODC activity measurements.
CIBERER is an initiative of Instituto de Salud Carlos III.
- PA
- polyamine
- AKT
- protein kinase B
- 2-DG
- 2-deoxy-d-glucose
- ETC
- electron transport chain
- L/E
- leak control ratio
- GSK3β
- glycogen synthase kinase-3β
- netR/E
- net routine control ratio
- ODC
- ornithine decarboxylase
- Put
- putrescine
- OXPHOS
- oxidative phosphorylation
- R/E
- routine control ratio
- SAMDC
- S-adenosylmethionine decarboxylase
- Spd
- spermidine
- SSAT
- spermidine/spermine N1-acetyltransferase
- Spm
- spermine
- BPTES
- bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- ETC
- electron transport chain
- qPCR
- quantitative PCR.
REFERENCES
- 1. Agostinelli E., Marques M. P., Calheiros R., Gil F. P., Tempera G., Viceconte N., Battaglia V., Grancara S., Toninello A. (2010) Polyamines: fundamental characters in chemistry and biology. Amino Acids 38, 393–403 [DOI] [PubMed] [Google Scholar]
- 2. Wolff E. C., Park M. H. (1999) Identification of lysine 350 of yeast deoxyhypusine synthase as the site of enzyme intermediate formation. Yeast 15, 43–50 [DOI] [PubMed] [Google Scholar]
- 3. Tabib A., Bachrach U. (1999) Role of polyamines in mediating malignant transformation and oncogene expression. Int. J. Biochem. Cell Biol. 31, 1289–1295 [DOI] [PubMed] [Google Scholar]
- 4. Coleman C. S., Hu G., Pegg A. E. (2004) Putrescine biosynthesis in mammalian tissues. Biochem. J. 379, 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghesquière B., Wong B. W., Kuchnio A., Carmeliet P. (2014) Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176 [DOI] [PubMed] [Google Scholar]
- 6. Nowotarski S. L., Woster P. M., Casero R. A., Jr. (2013) Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 15, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyskens F. L., Jr., Simoneau A. R., Gerner E. W. (2014) Chemoprevention of prostate cancer with the polyamine synthesis inhibitor difluoromethylornithine. Recent Results Cancer Res. 202, 115–120 [DOI] [PubMed] [Google Scholar]
- 8. Gerner E. W., Ignatenko N. A., Besselsen D. G. (2003) Preclinical models for chemoprevention of colon cancer. Recent Results Cancer Res. 163, 58–71; discussion 264–266 [DOI] [PubMed] [Google Scholar]
- 9. Bailey H. H., Kim K., Verma A. K., Sielaff K., Larson P. O., Snow S., Lenaghan T., Viner J. L., Douglas J., Dreckschmidt N. E., Hamielec M., Pomplun M., Sharata H. H., Puchalsky D., Berg E. R., Havighurst T. C., Carbone P. P. (2010) A randomized, double-blind, placebo-controlled phase 3 skin cancer prevention study of α-difluoromethylornithine in subjects with previous history of skin cancer. Cancer Prev. Res. (Phila.) 3, 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang C. V., Le A., Gao P. (2009) MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 15, 6479–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bello-Fernandez C., Packham G., Cleveland J. L. (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 90, 7804–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Z., Van Calcar S., Qu C., Cavenee W. K., Zhang M. Q., Ren B. (2003) A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 8164–8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forshell T. P., Rimpi S., Nilsson J. A. (2010) Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. (Phila.) 3, 140–147 [DOI] [PubMed] [Google Scholar]
- 14. Fernandez P. C., Frank S. R., Wang L., Schroeder M., Liu S., Greene J., Cocito A., Amati B. (2003) Genomic targets of the human c-Myc protein. Genes Dev. 17, 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerner E. W. (2010) Cancer chemoprevention locks onto a new polyamine metabolic target. Cancer Prev. Res. (Phila.) 3, 125–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Celano P., Baylin S. B., Casero R. A., Jr (1989) Polyamines differentially modulate the transcription of growth-associated genes in human colon carcinoma cells. J. Biol. Chem. 264, 8922–8927 [PubMed] [Google Scholar]
- 17. Brodeur G. M., Seeger R. C., Schwab M., Varmus H. E., Bishop J. M. (1984) Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121–1124 [DOI] [PubMed] [Google Scholar]
- 18. Maris J. M. (2010) Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lutz W., Stöhr M., Schürmann J., Wenzel A., Löhr A., Schwab M. (1996) Conditional expression of N-myc in human neuroblastoma cells increases expression of α-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 13, 803–812 [PubMed] [Google Scholar]
- 20. Geerts D., Koster J., Albert D., Koomoa D. L., Feith D. J., Pegg A. E., Volckmann R., Caron H., Versteeg R., Bachmann A. S. (2010) The polyamine metabolism genes ornithine decarboxylase and antizyme 2 predict aggressive behavior in neuroblastomas with and without MYCN amplification. Int. J. Cancer 126, 2012–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hogarty M. D., Norris M. D., Davis K., Liu X., Evageliou N. F., Hayes C. S., Pawel B., Guo R., Zhao H., Sekyere E., Keating J., Thomas W., Cheng N. C., Murray J., Smith J., Sutton R., Venn N., London W. B., Buxton A., Gilmour S. K., Marshall G. M., Haber M. (2008) ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 68, 9735–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun Y., Liu P. Y., Scarlett C. J., Malyukova A., Liu B., Marshall G. M., MacKenzie K. L., Biankin A. V., Liu T. (2014) Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene 33, 2987–2994 [DOI] [PubMed] [Google Scholar]
- 23. Schulze A., Harris A. L. (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373 [DOI] [PubMed] [Google Scholar]
- 24. Qing G., Skuli N., Mayes P. A., Pawel B., Martinez D., Maris J. M., Simon M. C. (2010) Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1α. Cancer Res. 70, 10351–10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dasso M. (2004) in Short Protocols in Cell Biology, 1st Ed. (Lippincott-Schwartz J., Yamada K. M., Harford J. B., Bonifacino J. S., Dasso M., eds) John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 26. Kunst A., Dreager B., Ziegenhorn J. (1984) Methods in Enzymatic Analysis, 3rd Ed. (Bergmeyer H. U., ed) pp. 178–185, Verlag Chemie, Weinheim, Germany [Google Scholar]
- 27. Noll F. (1984) in Methods of Enzymatic Analysis, Vol. IV, Metabolites 1: Carbohydrates, 3rd Ed (Bergmeyer H. U., ed) pp. 582–588, Verlag Chemie, Weinheim, Germany [Google Scholar]
- 28. Fajardo I., Urdiales J. L., Paz J. C., Chavarría T., Sánchez-Jiménez F., Medina M. A. (2001) Histamine prevents polyamine accumulation in mouse C57.1 mast cell cultures. Eur. J. Biochem. 268, 768–773 [DOI] [PubMed] [Google Scholar]
- 29. Zhong D., Xiong L., Liu T., Liu X., Liu X., Chen J., Sun S. Y., Khuri F. R., Zong Y., Zhou Q., Zhou W. (2009) The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J. Biol. Chem. 284, 23225–23233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urdiales J. L., Matés J. M., Núñez de Castro I., Sánchez-Jiménez F. M. (1992) Chlorpheniramine inhibits the ornithine decarboxylase induction of Ehrlich carcinoma growing in vivo. FEBS Lett. 305, 260–264 [DOI] [PubMed] [Google Scholar]
- 31. Gnaiger E. (2012) in Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis, 3rd Ed (Gnaiger E., ed) pp. 44–50, OROBOROS MiPNet Publications, Innsbruck [Google Scholar]
- 32. Schefe J. H., Lehmann K. E., Buschmann I. R., Unger T., Funke-Kaiser H. (2006) Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J. Mol. Med. 84, 901–910 [DOI] [PubMed] [Google Scholar]
- 33. Medina M. A., del Castillo-Olivares A., Schweigerer L. (1992) Plasma membrane redox activity correlates with N-myc expression in neuroblastoma cells. FEBS Lett. 311, 99–101 [DOI] [PubMed] [Google Scholar]
- 34. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell. Metab. 7, 11–20 [DOI] [PubMed] [Google Scholar]
- 35. Kurtoglu M., Gao N., Shang J., Maher J. C., Lehrman M. A., Wangpaichitr M., Savaraj N., Lane A. N., Lampidis T. J. (2007) Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer. Ther. 6, 3049–3058 [DOI] [PubMed] [Google Scholar]
- 36. Perez-Leal O., Merali S. (2012) Regulation of polyamine metabolism by translational control. Amino Acids 42, 611–617 [DOI] [PubMed] [Google Scholar]
- 37. Mandal S., Mandal A., Johansson H. E., Orjalo A. V., Park M. H. (2013) Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 2169–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulze-Lohoff E., Fees H., Zanner S., Brand K., Sterzel R. B. (1994) Inhibition of immediate-early-gene induction in renal mesangial cells by depletion of intracellular polyamines. Biochem. J. 298, 647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Auvinen M., Paasinen A., Andersson L. C., Hölttä E. (1992) Ornithine decarboxylase activity is critical for cell transformation. Nature 360, 355–358 [DOI] [PubMed] [Google Scholar]
- 40. Rodríguez-Caso C., Montañez R., Cascante M., Sánchez-Jiménez F., Medina M. A. (2006) Mathematical modeling of polyamine metabolism in mammals. J. Biol. Chem. 281, 21799–21812 [DOI] [PubMed] [Google Scholar]
- 41. Wallick C. J., Gamper I., Thorne M., Feith D. J., Takasaki K. Y., Wilson S. M., Seki J. A., Pegg A. E., Byus C. V., Bachmann A. S. (2005) Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene 24, 5606–5618 [DOI] [PubMed] [Google Scholar]
- 42. Koomoa D. L., Borsics T., Feith D. J., Coleman C. C., Wallick C. J., Gamper I., Pegg A. E., Bachmann A. S. (2009) Inhibition of S-adenosylmethionine decarboxylase by inhibitor SAM486A connects polyamine metabolism with p53-Mdm2-Akt/protein kinase B regulation and apoptosis in neuroblastoma. Mol. Cancer Ther. 8, 2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mazzio E., Soliman K. F. (2003) The role of glycolysis and gluconeogenesis in the cytoprotection of neuroblastoma cells against 1-methyl 4-phenylpyridinium ion toxicity. Neurotoxicology 24, 137–147 [DOI] [PubMed] [Google Scholar]
- 44. Passonneau J. V., Crites S. K. (1976) Regulation of glycogen metabolism in astrocytoma and neuroblastoma cells in culture. J. Biol. Chem. 251, 2015–2022 [PubMed] [Google Scholar]
- 45. Wang Q., Liang B., Shirwany N. A., Zou M. H. (2011) 2-Deoxy-d-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 6, e17234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ray R. M., Viar M. J., Johnson L. R. (2012) Amino acids regulate expression of antizyme-1 to modulate ornithine decarboxylase activity. J. Biol. Chem. 287, 3674–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gregory M. A., Hann S. R. (2000) c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells. Mol. Cell Biol. 20, 2423–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yeh E., Cunningham M., Arnold H., Chasse D., Monteith T., Ivaldi G., Hahn W. C., Stukenberg P. T., Shenolikar S., Uchida T., Counter C. M., Nevins J. R., Means A. R., Sears R. (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6, 308–318 [DOI] [PubMed] [Google Scholar]
- 49. Fell D. (1996) Understanding the Control of Metabolism (Fell D., ed) Portland Press Ltd., London [Google Scholar]
- 50. Sauer U. (2006) Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pegg A. E. (2014) The function of spermine. IUBMB Life 66, 8–18 [DOI] [PubMed] [Google Scholar]
- 52. Cascante M., Benito A., Zanuy M., Vizán P., Marín S., de Atauri P. (2010) Metabolic network adaptations in cancer as targets for novel therapies. Biochem. Soc. Trans. 38, 1302–1306 [DOI] [PubMed] [Google Scholar]
- 53. Ledford H. (2014) Metabolic quirks yield tumour hope. Nature 508, 158–159 [DOI] [PubMed] [Google Scholar]
- 54. Zhao Y., Butler E. B., Tan M. (2013) Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stein M., Lin H., Jeyamohan C., Dvorzhinski D., Gounder M., Bray K., Eddy S., Goodin S., White E., Dipaola R. S. (2010) Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 70, 1388–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]