

Background: The Golgi retention mechanism of core 2 N-acetylglucosaminyltransferase M (C2GnT-M) is not known.
Results: Keratin 1 retains C2GnT-M in the Golgi by interacting with its cytoplasmic tail via the rod domain.
Conclusion: Keratin 1 is the protein that helps retain C2GnT-M in the Golgi.
Significance: Keratin 1 plays a critical role in the regulation of O-glycosylation pathways.
Keywords: Glycobiology, Glycosyltransferase, Golgi, Keratin, Mucus, Trafficking
Abstract
Core 2 N-acetylglucosaminyltransferase 2/M (C2GnT-M) synthesizes all three β6GlcNAc branch structures found in secreted mucins. Loss of C2GnT-M leads to development of colitis and colon cancer. Recently we have shown that C2GnT-M targets the Golgi at the Giantin site and is recycled by binding to non-muscle myosin IIA, a motor protein, via the cytoplasmic tail (CT). But how this enzyme is retained in the Golgi is not known. Proteomics analysis identifies keratin type II cytoskeletal 1 (KRT1) as a protein pulled down with anti-c-Myc antibody or C2GnT-M CT from the lysate of Panc1 cells expressing bC2GnT-M tagged with c-Myc. Yeast two-hybrid analysis shows that the rod domain of KRT1 interacts directly with the WKR6 motif in the C2GnT-M CT. Knockdown of KRT1 does not affect Golgi morphology but increases the interaction of C2GnT-M with non-muscle myosin IIA and its transportation to the endoplasmic reticulum, ubiquitination, and degradation. During Golgi recovery after brefeldin A treatment, C2GnT-M forms a complex with Giantin before KRT1, demonstrating CT-mediated sequential events of Golgi targeting and retention of C2GnT-M. In HeLa cells transiently expressing C2GnT-M-GFP, knockdown of KRT1 does not affect Golgi morphology but leaves C2GnT-M outside of the Golgi, resulting in the formation of sialyl-T antigen. Interaction of C2GnT-M and KRT1 was also detected in the goblet cells of human colon epithelial tissue and primary culture of colonic epithelial cells. The results indicate that glycosylation and thus the function of glycoconjugates can be regulated by a protein that helps retain a glycosyltransferase in the Golgi.
Introduction
Glycoconjugates play important roles in many biological processes, such as protein folding, innate and acquired immunity, and mucus defense (1, 2). Loss of function of glycoconjugates resulted from altered glycans can lead to many human diseases, such as congenital deficiency in glycosylation, infection, cancer, etc. (1, 3, 4). To develop therapy for these diseased conditions, it is critically important to understand how these glycans are formed. Synthesis of the carbohydrates associated with membrane-bound and secreted glycoconjugates takes place primarily in the Golgi apparatus as catalyzed by glycosyltransferases (GTs)2 (5). In most cases each GT family contains several isozymes that exhibit similar substrate specificity when the activity is measured in vitro. But the results cannot be reproduced in vivo. Despite great progress made in elucidating the in vitro synthetic scheme for most GTs, much more remains to be learned about the in vivo biosynthetic scheme. This is the gap the current study is intended to fill.
GTs are type II membrane proteins consisting of a short cytoplasmic tail (CT) at the N terminus that is followed by a transmembrane domain, a short stem, and then a large catalytic region (5–9). The CT, the only part of the GT molecule exposed to the cytosol, is involved in every intracellular trafficking step of GTs, including endoplasmic reticulum (ER) exit (10), Golgi targeting (11), Golgi retention (12–15), and recycling (16–18). It is also involved in the morphological changes of the Golgi apparatus in response to stress (19, 20). The (R/K)X(R/K) sequence in the CT has been identified as the Sar1-dependent ER exit signal for many Golgi GTs (10). Also, the Golgi targeting of GTs was originally proposed to be a COPII-dependent process (21). However, a recent report showed that this process is independent of COPII (11). This study also demonstrated that Giantin, GM130-GRASP65, and GM130-Giantin are the Golgi targeting sites for GTs in a GT-specific manner. Recently, we also show that in advanced prostate cancer cells, differential Golgi targeting of Galβ1–3GalNAcαSer/Thr:α2–3-sialyltransferase 1 (ST3Gal1) and core 2 N-acetylglucosaminyltransferase-L/1 (C2GnT-L) determines the susceptibility of these cells to Galectin-1-induced apoptosis (22).
The CT-specific Golgi retention proteins have been reported for only few GTs (12–14, 23–25). It has been generally thought that Golgi-to-ER retrograde transport of GTs is a COPI-dependent process (26, 27). However, other reports show that this process is COPI-independent (11, 28–30). We have shown that this step is mediated by non-muscle myosin IIA (NMIIA) (30), and the interaction of the C-terminal cargo binding region of NMIIA with the CT of GTs is responsible for not only the recycling of GTs and the remodeling of the Golgi under normal physiological conditions but also Golgi fragmentation in stressed cells (19, 20) and advanced cancer cells (22).
To date, despite only limited knowledge about the mechanisms underlying the ER exit, Golgi targeting, retention, and recycling of GTs (31), there are even disagreements among them. To gain a comprehensive understanding of these important biological processes and help clarify some of the mechanisms, characterization of the intracellular transportation pathways of more GTs is needed.
Mucins are high molecular weight glycoproteins that serve as a protector of the mucus secretory tissues and a mediator of leukocyte trafficking (2, 32). The functions of mucins reside primarily in the conjugated glycans. Synthesis of these glycans occurs in a template-independent, stepwise manner. Among the many enzymatic steps, the ones that are specified by the core enzymes play a critical regulatory role because many biologically important carbohydrate epitopes are extended from these core structures. For example, the leukocyte trafficking and cancer metastasis are mediated by selectin ligands decorated on core 2 generated by C2GnT-L (33–35). Recently, we extended this concept by showing that cancer metastatic potential also can be regulated by Golgi phosphoprotein 3 (GOLPH3), which is needed for the Golgi retention of C2GnT-L (24). To expand this study, we set out to identify the protein that helps retain in the Golgi another core 2 enzyme C2GnT-M, which is an important GT for the functions of secreted mucins because this enzyme is responsible for the synthesis of all three β6GnTs, including core 2, core 4, and blood group I antigen in secreted mucins (36–39). C2GnT-M deficiency is accompanied by impaired mucosal barrier function, thereby increasing susceptibility to colitis (40) and growth of colon tumor (41).
In the past decade studies have shown that a number of cytoskeleton proteins interact with Golgi membranes and participate in the maintenance of Golgi architecture. Disruption of microtubules or actin filaments leads to Golgi fragmentation and substantial impairment of intracellular trafficking (42, 43). Notably, Golgi retention of some galactosyltransferases is determined by direct link between their CT and cytoskeleton proteins (14, 23). Another interesting aspect is the relationship between the Golgi and intermediate filaments (IFs). For example, the tight association of vimentin with Golgi proteins has been found to be critical for intracellular transport of cholesterol (44, 45). The cytokeratins, which include type I, acidic keratins (9–20), and type II, neutral-basic keratins (1–8), comprise the largest epithelium-specific IFs. The presence of cytokeratins, the most abundant members of IFs, in the Golgi has been shown to regulate Golgi stability, specifically in cooperation with F-actin (46, 47). In fibroblasts, keratin 6a is detected in the complex with Golgi-specific protein kinase C (PKCϵ) in the common assembly with NMIIA, actin, and β-COP (48).
In this study we report the identification of keratin 1 (KRT1) as the protein that helps retain C2GnT-M in the Golgi by interacting directly with the CT of this enzyme. Loss of KRT1 leads to NMIIA-mediated redistribution of C2GnT-M from Golgi to ER followed by proteasomal degradation. As a result, T-antigen is replaced with sialyl-T antigen.
EXPERIMENTAL PROCEDURES
Cell Culture, Drug Treatment, and Antibodies (Abs)
Panc1 cells expressing bC2GnT-M tagged with c-Myc at the C terminus (Panc1-bC2GnT-M-c-Myc) were prepared as previously described (49). Human colon epithelial cells obtained from a 45-year old white male at passage 2 (Celprogen, Torrance, CA; lot #1314111-016) were cultured on appropriate Human Colon Primary Cell Culture Extracellular Matrix in the Human Colon Complete growth medium containing 10% fetal bovine serum according to the manufacturer's instructions (Celprogen). Cells were grown at 37 °C under a humidified 5% CO2 atmosphere. These primary cells were 95% positive for the epithelial marker, keratin 19 (Celprogen), and expanded 1:5 before being used for the experiments at ∼60% confluence. Brefeldin A (EMD Chemicals) was added to the culture cells at a final concentration of 36 μm, which was followed by incubation at 37 °C for the time periods given in each experiment. Cells treated with a corresponding amount of DMSO served as controls. To study Golgi recovery after brefeldin A treatment, cells were washed at least 3 times with prewarmed drug-free medium followed by incubation under regular culture conditions for various durations as described in each experiment. The primary Abs were (a) rabbit polyclonal non-muscle myosin IIA (Sigma, M8064), c-Myc (Santa Cruz Biotechnology; sc-789), Giantin, protein disulfide isomerase (PDI), ubiquitin, and KRT1 (Abcam; ab24586, ab13507, ab7780, and ab93652, respectively), (b) mouse monoclonal c-Myc, cytokeratin 7, 8, and 10 (Santa Cruz Biotechnology; sc-40, sc-23876, sc-374274, and sc-53252, respectively), β-actin (Sigma, M4821), Giantin, GFP, and core 1 β3-galactosyltransferase ( (C1GalT1) (Abcam; ab37266, ab1218, and ab57492, respectively); mouse polyclonal ST3Gal1 (Abnova, H00006482-B01P), mannosyl α-1,3-glycoprotein β-1,2-N-acetyl-glucosaminyltransferase (MGAT1) (Abcam, ab167365), and (c) goat polyclonal γ-tubulin (Santa Cruz Biotechnology, sc-7396), C2GnT-M (Everest, EB08257). The secondary antibodies were (a) HRP-conjugated donkey anti-rabbit, donkey anti-mouse and donkey anti-goat Abs for Western blotting (Jackson ImmunoResearch) and (b) donkey anti-goat DyLight594, donkey anti-mouse DyLight488, donkey anti-rabbit DyLight594 and DyLight594-conjugated IgG fraction monoclonal mouse anti-biotin (Jackson ImmunoResearch) for immunofluorescence.
Co-immunoprecipitation (Co-IP) and Transfection
For identification of proteins in the complexes pulled down by co-IP, confluent cells grown in a T75 flask were washed 3 times with 6 ml of PBS each, harvested by trypsinization, and neutralized with soybean trypsin inhibitor at a 2× weight of trypsin. After washing 3 times with PBS, the cells were lysed with 1.5 ml of a lysis buffer that contained 50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40 (w/w), and 1% (v/v) of a mammalian protease inhibitor mixture (Sigma). Co-IP was performed on cell lysate using the Pierce co-immunoprecipitation kit (Pierce) according to the manufacturer's instructions. Briefly, 2.5 mg of protein were precleared by incubation with 100 μl of control agarose resin to minimize nonspecific binding. Fifty μg of antigen-specific Ab were covalently coupled onto an amine-reactive resin. The precleared lysates were subsequently incubated with antibody-coupled beads overnight at 4 °C. The immunoprecipitates were collected by centrifugation followed by boiling under high (5% of β-mercaptoethanol) reducing conditions. Non-specific IgG antibodies were used as a nonspecific control. Pools of three siRNAs targeting KRT1, -7, -8, and 10 (keratin 1, 7, 8, and 10, respectively), ST3Gal1, or scrambled ON-TARGETplus smartpool siRNAs were obtained from Santa Cruz Biotechnology. Cells were transfected with 75–100 nm siRNAs using Lipofectamine RNAi MAX reagent (Life Technologies). After culturing for 2–3 days, cells were analyzed for specific proteins by Western blotting. Proteins were separated on SDS-PAGE on mini-gels with various % gel specified for each experiment (Bio-Rad). Western blotting was developed using HRP-coupled antibodies and Thermo Scientific SuperSignal West Pico Chemiluminescent Substrate reagents and then exposed to BioExpress Blue Basic Autorad chemiluminescence film. The bands on the autoradiography films were digitized by scanning with ScanJet 6200C (Hewlett Packard) driven by Adobe Photoshop.
Proteomics Analysis of Electrophoretically Separated Proteins
The SDS-PAGE separated and Coomassie Blue-stained bands obtained from the immunoprecipitates were excised, trypsinized, and treated with 60% acetonitrile containing 0.1% trifluoroacetic acid to extract the peptides. The peptides were analyzed by the Thermo Fisher LCQ Deca Plus system liquid chromatograph ion trap mass spectrometry with peaks subjected to MS/MS fragmentation by collision-induced dissociation. Full mass spectra of parent ions and MS/MS fragmentation data were processed using BioWorks 3.2 software based on SEAQUEST algorithm. Proteomics analysis of these protein bands was carried out by the Mass Spectrometry and Proteomics Core Facility at the University of Nebraska Medical Center.
Plasmid Construction and Transient Transfection in HeLa Cells
The wild-type and mutant hC2GnT-M cDNAs were cloned by PCR and inserted into the EGFP-N1 eukaryotic expression vector (Clontech). The coding region of the hC2GnT-M gene (GenBankTM accession number NM_004751) was PCR-amplified using the primer set of 5′-ATctcgagCGCCACCATGGTTCAATGGAAGAGACTCTGCCAGCTGCATTACTTG-3′and 5′-ATagatctCCTCCAAGTTCAGTCCCATAGATGGCCTTATAAC-3′ and ligated into XhoI and BamHI sites of the EGFP-N1 vector to generate hC2GnT-M-pEFGP-N1. The integrity and orientation of the plasmids were confirmed by restriction digestion and sequencing. Transfections of HeLa cells were carried out using the Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol and analyzed after 2–3 days culture in DMEM (Sigma) plus 10% FBS and antibiotics.
Isolation of Golgi Membrane Fractions by Sucrose Density Gradient Centrifugation
Preparation of Golgi membranes was performed by the methods described previously (19). Panc1-bC2GnT-M-c-Myc cells from 10–12 75-cm2 culture flasks were harvested by PBS containing 0.5× protease inhibitors (1.2 ml per flask). After centrifugation for 5 min at 1000 rpm and 4 °C, the pellet was resuspended in 3 ml of homogenization buffer (0.25 m sucrose, 3 mm imidazole, 1 mm Tris-Cl, pH 7.4, 1 mm EDTA). The cells were homogenized by drawing ∼30 times through a 25-gauge needle until the ratio between unbroken cells and free nuclei reached 20%:80%. The postnuclear supernatant was obtained by centrifugation at 2500 rpm and 4 °C for 3 min, and then the supernatant was adjusted to 1.4 m sucrose by the addition of ice-cold 2.3 m sucrose containing 10 mm Tris-HCl, pH 7.4. Next, 1.2 ml of 2.3 m sucrose was placed at the bottom of tube, which was sequentially overlaid with 1.2 ml of the adjusted supernatant, 1.2 ml of 1.2 m, and 0.5 ml of 0.8 m sucrose (10 mm Tris-HCl, pH 7.4). The gradients were centrifuged for 3 h at 36,000 rpm (4 °C) in a SW40 rotor (Beckman Coulter). The turbid band at the 0.8 m/1.2 m sucrose interface, which contains Golgi membranes, was harvested in ∼500-μl fractions by syringe puncture. The fraction at ∼1.0–1.4 mg of protein/ml was used for the experiments described under “Results.”
Confocal Immunofluorescence Microscopy
Cells were grown overnight on coverslips placed in a 6-well plate, washed twice with PBS, and immediately fixed with 4% paraformaldehyde, PBS for 30 min at room temperature. After incubation with primary Abs at 37 °C for 1 h followed by washing with PBST three times, cells were stained with DyLight secondary Abs. After the final wash in PBST, cells were mounted in ProLong Gold antifade reagent with or without DAPI (Invitrogen).
For peanut agglutinin (PNA)-lectin staining using biotinylated PNA, before the addition of the lectin, the cells were incubated for 15 min at room temperature with 1.7 μm streptavidin (Sigma) diluted in PBST. After washing, cells were treated for 1 h at room temperature with 2 mm biotin (Sigma) dissolved in PBST followed by washing and incubation for 3 h at room temperature with 10 μg/ml of biotinylated PNA-lectins (EY Laboratories). After washing, cells were incubated for another hour at room temperature with DyLight594 anti-biotin Abs.
Normal human colon tissue arrays were purchased from US Biomax (Rockville, MD). After deparaffinization, sections were rinsed with water and then treated with 10 mm Tris, 1 mm EDTA, 0.05% Tween 20 at pH 9 for 20 min in a microwave for 20 min to retrieve the antigen. After blocking with 1% donkey serum for 1 h at room temperature, the sections were incubated with anti-C2GnT-M and anti-KRT1 Abs for 1 h at room temperature followed by treatment for 1 h at room temperature with DyLight secondary Abs.
All slides were examined under a Zeiss 510 Meta Confocal Laser Scanning Microscope performed at the Advanced Microscopy Core Facility of the University of Nebraska Medical Center. Fluorescence signal was detected using an emission filter of a 505–550-nm band pass for green and a 575–615-nm band pass for red. Images were analyzed using Zeiss 510 and ZEN 2009 software. For some figures, image analysis was performed using Adobe Photoshop and ImageJ. The average fluorescence intensity was measured as the mean ± S.D. of integrated fluorescence intensity (in arbitrary units (a.u.)) or total fluorescence intensity (in pixels) measured for every cell individually. The Golgi and ER regions were determined using the intensity of Giantin and PDI staining, respectively.
Cloning, Expression, and Purification of Recombinant Human KRT1 in HEK293 Cells
Human RNAs isolated from HEK293 cells were used for PCR cloning of the cDNA of KRT1 (GenBankTM accession no. NM_006121). The forward primer 5′-ATAGAATTCAGCAGCGAGAACCTGTACTTTCAGGGCATGAGTCGACAGTTTAGTTCCAGG-3′ and the reverse primer 5′-ATAAAGCTTATCATCTGGTTACTCCGGAATAAGTGG-3′ were used. The EcoRI and HindIII restriction sites are underlined. The purified PCR product (1982 bp) was cloned into the 3′ end of the GFP cDNA flanked by a tobacco etch virus (TEV) protease cleavage site in the pXLGNterm-TCMhisAviGfpTEV (a gift from the Kelley Moremen Laboratory, Complex Carbohydrate Research Center) at these two sites. pXLGNterm-TCMhisAviGfpTEV is a mammalian expression vector with a CMV promoter and an N-terminal TEV-cleavable signal sequence, His, Avi, and GFP tags. The final coding region encompasses the N-terminal signal sequence from the Trypanosoma cruzi lysosomal α-mannosidase, His tag, AviTag, GFP, the recognition sequence of TEV protease, and KRT1 coding sequences. The TEV protease cleavage site (/) was introduced into a nucleotide sequence (italicized above), GAGAACCTGTACTTTCAGGGC, that encodes Glu-Asn-Leu-Tyr-Phe-Gln/Gly downstream of GFP. This pXLG-NtermTCMHisAviGfp TEV vector carrying the 1982-bp insert was transformed into high efficiency competent Escherichia coli DH5 cells (New England Biolabs, Ipswich, MA). The integrity and orientation of the construct were confirmed by restriction digestion and sequencing.
Human GFP-KRT1 was expressed as a soluble, secreted fusion protein by transient transfection of HEK293 cells. The HEK293 cell line was maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. Cells were transfected with sequence-verified vector and the construct containing the 1982 bp KRT1 cDNA using Lipofectamine 2000 (Life Technologies). Transfection was performed according to the manufacturer's protocol. Culture medium was collected every 48 h and was clarified by centrifugation at 4000 rpm for 5 min. The medium was adjusted to contain 20 mm imidazole and loaded onto a column containing 1 ml of Ni-NTA resin (Qiagen, Valencia, CA) equilibrated with phosphate buffer saline containing 20 mm imidazole, pH 7.4. After the sample was loaded, the column was washed with 15 ml of the equilibration buffer and eluted with 1 ml of equilibration buffer containing 250 mm imidazole. About 150-μl fractions were collected, and fractions containing high protein concentrations, verified by A280 and PAGE/Coomassie, were pooled. A 10-ml initial culture medium collected yielded 900 μg of purified protein at a final concentration of 2 μg/μl.
We then utilized a His-tagged TEV protease (a gift from Tahir Tahirov's Laboratory, University of Nebraska Medical Center) to cleave the purified GFP-KRT1 and remove both the His-tagged TEV protease and the TCM-Histag-AviTag-GFP domains by absorption to the Ni-NTA resin. GFP KRT1 eluted in PBS containing 250 mm imidazole was subsequently passed through a Sephadex-G50 gel filtration column (GE Healthcare) equilibrated in PBS. For cleaving 500 μg of the purified GFP-KRT1 protein, 25 μg of recombinant His6-TEV protease were used in 500 μl of phosphate buffer saline containing 2 mm 2-mercaptoethanol and 0.2 mm EDTA overnight at 4 °C followed by a second passage through a Ni-NTA column. The fractions containing the protein were verified by PAGE/Coomassie staining and Western blotting.
Yeast Two-hybrid Analysis of the Interactions between KRT1 Head, Tail, or Rod Domain and the Cytoplasmic Peptides of hC2GnT-M
The plasmids encoding GAL4(DBD)-C2GnT-M(1–14) (M1VQWKRLCQLHYLW14), GAL4 (DBD)-C2GnT-M(Δ2,3), GAL4(DBD)-C2GnT-M (Δ4–6), GAL4(DBD)-C2GnT-M(Δ7–9), GAL4 (DBD)-C2GnT-M(Δ10–12), or GAL4(DBD)-C2GnT-M(Δ13,14) were co-transformed with GAL4(AD)-KRT1(1–644; full-length), GAL4(AD)-KRT1(1–179;head domain), GAL4(AD)-KRT1(180–489; rod domain), or GAL4(AD)-KRT1(490–644; tail domain into yeast Y2HGold). Interaction of GAL4(DBD) and GAL4(AD) fusion proteins enables the cells to grow in the absence of His and in the presence of Aureobasidin A antibiotic. Cotransformants that survive Leu(−) and Try(−) conditions were spotted at 1.5 × 107 cells/10-μl concentration onto plates lacking His and containing Aureobasidin A. The plates were incubated at 30 °C for 5–7 days before being photographed. The Gal4(DBD)-p53 and Gal4(AD)-SV40 large T antigen pair was used as a positive control, whereas Gal4(DBD)-Lamin and Gal4(AD)-SV40 large T antigen pair was used as a negative control.
Isolation of KRT1 in Panc1-bC2GnT-M-c-Myc Cell Lysates or Recombinant KRT1 with Biotinylated N-terminal 21- and 7-Amino Acid (aa) Peptides of hC2GnT-M
A 21-aa peptide, which includes the cytoplasmic tail and a portion of the transmembrane domain located at the N terminus of hC2GnT-M, was tagged with a biotin at the C terminus through a lysine (LifeTein LLC, South Plainfield, NJ). An N-terminal 7-aa peptide (MVQWKRL7) was tagged with biotin at the Met (AAPPTec LLC, Louisville, KY). A 15-aa peptide (LCQLHYLWALGCYMK21) was prepared by tagging with biotin at the C terminus. Control peptide biotin-GHGTGSTGSGSMLRTLLRRRL was purchased from LifeTein LLC. To isolate KRT1, 20 μl of hC2GnT-M N-terminal biotinylated peptide in 25% acetic acid (0.1 mg/ml) was mixed with 20–40 μl of cell lysate (1.5–3.5 mg/ml of protein) or 100 μl of purified KRT1 (0.5 mg/ml). After incubation at 37 °C for 1 h, 100 μl of Dynabeads M-280 streptavidin (Dynal, Oslo, Norway) was added. Following gentle rotation for an additional 30 min, the beads with immobilized complexes were trapped with a magnet. The captured proteins were separated on 8% SDS-PAGE followed by Western blotting with rabbit anti-KRT1 Ab.
Miscellaneous
Protein concentrations were determined with the Coomassie Plus Protein Assay (Pierce) using BSA as the standard. Data are expressed as the mean ± S.D. Analysis was performed using two-sided t test. A value of p < 0.05 was considered statistically significant.
RESULTS
C2GnT-M Interacts with KRT1 via Its Cytoplasmic Tail
As a classical member of IFs, the polar dimer subunits of KRT1 form staggered antiparallel tetramers in a head-to-tail fashion that associate longitudinally and laterally into apolar protofilaments. Two protofilaments form a protofibril, and three to four protofibrils intertwine to produce an apolar intermediate filament 10 nm in diameter (50, 51). Each monomer of KRT1 was composed of N- and C-terminal glycine-rich regions (head and tail domains, respectively) and a central helical rod domain (Fig. 1A). By mass spectrometry analysis, we identified 14 peptide fragments that matched to the KRT1 rod domain sequence from a prominent protein band between 55 and 72 kDa among the proteins immunoprecipitated from the homogenates of Panc1-C2GnT-M-c-Myc cells with c-myc antibody (Fig. 1, B and C). The result was confirmed by a two-way co-IP (Fig. 1D). Next, in c-Myc co-IP samples treated with low (1%) β-mercaptoethanol, we detected many KRT1-containing bands in addition to KRT1 monomer and dimer (Fig. 1E), confirming the essential role the disulfide bonds play in the function of KRT1 including KRT1 polymerization (52) and formation of complex with other proteins including C2GnT-M.
FIGURE 1.

KRT1 interacts with the CT of C2GnT-M through its rod domain. A, head, rod, and tail domains of KRT1 monomer. B and C, identification of KRT1 by proteomics analysis of the 55–72-kDa bands (B, left lane, indicated by the arrow) in the complexes pulled down from the lysate of Panc1-bC2GnT-M-c-Myc cells with anti-c-Myc Abs. The immunoprecipitates were analyzed on 10% SDS-PAGE under reducing condition. Fourteen peptides (C, underlined) were matched to the amino acid sequence of the keratin type II cytoskeletal 1 (67-kDa cytokeratin 1). The KRT1-specific band was absent in control IgG Co-IP (B, right lane). D, the complexes pulled down with anti-KRT1 and anti-c-Myc Abs from the lysate of Panc1-bC2GnT-M-c-Myc cells were blotted with anti-c-Myc and anti-KRT1 Abs, respectively. Cell lysate exposed to nonspecific IgG served as the negative control. W-B, Western blot. E, the c-Myc co-IP samples were reduced with 5% or 1% β-mercaptoethanol (ME) before loading and blotted with anti-KRT1 Ab. F, Western blot of complexes pulled down from Panc1-bC2GnT-M-c-Myc cell lysate with biotinylated control and hC2GnT-M N-terminal peptide (1–7 aa), cell lysate input, and complex of cell lysate proteins bound by magnetic beads without peptide. The samples were run on 8% SDS-PAGE gel and blotted with KRT1 Ab. G, KRT1 Western blot of the complexes pulled down from Panc1-bC2GnT-M-c-Myc cell lysate with biotinylated hC2GnT-M N-terminal peptide (1–21, 1–7, and 7–21 aa) after separation on 8% SDS-PAGE gel. H, KRT1 Western blot of the recombinant KRT1 (1–644 aa) pulled down with biotinylated hC2GnT-M (1–7 aa) peptide, and control peptide. Beads exposed to recombinant KRT1 was used as a control. I, yeast-two-hybrid analysis of the interaction of hC2GnT-M peptide (1–14 aa) and KRT1 peptides: (1–644 aa), the N-terminal head domain (1–179 aa), rod domain (180–489 aa), and C-terminal tail domain (490–644 aa). Yeast cell suspensions (1.5 × 107cells/10 μl) were plated on growth medium lacking leucine, tryptophan, and histidine in the presence of 100 ng/ml Aureobasidin A. SV40-T antigen + p53 and SV40-T + Lamin are positive and negative controls, respectively. J, yeast-two-hybrid analysis of the interaction of the rod domain of recombinant KRT1 with hC2GnT-M peptide (1–14 aa) deletion mutants: Δ2,3; Δ4–6; Δ7–9; Δ10–12.
KRT1 also was pulled down from the cell lysate with a biotinylated CT peptide MVQWKRL of C2GnT-M (Fig. 1F) but not an N-terminal peptide devoid of the CT peptide sequence (Fig. 1G). To determine whether this interaction is specific, we performed a C2GnT-M CT pulldown of recombinant KRT1. As shown in Fig. 1H, the KRT1 protein was pulled down by C2GnT-M CT but only minimally by a control peptide, indicating that the C2GnT-M CT interacts directly with KRT1.
Next, we performed a series of yeast two-hybrid analyses to identify the critical region of KRT1 and the amino acids in the C2GnT-M CT that are involved in the interaction. The prey for the first set of the experiment included (a) a full-length KRT1 molecule (1–644 aa), (b) the N-terminal head region (1–179 aa), (c) the rod domain (180–489 aa), and (d) the C-terminal tail region (490–644 aa). The 1–14-aa peptide of hC2GnT-M was used as bait. A strong interaction was detected for only KRT1 (1–644 aa) and KRT1 (180–489 aa) peptides, suggesting that the C2GnT-M CT-binding site resides in the rod domain of KRT1 (Fig. 1I). Subsequent experiments were carried out using the KRT1 rod domain as the prey and a series of deletion mutants of the C2GnT-M N-terminal region as bait. As shown in Fig. 1J, the WKR6 deletion mutant, C2GnT-MΔ4–6, failed to bind KRT1, whereas other mutants, including C2GnT-MΔ2,3, C2GnT-MΔ7–9, or C2GnT-MΔ10–12, did bind. We recently found that the AAA6 mutant of C2GnT-M was in the ER (19). Taken together, these results indicate that the WKR6 sequence in the C2GnT-M CT is not only critical for KRT1 binding but also necessary for Golgi localization of this enzyme.
Knockdown of KRT1 Enhances C2GnT-M Binding to NMIIA and C2GnT-M Degradation by Proteasome
To determine the biological significance of the interaction of the C2GnT-M CT with KRT1, we monitored the intracellular distribution of C2GnT-M after KD of KRT1. We found that in control cells the bulk (92%) of C2GnT-M was in the Golgi (Fig. 2A) and co-localized with KRT1 at the cytoplasmic boundary of the Golgi stacks (Fig. 2B, white arrowheads). To validate this observation, we isolated the Golgi membranes from Panc1-bC2GnT-M-c-Myc cells. KRT1 was pulled down from the Golgi fraction with c-Myc Ab, suggesting that C2GnT-M and KRT1 are associated with the Golgi membrane (Fig. 2C). In cells treated with KRT1 siRNAs for 48 h, the KRT1 protein level was reduced by 70%, but C2GnT-M level was reduced by only 15%. However, 80% of C2GnT-M were outside the Golgi and colocalized with an ER marker, PDI, and NMIIA protein level was increased by 90% (Fig. 2, A, B, D, and E). Treatment with KRT1 siRNAs for 72 h resulted in a 90% depletion of KRT1 protein and an 85% reduction of C2GnT-M protein but a further increase of NMIIA protein level (Fig. 2, A, B, D, and E). We also found that KRT1 siRNA treatment enhanced the association of C2GnT-M with NMIIA (Fig. 2F) and ubiquitination of C2GnT-M (Fig. 2G). It was noted that treatment with KRT1 siRNAs for 48 h and 72 h did not have an apparent effect on Golgi morphology (Fig. 2A). Colocalization of C2GnT-M and KRT1 was also detected in the goblet cells of human normal colon tissue (Fig. 2, H and I). KD of KRT1 in the primary culture of human colonic epithelial cells resulted in shifting of the distribution of C2GnT-M from Golgi to ER (Fig. 2, J–L). Furthermore, formation of the C2GnT-M-KRT1 complex in these cells was also shown by two-way co-IP (Fig. 2M). The results suggest that C2GnT-M is localized to the Golgi by binding to the KRT1, and without KRT1, C2GnT-M is ER-bound as mediated by NMIIA to undergo proteasomal degradation as we previously described (19, 30). To address the question of whether KRT1 is also required for the Golgi retention of other GTs, we examined Golgi localization of three other GTs after siRNA KD of KRT1. These enzymes include two key O-glycosylation GTs, C1GalT1 and ST3Gal1, and another important enzyme, MGAT1, which initiates formation of complex-type N-linked carbohydrate. We found that after knockdown of KRT1, all three GTs were still in the Golgi, suggesting that KRT1 is dispensable for Golgi localization of C1GalT1, ST3Gal1, and MGAT1 (Fig. 3, A–D).
FIGURE 2.

Knockdown of KRT1 resulted in NMIIA-mediated ER redistribution of C2GnT-M in Panc1-bC2GnT-M-c-Myc cells. A, confocal immunofluorescence images of C2GnT-M and Giantin and of C2GnT-M and PDI in cells treated with scramble siRNAs or KRT1 siRNAs for 48 h and 72 h. B, confocal immunofluorescence images of C2GnT-M and KRT1 in cells treated with scramble or KRT1 siRNAs for 72 h. White boxes in each panel are enlarged and shown at the right side as green, red, and merged images. Nuclei were counterstained with DAPI (blue). All confocal images were acquired with same imaging parameters; bars, 10 μm. C, C2GnT-M and KRT1 were associated with the Golgi. Golgi membranes were isolated from control cells as described under “Experimental Procedures,” and 30 μg of total protein of Golgi or postnuclear supernatant (PS) was immunoblotted for Giantin and KRT1. The Golgi fraction was subjected to co-IP with c-Myc Ab. D, NMIIA, KRT1, and c-Myc Western blots of the lysates of cells treated with scramble or KRT1 siRNAs for 48 h and 72 h; β-actin was a loading control. E, quantification of relative fluorescence intensity (in a.u.) of C2GnT-M in ER versus Golgi in cells presented in A; *, p < 0.001. F and G, NMIIA and ubiquitin Western blot (W-B) of complexes pulled down with anti-c-Myc Ab from the lysate of cells treated with scramble or KRT1 siRNAs for 48 h. Lysates containing equal amounts of NMIIA (F) or C2GnT-M (G), respectively, were used for co-IP. H, KRT1 and C2GnT-M distribution in the goblet cells of human normal colon tissues. The paraffin sections were immunofluorescently stained for KRT1 (green) and C2GnT-M (red) and then analyzed by confocal fluorescence microscopy. White boxes indicate an area enlarged in the bottom panel that represents merged channels; bar, 10 μm. I, quantification of Mander's coefficient of KRT1 and C2GnT-M colocalization of cells presented in H. J, colocalization of C2GnT-M with Giantin and PDI in the primary human colonic epithelial cells. White boxes in each panel are enlarged and shown at the right side as green, red, and merged images. Nuclei were counterstained with DAPI (blue). All confocal images were acquired with same imaging parameters; bars, 10 μm. K, KRT1 Western blot of the lysate of primary human colonic epithelial cells treated with scramble or KRT1 siRNAs for 72 h; β-actin was a loading control. L, quantification of relative fluorescence intensity (in a.u.) of C2GnT-M in ER versus Golgi in cells presented in J; *, p < 0.001. M, the complexes pulled down with anti-KRT1 and anti-C2GnT-M Abs from the lysate of primary human colonic epithelial cells were blotted with anti-C2GnT-M and anti-KRT1 Abs, respectively. Cell lysate exposed to nonspecific IgG served as the negative control.
FIGURE 3.

KRT1 was dispensable for Golgi localization of C1GalT1, ST3Gal1, and MGAT1. Shown are confocal immunofluorescence images of C1GalT1 (A), ST3Gal1 (B), MGAT1 (C), and KRT1 in Panc1-bC2GnT-M-c-Myc cells treated with scramble or KRT1 siRNAs for 72 h. Nuclei were counterstained with DAPI (blue). All confocal images were acquired with same imaging parameters; bars, 10 μm. D, quantification of the fluorescence intensity of cytoplasm/Golgi for indicated GTs in cells shown in A–C.
Next, we examined whether other keratins are involved in the Golgi localization of C2GnT-M because of the following reports: (a) KRT1 forms a tight complex with KRT10 during normal keratinocyte differentiation (53) even though the same phenomenon was not observed in non-epidermal epithelial cells (54), (b) KRT8 is involved in the injury of the pancreas and pathogenesis of pancreatitis (55), and (c) KRT7 is highly expressed in the adenocarcinoma of the pancreas (56). We found that siRNA KD of KRT10 (Fig. 4, A, D, and G), KRT8 (Fig. 4, A, C, and F), or KRT7 (Fig. 4, A, B, and E) did not affect KRT1 level, Golgi morphology, and Golgi localization of C2GnT-M. The results suggest that KRT1 is, and KRT7, -8, and -10 are not required for Golgi localization of C2GnT-M. Furthermore, none of these KRTs is involved in maintaining the compact Golgi morphology.
FIGURE 4.

Knockdown of KRT7, -8, or -10 had no effect on level of KRT1 and intra-Golgi localization of C2GnT-M. A, KRT1 Western blot (W-B) of the lysates of cells treated with scramble, KRT1, KRT7, KRT8, or KRT10 siRNAs for 72 h; γ-tubulin was a loading control. B–D, confocal immunofluorescence images of C2GnT-M, KRT7, KRT8, and KRT10, in Panc1-bC2GnT-M-c-Myc cells treated with scramble, KRT7 (B), KRT8 (C), or KRT10 (D) siRNAs for 72 h, respectively. Nuclei were counterstained with DAPI (blue). All confocal images were acquired with same imaging parameters; bars, 10 μm. E–G, KRT7, KRT8, and KRT10 Western blots of the lysates of cells treated with scramble, KRT7 (E), KRT8 (F), or KRT10 (G) siRNAs for 72 h, respectively.
Golgi Localization of C2GnT-M follows Its Giantin-dependent Targeting
To further confirm that KRT1 is a retention partner for C2GnT-M, we employed HeLa cells transiently expressing wild-type hC2GnT-M tagged with GFP. HeLa cells were chosen for this experiment because of their high transfection efficiency and lack of endogenous C2GnT-M. Predictably, in control cells, C2GnT-M-WT (wild-type)-GFP was in the Golgi and co-stained with KRT1 at the Golgi periphery (Fig. 5A, white arrowheads). However, in cells with depleted KRT1, C2GnT-M-WT-GFP was found predominantly in the cytoplasm, whereas the Golgi morphology remained unaffected (Fig. 5, A–C). Next, we monitored the role of KRT1 in the Golgi localization of C2GnT-M during Golgi biogenesis. For this purpose we took advantage of the well established Golgi morphology restoration property during the wash out (WO) phase of the brefeldin A (BFA)-induced Golgi dissolution (57, 58). In cells treated with 36 μm BFA for 30 min, both C2GnT-M-WT-GFP and Giantin were detected mostly in the cytoplasm, but after a 45-min WO, Golgi morphology was restored, and C2GnT-M-WT-GFP was colocalized with Giantin in the Golgi (Fig. 5, D and E). In cells lacking KRT1, Golgi was recovered and phenotypically closed to the control; however, C2GnT-M-WT-GFP was in the cytosol (Fig. 5, D and E). These data suggest that KRT1 is not essential for maintaining Golgi morphology but necessary for Golgi localization of C2GnT-M.
FIGURE 5.

KRT1 was not required for Golgi biogenesis but was critical for Golgi localization of C2GnT-M in HeLa cells. A and B, confocal immunofluorescence images of wild-type C2GnT-M (WT)-GFP colocalization with KRT1 (A) and Giantin (B) in cells treated with scramble or KRT1 siRNAs for 72 h. Arrowheads indicate peri-Golgi colocalization of proteins in control cells. White boxes in the images indicate the area enlarged in the bottom panel that represents merged channels. The results shown are representative of three independent experiments. All confocal images were acquired with same imaging parameters; bars, 10 μm. Nuclei were counterstained with DAPI (blue). C, quantification of relative fluorescence intensity (in a.u.) of C2GnT-M in cytoplasm versus Golgi in cells presented in A and B; *, p < 0.001. D, colocalization of C2GnT-M-WT-GFP and Giantin in cells treated with scramble or KRT1 siRNAs. Next, cells were treated with BFA followed by recovery for 45 min after initiation of BFA WO. E, quantification of relative fluorescence intensity of C2GnT-M in cytoplasm versus Golgi in cells presented in D; *, p < 0.001. F and G, Giantin and KRT1 Western blot (W-B) of complexes pulled down with anti-GFP Ab from the lysates of HeLa cells expressing C2GnT-M (WT)-GFP and treated with BFA followed by WO for 45 and 60 min.
We recently reported that C2GnT-M targeted the Golgi using the Giantin site exclusively (11). To determine whether Giantin and KRT1 act simultaneously or sequentially during the Golgi relocalization process of C2GnT-M, we carried out a series of co-IP experiments during the BFA WO phase. We compared the amount of C2GnT-M-WT-GFP associated with Giantin or KRT1 before and after BFA WO for 45 and 60 min. As shown in Fig. 5, F and G, before BFA WO was initiated, a low level of the complex was detected between C2GnT-M and Giantin (40% of the control) or KRT1 (34% of the control). At 45 min of Golgi recovery, a huge increase in the amounts of Giantin (145% of the control) but only a moderated increase in the amount of KRT1 (52% of the control) pulled down with anti-GFP antibody was observed. At 60 min of Golgi recovery, the amounts of Giantin pulled down with anti-GFP antibody returned to that in the control (96% of the control), and the fraction of KRT1 also approached the level in the untreated cells (97% of the control). The data indicate that Giantin-mediated Golgi targeting of C2GnT-M precedes the Golgi retention of this enzyme by KRT1.
KRT1 Regulates the Glycosylation Function of C2GnT-M
Recently, we showed that the glycosylation function of C2GnT-L was greatly reduced in the absence of its retention partner, GOLPH3 (24). In this study we examined the effect of the presence or absence of KRT1 on the appearance of T-antigen as detected by PNA lectin staining. The presence of C2GnT-M in the Golgi, which is KRT1-dependent, is expected to prevent the masking of the T-antigen with α2–3Sia (α2,3-linked sialic acid) by out-competing ST3Gal1 for the T-antigen (22, 59) (Fig. 6A). Therefore, we reasoned that overexpression of C2GnT-M in HeLa cells, which express endogenous ST3Gal1, would result in increased production of PNA-specific signal. As shown in Fig. 6, B–D, in cells lacking ST3Gal1 after KD with siRNAs, the PNA reactivity was enhanced. Furthermore, PNA-specific fluorescence intensity was dramatically increased after C2GnT-M-WT-GFP cDNA transfection, suggesting that core 2 extension pathway dominates the ST3Gal1-mediated glycosylation. Also, KD of KRT1, which prevents the Golgi localization of C2GnT-M, reduced the PNA binding signal, indicating that the absence of C2GnT-M in the Golgi is accompanied by loss of T-antigen (Fig. 6, E and F). To further confirm this result, we compared the PNA-specific fluorescence signal associated with the proteins in the cell lysate. The results showed that the PNA-detectable T-antigen associated with glycoproteins in control cells and cells transfected with C2GnT-M-WT-GFP cDNA plus KRT1 siRNAs were substantially lower than that in cells transfected with only C2GnT-M-WT-GFP cDNA or ST3Gal1 siRNAs (Fig. 6G).
FIGURE 6.

Loss of C2GnT-M Golgi localization was accompanied by the production of sialyl-T antigen. A, schema of selective mucin core 1 and core 2-associated O-glycans in mammalian cells. The core 1 structure may be extended by core 2 as catalyzed by C2GnT-M or alternatively shortened by formation of Siaα2–3Galβ1–3GalNAc (Sialyl-T) antigen as catalyzed by ST3Gal1. The PNA lectin binds non-sialylated core 1 O-glycans, Gal-(±GlcNAc)GalNAc. B, confocal immunofluorescence images of PNA lectin-staining in HeLa cells treated with scramble or ST3Gal1 siRNAs. C, ST3Gal1 Western blot of the lysate of HeLa cells treated with scramble or ST3Gal1 siRNAs for 72 h; β-actin was a loading control. D, quantification of PNA-specific average integrated fluorescence (in a.u.) in the cells shown in B; *, p < 0.001. E, confocal immunofluorescence images of C2GnT-M-WT-GFP and PNA lectin-staining in HeLa cells expressing C2GnT-M-WT-GFP and treated with scramble or KRT1 siRNAs. All confocal images were acquired with the same imaging parameters; bars, 10 μm. Nuclei were counterstained with DAPI (blue). F, quantification of PNA-specific average integrated fluorescence (in a.u.) in the cells shown in E; *, p < 0.001. G, Coomassie Blue staining of the 8% SDS-PAGE for separating the glycoproteins pulled down with biotinylated PNA and then streptavidin magnet beads from the lysates of HeLa cells transiently expressing C2GnT-M-GFP and then treated with scramble or KRT1 siRNAs or wild-type HeLa cells treated with scramble or ST3Gal1 siRNAs.
DISCUSSION
In comparison to microtubules and actin filaments, IFs are considered more stable and elastic because they do not exhibit polarity and their monomer units are presented as a filamentous structure and not globular as in the case of actins or microtubules. These mechanical properties allow them to maintain cell and tissue integrity and serve as an ideal cytoplasmic partner for dynamic Golgi (60, 61). Here, we observe that the CT of C2GnT-M interacts with the rod domain of KRT1. Given that KRT1 forms a polymer structure through a head-to-tail complex (50, 51), the above-mentioned interaction suggests that C2GnT-M may serve as a bridge to link Golgi with the IFs. This mutual cooperation may play a significant role in not only the Golgi localization of C2GnT-M but also the integrity and stability of cytokeratin filaments. Our data indicate that KRT1 binds specifically to the CT of C2GnT-M via the rod domain, and in the absence of KRT1, C2GnT-M, contrary to the C1GalT1 and ST3Gal1, is no longer retained in the Golgi. Thus, KRT1 indirectly regulates the formation of O-glycan because the reduced KRT1 expression is correlated with enhanced ST3Gal1-mediated pathway. On the other hand, the finding that KRT1 is dispensable for Golgi localization of MGAT1 suggests that formation of complex-type N-glycans is not regulated by KRT1.
Importantly, the interaction between C2GnT-M and KRT1 is not restricted to transfected cell lines because it is also observed in the human goblet cells of colonic tissue and the human primary colonic epithelial cells (Fig. 2, H–M), suggesting that this interaction represents a process of physiological significance. Several KRT1 gene mutations were found in patients with epidermolytic hyperkeratosis characterized by skin erosions and immune barrier defects (62–65). Intriguingly, most of the KRT1 mutants still form filamental network; however, the number of KRT1/KRT1 interactions is substantially decreased, resulting in weaker IF architecture. Although the majority of the reported point mutations in heterozygotes were found in the rod domain of KRT1, it is not known which of these mutations affect Golgi localization of C2GnT-M. This possibility will be the subject of future investigation. Furthermore, Krt−/− mice exhibit crucial skin inflammation and suffer from perinatal lethality (66). It would be of great interest to see if postnatally induced Krt1−/− mouse would develop a similar phenotypical defect, such as colitis, exhibited by mouse devoid of C2GnT-M (40).
The Golgi residential GTs are distributed across the Golgi stacks according to the glycosylation steps in which they participate. The enzymes that are localized at the early Golgi cisternae have a clear advantage over the enzymes located at the later Golgi cisternae in determining the products to be generated. For example, C2GnTs are localized at cis-medial Golgi, whereas ST3Gal1 is at medial-trans Golgi (67, 68). Under basal conditions, leukocytes produce sialyl-T antigen because of the low level of C2GnT-L. After activation of these cells, the level of C2GnT-L is increased, which shifts the glycan structure from core 1 to core 2 on which the selectin ligands are decorated to help direct these cells to the injured site (69). This is how leukocytes take advantage of the differential Golgi localization of these two enzymes, which compete for same substrate to acquire their trafficking property by regulating the expression of C2GnT-L gene. However, increased gene expression is only one part of the equation because for these enzymes to perform their glycosylation functions they have to be localized to the Golgi. To accomplish that, these enzymes have to target the Golgi and then be retained. These two steps are regulated by different mechanisms as described next.
Recently, we have identified the Golgi targeting sites for four different GTs, including core 1 synthase, C2GnT-M (11), C2GnT-L, and ST3Gal1 (22). Core 1 synthase uses GM130-GRASP65 or GM130-Giantin, and C2GnT-M and C2GnT-L use Giantin exclusively, whereas ST3Gal1 employs Giantin and GM130-GRASP65. Interestingly, in cells with defective Giantin as is found in advanced prostate cancer cells, C2GnT-L is outside of the Golgi, which results in the production of sialyl-T antigen because ST3Gal1 still can reach the Golgi using GM130-GRASP65 site (22). The sialyl-T antigen disappears after the Giantin defect is corrected by inhibition or KD of NMIIA, and C2GnT-L is redistributed to the Golgi. Therefore, the Golgi targeting of GTs is an important step in regulating the synthesis of mucin O-glycans. In the current study we also show that during Golgi biogenesis, C2GnT-M forms a complex with Giantin before the formation of C2GnT-M-KRT1 complex, indicating that Golgi targeting of C2GnT-M is followed by its retention in the Golgi. Our preliminary screening shows that CTs of both C2GnT-M and C2GnT-L interact with Giantin,3 suggesting another diversified function of the cytoplasmic domain.
Previously, we have also shown that GOLPH3 is the protein that helps retain C2GnT-L in the Golgi by binding to its CT (24), confirming that C2GnT-CT can function as a Golgi localization determinant (70). KD of GOLPH3 prevents this enzyme from being retained in the Golgi and formation of core 2-associated selectin ligand sialyl Lewis x. As a result, the metastatic potential of these cancer cells is severely dampened. Our current study shows that KRT1 is the protein that helps retain C2GnT-M in the Golgi. Depletion of KRT1 leaves C2GnT-M outside of the Golgi, which results in the synthesis of sialyl-T antigen, one of the tumor-associated carbohydrate antigens (71). This result resembles the mucin O-glycan phenotype of advanced prostate cancer cells in which C2GnT-L was found to be outside of the Golgi (22).
Our group previously reported that WKR6 in the CT of C2GnT-M is the critical amino acid sequence for its interaction with NMIIA and thus the Golgi-to-ER retrograde transportation of C2GnT-M (30). Strikingly, the same motif also determines the interaction of CT of this enzyme with KRT1 and helps retains the enzyme in the Golgi.
The data described in this communication are filling the gap between two known mechanisms, ER-to-Golgi anterograde trafficking and Golgi-to-ER retrograde transportation of Core 2 enzymes (11, 19, 20). We favor a mechanism in which GTs are delivered from the ER to Golgi by a COPI- and COPII-independent process (11). Furthermore, to the best of our knowledge, what we describe here represents the first evidence of a direct interaction of IFs with a Golgi residential protein. We show here that Golgi biogenesis is accompanied by an increase of Giantin-C2GnT-M complex, which is replaced by the KRT1-C2GnT-M complex after the Golgi is completely recovered. The presence of KRT1 is critical for intra-Golgi localization of C2GnT-M. Furthermore, in cells lacking KRT1, we have detected an increase of C2GnT-M-NMIIA complex (Fig. 7). Such an interaction results in ER-directed transportation of the enzyme followed by its proteasomal degradation, a phenomenon consistent with the function of NMIIA in the recycling of GTs we described previously (30). Given that GTs are dynamically recycled (72), they are distributed among several intracellular pools, including (a) vesicles that transport the enzyme from the ER to the Giantin site in the Golgi (11), (b) KRT1-associated for Golgi localization, (c) COPI encapsulated for intra-Golgi retrograde transport (73), and (d) vesicles that transport the NMIIA-bound enzyme from Golgi to proteasomes for degradation (30). In sum, we believe that the CTs of GTs sequentially interact with various protein partners for (a) ER exit, (b) Golgi targeting, (c) Golgi retention, (d) intracellular Golgi transport, and (e) recycling (Fig. 7). One fascinating aspect of the intracellular trafficking of GTs is the dynamic association of GTs with their interacting partners at every intracellular trafficking step. During shuttling between the donor vesicles carrying the enzyme and the acceptor membrane, the GT is expected to be temporally freed from interaction with their retention protein. The question of how these interacting partners position themselves in the right place at the right time awaits further study.
FIGURE 7.
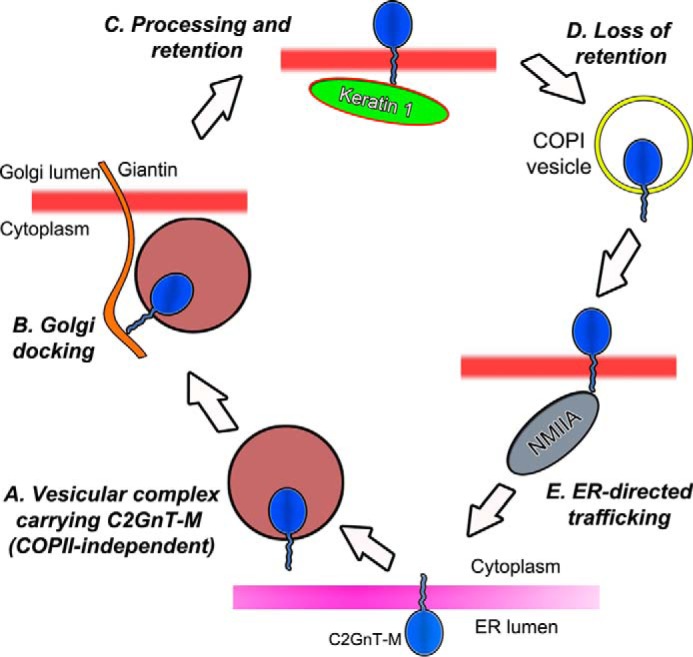
Proposed model of the role of cytoplasmic tail of C2GnT-M in its intracellular trafficking. After its synthesis in the ER, C2GnT-M exits the ER mediated by a CT-dependent process (A) and is transported to the Golgi where it interacts with Giantin via the CT (B). After completion of the processing of its N-glycan, the enzyme is retained in the Golgi by binding to the rod domain of KRT1 via its CT (C). For recycling, the enzyme presumably is transported back to the early Golgi stacks by COPI vesicles (D) and then to the proteasomes by binding to the C-terminal portion (1682–1960 aa) of NMIIA heavy chain (30) via the WKR6 motif of the CT (E). Loss of KRT1 results in increased NMIIA and C2GnT-M mutual cooperation and leads to redistribution of C2GnT-M from Golgi to ER for proteasomal degradation.
The requirement of KRT1 for the Golgi retention of C2GnT-M and the synthesis of glycans extended from core 2 clearly demonstrate that the protein that helps retain a GT in the Golgi can modulate the glycan structure by regulating the retention of its cognate GT in the Golgi. Finally, KRT1 could play a critical role in the mucus function by controlling the Golgi localization of an important enzyme involved in the production of disease-prevention glycans in secreted mucins.
This work was supported, in whole or in part, by the Office of Research and Development, Department of Veterans Affairs (VA 1I1BX000985), the National Institutes of Health Grant R21 HL097238, and the State of Nebraska (LB506).
A. Petrosyan, M. F. Ali, and P.-W. Cheng, unpublished observation.
- GT
- glycosyltransferase
- KD
- knockdown
- C2GnT-M
- core 2 N-acetylglucosaminyltransferase M
- C2GnT-L
- core 2 N-acetylglucosaminyl-transferase-L/1
- Panc1-bC2GnT-M-c-Myc cells
- Panc1 cells expressing bC2GnT-M tagged with c-Myc at the C terminus
- IF
- intermediate filament
- KRT1
- keratin 1
- NMIIA
- non-muscle myosin IIA
- CT
- cytoplasmic tail
- ER
- endoplasmic reticulum
- BFA
- brefeldin A
- WO
- wash out
- ST3Gal1
- Galβ1–3-GalNAcαSer/Thr:α2–3sialyltransferase 1
- PDI
- protein disulfide isomerase
- GOLPH3
- Golgi phosphoprotein 3
- Ab
- antibodies
- co-IP
- co-immunoprecipitation
- PNA
- peanut agglutinin
- a.u.
- arbitrary units
- TEV
- tobacco etch virus
- Ni-NTA
- nickel-nitrilotriacetic acid
- C1GalT1
- core 1 β3galactosyltransferase
- COP
- coat-protein complex
- MGAT1
- mannosyl α-1,3-glycoprotein β-1,2-N-acetyl-glucosaminyltransferase
- aa
- amino acid(s)
- Ab
- antibody.
REFERENCES
- 1. Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds (2009) Essentials of Glycobiology, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York: [PubMed] [Google Scholar]
- 2. Cheng P. W., Radhakrishnan P. (2011) Mucin O-glycan branching enzymes: structure, function, and gene regulation. Adv. Exp. Med. Biol. 705, 465–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eklund E. A., Freeze H. H. (2006) The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx. 3, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hakomori S. (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U.S.A. 99, 10231–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colley K. J. (1997) Golgi localization of glycosyltransferases: more questions than answers. Glycobiology 7, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoki D., Lee N., Yamaguchi N., Dubois C., Fukuda M. N. (1992) Golgi retention of a trans-Golgi membrane protein, galactosyltransferase, requires cysteine and histidine residues within the membrane-anchoring domain. Proc. Natl. Acad. Sci. U.S.A. 89, 4319–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burke J., Pettitt J. M., Humphris D., Gleeson P. A. (1994) Medial-Golgi retention of N-acetylglucosaminyltransferase I. Contribution from all domains of the enzyme. J. Biol. Chem. 269, 12049–12059 [PubMed] [Google Scholar]
- 8. El-Battari A. (2006) Autofluorescent proteins for monitoring the intracellular distribution of glycosyltransferases. Methods Enzymol. 416, 102–120 [DOI] [PubMed] [Google Scholar]
- 9. Nilsson T., Rabouille C., Hui N., Watson R., Warren G. (1996) The role of the membrane-spanning domain and stalk region of N-acetylglucosaminyltransferase I in retention, kin recognition and structural maintenance of the Golgi apparatus in HeLa cells. J. Cell Sci. 109, 1975–1989 [DOI] [PubMed] [Google Scholar]
- 10. Giraudo C. G., Maccioni H. J. (2003) Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol. Biol. Cell 14, 3753–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petrosyan A., Ali M. F., Cheng P. W. (2012) Glycosyltransferase-specific Golgi-targeting mechanisms. J. Biol. Chem. 287, 37621–37627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Osman N., McKenzie I. F., Mouhtouris E., Sandrin M. S. (1996) Switching amino-terminal cytoplasmic domains of α-1,2-fucosyltransferase and α1,3-galactosyltransferase alters the expression of H substance and Galα1,3Gal. J. Biol. Chem. 271, 33105–33109 [DOI] [PubMed] [Google Scholar]
- 13. Quintero C. A., Valdez-Taubas J., Ferrari M. L., Haedo S. D., Maccioni H. J. (2008) Calsenilin and CALP interact with the cytoplasmic tail of UDP-Gal:GA2/GM2/GD2 β1,3-galactosyltransferase. Biochem. J. 412, 19–26 [DOI] [PubMed] [Google Scholar]
- 14. Wassler M. J., Foote C. I., Gelman I. H., Shur B. D. (2001) Functional interaction between the SSeCKS scaffolding protein and the cytoplasmic domain of β1,4-galactosyltransferase. J. Cell Sci. 114, 2291–2300 [DOI] [PubMed] [Google Scholar]
- 15. Schmitz K. R., Liu J., Li S., Setty T. G., Wood C. S., Burd C. G., Ferguson K. M. (2008) Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 14, 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tu L., Tai W. C., Chen L., Banfield D. K. (2008) Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 321, 404–407 [DOI] [PubMed] [Google Scholar]
- 17. Okamoto M., Yoko-o T., Miyakawa T., Jigami Y. (2008) The cytoplasmic region of α1,6-mannosyltransferase Mnn9p is crucial for retrograde transport from the Golgi apparatus to the endoplasmic reticulum in Saccharomyces cerevisiae. Eukaryot. Cell 7, 310–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Todorow Z., Spang A., Carmack E., Yates J., Schekman R. (2000) Active recycling of yeast golgi mannosyltransferase complexes through the endoplasmic reticulum. Proc. Natl. Acad. Sci. 97, 13643–13648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petrosyan A., Cheng P. W. (2013) A non-enzymatic function of Golgi glycosyltransferases: mediation of Golgi fragmentation by interaction with non-muscle myosin IIA. Glycobiology 23, 690–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petrosyan A., Cheng P. W. (2014) Golgi fragmentation induced by heat shock or inhibition of heat shock proteins is mediated by non-muscle myosin IIA via its interaction with glycosyltransferases. Cell Stress Chaperones 19, 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quintero C. A., Giraudo C. G., Villarreal M., Montich G., Maccioni H. J. (2010) Identification of a site in Sar1 involved in the interaction with the cytoplasmic tail of glycolipid glycosyltransferases. J. Biol. Chem. 285, 30340–30346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petrosyan A., Holzapfel M. S., Muirhead D. E., Cheng P. W. (2014) Restoration of compact Golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin O-glycan synthesis. Mol. Cancer Res. 12, 1704–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi N., Fukuda M. N. (1995) Golgi retention mechanism of β-1,4-galactosyltransferase. Membrane-spanning domain-dependent homodimerization and association with α- and β-tubulins. J. Biol. Chem. 270, 12170–12176 [DOI] [PubMed] [Google Scholar]
- 24. Ali M. F., Chachadi V. B., Petrosyan A., Cheng P. W. (2012) Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of Core 2 N-acetylglucosaminyltransferase 1. J. Biol. Chem. 287, 39564–39577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pereira N. A., Pu H. X., Goh H., Song Z. (2014) Golgi phosphoprotein 3 mediates the Golgi localization and function of protein O-linked mannose β-1,2-N-acetlyglucosaminyltransferase 1. J. Biol. Chem. 289, 14762–14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee M. C., Miller E. A., Goldberg J., Orci L., Schekman R. (2004) Bidirectional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20, 87–123 [DOI] [PubMed] [Google Scholar]
- 27. Gill D. J., Chia J., Senewiratne J., Bard F. (2010) Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 189, 843–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Girod A., Storrie B., Simpson J. C., Johannes L., Goud B., Roberts L. M., Lord J. M., Nilsson T., Pepperkok R. (1999) Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1, 423–430 [DOI] [PubMed] [Google Scholar]
- 29. Jiang S., Storrie B. (2005) Cisternal rab proteins regulate Golgi apparatus redistribution in response to hypotonic stress. Mol. Biol. Cell 16, 2586–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petrosyan A., Ali M. F., Verma S. K., Cheng H., Cheng P. W. (2012) Non-muscle myosin IIA transports a Golgi glycosyltransferase to the endoplasmic reticulum by binding to its cytoplasmic tail. Int. J. Biochem. Cell Biol. 44, 1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tu L., Banfield D. K. (2010) Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. 67, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Linden S. K., Sutton P., Karlsson N. G., Korolik V., McGuckin M. A. (2008) Mucins in the mucosal barrier to infection. Mucosal Immunol. 1, 183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakamura M., Kudo T., Narimatsu H., Furukawa Y., Kikuchi J., Asakura S., Yang W., Iwase S., Hatake K., Miura Y. (1998) Single glycosyltransferase, core 2136-N-acetylglucosaminyltransferase, regulates cell surface sialyl-Lex expression level in human pre-B lymphocytic leukemia cell line KM3 treated with phorbol ester. J. Biol. Chem. 273, 26779–26789 [DOI] [PubMed] [Google Scholar]
- 34. Kumar R., Camphausen R. T., Sullivan F. X., Cumming D. A. (1996) Core 2 1,6-N-acetylglucosaminyltransferase enzyme activity is critical for P-selectin glycoprotein ligand-1 binding to P-selectin. Blood 88, 3872–3879 [PubMed] [Google Scholar]
- 35. St. Hill C. A., Baharo-Hassan D., Farooqui M. (2011) C2-OsLeX glycoproteins are E-selectin ligands that regulate invasion of human colon and hepatic carcinoma cells. PLoS ONE 6, e16281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ropp P. A., Little M. R., Cheng P. W. (1991) Mucin biosynthesis: purification and characterization of a mucin N-acetylglucosaminyltransferase. J. Biol. Chem. 266, 23863–23871 [PubMed] [Google Scholar]
- 37. Schwientek T., Yeh J. C., Levery S. B., Keck B., Merkx G., van Kessel A. G., Fukuda M., Clausen H. (2000) Control of O-glycan branch formation: molecular cloning and characterization of a novel thymus-associated core 2 β1,6-N-acetylglucosaminyltransferase. J. Biol. Chem. 275, 11106–11113 [DOI] [PubMed] [Google Scholar]
- 38. Yeh J. C., Ong E., Fukuda M. (1999) Molecular cloning and expression of a novel β-1,6-N-acetylglucosaminyltransferase that forms core 2, core 4, and I branches. J. Biol. Chem. 274, 3215–3221 [DOI] [PubMed] [Google Scholar]
- 39. Choi K. H., Osorio F. A., Cheng P. W. (2004) Mucin biosynthesis: bovine C2GnT-M gene, tissue-specific expression, and herpes virus-4 homologue. Am. J. Respir. Cell Mol. Biol. 30, 710–719 [DOI] [PubMed] [Google Scholar]
- 40. Stone E. L., Ismail M. N., Lee S. H., Luu Y., Ramirez K., Haslam S. M., Ho S. B., Dell A., Fukuda M., Marth J. D. (2009) Characterization of mice with targeted deletion of the gene encoding core 2 β1,6-N-acetylglucosaminyltransferase-2. Mol. Cell. Biol. 29, 3770–378219349303 [Google Scholar]
- 41. Huang M. C., Chen H. Y., Huang H. C., Huang J., Liang J. T., Shen T. L., Lin N. Y., Ho C. C., Cho I. M., Hsu S. M. (2006) C2GnT-M is down-regulated in colorectal cancer and its re-expression causes growth inhibition of colon cancer cells. Oncogene 25, 3267–3276 [DOI] [PubMed] [Google Scholar]
- 42. May J. A., Ratan H., Glenn J. R., Lösche W., Spangenberg P., Heptinstall S. (1998) GPIIb-IIIa antagonists cause rapid disaggregation of platelets pre-treated with cytochalasin D. Evidence that the stability of platelet aggregates depends on normal cytoskeletal assembly. Platelets 9, 227–232 [DOI] [PubMed] [Google Scholar]
- 43. Turner J. R., Tartakoff A. M. (1989) The response of the Golgi complex to microtubule alterations: the roles of metabolic energy and membrane traffic in Golgi complex organization. J. Cell Biol. 109, 2081–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao Y., Sztul E. (2001) A novel interaction of the Golgi complex with the vimentin intermediate filament cytoskeleton. J. Cell Biol. 152, 877–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C., JeBailey L., Ridgway N. D. (2002) Oxysterol-binding-protein (OSBP)-related protein 4 binds 25-hydroxycholesterol and interacts with vimentin intermediate filaments. Biochem. J. 361, 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumemura H., Harada M., Omary M. B., Sakisaka S., Suganuma T., Namba M., Sata M. (2004) Aggregation and loss of cytokeratin filament networks inhibit Golgi organization in liver-derived epithelial cell lines. Cell Motil. Cytoskeleton 57, 37–52 [DOI] [PubMed] [Google Scholar]
- 47. Weber K. L., Bement W. M. (2002) F-actin serves as a template for cytokeratin organization in cell free extracts. J. Cell Sci. 115, 1373–1382 [DOI] [PubMed] [Google Scholar]
- 48. England K., Ashford D., Kidd D., Rumsby M. (2002) PKCϵ is associated with myosin IIA and actin in fibroblasts. Cell. Signal. 14, 529–536 [DOI] [PubMed] [Google Scholar]
- 49. Choi K. H., Basma H., Singh J., Cheng P. W. (2005) Activation of CMV promoter-controlled glycosyltransferase and-galactosidase glycogenes by butyrate, tricostatin A, and 5-aza-2′-deoxycytidine. Glycoconj. J. 22, 63–69 [DOI] [PubMed] [Google Scholar]
- 50. Johnson L. D., Idler W. W., Zhou X. M., Roop D. R., Steinert P. M. (1985) Structure of a gene for the human epidermal 67-kDa keratin. Proc. Natl. Acad. Sci. U.S.A. 82, 1896–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fuchs E., Cleveland D. W. (1998) A structural scaffolding of intermediate filaments in health and disease. Science 279, 514–519 [DOI] [PubMed] [Google Scholar]
- 52. Hatzfeld M., Weber K. (1990) The coiled coil of in vitro assembled keratin filaments is a heterodimer of type I and II keratins: use of site-specific mutagenesis and recombinant protein expression. J. Cell Biol. 110, 1199–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hatzfeld M., Franke W. W. (1985) Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J. Cell Biol. 101, 1826–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kartasova T., Roop D. R., Holbrook K. A., Yuspa S. H. (1993) Mouse differentiation-specific keratins 1 and 10 require a preexisting keratin scaffold to form a filament network. J. Cell Biol. 120, 1251–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Toivola D. M., Nakamichi I., Strnad P., Michie S. A., Ghori N., Harada M., Zeh K., Oshima R. G., Baribault H., Omary M. B. (2008) Keratin overexpression levels correlate with the extent of spontaneous pancreatic injury. Am. J. Pathol. 172, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chu P., Wu E., Weiss L. M. (2000) Cytokeratin 7 and cytokeratin 20 expression in epithelial neoplasms: a survey of 435 cases. Mod. Pathol. 13, 962–972 [DOI] [PubMed] [Google Scholar]
- 57. Burgess T. L., Skoufias D. A., Wilson L. (1991) Disruption of the Golgi apparatus with brefeldin A does not destabilize the associated detyrosinated microtubule network. Cell Motil. Cytoskeleton 20, 289–300 [DOI] [PubMed] [Google Scholar]
- 58. Hendricks L. C., McClanahan S. L., Palade G. E., Farquhar M. G. (1992) Brefeldin A affects early events but does not affect late events along the exocytic pathway in pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 89, 7242–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lin Y. R., Reddy B. V., Irvine K. D. (2008) Requirement for a core 1 galactosyltransferase in the Drosophila nervous system. Dev. Dyn. 237, 3703–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kreplak L., Fudge D. (2007) Biomechanical properties of intermediate filaments: from tissues to single filaments and back. Bioessays 29, 26–35 [DOI] [PubMed] [Google Scholar]
- 61. Styers M. L., Kowalczyk A. P., Faundez V. (2005) Intermediate filaments and vesicular membrane traffic: the odd couple's first dance? Traffic 6, 359–365 [DOI] [PubMed] [Google Scholar]
- 62. Chipev C. C., Korge B. P., Markova N., Bale S. J., DiGiovanna J. J., Compton J. G., Steinert P. M. (1992) A leucine-proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 70, 821–828 [DOI] [PubMed] [Google Scholar]
- 63. Yang J. M., Chipev C. C., DiGiovanna J. J., Bale S. J., Marekov L. N., Steinert P. M., Compton J. G. (1994) Mutations in the H1 and 1A domains in the keratin 1 gene in epidermolytic hyperkeratosis. J. Invest. Dermatol. 102, 17–23 [DOI] [PubMed] [Google Scholar]
- 64. Tal O., Bergman R., Alcalay J., Indelman M., Sprecher E. (2005) Epidermolytic hyperkeratosis type PS-1 caused by aberrant splicing of KRT1. Clin. Exp. Dermatol. 30, 64–67 [DOI] [PubMed] [Google Scholar]
- 65. Virtanen M., Smith S. K., Gedde-Dahl T., Jr., Vahlquist A., Bowden P. E. (2003) Splice site and deletion mutations in keratin (KRT1 and KRT10) genes: unusual phenotypic alterations in Scandinavian patients with epidermolytic hyperkeratosis. J. Invest. Dermatol. 121, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 66. Roth W., Kumar V., Beer H. D., Richter M., Wohlenberg C., Reuter U., Thiering S., Staratschek-Jox A., Hofmann A., Kreusch F., Schultze J. L., Vogl T., Roth J., Reichelt J., Hausser I., Magin T. M. (2012) Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J. Cell Sci. 125, 5269–5279 [DOI] [PubMed] [Google Scholar]
- 67. Whitehouse C., Burchell J., Gschmeissner S., Brockhausen I., Lloyd K. O., Taylor-Papadimitriou J. (1997) A transfected sialyltransferases that is elevated in breast cancer and localizes to the medial/trans-Golgi apparatus inhibits the development of core-2-based O-glycans. J. Cell Biol. 137, 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dalziel M., Whitehouse C., McFarlane I., Brockhausen I., Gschmeissner S., Schwientek T. (2001) The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tumor-associated epitope on MUC1. J. Biol. Chem. 276, 11007–11015 [DOI] [PubMed] [Google Scholar]
- 69. Ellies L. G., Tsuboi S., Petryniak B., Lowe J. B., Fukuda M., Marth J. D. (1998) Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity 9, 881–890 [DOI] [PubMed] [Google Scholar]
- 70. Zerfaoui M., Fukuda M., Langlet C., Mathieu S., Suzuki M., Lombardo D., El-Battari A. (2002) The cytosolic and transmembrane domains of the β1,6-N-acetylglucosaminyltransferase (C2GnT) function as a cis to medial/Golgi-targeting determinant. Glycobiology 12, 15–24 [DOI] [PubMed] [Google Scholar]
- 71. Dabelsteen E. (1996) Cell surface carbohydrates as prognostic markers in human carcinomas. J. Pathol. 179, 358–369 [DOI] [PubMed] [Google Scholar]
- 72. Rhee S. W., Starr T., Forsten-Williams K., Storrie B. (2005) The steady-state distribution of glycosyltransferases between the Golgi apparatus and the endoplasmic reticulum is approximately 90:10. Traffic 6, 978–990 [DOI] [PubMed] [Google Scholar]
- 73. Pellett P. A., Dietrich F., Bewersdorf J., Rothman J. E., Lavieu G. (2013) Inter-Golgi transport mediated by COPI-containing vesicles carrying small cargoes. Elife 2, e01296. [DOI] [PMC free article] [PubMed] [Google Scholar]