

Background: Kindlin-3 regulates integrin activation in hematopoietic cells.
Results: Stimulation of cells bearing integrin αIIbβ3 leads to enhanced kindlin-3 phosphorylation and prevention of phosphorylation-inhibited integrin-mediated cellular responses.
Conclusion: Phosphorylation is integral to the capacity of kindlin-3 to regulate integrin function.
Significance: A post-translational modification has been identified as a key event in the mechanism by which kindlin-3 regulates integrin-dependent responses.
Keywords: Hematopoiesis, Integrin, Kindlin, Phosphorylation, Platelet
Abstract
The contributions of integrins to cellular responses depend upon their activation, which is regulated by binding of proteins to their cytoplasmic tails. Kindlins are integrin cytoplasmic tail binding partners and are essential for optimal integrin activation, and kindlin-3 fulfills this role in hematopoietic cells. Here, we used human platelets and human erythroleukemia (HEL) cells, which express integrin αIIbβ3, to investigate whether phosphorylation of kindlin-3 regulates integrin activation. When HEL cells were stimulated with thrombopoietin or phorbol 12-myristate 13-acetate (PMA), αIIbβ3 became activated as evidenced by binding of an activation-specific monoclonal antibody and soluble fibrinogen, adherence and spreading on fibrinogen, colocalization of β3 integrin and kindlin-3 in focal adhesions, and enhanced β3 integrin-kindlin-3 association in immunoprecipitates. Kindlin-3 knockdown impaired adhesion and spreading on fibrinogen. Stimulation of HEL cells with agonists significantly increased kindlin-3 phosphorylation as detected by mass spectrometric sequencing. Thr482 or Ser484 was identified as a phosphorylation site, which resides in a sequence not conserved in kindlin-1 or kindlin-2. These same residues were phosphorylated in kindlin-3 when platelets were stimulated with thrombin. When expressed in HEL cells, T482A/S484A kindlin-3 decreased soluble ligand binding and cell spreading on fibrinogen compared with wild-type kindlin-3. A membrane-permeable peptide containing residues 476–485 of kindlin-3 was introduced into HEL cells and platelets; adhesion and spreading of both cell types were blunted compared with a scrambled control peptide. These data identify a role of kindlin-3 phosphorylation in integrin β3 activation and provide a basis for functional differences between kindlin-3 and the two other kindlin paralogs.
Introduction
The ligand-binding function of integrin adhesion receptors, particularly on circulating blood cells, is tightly regulated. Under most conditions, the integrins exist on the cell surface in a quiescent state, incapable of binding cognate ligands with high affinity. However, when the cells are confronted with a stimulatory agonist, the integrins rapidly adopt active, ligand-competent conformations. Such integrin activation is regulated by engagement of binding partners to the short cytoplasmic tails of integrins (1–3), which elicit inside-out signals across the transmembrane domains of the integrin to the ligand binding domain within their extracellular region. Ligand binding can, in turn, elicit outside-in signaling that triggers intracellular responses. Kindlins are a family of intracellular proteins that have been implicated in bidirectional signaling across integrins. Kindlins link integrin signaling to the actin cytoskeleton and localize to integrin adhesion sites (4–6). They are evolutionarily conserved with an ortholog, UNC-112, present in Caenorhabditis elegans (7). In mammals, there are three kindlin family members, each characterized by a FERM domain bisected by a pleckstrin homology domain. The FERM domains of kindlins are most closely related to the FERM domain of talin, which is also involved in regulation of integrin signaling (8–12). Kindlins and talin bind to the cytoplasmic tails of integrin β subunits via their F3 (phosphotyrosine binding) subdomains within their FERM domains. However, the binding sites of talin and kindlins within the β-cytoplasmic tails do not overlap (6, 13), and the two interactions appear to act cooperatively to optimize integrin activation (13, 14). Hence, cells or mice with decreased kindlin expression levels are unable to properly activate their integrins. Kindlin-1 is expressed predominantly in epithelial cells, and mutation in the kindlin-1 gene causes Kindler syndrome in humans (15, 16), a rare disease characterized by skin blistering, poikiloderma with frequent intestinal complications. These phenotypes are recapitulated in mice in which the kindlin-1 gene has been inactivated (17). Kindlin-2 is expressed in most tissues and in many different cell types, and knock-out of kindlin-2 is lethal in mice and zebrafish during embryonic development (14, 18). Mice in which the kindlin-3 gene has been inactivated display defects in platelet (19) and leukocyte (20) responses dependent on integrin activation, and the mice die by day 7 postnatally (19). Kindlin-3 mutations in have been identified in humans with a rare syndrome referred to as LADIII (21–25) with manifestations that include episodic bleeding, susceptibility to frequent infections and osteopetrosis, which are consequences of an inability to activate β1, β2 and β3 integrin (22, 23, 25), and variably in abnormal red cell shapes (25). Kindlin-3 is also present and functional in endothelial cells (26) and breast cancer cells (27), where it acts as a tumor promoter (27). Despite this ample evidence emphasizing the role of kindlin-3 in integrin function in variety of cells, the mechanisms underlying kindlin-mediated integrin activation are largely unknown. Recently, it has been established that the calpain I cleavage of kindlin-3 at Tyr373 controls the dynamics of integrin/kindlin-3 interaction and, in turn, integrin-dependent adhesion and migration of hematopoietic cells (28). The other known functional site in kindlin-3 is its integrin-binding site in its F3 domain that centers at Gln597/Trp598. Mice in which these two residues have been mutated to alanines are unable to stop bleeding upon tail resection or form arterial occlusions but are viable for least 6 months (29).
Here, we sought to identify possible post-translational modification(s) that regulate kindlin-3 functions in hematopoietic cells. One of the possible mechanisms regulating the ability of kindlins to activate integrins is phosphorylation. Previous work has shown that phosphorylation on the integrin β3 CT2 regulates kindlin-2 binding (30). In this work, we have considered how this post-translational modification might regulate the function of kindlin-3 and have utilized HEL cells and human platelets as model systems. HEL cells are suspension cells that express high levels of integrin αIIbβ3 (31, 32) and have served as a model of analyses of platelet responses (33, 34). When stimulated with PMA or TPO, HEL cells adhere to immobilized fibrinogen via their integrin αIIbβ3 (35, 36). In this study, using a variety of approaches, we confirm that kindlin-3 is crucial for integrin activation in hematopoietic cells, and this agonist-induced function of kindlin-3 depends on β3 integrin cytoplasmic tail/kindlin-3 interaction and phosphorylation in a variable region of kindlin-3. These results establish a previously unknown role of kindlin-3 phosphorylation in the regulation of integrin function in hematopoietic cells, and they provide a molecular basis for the difference between kindlin-3 and other two kindlin family members.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Mouse monoclonal antibody (mAb) against EGFP (JL-8) was from Clontech; mouse monoclonal antibody against human β3 integrin (RUU-PL7F12) was from BD Biosciences; mouse mAb against GAP DH was from Thermo Fisher Scientific (Waltham, MA); mouse mAb against kindlin-2 was from EMD Millipore (Billerica, MA); mouse mAb 8d4 against talin was from Sigma; mouse mAb PAC-1 against activated αIIbβ3 integrin was from BD Biosciences; mouse PerCP-eFluor 710-labeled mAb against CD162 (PSGL-1) was from eBioscience (San Diego); rabbit mAb against β3 integrin was from Origene (Rockville, MD); mouse PE-labeled mAb against CD62 (P-selectin) was from BD Biosciences; and rat mAb 9EG7 against activated mouse β1 integrin was from BD Biosciences. Rabbit anti-kindlin-3 antibodies were prepared as described previously (26). Mouse mAb 2G12 against human αIIbβ3 integrin has been described (37). Alexa and (R)-PE-coupled secondary antibodies, Alexa-coupled phalloidins, and Alexa 647-coupled human fibrinogen were from Thermo Fisher Scientific. Horseradish peroxidase-conjugated secondary antibodies and A/G protein-agarose were from Santa Cruz Biotechnology (Dallas); ECL reagent was from Roche Applied Science; and human fibrinogen was from Enzyme Research Laboratories (South Bend, IN). Thrombin was from Sigma, and PMA and membrane-permeable protein kinase C inhibitor were from EMD Millipore. Kindlin 3-specific and nontargeting siRNAs from Thermo Fisher Scientific and QuikChange site-directed mutagenesis kit were from Agilent Technologies (Santa Clara, CA). Iscove's modified Dulbecco's medium, RPMI 1640 medium, DMEM/F-12, penicillin/streptomycin, and l-glutamine were from Media Lab (Cleveland Clinic); EGM® endothelial cell growth medium was from Lonza, and fetal calf serum was from Atlanta Biologicals. 9R and fluorescein-tagged peptide corresponding to residues 476–485 of kindlin-3 (FLSLQRTGSG) and a scrambled peptide (SRGLSQFGTL) were synthesized in the Biotechnology Core of the Cleveland Clinic.
cDNA Constructs
EGFP-tagged kindlin-3 was created by cloning full-length kindlin-3 in-frame with EGFP into the pEGFP-C2 vector (Clontech) as described previously (23). The PSGL-1/β3 chimera was constructed in pcDNA3.1 vector in which the N terminus (1–91 amino acids) of human PSGL-1 was fused onto the C terminus (468–762 amino acids) of human β3 subunit as described previously (13). All the indicated mutations of kindlin-3 and PSGL-1 chimera were introduced into the respective constructs using QuikChange site-directed mutagenesis kit (Agilent Technologies) and confirmed by gene sequencing.
Cells and Transfections
HEL cells and RAW 264.7 cells were from American Type Culture Collection (Manassas, VA). Human umbilical vein endothelial cells were provided by Dr. Paul DiCorleto (Cleveland Clinic) and maintained in EGM® endothelial cell growth medium. K562 αIIbβ3 cells were kindly provided by Scott Blystone (State University of New York Upstate Medical University, Syracuse). Chinese hamster ovary cell line stably expressing αIIbβ3 (CHO-A5) was described before (13). Transient transfections of A5 cells were performed using Lipofectamine 2000, accordingly to the manufacturer's protocol (Thermo Fisher Scientific). Transient nucleofections of cells were performed using nucleofection kit V from Lonza (Walkersville, MD), according to the manufacturer instructions and program X-005 for HEL cells, T-016 for K562 cells, and D-032 for RAW 264.7 cells with 3 μg of DNA per sample.
Ethics Statement
All experiments done with human platelets were approved by the Cleveland Clinic's Institutional Review Board. Written informed consent was obtained in accordance with the Declaration of Helsinki.
Isolation of Platelets from Human Blood
Platelets were isolated as described previously (39). Briefly, blood was drawn from healthy volunteers into acid/citrate/dextrose (ACD: 65 mm citric acid, 85 mm sodium citrate, 111 mm dextrose, pH 4.61) and 2 μm prostaglandin E1. Blood was centrifuged to obtain platelet-rich plasma, which was then further centrifuged, and the platelet pellet was suspended in Ca2+- and Mg2+-free Tyrode's buffer (138 mm NaCl, 12 mm NaHCO3, 0.36 mm Na2HPO4, 2.9 mm KCl, 10 mm HEPES, 0.1% glucose, 0.1% bovine serum albumin, pH 7.1) and gel-filtered through a Sepharose CL-4B (GE Healthcare) column in Tyrode's buffer to obtain the platelet preparations. Platelet count was determined using a hemocytometer.
Western Blotting
Cells were solubilized in a Laemmli buffer containing 62.5 mm Tris-HCl, pH 7.4, 2% SDS, 5% 2-mercaptoethanol, and 10% glycerol. Pellets were solubilized by addition of 2× Laemmli buffer and subjected to SDS-PAGE. Proteins were transferred to PVDF, and blots were probed with antibodies of interest, using 5% BSA as a blocking agent.
Immunoprecipitation
For antibody-mediated precipitation of endogenous kindlin-3, HEL cells were stimulated with PMA or TPO, and platelets were stimulated with thrombin or PMA. Samples were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm sodium chloride, 1% Nonidet P-40, 1 mm calcium chloride, containing protease and phosphatase inhibitors. Lysates were held on ice for 30 min prior to centrifugation at 12,000 × g for 15 min. Aliquots of the detergent-soluble material were precleared on A/G protein-agarose for 1 h at 4 °C. Precleared lysates were incubated with 2 μl of antibodies or preimmune serum and protein A/G-agarose for 16 h at 4 °C. Immunoprecipitated proteins were solubilized in Laemmli buffer and analyzed on Western blots with β3 integrin antibodies and kindlin-3 antibodies.
Mass Spectrometry
Endogenous kindlin-3 was immunoprecipitated from HEL cells and platelets as described above. Polyacrylamide gels were stained with Coomassie Blue to visualize bands. Proteins were identified by sequencing tryptic peptides by tandem in the mass spectrometry laboratory (Cleveland Clinic). The data were analyzed using the Mascot program to analyze the collision-induced dissociation spectra collected from the LC-MS.
Immunostaining
HEL cells were stimulated with 800 nm PMA and plated on 20 μg/ml fibrinogen for the times indicated, fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, blocked in horse serum, and stained with the indicated antibodies for ∼18 h. Antigen-antibody complexes were detected by staining with Alexa-coupled secondary antibodies for 1 h. Stained cells were visualized with a ×40 or ×63 1.4 oil objective using a Leica TCS-NT laser scanning confocal microscope (Imaging Core, Cleveland Clinic). Laser intensities were adjusted to eliminate cross-talk between channels, and images were collected using Leica confocal software (version 2.5 build 1227).
Total Internal Reflection Fluorescence (TIRF)
For cell imaging by TIRF microscopy, HEL cells were nucleofected with kindlin-3 constructs, after 24 h treated with 800 nm PMA, and allowed to spread for 1 h on glass-bottom dishes (MatTek Corp., Ashland, MA) coated with 20 μg/ml fibrinogen. Cells were fixed with 4% PFA, permeabilized with 0.1% Triton X-100, blocked in horse serum, and stained with β3 integrin antibodies for ∼18 h. Antigen-antibody complexes were detected by staining with Alexa 568-coupled secondary antibodies for 1 h. Cells were imaged with a 100 × 1.49 NA TIRF objective on a Leica AM TIRF inverted microscope equipped with Dual Hamamatsu cameras (ImageEM and Orca-R2).
siRNA Interference
To knock down endogenous kindlin-3 in HEL cells, 100 nm kindlin-3, or control, nontargeting siRNAs were transfected into cells using nucleofection kit V according to the manufacturer's protocol. The level of suppression and specificity of kindlin-3 siRNA was evaluated by Western blotting with antibodies against kindlin-3 or against GAPDH as a loading control.
Platelet Aggregation
Effect of kindlin-3 and scramble peptides on platelet aggregation was examined using an aggregometer (Chrono-log Corp., Havertown, PA). Platelets (3 × 108/ml) were equilibrated at 37 °C for 5 min. Then 10 μm peptides or agonist was added, and aggregation was monitored for 10 min at 37 °C with stirring. The final volume in each cuvette was 500 μl. 1 unit of thrombin was used as a positive control, and baseline for 100% aggregation was set with Tyrode's buffer.
Isolation and Analysis of Platelet Cytoskeletons
Platelet suspensions were stimulated with thrombin for the times indicated. Aggregation was terminated by addition of an equal volume of ice-cold lysis buffer containing 2% Triton X-100, 10 mm EGTA, 100 mm Tris-HCl, pH 7.4, with protease and phosphatase inhibitors (40). Lysates were immediately centrifuged at 15,600 × g at 4 °C for 4 min to obtain the low speed detergent-insoluble pellet. Detergent-insoluble and -soluble fractions were solubilized by addition of an SDS-containing buffer in the presence of reducing agent (40), denatured at 95 °C, and electrophoresed on 4–20% gradient acrylamide gels in SDS. Proteins present in the fractions were detected by transferring samples to PVDF and developing the Western blots with antibodies against β3 integrin and kindlin-3.
Flow Cytometry and Soluble Ligand Binding
To assess changes in P-selectin surface expression, platelet suspensions were stimulated with kindlin-3 peptides or 1 unit of thrombin for 30 min in the presence of 20 μl of PE-labeled P-selectin antibody and fixed with 2% PFA for 10 min. To assess fibrinogen binding to HEL cells, cells were stimulated with 800 nm PMA for 10 min, and cells were fixed with 1% PFA for 10 min in room temperature. Fixed cells were incubated with 20 μg/ml of soluble Alexa 647-labeled fibrinogen for 1 h at room temperature. To address the functional significance of kindlin-3 phosphorylation in integrin-mediated signaling, HEL cells were transfected with the indicated constructs. PAC-1 binding to the different transfectants was analyzed by gating only on the EGFP-positive cells. Mean fluorescence intensities (MFI) of PAC-1 binding were normalized based on the basal level of PAC-1 binding to cells transfected with the EGFP vector alone to obtain relative MFI values. 2 × 105 cells/sample were stimulated with 800 nm PMA for 10 min in the presence of 10 μg/ml PAC-1 in Hanks' buffered saline solution containing 0.1% BSA, 0.5 mm CaCl2, MgCl2. After incubation, cells were fixed with 1% PFA for 10 min in room temperature. After washing, cells were incubated with 10 μg/ml (R)-PE-labeled secondary antibody for 30 min in room temperature. PAC-1 binding was normalized by αIIbβ3 expression level on the cell surfaces measured by 2G12 mAb, which reacts with αIIbβ3 independent of its activation state (37). RAW 264.7 cells were transfected, and 2 × 105 cells were incubated with 10 μg/ml 9EG7 antibody as described for PAC-1. K562 αIIbβ3 cells were transfected; 2 × 105 cells/sample were stimulated with 800 nm PMA for 10 min, and cells were fixed with 1% PFA for 10 min in room temperature. Fixed cells were incubated with 20 μg/ml of soluble Alexa 647-labeled fibrinogen for 1 h at room temperature. Ligand binding was analyzed using LSRFortessa flow cytometer and FlowJo software (BD Biosciences) in Cleveland Clinic Flow Core.
Adhesion and Spreading Assays
For knockdown studies, nontransfected HEL cells or HEL cells transfected with kindlin-3 or control siRNA were stimulated with 800 nm PMA for 5 min and incubated with immobilized fibrinogen in triplicate for 15, 30, or 60 min at 37 °C. After extensive washing with PBS, the adherent cells were fixed with 4% PFA and stained with Alexa 488 phalloidin. The cells were photographed at ×40, and the adherent cells were counted from 20 randomly selected fields; the cell area was measured for 300 cells. To define the β3 CT core for kindlin-3, nontransfected HEL cells or HEL cells transfected with kindlin-3 and/or β3 CT fused to PSGL-1 constructs were stimulated with 800 nm PMA for 5 min and incubated with immobilized fibrinogen in triplicate, for 30 min at 37 °C. After extensive washing with PBS, the adherent cells were fixed with 4% paraformaldehyde and stained with Alexa 568 phalloidin. The cells were photographed with a ×40 objective, and the adherent cells were counted from 20 randomly selected fields; the cell area was measured for 300 cells. For peptide import experiments, HEL cells were incubated with 10 μm kindlin-3 or scramble peptide for 5 min, then stimulated with 800 nm PMA for 5 min, and incubated with immobilized fibrinogen for 30 or 60 min at 37 °C. The adherent cells were fixed with 4% PFA and stained with Alexa 568 phalloidin. The cells were photographed with a ×40 objective, and the adherent cells were enumerated as described above. For peptide import studies with human platelets, isolated human platelets as described above were incubated with 10 μm kindlin-3 or scramble peptide for 5 min and plated on fibrinogen for 30 min. The adherent cells were fixed with 4% PFA and stained with Alexa 568 phalloidin. Platelets were photographed with a ×100 objective, and the adherent platelets were counted from 15 randomly selected fields.
Statistical Analysis
Two-tailed Student's t tests were performed where indicated in the text using SigmaPlot 11. Differences were considered to be significant at p < 0.05.
RESULTS
HEL Cells as a Model for Kindlin-3-mediated Regulation of Integrin αIIbβ3 Function
To consider whether post-translational modification(s) might regulate the integrin-activating kindlin-3 function, we first sought to establish HEL cells as a transfectable model cell system. First, we examined the levels of kindlins and talin in HEL cells as compared with K562 cells, another erythroblastoid cell line, which had been transfected to stably express αIIbβ3 integrin. Anti-kindlin-3 polyclonal antibodies (26) readily recognized the ∼72-kDa band in whole HEL cell lysate, and this band was absent from K562 cells (Fig. 1A, left-hand panel). Kindlin-2 was absent from HEL cells and K562 cells but was readily detected in whole human umbilical vein endothelial cell lysates (Fig. 1B, middle panel). HEL cells and K562 cells expressed high amounts of talin (Fig. 1A, right-hand panel). Thus, kindlin-3 is a major kindlin in HEL cells. Levels of αIIbβ3 integrin in HEL cells (Fig. 1B) were assessed with flow cytometry, and monoclonal antibody 2G12, which recognizes the αIIbβ3 integrin complex (37), reacted well with the cells. HEL cells are suspension cells. Upon agonist stimulation, they increase their binding of soluble ligand (Fig. 1C) and require such stimulation to adhere and spread efficiently on immobilized ligands. HEL cells stimulated for 10 min with 800 nm PMA increased their binding of Alexa 647-labeled soluble fibrinogen when compared with nonstimulated HEL cells, and this interaction was completely abolished with 10 mm EDTA (Fig. 1C) as would be expected for an integrin-mediated binding interaction. Although the majority of HEL cells showed basal levels of fibrinogen binding in the absence of added agonist, PMA stimulation increased the extent of fibrinogen binding to the entire cell population (Fig. 1D). Similar increases in soluble ligand binding were observed when integrin activation was monitored with PAC-1 or when TPO was used as an agonist (data not shown).
FIGURE 1.

Kindlin-3 is a major kindlin in HEL cells. A, Western blots of HEL cells, K562 αIIbβ3 cells, and human umbilical vein endothelial cell (HUVEC) lysates were probed with antibodies to kindlin-3 (left side panel), kindlin-2 (middle panel), or talin (right side panel). B, αIIbβ3 integrin expression levels in HEL cells were determined by flow cytometry with 2G12 monoclonal antibody, and goat anti-mouse Fab conjugated to Alexa 488 was used as a secondary detection. C, Alexa 647-labeled soluble fibrinogen (20 μg/ml) binding to control and PMA stimulated with or without 10 mm EDTA HEL cells (single asterisk p = 0.019 and double asterisk p < 0.001). D, representative histogram showing the binding of Alexa 647-labeled soluble fibrinogen to control and PMA-stimulated with or without 10 mm EDTA HEL cells. The error bars represent means ± S.E.
It has been shown previously (41, 42) that HEL cells localize αIIbβ3 integrin to focal adhesions when spread on an integrin substrate. Knowing that kindlin-3 is a major kindlin in HEL cells, we sought to determine whether kindlin-3 colocalizes with integrin β3 in HEL cells spreading on substrate for this integrin. It has been reported previously in several studies (36, 41–43) that HEL cell adhesion and spreading on integrin substrates, including fibronectin, vitronectin, and fibrinogen, requires stimulation with agonists such as PMA or thrombin, thereby distinguishing these cells from platelets that adhere to these substrates in the absence of exogenously added agonists. It has been suggested that PMA might increase αIIbβ3 integrin-dependent adhesion to immobilized ligand by increasing the local concentration of integrin, resulting in an increased avidity and changing the affinity state of αIIbβ3 integrin, or both (36). Under the conditions we used and consistent with these prior reports, in the absence of agonist stimulation, HEL cells displayed little adhesion, and they did not spread on fibrinogen. To determine the kindlin-3 distribution in HEL cells, cells were pretreated with 800 nm PMA for 10 min, allowed to spread on fibrinogen for 60 min, fixed, and stained with kindlin-3 and integrin-specific antibodies. At the 60-min time point, the cells were fully spread and β3 integrin was present in focal adhesions (Fig. 2, upper panels). Staining for kindlin-3 revealed that kindlin-3 colocalized with β3 integrin in these structures (Fig. 2, upper panels, sites of colocalization are indicated with the arrows in merge panel). To assess whether αIIbβ3 integrin is a major integrin present in kindlin-3-containing focal adhesions, HEL cells were stained with antibodies that specifically recognize this integrin. The staining revealed that αIIbβ3, detected with 2G12, was present in kindlin-3-containing focal adhesions (Fig. 2, lower panels, sites of colocalization are indicated with the arrows in merge panel).
FIGURE 2.

Kindlin-3 distribution in HEL cell spreading on fibrinogen. HEL cells were treated with 800 nm PMA and spread on fibrinogen for 60 min. Cells were fixed, permeabilized, and stained with antibodies against kindlin-3 (26), total β3 integrin (RUU-PL7F12), and αIIbβ3 integrin (2G12) followed by Alexa 488 anti-mouse IgG and Alexa 568 anti-rabbit IgG. Kindlin-3 colocalized with integrins in focal adhesions (indicated with arrows). Bar, 20 μm. The images shown are representative of 12–15 independent fields with each antibody.
Kindlin-3 Knockdown Blunts Integrin-dependent Adhesion and Spreading in HEL Cells
To determine whether kindlin-3 deficiency blunts integrin responses in HEL cells, we performed RNA interference experiments. HEL cells were transfected with siRNA specific to kindlin-3 or a nontargeting siRNA as a control. The levels of expression of kindlin-3 were analyzed on Western blots (Fig. 3A). Control siRNA did not have an effect on expression of kindlin-3 (Fig. 3A, lane 2 of kindlin-3 blot). Kindlin-3 siRNA effectively inhibited the expression of kindlin-3 protein (Fig. 3A, lane 3 of kindlin-3 blot); in three independent experiments, 60–80% knockdown was achieved. None of the siRNAs used affected β3 integrin expression as assessed by flow cytometry with β3 integrin-specific antibody (data not shown). To assess the function of kindlin-3 in integrin-mediated cell adhesion, nontransfected and siRNA-transfected HEL cells were stimulated with 800 nm PMA for 10 min and were plated on fibrinogen for 15, 30, or 60 min. Cells were fixed, and actin was visualized with Alexa 488 phalloidin. Cell adhesion was blunted significantly at the 30- and 60-min time points (50–60% decrease in adhesion, p < 0.001) in cells transfected with kindlin-3 siRNA but not with nontargeting siRNA when compared with control cells (Fig. 3B). Cell area was determined by measuring 300 cells at the 15- and 30-min time points. Compared with control HEL cells and cells transfected with nontargeting siRNA, knockdown of kindlin-3 inhibited β3-mediated cell spreading by ∼50% at the 15-min (p < 0.001) and by ∼40% at the 30-min time point (p < 0.001) (Fig. 3C).
FIGURE 3.

Kindlin-3 knockdown inhibits integrin-mediated adhesion and spreading of HEL cells. A, siRNA suppression of kindlin-3 expression in HEL cells. Expression of kindlin-3 in nontransfected cells, nontargeting siRNA, and kindlin-3 siRNA was analyzed by Western blotting with kindlin-3 and GAPDH antibodies. B, nontransfected HEL cells or HEL cell-transfected kindlin-3 or nontargeting siRNA were used in adhesion assays. The cells were treated with 800 nm PMA, allowed to adhere for 30 or 60 min, fixed, stained with Alexa 488-phalloidin, and counted (*, p < 0.001). The error bars are means ± S.E. of three independent experiments. C, HEL cells were treated with 800 nm PMA and allowed to adhere to fibrinogen-coated coverslips, and cell spreading was measured after 15 and 30 min. The adherent cells were fixed and stained with Alexa 488 phalloidin. The areas of cells were measured using ImageJ software, and 300 cells were quantified in each experiment (*, p < 0.001). The error bars represent means ± S.E. of three independent experiments.
Kindlin-binding Sites in Integrin β3 Cytoplasmic Tail Inhibit HEL Cell Spreading on Immobilized Fibrinogen
In a prior study, the kindlin-2 binding core in integrin β3 CT was established as Ala750 to Gly761 (44). The strategy involved expression of various β3 integrin CT as fusion proteins linked to extracellular and transmembrane portions of PSGL-1 and with the β3 CT replacing the PSGL-1 CT in CHO cell line stably expressing αIIbβ3 integrin (13). This strategy allowed the competition of chimeric proteins with endogenous β3 CT for integrin binding partners, kindlin-2 and talin. In this study, we implemented a similar strategy in HEL cells to map the kindlin-3-binding site on β3 CT. PSGL-1 alone and chimeric constructs were introduced into HEL cells by nucleofection. After 24 h, expression of the PSGL-1/β3CT constructs on the cell surface as assessed by flow cytometry with anti PSGL-1 antibody differed by <10%. We then evaluated the effects of the various β3 CT on β3-mediated HEL cell spreading on immobilized fibrinogen. HEL cells were stimulated with 800 nm PMA for 10 min, and 2 × 105 cells were plated on immobilized fibrinogen for 30 min. Cells were fixed, and actin was visualized with Alexa 488 phalloidin. Cell area was measured for 300 cells/construct. Compared with control HEL cells and cells expressing PSGL-1 alone, expression of the wild-type β3CT chimera inhibited β3-mediated cell spreading by 50% (p < 0.001) (Fig. 4). As a specificity control, the Y747A mutation, which interferes with talin binding, resulted in a loss of inhibitory activity (p < 0.001), whereas Y747F mutation, which still allows talin binding, had an inhibitory effect on cell spreading. Mutations that abolished kindlin-2 binding, S752P and Y759A, also led to loss of competitive activity (p < 0.001) (Fig. 4). This loss was not the observed S752A substitution, which would retain kindlin binding to β3 CT (Fig. 4). The β3CT kindlin-2 core (Ala750–Gly761) (44) was also sufficient to significantly suppress cell spreading (p < 0.001). The β3CT Δ748, which only expresses the talin and not the kindlin-binding site (44), also significantly inhibited HEL cell spreading (p < 0.001) (Fig. 4). Thus, these data establish that both kindlin-3 and talin contributed to integrin-mediated functions in HEL cells, and kindlin-3 utilizes the same binding core on integrin β3CT as kindlin-2 to regulate integrin function in HEL cells.
FIGURE 4.
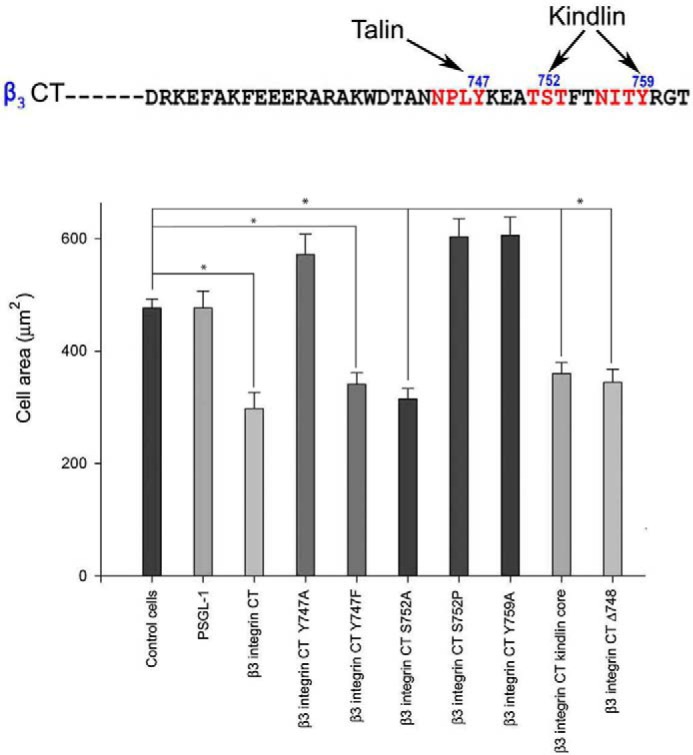
Kindlin-binding site residues in integrin β3 cytoplasmic tail influence HEL cell spreading. HEL cells were stimulated with 800 nm PMA for 5 min, and cells were plated on fibrinogen for 30 min. Cells were fixed, and actin was visualized with Alexa 488 phalloidin. Compared with control HEL cells and cells expressing PSGL-1 alone, expression of the wild-type β3CT chimera inhibited β3-mediated cell spreading by 50%. Y747A mutation, which interferes with talin binding, resulted in a loss of inhibitory activity, whereas Y747F mutation, which still allows talin binding, had an inhibitory effect on spreading. S752P and Y759A, which block kindlin-2 binding, also led to loss of competitive activity. This loss was not observed in S752A substitution, which would retain kindlin binding to β3 CT. The β3 CT kindlin-2 core (Ala750 to Gly761) was also sufficient to significantly suppress cell spreading. The β3CT Δ748, which only expresses the talin but not the kindlin-binding site, also significantly inhibited HEL cell spreading. The areas of cells were measured using ImageJ software, and 300 cells were quantified in each experiment (*, p < 0.001). The error bars represent means ± S.E. of three independent experiments.
Kindlin-3 Is Phosphorylated in HEL Cells and Platelets upon Agonist Stimulation
Knowing that kindlin-3 regulates β3 integrin functions in HEL cells upon agonist stimulation, we sought to determine whether the association of kindlin-3 with β3 integrin is stimulus-dependent in HEL cells. and we extended this analysis to platelets. First, HEL cells or platelets were stimulated with agonists, and kindlin-3 was immunoprecipitated with anti-kindlin-3. The immunoprecipitates were then analyzed on Western blots with antibodies against β3 integrin and kindlin-3. Anti-kindlin-3, but not preimmune serum, used as a negative control, immunoprecipitated a single ∼72-kDa band (Fig. 5, A and B, kindlin-3 blots). Anti-β3 integrin antibody detected a single ∼100-kDa band in immunoprecipitates of cells stimulated with agonists. β3 integrin associated with kindlin-3 in HEL cells stimulated with 800 nm PMA (Fig. 5A, β3 blot) and in thrombin-stimulated platelets (Fig. 5B, β3 blot), but this interaction was not detected in nonstimulated HEL cells or in resting platelets. MnCl2, which activates integrin externally, failed to promote integrin-kindlin-3 association alone and did not increase PMA-induced integrin-kindlin-3 association (Fig. 5A).
FIGURE 5.

Kindlin-3 interacts with β3 integrin in HEL cells and platelets upon agonist stimulation. A, total lysates of nonstimulated HEL cells and HEL cells treated with 1 mm MnCl2 or 800 nm PMA were immunoprecipitated with control serum (preimmune) and antibodies against kindlin-3. The immunoprecipitates were subjected to Western blotting with antibodies against β3 integrin and kindlin-3. Kindlin-3 coimmunoprecipitated β3 integrin from HEL cells treated with 800 nm PMA but not from nonstimulated cells or MnCl2-treated cells. B, total lysates of resting human platelets and platelets treated with thrombin were immunoprecipitated with control serum (preimmune) and antibodies against kindlin-3. The immunoprecipitates were subjected to Western blotting with antibodies against β3 integrin and kindlin-3. Kindlin-3 coimmunoprecipitated β3 integrin from platelets treated with thrombin but not from resting platelets. C, redistribution of β3 integrin and kindlin-3 to the cytoskeletal fraction following thrombin-induced aggregation of platelets. Platelet suspensions were stirred in the presence of 1 unit of thrombin for the times indicated. Incubations were terminated by addition of Triton X-100-containing buffer. Kindlin-3 and β3 integrin in the low speed pellets and supernatants (separated by centrifugation at 15,600 × g for 4 min) were detected by Western blots.
Additional evidence for the stimulus dependence of the association of kindlin-3 with integrin was obtained from analysis of the detergent-insoluble cytoskeletal fraction of thrombin-stimulated platelets. It has been shown previously (45, 46), upon binding of fibrinogen to aggregating platelets, that a portion of the αIIbβ3 integrin associates with detergent-insoluble cytoskeletal fraction, which can be recovered by low speed centrifugation. Fibrinogen binding also stimulates redistribution of numerous cytoskeletal and signaling proteins, including αIIbβ3 integrin-binding proteins, such as c-Src and talin, to the low speed cytoskeletal fraction (40). We sought to determine whether kindlin-3 redistributes together with the β3 subunit to detergent-insoluble fraction of thrombin-stimulated platelets. Platelets were stirred with the addition of 1 unit of thrombin for the times indicated, and detergent-insoluble and detergent-soluble fractions were separated and analyzed by Western blotting for the presence of β3 integrin and kindlin-3 following standard protocols (40). As expected, thrombin stimulation resulted in redistribution of β3 to the detergent-insoluble fraction (Fig. 5C, lane 1 and lanes 2–4 are the detergent-insoluble fractions from resting platelets and thrombin-stimulated platelets, respectively; lanes 5 and 6–8 are the detergent-soluble fractions from resting and thrombin-stimulated platelets, respectively). Thrombin-induced redistribution of β3 to the detergent-insoluble pellet was accompanied by kindlin-3 redistribution, whereas more of the kindlin-3 remained in detergent-soluble supernatant from nonstimulated platelets.
With evidence that the kindlin-3/β3 integrin interaction was regulated, we sought to determine whether post-translational modification of kindlin-3 might play a regulatory role. HEL cells and platelets were treated with agonists to promote kindlin-3 and β3 integrin association, and kindlin-3 was immunoprecipitated from cell lysates and resolved by SDS-PAGE. Protein bands corresponding to kindlin-3 were excised from the gel and digested with trypsin, and the tryptic peptides were subjected to tandem mass spectrometry sequencing. The stimulation of HEL cells with PMA and TPO led to increased kindlin-3 phosphorylation (Fig. 6, A and B). Thr482 or Ser484 was identified as the phosphorylation site in PMA- and TPO-stimulated HEL cells. Mass analysis of the spectra suggested a single phosphorylation site in kindlin-3 from cells stimulated with either agonist, but the identity of the site, Thr482 or Ser484, could not be distinguished. PMA led to an ∼8-fold increase in phosphorylation. TPO-induced kindlin-3 phosphorylation was dose-dependent with 600 nm TPO having a more pronounced effect than 400 nm TPO (Fig. 6B).
FIGURE 6.

Kindlin-3 is phosphorylated in HEL cells and platelets upon agonist stimulation. A, HEL cells were treated with PMA, and endogenous kindlin-3 was immunoprecipitated with kindlin-3 antibodies. The 72-kDa kindlin-3 band was excised from the gel and subjected to mass spectrometry sequencing. Thr482 and/or Ser484 were identified as a phosphosite in three independent experiments. B, HEL cells were treated with TPO, and endogenous kindlin-3 was immunoprecipitated with kindlin-3 antibodies. The 72-kDa kindlin-3 band was excised from the gel and subjected to mass spectrometry sequencing. Thr482 and/or Ser484 were identified as a phosphosite in two independent experiments. C, HEL cells were treated with PMA in the absence or presence of membrane-permeable PKC inhibitor, and endogenous kindlin-3 was immunoprecipitated with kindlin-3 antibodies. The 72-kDa kindlin-3 band was excised from the gel and subjected to mass spectrometry sequencing. Thr482 and/or Ser484 were identified as a phosphosite in two independent experiments. D, human platelets were treated with thrombin or PMA, and endogenous kindlin-3 was immunoprecipitated with kindlin-3 antibodies. The 72-kDa band was excised from the gels and subjected to mass spectrometry sequencing. Mass analysis of the spectra suggested a single phosphorylation site in kindlin-3 from both cell types, but the identity of the site, Thr482 or Ser484, could not be distinguished. These phosphosites reside in a hyper-variable region of kindlin-3 region, i.e. it is not present in either kindlin-1 or kindlin-2.
Protein kinase C (PKCs) is a family of related serine/threonine kinases that have been suggested to play indispensable roles in αIIbβ3 integrin activation and outside-in signaling (47–50). Because kindlin-3 is essential for integrin activation, we wished to establish whether PKC is involved in agonist-induced kindlin-3 phosphorylation. HEL cells were treated with 800 nm PMA in the absence or in the presence of membrane-permeable myristoylated PKC inhibitor (51), and kindlin-3 was immunoprecipitated from cell lysates and resolved by SDS-PAGE as described above. Mass spectrometry analysis revealed that the PKC inhibitor blocked PMA-induced kindlin-3 phosphorylation in the dose-dependent manner (Fig. 6C), suggesting a direct or indirect role of PKC in kindlin-3 phosphorylation.
Identical phosphorylation sites, Thr482 or Ser484, were identified in kindlin-3 from thrombin- or PMA-stimulated platelets (Fig. 6D). Interestingly, platelets achieved maximal kindlin-3 phosphorylation in the presence of EDTA, which prevents platelet aggregation, but it still allows integrin activation, suggesting that kindlin-3 phosphorylation in platelets may be a dynamic process (Fig. 6D). These phosphosites reside in a hyper-variable region of kindlin-3 region, i.e. it is not present in either kindlin-1 or kindlin-2 (Fig. 7A). To determine whether these phosphoresidues are conserved among the species, we aligned the human kindlin-3 peptide sequence in question with kindlin-3 from several other species (Fig. 7B). The phosphorylation site(s) were found to be evolutionarily conserved among the species, with at least one residue present in all sequences analyzed (Fig. 6B).
FIGURE 7.
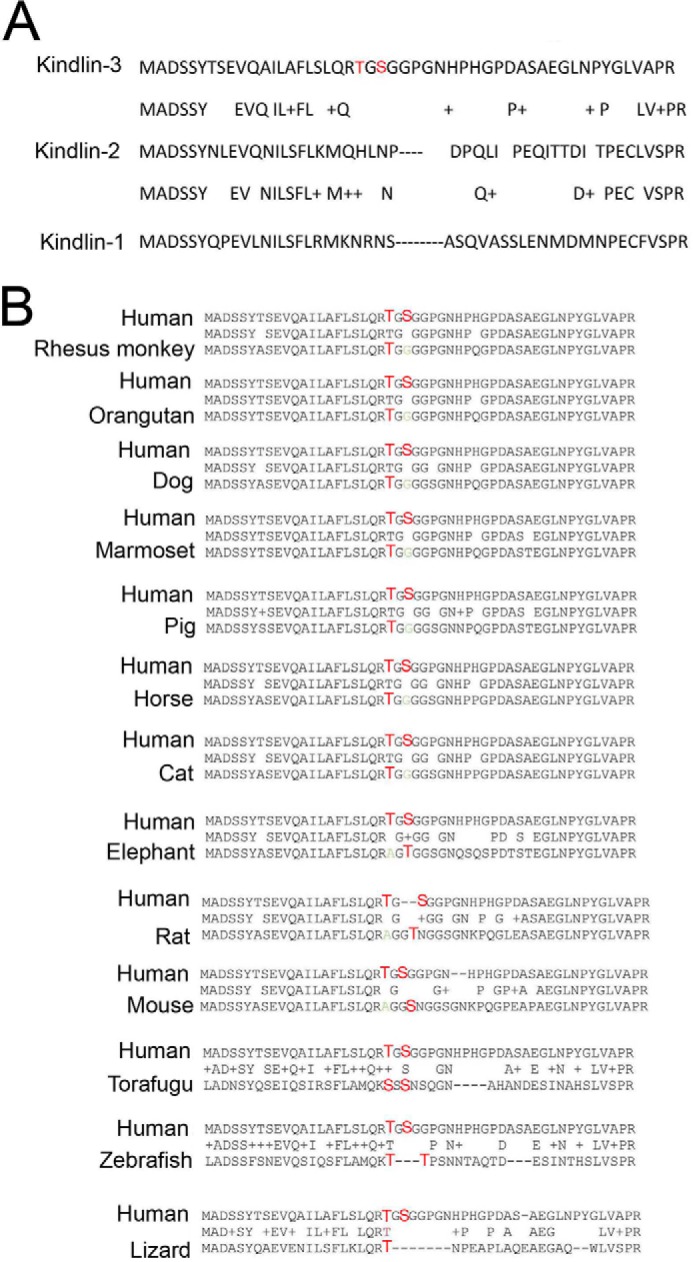
Kindlin-3 phosphosites are evolutionarily conserved. A, identified kindlin-3 phosphosites reside in a hypervariable region of kindlin-3 compared with kindlin-2 and kindlin-1. B, comparison of the human kindlin-3 sequence with kindlin-3 from several other species. The phosphorylation site(s) were found to be evolutionarily conserved among the species (conserved residues depicted in red), including very distant species of T. rubripes (torafugu) and D. rerio (zebrafish).
T482A/S484A Kindlin-3 Mutant Impairs Integrin-mediated Cell Responses
To address the functional significance of kindlin-3 phosphorylation in integrin-mediated signaling, site-directed mutagenesis was performed in which Thr482 and Ser484 were replaced with alanines, and the effects of these mutations on kindlin-3 localization, integrin-induced spreading, and soluble ligand binding were explored in HEL cells. EGFP alone or EGFP-tagged kindlin-3 constructs were introduced into HEL cells by nucleofection. Wild-type kindlin-3 was used as a positive control, and Q597A/W598A kindlin-3, which disables the primary integrin-binding site in the F3 subdomain of kindlin-3 (29), was a negative control. By Western blotting, we confirmed that kindlin-3 mutants were of the same size as wild-type kindlin-3 (data not shown). First, we investigated localization of the kindlin-3 mutant in PMA-treated HEL cells spread on fibrinogen. Because the expression level of EGFP-tagged kindlin-3 in HEL cells is so high as to obscure subcellular localization of its fluorescence, we implemented TIRF microscopy that provides a means to selectively excite fluorophores near the adherent cell surface while minimizing fluorescence from intracellular regions. Because TIRF illuminates only fluorophores very close (e.g. within 100 nm) to the coverslip-sample interface, this serves to increase the signal-to-noise ratio (52). We have previously used TIRF to visualize EGFP-tagged kindlin-3 at contact sites of the adherent cells with the substrate in rescue experiments in transformed lymphocytes from the human subjects lacking kindlin-3 expression (23). HEL cells were allowed to spread on fibrinogen for 60 min, fixed, and stained with kindlin-3 and integrin-specific antibodies. At the 60-min time point, the cells were fully spread, and β3 integrin was present in focal adhesions (Fig. 8, A and B, middle panels). Wild-type kindlin-3 (Fig. 8A, left-hand panels), identified with EGFP fluorescence, colocalized with β3 integrin in these structures (Fig. 8A, right-hand panels, sites of colocalization are indicated with arrows). T482A/S484A kindlin-3 mutant (Fig. 8A, left-hand panel) also colocalized with β3 integrin in focal adhesions (Fig. 8A, left-hand panels, sites of colocalization indicated with arrows); however, the majority of T482A/S484A kindlin-3 mutant transfected cells were less spread than nontransfected cells in the same experiment. Overexpression of Q597A/W598A kindlin-3, a mutant that binds poorly to β3 integrin (29), impaired cell spreading on fibrinogen as well; however, we were able to identify some partially spread cells expressing Q597A/W598A mutant. In these cells, the Q597A/W598A kindlin-3 mutant (Fig. 8B, left-hand upper panel) did not colocalize with β3 integrin in focal adhesions close to the cell surface (Fig. 8B, right-hand upper panel, transfected cells indicated with arrows). EGFP alone (Fig. 8B, left-hand lower panel) also did not localize to β3-containing focal adhesions (Fig. 8B, right-hand lower panel, transfected cells indicated with arrows). Knowing that T482A/S484A kindlin-3 mutant is still present in β3-containing focal adhesions, we sought to determine whether the kindlin-3 mutant still associates with β3 when compared with wild-type kindlin-3. EGFP alone, EGFP-tagged wild-type kindlin-3, and EGFP-tagged T482A/S484A kindlin-3 mutant were expressed in CHO-A5 cells (expressing human αIIbβ3 integrin), and the capacity of anti-EGFP to pull down β3 integrin was evaluated by SDS-PAGE and Western blotting using anti-β3 integrin antibody (Fig. 8C, upper blot). Expression levels of EGFP constructs were evaluated with EGFP antibody. Wild-type and mutant kindlin-3 expressed at similar levels, and EGFP alone expressed at much higher level (Fig. 8C, lower blot). Despite the high level of expression, EGFP alone did not bring down β3 integrin; however, the kindlin-3 constructs coimmunoprecipitated β3 equally well (Fig. 8C). These results are consistent with the TIRF data and support the notion that kindlin-3 phosphorylation does not affect integrin binding directly.
FIGURE 8.

Kindlin-3 phosphomutant (S482A/T484A (S482T484/AA) colocalizes with β3 integrin in focal adhesions as assessed by TIRF and binds to β3 integrin. A and B, Hel cells were transiently transfected with plasmids encoding EGFP alone and EGFP-kindlin-3 constructs. Transfected HEL cells were treated with PMA and allowed to adhere to fibrinogen for 1 h. The adherent cells were fixed and stained with β3 integrin antibody followed with Alexa 568-conjugated anti-mouse secondary antibody. Kindlin-3 distribution was visualized with EGFP fluorescence. A, wild-type and phosphomutant kindlin-3 colocalized with integrins in focal adhesions (indicated with arrows). B, Q597A/W598A (QW/AA) kindlin-3 (mutation of the primary integrin-binding site) and EGFP alone were absent from β3-containing focal adhesions (transfected cells indicated with arrows). Bar, 20 μm. C, lysates of CHO-A5 cells transfected with EGFP-tagged wild-type kindlin-3, S482A/T484A kindlin-3, or EGFP alone were used for coimmunoprecipitation assays. After incubating with A/G-agarose and EGFP antibody (Ab), β3 integrin bound to kindlin-3 constructs was evaluated by SDS-PAGE and Western blotting using anti-β3 integrin antibody. Kindlin-3 and EGFP levels in immunoprecipitates are also shown as detected with an EGFP antibody. C, control.
To assess the function of kindlin-3 phosphomutant in integrin-mediated cell spreading, transfected HEL cells were stimulated with 800 nm PMA for 10 min and plated onto fibrinogen for 30 min. Cells were fixed, and actin was visualized with Alexa 568 phalloidin. Cell area was determined by measuring 300 cells/construct. Compared with control HEL cells and cells transfected with wild-type kindlin-3, the phosphomutant inhibited β3-mediated cell spreading by 40%, which was similar to the effect of Q597A/W598A mutant kindlin-3, suggesting the importance of phosphoresidues for integrin-induced cell spreading (Fig. 9A). Because the T482A/S484A mutant still localized in β3-containing focal adhesions (Fig. 8A) and bound to β3 integrin (Fig. 8C), these data suggest that Thr482 and/or Ser484 phosphorylation might facilitate kindlin-3 binding to a still unidentified protein that is involved in integrin-induced outside-in signaling and cell spreading. Next, we assessed the effect of kindlin-3 phosphomutant overexpression on the activation of endogenous αIIbβ3 integrin in HEL cells. The cells were nucleofected with EGFP alone or with EGFP-tagged kindlin-3 constructs. After 24 h, cells were stimulated with 800 nm PMA for 10 min and subsequently incubated with PAC-1. The populations of cells expressing EGFP-tagged proteins were gated; 60–80% of the cells were EGFP-positive cells, and binding of PAC-1 antibody to these populations was measured. Expression of wild-type kindlin-3 significantly increased activation of αIIbβ3 integrin (Fig. 9B). HEL cells express endogenous kindlin-3 (Fig. 1A). However, it has been shown previously (53) that in another hematopoietic cell line, CMK megakaryocytic cells, which express high levels of endogenous kindlin-3, transient expression of kindlin-3 can augment αIIbβ3 integrin activation in the presence but not in the absence of agonist, as measured by PAC-1 binding (53). Thus, despite the high levels of endogenous kindlin-3, overexpression of WT kindlin-3 increased agonist-induced integrin activation. In contrast, T482A/S484A kindlin-3 mutant failed to stimulate integrin activation in HEL cells as compared with EGFP-expressing cells (Fig. 9B). Next, we extended our studies to other cell models, K562 cells expressing exogenous αIIbβ3 integrin and RAW 264.7 mouse macrophage cell line, which expresses high levels of endogenous α5β1 integrin. The K562-αIIbβ3 cell line we use has undetectable levels of kindlin-2 and kindlin-3 (see Fig. 1A); thus, kindlin-dependent responses are entirely dependent on expressed proteins. (We are aware that the K562-αIIbβ3 cells we used differ from those reported by others (28), who found substantial amounts of kindlin-3 in their K562 cells and attribute this to subline variability.) K562-αIIbβ3 cells are suspension cells, and they activate integrins upon agonist stimulation. K562 cells were nucleofected with EGFP constructs, and binding of soluble Alexa 647-labeled fibrinogen was measured. When EGFP-tagged kindlin-3 constructs were expressed in K562 cells, wild-type kindlin-3 increased binding of soluble fibrinogen significantly (Fig. 9C), whereas T482A/S484A kindlin-3 was as ineffective in increasing integrin activation as the Q597A/W598A kindlin-3 (Fig. 9C). RAW 264.7 mouse macrophage cell line, expressing endogenous kindlin-3, was previously shown to activate α5β1 integrin when wild-type but not Q597A/W598A kindlin-3 was expressed (19). In our assay, we used 9EG7 monoclonal antibody that recognizes activated mouse β1 integrin (54). Wild-type kindlin-3 expression in RAW 264.7 cells yielded a significant increase of 9EG7 binding when compared with EGFP control (Fig. 9D). Expression of T482A/S484A kindlin-3 did not trigger integrin activation, and 9EG7 binding was comparable with EGFP, and Q597A/W598A kindlin-3 was used as a negative control (Fig. 9D). Taken together, these data indicate that kindlin-3 is capable of inducing integrin activation in various cell models, and Thr482and/or Ser484 are required for this activity.
FIGURE 9.

Kindlin-3 phosphomutant (S482A/T484A (S482T484/AA)) blunts integrin-induced cell spreading and soluble ligand binding. A, HEL cells were transiently transfected with plasmids encoding EGFP alone and EGFP-kindlin-3 constructs. EGFP expression levels in HEL cells were determined by flow cytometry; transfection efficiency was 70–80%. Transfected HEL cells were treated with PMA and allowed to adhere to fibrinogen coated coverslips, and cell spreading was measured after 30 min. The adherent cells were fixed and stained with Alexa 568 phalloidin. The areas of cells were measured using ImageJ software, and 300 cells were quantified in each experiment. The error bars represent means ± S.D. of three independent experiments (*, p < 0.001). B, plasmids encoding EGFP alone or EGFP-kindlin-3 constructs were transiently transfected to HEL cells. HEL cells were stimulated with 800 nm PMA, and the transfected cells were stained with PAC-1 to assess αIIbβ3 activation. Flow cytometry was used to measure the MFI of PAC-1 binding. Integrin expression of transfected cells was measured in parallel with 2G12 antibody that binds αIIbβ3 integrin in an activation-independent manner. The error bars represent means ± S.D. of three independent experiments (*, p < 0.001). C, plasmids encoding EGFP alone or EGFP-kindlin-3 constructs were transiently transfected to K562 αIIbβ3 cells. EGFP expression levels were determined by flow cytometry with a transfection efficiency of 70–80%. The transfected cells were stimulated with 800 nm PMA and incubated with 20 μg/ml human fibrinogen, and the flow cytometry was used to measure the MFI of fibrinogen binding. The error bars represent means ± S.E. of three independent experiments (*, p < 0.001). D, mouse RAW 264.7 cells were transiently transfected with plasmids encoding EGFP alone or EGFP-kindlin-3 constructs. The transfected cells were stained with 9EG7 monoclonal antibody to assess β1 integrin activation. Flow cytometry was used to measure the MFI of 9EG7 binding. The error bars represent means ± S.E. of three independent experiments (*, p < 0.001).
Kindlin-3-derived Peptide Inhibits Adhesion and Spreading of HEL Cells and Platelets
A peptide corresponding to the kindlin-3 peptide Phe476–Gly485, designated WT-K3 peptide, and containing the candidate phosphosites and a scrambled peptide, designated Scr-K3, were synthesized with a nine arginine (9R) membrane-penetrating tag at their C terminus to support intracellular delivery (54, 55). To visualize the uptake of WT-K3 and Scr-K3 peptides by cells, each peptide was labeled with fluorescein (Fig. 10A). The capacity of these peptides to penetrate intact HEL cells was evaluated by flow cytometry. Trypan blue quenching was performed to distinguish between extracellular (quenched) and intracellular pools of peptides. WT-K3 and Scr-K3 peptides (10 μm) were incubated with HEL cells for 10 min, and then cells were left untreated or treated with 0.2 mg/ml trypan blue for 5 min. After fixation, samples were analyzed by flow cytometry. The fluorescence signal was detected for all cells analyzed, and it was reduced after quenching by ∼50% (Fig. 10A, blue lines) when compared with unquenched cells (Fig. 10A, green lines). These results indicated efficient and comparable uptake of 9R WT-K3 and Scr-K3 peptides into the HEL cells. Their uptake was monitored by flow cytometry, and we first examined the effects of the peptides on integrin-mediated adhesion and spreading in HEL cells plated on immobilized fibrinogen. HEL cells were incubated with 10 μm peptides in the presence of 800 nm PMA, and cells were plated on fibrinogen. After 30 min, the cells were fixed, and actin was visualized with Alexa 568 phalloidin. Cell adhesion was dramatically blunted at 30 min in cells treated with WT-K3 peptide (Fig. 10B), whereas Scr-K3 peptide did not have an effect on adhesion when compared with control cells (Fig. 10B). Cell area was determined by measuring 300 cells/peptide at 30 min and WT-K3 peptide inhibited cell spreading by ∼50% when compared with cells treated with Scr-K3 peptide and control cells (Fig. 10C).
FIGURE 10.

Kindlin-3 peptide blunts adhesion and spreading in HEL cells and platelets. A, ability of peptides to enter HEL cells was evaluated by flow cytometry with trypan blue used to distinguish between extracellular and intracellular pools of peptides. Peptides (10 μm) were incubated with HEL cells for 10 min, and the fluorescence signal was reduced after quenching by ∼50% (blue lines) compared with the unquenched signal (green lines). B, HEL cells were treated with 10 μm Scr-K3 peptide or WT-K3 peptide for 5 min. The cells were treated with 800 nm PMA, allowed to adhere to immobilized fibrinogen for 30 min, stained with and Alexa 568-phalloidin, and counted (*, p < 0.001). The error bars are means ± S.E. of three independent experiments. C, HEL cells were treated with 10 μm peptides followed by 800 nm PMA and allowed to adhere to fibrinogen-coated coverslips, and cell spreading was measured after 30 min. The adherent cells were fixed and stained with Alexa 568 phalloidin. The cell areas were measured using ImageJ software, and 300 cells were quantified in each experiment (*, p < 0.001). The error bars represent means ± S.E. of three independent experiments. D, representative traces showing the aggregation kinetics of human platelets treated with 10 μm kindlin-3 peptides in the absence or presence of apyrase. 1 unit of human thrombin was used as a positive control. Platelets were aggregated for 7 min in 37 °C. E, surface expression levels of P-selectin were evaluated in human platelets treated with kindlin-3 peptides. Human platelets were left untreated (resting platelets) or treated with 10 μm kindlin-3 peptides. 1 unit of thrombin was used as a positive control. Flow cytometry was used to measure the MFI of P-selectin antibody binding (*, p < 0.001). F, human platelets were pretreated with 10 μm peptides in the absence or presence of apyrase and plated on fibrinogen for 30 min, then fixed and stained with Alexa 568 phalloidin and counted (*, p < 0.001). The error bars are means ± S.E. of two independent experiments.
Next, we sought to determine whether the WT-K3 peptide affected platelet responses. Evidence for the functional activity of WT K3 peptide was first sought in platelet aggregation studies. Surprisingly, addition of 10 μm WT-K3 peptide added to resting platelets induced significant platelet aggregation, comparable with that induced by thrombin, whereas Scr-K3 peptide did not have any effect (Fig. 10D). These differential effects of the WT-K3 peptide and Scr-K3 peptide on platelet aggregation were reproducible with three different donors. However, the observed response was blocked by the ADP scavenger, apyrase, indicating that the effects arose from released ADP, either by induction of secretion or by lysis of some platelets (Fig. 10D). To assess whether WT-K3 peptide induced α-granule secretion in platelets, P-selectin surface expression upon treatment with membrane-permeable peptide platelet suspensions was measured with fluorescently labeled P-selectin antibody. Treatment with 1 unit of human thrombin served as a positive control and stimulated an ∼8-fold increase in P-selectin surface expression when compared with unstimulated platelets (Fig. 10E). Scr-K3 (10 μm) peptide induced a slight but not statistically significant increase in P-selectin surface expression. However, 10 μm WT-K3 peptide increased P-selectin surface expression ∼4-fold when compared with resting platelets. Accordingly, in testing the effects of the peptides in platelet adhesion assays, we also included 5 μm apyrase to degrade released ADP. Platelets were pretreated with 10 μm WT-K3 or Scr-K3 peptide in the absence or presence of apyrase, were plated on fibrinogen for 30 min, and then fixed and stained with Alexa 568 phalloidin. There was no effect on platelet adhesion upon treatment with Scr-K3 peptide when compared with control platelets (Fig. 10F). Treatment with apyrase had no effect on adhesion of platelets incubated with the scrambled peptide (Fig. 10F); adhesion of platelets treated with WT-K3 peptide in the presence of apyrase was significantly blunted when compared with control or Scr-K3 peptide-treated platelets (Fig. 10F).
DISCUSSION
Kindlins play an indispensable role in integrin activation, but the mechanisms by which they have an impact on integrin activation remain unresolved. Despite their sequence similarity, kindlin-3 fails to activate αIIbβ3 in CHO cells where kindlin-1 and kindlin-2 exhibit coactivator function. Recently, Kasirer-Friede et al. (56) were able to show an effect of kindlin-3 on integrin activation in CHO cells coexpressing αIIbβ3 integrin, PAR1, adhesion and degranulation adaptor protein, mouse talin, and kindlin-3. Association of the adaptor protein with αIIbβ3 integrin and talin enabled agonist-dependent, kindlin-3-induced modest αIIbβ3 integrin activation (56). In this study, we have developed HEL cells as a transfectable model and have used human platelets to show that a post-translational modification of kindlin-3 might regulate its agonist-induced activity toward integrins. HEL cells are well established hematopoietic cell model to study integrin αIIbβ3 function and signaling (33, 34). The work from different laboratories established agonist-dependent αIIbβ3 integrin activation in HEL cells, measured by soluble ligand binding and increased adhesion and spreading on integrin substrates, with αIIbβ3 integrin being a prominent component of focal adhesions (35, 36). Although HEL cells have been used broadly and are supported by our studies as an excellent model for αIIbβ3-dependent functions, it is noted that, unlike platelets that adhere to fibrinogen in the absence of agonist stimulation, HEL cells require agonist stimulation to adhere to substratum (this study and see Refs. 36, 41–43). This difference may depend on the high density of αIIbβ3 on platelets (57) and the release of ADP from dense granules, which activates the integrin (58).
Here, we show that kindlin-3 is highly expressed in HEL cells (Fig. 1), kindlin-3 colocalizes with β3 integrin in focal adhesions during the cell spreading on fibrinogen (Fig. 2), kindlin-3 knockdown inhibits adhesion and spreading (Fig. 3), and both kindlin-3 and talin contribute to agonist-induced integrin function in HEL cells (Fig. 4). Of note, using PSGL-1 chimeras, kindlin-3 was found to utilize the same residues in the integrin β3 subunit as kindlin-2 uses in CHO cells (44). Point mutations and kindlin core spanning residues 749–762 of β3 integrin CT (44) were equally effective in blunting cell spreading. The β3 CT (Δ748), which only expresses the talin but not the kindlin-binding site, also significantly inhibited cell spreading. These data are consistent with the contribution of both talin and kindlin-3 to integrin-mediated functions in HEL cells.
In vitro studies using purified kindlins and integrin cytoplasmic tails have showed their direct interaction by various approaches (30, 44, 59, 60). However, in both HEL cells and platelets, the agonist stimulated was needed to demonstrate substantial interaction by coimmunoprecipitation approaches. Agonist stimulation was found to be required for the association of kindlin-3 with β2-containing integrins in T cells (28, 61). Taken together, these data suggest agonist stimulation is a common mechanism in promoting association of kindlin-3 with integrins.
Phosphorylation of talin and β3 CT itself has been proposed to serve as a key regulator of integrin-mediated functions (30, 62, 63), with β3 phosphorylation decreasing integrin/talin and integrin/kindlin interactions and phosphorylation of talin controlling talin turnover and cell migration. In our study using mass spectrometry sequencing, we identified agonist-induced kindlin-3 phosphorylation in HEL cells and platelets. PMA in HEL cells and thrombin in platelets promoted kindlin-3-β3 integrin association and at the same time promoted kindlin-3 phosphorylation on Thr482 and/or Ser484 (Fig. 5).
The identified phosphosite resides in a highly variable region of kindlin-3, and at least one of the residues, Thr482 or Ser484, is evolutionarily conserved in all kindlin-3 orthologs analyzed (Fig. 7), including very distant species of Takifugu rubripes (torafugu) and Danio rerio (zebrafish), consistent with the importance of these phosphosites in kindlin-3 function. T482A/S484A kindlin-3 mutant impeded soluble ligand binding and cell spreading in HEL cells to a level comparable with Q597A/W598A kindlin-3 mutant, which is unable to bind integrin with high affinity (Fig. 9). Comparison of T482A/S484A mutant and wild-type kindlin-3 in other cell types, namely K562 αIIbβ3 cells and mouse macrophage cell line RAW 267.4, allowed us to extend our findings to another model cell type expressing αIIbβ3 integrin and to the regulation of β1 integrin activation. The K562 αIIbβ3 cells that we used express talin (Fig. 1) but can only activate their integrin upon PMA stimulation when kindlin-3 is exogenously expressed. In this kindlin-3 null background, T482A/S484A kindlin-3 failed to activate integrin (Fig. 9), despite the expression levels comparable with wild-type kindlin-3, emphasizing further the pivotal role of phosphosites. RAW 267.4 cells had been used previously to demonstrate a role for kindlin-3 in β1 integrin activation (19). In our experiments, we used 9EG7 monoclonal antibody, which recognizes only activated β1 integrin and showed that T482A/S484A mutant-transfected RAW cells showed virtually no increase in 9EG7 antibody binding, comparable with Q597A/W598A kindlin-3 and vector control, whereas wild-type kindlin-3 was effective in β1 integrin activation. These data indicate the importance of Thr482 and Ser484 also in β1 integrin activation, suggesting a general mechanism for phosphorylation of kindlin-3-induced integrin activation. Supporting this conclusion, recent phosphoproteomic analyses identified Thr482 phosphorylation in T cells upon T cell receptor stimulation with anti-CD3 antibody (64) and Ser484 as a target of phosphorylation upon platelet activation by thrombin (65). In rat kindlin-3, Thr but not an Ser is present, whereas in mouse kindlin-3, Ser but not Thr is present. Thus, phosphorylation may be dependent on the species and cell type.
Mutation of Thr482 and Ser484 affects both inside-out (integrin activation) and outside-in (cell spreading) signaling, but it does not affect the binding of kindlin-3 to the β3 integrin. One possible explanation is that phosphorylation of Thr482 or Ser484 in this sequence may create a binding site(s) for yet unidentified kindlin-3 partners. The T482A/S484A mutant acted as dominant negative in adhesion and spreading assays, suggesting that it may compete with endogenous kindlin-3 for integrin binding and at the same time prevent an endogenous protein from binding to kindlin-3. In peptide-based functional assays, where cell-permeable peptides were introduced into HEL cells and platelets, the kindlin-3-derived peptide was very effective in inhibiting cell spreading and adhesion (Fig. 10), consistent with this hypothetical kindlin-3 binding partner. Interestingly, kindlin-3 peptide caused α-granule secretion, as assessed by an increase in surface expression of P-selectin and subsequent platelet aggregation, and this effect was abolished with apyrase treatment, an ADP scavenger (Fig. 10, D and E). The kindlin-3 peptide significantly increased platelet adhesion to immobilized fibrinogen, and this effect was abolished completely with apyrase treatment, with very few platelets adhering, when compared with control or scramble peptide. This observation suggests that this region of kindlin-3 may be involved in linking kindlin-3 to the cytoskeleton of HEL cells and platelets, and this interaction may regulate granule secretion in activated platelets.
We noted that thrombin induced phosphorylation of kindlin-3, and the extent of this phosphorylation was increased in the presence of EDTA (Fig. 6D), which would inhibit platelet aggregation. This effect is somewhat surprising, but several explanations can be considered. Most likely, the phosphorylation of kindlin-3 may be followed by rapid dephosphorylation. PKC-induced phosphorylation events, as we have shown kindlin-3 to be a PKC phosphorylation target (Fig. 6C), are often followed by rapid activation of phosphatases (38). Time course experiments will need to be undertaken to test this possibility. Another possibility is that binding of fibrinogen released from platelets to αIIbβ3 that occurs in the presence of divalent ions, but is prevented by EDTA, may blunt the phosphorylation of kindlin-3. There may also be decreased recovery of kindlin-3 from aggregated platelets, perhaps due to incorporation into the insoluble cytoskeleton, and we present some evidence consistent with this possibility in Fig. 5C, although we do not yet know whether it is phosphorylated kindlin-3 that selectively associates with the cytoskeleton.
In summary, we identified a novel mechanism of regulating kindlin-3 function and integrin activation in hematopoietic cells. Our combined data provide evidence that kindlin-3 phosphorylation is crucial for effective integrin activation in different cell models and platelets and provide an explanation for functional differences between kindlin-3 and the two other kindlin family members.
Acknowledgments
We thank Judy Drazba from Cleveland Clinic Imaging Core for assistance with confocal microscopy, Cathy Shemo from Cleveland Clinic Flow Core for help with flow cytometry analysis, and Belinda Willard from Cleveland Clinic Mass Spectrometry Core for help with sequencing and analysis of kindlin-3 modifications.
This work was supported, in whole or in part, by National Institutes of Health Grants P01HL073311 and R01 HL096062 from the NHLBI.
- CT
- cytoplasmic tail
- EGFP
- enhanced green fluorescent protein
- HEL
- human erythroleukemia
- PFA
- paraformaldehyde
- PMA
- phorbol 12-myristate 13-acetate
- PSGL-1
- P-selectin glycoprotein ligand-1
- TIRF
- total internal reflection fluorescence
- TPO
- thrombopoietin
- MFI
- mean fluorescence intensity
- PE
- phycoerythrin.
REFERENCES
- 1. Qin J., Vinogradova O., Plow E. F. (2004) Integrin bidirectional signaling: a molecular view. PLoS Biol. 2, e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ma Y. Q., Qin J., Plow E. F. (2007) Platelet integrin α(IIb)β(3): activation mechanisms. J. Thromb. Haemost. 5, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 3. Morse E. M., Brahme N. N., Calderwood D. A. (2014) Integrin cytoplasmic tail interactions. Biochemistry 53, 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tu Y., Wu S., Shi X., Chen K., Wu C. (2003) Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 113, 37–47 [DOI] [PubMed] [Google Scholar]
- 5. Ussar S., Wang H. V., Linder S., Fässler R., Moser M. (2006) The kindlins: subcellular localization and expression during murine development. Exp. Cell Res. 312, 3142–3151 [DOI] [PubMed] [Google Scholar]
- 6. Shi X., Ma Y. Q., Tu Y., Chen K., Wu S., Fukuda K., Qin J., Plow E. F., Wu C. (2007) The MIG-2/integrin interaction strengthens cell-matrix adhesion and modulates cell motility. J. Biol. Chem. 282, 20455–20466 [DOI] [PubMed] [Google Scholar]
- 7. Rogalski T. M., Mullen G. P., Gilbert M. M., Williams B. D., Moerman D. G. (2000) The UNC-112 gene in Caenorhabditis elegans encodes a novel component of cell-matrix adhesion structures required for integrin localization in the muscle cell membrane. J. Cell Biol. 150, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calderwood D. A., Zent R., Grant R., Rees D. J., Hynes R. O., Ginsberg M. H. (1999) The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071–28074 [DOI] [PubMed] [Google Scholar]
- 9. Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T., Plow E., Qin J. (2002) A structural mechanism of integrin α(IIb)β(3) “inside-out” activation as regulated by its cytoplasmic face. Cell 110, 587–597 [DOI] [PubMed] [Google Scholar]
- 10. García-Alvarez B., de Pereda J. M., Calderwood D. A., Ulmer T. S., Critchley D., Campbell I. D., Ginsberg M. H., Liddington R. C. (2003) Structural determinants of integrin recognition by talin. Mol. Cell 11, 49–58 [DOI] [PubMed] [Google Scholar]
- 11. Tadokoro S., Shattil S. J., Eto K., Tai V., Liddington R. C., de Pereda J. M., Ginsberg M. H., Calderwood D. A. (2003) Talin binding to integrin β tails: a final common step in integrin activation. Science 302, 103–106 [DOI] [PubMed] [Google Scholar]
- 12. Nieswandt B., Moser M., Pleines I., Varga-Szabo D., Monkley S., Critchley D., Fässler R. (2007) Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med. 204, 3113–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma Y. Q., Qin J., Wu C., Plow E. F. (2008) Kindlin-2 (Mig-2): a co-activator of β3 integrins. J. Cell Biol. 181, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montanez E., Ussar S., Schifferer M., Bösl M., Zent R., Moser M., Fässler R. (2008) Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 22, 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jobard F., Bouadjar B., Caux F., Hadj-Rabia S., Has C., Matsuda F., Weissenbach J., Lathrop M., Prud'homme J. F., Fischer J. (2003) Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum. Mol. Genet. 12, 925–935 [DOI] [PubMed] [Google Scholar]
- 16. Siegel D. H., Ashton G. H., Penagos H. G., Lee J. V., Feiler H. S., Wilhelmsen K. C., South A. P., Smith F. J., Prescott A. R., Wessagowit V., Oyama N., Akiyama M., Al Aboud D., Al Aboud K., Al Githami A., et al. (2003) Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am. J. Hum. Genet. 73, 174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ussar S., Moser M., Widmaier M., Rognoni E., Harrer C., Genzel-Boroviczeny O., Fässler R. (2008) Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 4, e1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dowling J. J., Gibbs E., Russell M., Goldman D., Minarcik J., Golden J. A., Feldman E. L. (2008) Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ. Res. 4, 423–431 [DOI] [PubMed] [Google Scholar]
- 19. Moser M., Nieswandt B., Ussar S., Pozgajova M., Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 20. Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M., Fässler R. (2009) Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305 [DOI] [PubMed] [Google Scholar]
- 21. Kuijpers T. W., van de Vijver E., Weterman M. A., de Boer M., Tool A. T., van den Berg T. K., Moser M., Jakobs M. E., Seeger K., Sanal O., Unal S., Cetin M., Roos D., Verhoeven A. J., Baas F. (2009) LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 113, 4740–4746 [DOI] [PubMed] [Google Scholar]
- 22. Svensson L., Howarth K., McDowall A., Patzak I., Evans R., Ussar S., Moser M., Metin A., Fried M., Tomlinson I., Hogg N. (2009) Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Malinin N. L., Zhang L., Choi J., Ciocea A., Razorenova O., Ma Y.-Q., Podrez E. A., Tosi M., Lennon D. P., Caplan A. I., Shurin S. B., Plow E. F., Byzova T. V. (2009) A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ehlers R., Ustinov V., Chen Z., Zhang X., Rao R., Luscinskas F. W., Lopez J., Plow E., Simon D. I. (2003) Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibα. J. Exp. Med. 198, 1077–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meller J., Malinin N. L., Panigrahi S., Kerr B. A., Patil A., Ma Y., Venkateswaran L., Rogozin I. B., Mohandas N., Ehlayel M. S., Podrez E. A., Chinen J., Byzova T. V. (2012) Novel aspects of Kindlin-3 function in humans based on a new case of leukocyte adhesion deficiency III. J. Thromb. Haemost. 10, 1397–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bialkowska K., Ma Y. Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X., Malinin N., Qin J., Byzova T., Plow E. F. (2010) The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sossey-Alaoui K., Pluskota E., Davuluri G., Bialkowska K., Das M., Szpak D., Lindner D. J., Downs-Kelly E., Thompson C. L., Plow E. F. (2014) Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis. FASEB J. 28, 2260–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y., Malinin N. L., Meller J., Ma Y., West X. Z., Bledzka K., Qin J., Podrez E. A., Byzova T. V. (2012) Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J. Biol. Chem. 287, 40012–40020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Z., Chen X., Zhi H., Gao J., Bialkowska K., Byzova T. V., Pluskota E., White G. C., 2nd, Liu J., Plow E. F., Ma Y. Q. (2014) Direct interaction of kindlin-3 with integrin αIIbβ3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 1961–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bledzka K., Bialkowska K., Nie H., Qin J., Byzova T., Wu C., Plow E. F., Ma Y. Q. (2010) Tyrosine phosphorylation of integrin β3 regulates kindlin-2 binding and integrin activation. J. Biol. Chem. 285, 30370–30374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimrin A. B., Eisman R., Vilaire G., Schwartz E., Bennett J. S., Poncz M. (1988) Structure of platelet glycoprotein IIIa. A common subunit for two different membrane receptors. J. Clin. Invest. 81, 1470–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poncz M., Eisman R., Heidenreich R., Silver S. M., Vilaire G., Surrey S., Schwartz E., Bennett J. S. (1987) Structure of the platelet membrane glycoprotein IIb. Homology to the α subunits of the vitronectin and fibronectin membrane receptors. J. Biol. Chem. 262, 8476–8482 [PubMed] [Google Scholar]
- 33. Williams A. G., Woolkalis M. J., Poncz M., Manning D. R., Gewirtz A. M., Brass L. F. (1990) Identification of the pertussis toxin-sensitive G proteins in platelets, megakaryocytes, and human erythroleukemia cells. Blood 76, 721–730 [PubMed] [Google Scholar]
- 34. Brass L. F., Manning D. R., Williams A. G., Woolkalis M. J., Poncz M. (1991) Receptor and G protein-mediated responses to thrombin in HEL cells. J. Biol. Chem. 266, 958–965 [PubMed] [Google Scholar]
- 35. Zauli G., Bassini A., Vitale M., Gibellini D., Celeghini C., Caramelli E., Pierpaoli S., Guidotti L., Capitani S. (1997) Thrombopoietin enhances the αIIbβ3-dependent adhesion of megakaryocytic cells to fibrinogen or fibronectin through PI 3 kinase. Blood 89, 883–895 [PubMed] [Google Scholar]
- 36. Boudignon-Proudhon C., Patel P. M., Parise L. V. (1996) Phorbol ester enhances integrin α IIb β3-dependent adhesion of human erythroleukemic cells to activation-dependent monoclonal antibodies. Blood 87, 968–976 [PubMed] [Google Scholar]
- 37. Woods V. L., Jr., Oh E. H., Mason D., McMillan R. (1984) Autoantibodies against the platelet glycoprotein IIb/IIIa complex in patients with chronic ITP. Blood 63, 368–375 [PubMed] [Google Scholar]
- 38. Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 [DOI] [PubMed] [Google Scholar]
- 39. Plow E. F., Ginsberg M. H. (1981) Specific and saturable binding of plasma fibronectin to thrombin-stimulated human platelets. J. Biol. Chem. 256, 9477–9482 [PubMed] [Google Scholar]
- 40. Fox J. E., Lipfert L., Clark E. A., Reynolds C. C., Austin C. D., Brugge J. S. (1993) On the role of the platelet membrane skeleton in mediating signal transduction. Association of GP IIb-IIIa, pp60c-src, pp62c-yes, and the p21ras GTPase-activating protein with the membrane skeleton. J. Biol. Chem. 268, 25973–25984 [PubMed] [Google Scholar]
- 41. Ylänne J., Cheresh D. A., Virtanen I. (1990) Localization of β1, β3, α5, αv, and αIIb subunits of the integrin family in spreading human erythroleukemia cells. Blood 76, 570–577 [PubMed] [Google Scholar]
- 42. Ylänne J., Hormia M., Järvinen M., Vartio T., Virtanen I. (1988) Platelet glycoprotein IIb/IIIa complex in cultured cells. Localization in focal adhesion sites in spreading HEL cells. Blood 72, 1478–1486 [PubMed] [Google Scholar]
- 43. Järvinen M., Ylänne J., Vartio T., Virtanen I. (1987) Tumor promoter and fibronectin induce actin stress fibers and focal adhesion sites in spreading human erythroleukemia (HEL) cells. Eur. J. Cell Biol. 44, 238–246 [PubMed] [Google Scholar]
- 44. Bledzka K., Liu J., Xu Z., Perera H. D., Yadav S. P., Bialkowska K., Qin J., Ma Y. Q., Plow E. F. (2012) Spatial coordination of kindlin-2 with talin head domain in interaction with integrin β cytoplasmic tails. J. Biol. Chem. 287, 24585–24594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phillips D. R., Jennings L. K., Edwards H. H. (1980) Identification of membrane proteins mediating the interaction of human platelets. J. Cell Biol. 86, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kouns W. C., Fox C. F., Lamoreaux W. J., Coons L. B., Jennings L. K. (1991) The effect of glycoprotein IIb-IIIa receptor occupancy on the cytoskeleton of resting and activated platelets. J. Biol. Chem. 266, 13891–13900 [PubMed] [Google Scholar]
- 47. Yacoub D., Théorêt J. F., Villeneuve L., Abou-Saleh H., Mourad W., Allen B. G., Merhi Y. (2006) Essential role of protein kinase Cδ in platelet signaling, αIIbβ3 activation, and thromboxane A2 release. J. Biol. Chem. 281, 30024–30035 [DOI] [PubMed] [Google Scholar]
- 48. Buensuceso C. S., Obergfell A., Soriani A., Eto K., Kiosses W. B., Arias-Salgado E. G., Kawakami T., Shattil S. J. (2005) Regulation of outside-in signaling in platelets by integrin-associated protein kinase Cβ. J. Biol. Chem. 280, 644–653 [DOI] [PubMed] [Google Scholar]
- 49. Soriani A., Moran B., de Virgilio M., Kawakami T., Altman A., Lowell C., Eto K., Shattil S. J. (2006) A role for PKCθ in outside-in α(IIb)β3 signaling. J. Thromb. Haemost. 4, 648–655 [DOI] [PubMed] [Google Scholar]
- 50. Cifuni S. M., Wagner D. D., Bergmeier W. (2008) CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin αIIbβ3 in platelets. Blood 112, 1696–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ward N. E., O'Brian C. A. (1993) Inhibition of protein kinase C by N-myristoylated peptide substrate analogs. Biochemistry 32, 11903–11909 [DOI] [PubMed] [Google Scholar]
- 52. Mattheyses A. L., Simon S. M., Rappoport J. Z. (2010) Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621–3628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakazawa T., Tadokoro S., Kamae T., Kiyomizu K., Kashiwagi H., Honda S., Kanakura Y., Tomiyama Y. (2013) Agonist stimulation, talin-1, and kindlin-3 are crucial for α(IIb)β(3) activation in a human megakaryoblastic cell line, CMK. Exp. Hematol. 41, 79–90 [DOI] [PubMed] [Google Scholar]
- 54. Rich S., Van Nood N., Lee H. M. (1996) Role of α5β1 integrin in TGF-β1-costimulated CD8+ T cell growth and apoptosis. J. Immunol. 157, 2916–2923 [PubMed] [Google Scholar]
- 55. David T., Ohlmann P., Eckly A., Moog S., Cazenave J. P., Gachet C., Lanza F. (2006) Inhibition of adhesive and signaling functions of the platelet GPIb-V-IX complex by a cell penetrating GPIbα peptide. J. Thromb. Haemost. 4, 2645–2655 [DOI] [PubMed] [Google Scholar]
- 56. Kasirer-Friede A., Kang J., Kahner B., Ye F., Ginsberg M. H., Shattil S. J. (2014) ADAP interactions with talin and kindlin promote platelet integrin αIIbβ3 activation and stable fibrinogen binding. Blood 123, 3156–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wagner C. L., Mascelli M. A., Neblock D. S., Weisman H. F., Coller B. S., Jordan R. E. (1996) Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood 88, 907–914 [PubMed] [Google Scholar]
- 58. Gachet C. (2006) Regulation of platelet functions by P2 receptors. Annu. Rev. Pharmacol. Toxicol. 46, 277–300 [DOI] [PubMed] [Google Scholar]
- 59. Kahner B. N., Kato H., Banno A., Ginsberg M. H., Shattil S. J., Ye F. (2012) Kindlins, integrin activation and the regulation of talin recruitment to αIIbβ3. PLoS One 7, e34056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yates L. A., Füzéry A. K., Bonet R., Campbell I. D., Gilbert R. J. (2012) Biophysical analysis of Kindlin-3 reveals an elongated conformation and maps integrin binding to the membrane-distal β-subunit NPXY motif. J. Biol. Chem. 287, 37715–37731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kliche S., Worbs T., Wang X., Degen J., Patzak I., Meineke B., Togni M., Moser M., Reinhold A., Kiefer F., Freund C., Förster R., Schraven B. (2012) CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood 119, 777–785 [DOI] [PubMed] [Google Scholar]
- 62. Huang C., Rajfur Z., Yousefi N., Chen Z., Jacobson K., Ginsberg M. H. (2009) Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 11, 624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Anthis N. J., Haling J. R., Oxley C. L., Memo M., Wegener K. L., Lim C. J., Ginsberg M. H., Campbell I. D. (2009) β integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. J. Biol. Chem. 284, 36700–36710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruperez P., Gago-Martinez A., Burlingame A. L., Oses-Prieto J. A. (2012) Quantitative phosphoproteomic analysis reveals a role for serine and threonine kinases in the cytoskeletal reorganization in early T cell receptor activation in human primary T cells. Mol. Cell. Proteomics 11, 171–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zimman A., Titz B., Komisopoulou E., Biswas S., Graeber T. G., Podrez E. A. (2014) Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS One 9, e84488. [DOI] [PMC free article] [PubMed] [Google Scholar]