

Background: The Spir·formin actin nucleator complex organizes actin structures at vesicle membranes.
Results: The Spir FYVE-type domain binds nonspecifically to negatively charged membranes and to the N-terminal KIND domain (in cis).
Conclusion: Conformational states of Spir restrict recruitment of formin to the membrane surface.
Significance: Studying the vesicle targeting mechanisms of actin nucleators is essential for our understanding of intracellular membrane trafficking.
Keywords: Actin, Formin, Membrane Trafficking, Protein Complex, Vesicles, FYVE, Spir
Abstract
Spir and formin (FMN)-type actin nucleators initiate actin polymerization at vesicular membranes necessary for long range vesicular transport processes. Here we studied in detail the membrane binding properties and protein/protein interactions that govern the assembly of the membrane-associated Spir·FMN complex. Using biomimetic membrane models we show that binding of the C-terminal Spir-2 FYVE-type zinc finger involves both the presence of negatively charged lipids and hydrophobic contributions from the turret loop that intrudes the lipid bilayer. In solution, we uncovered a yet unknown intramolecular interaction between the Spir-2 FYVE-type domain and the N-terminal kinase non-catalytic C-lobe domain (KIND) that could not be detected in the membrane-bound state. Interestingly, we found that the intramolecular Spir-2 FYVE/KIND and the trans-regulatory Fmn-2-FSI/Spir-2-KIND interactions are competitive. We therefore characterized co-expressed Spir-2 and Fmn-2 fluorescent protein fusions in living cells by fluorescence cross-correlation spectroscopy. The data corroborate a model according to which Spir-2 exists in two different states, a cytosolic monomeric conformation and a membrane-bound state in which the KIND domain is released and accessible for subsequent Fmn-2 recruitment. This sequence of interactions mechanistically couples membrane binding of Spir to the recruitment of FMN, a pivotal step for initiating actin nucleation at vesicular membranes.
Introduction
The multiple cellular functions of the dynamic actin cytoskeleton are achieved by targeting actin nucleator complexes at distinct subcellular sites to locally initiate the polymerization of filaments. The organization and regulation of cortical actin at the cell membrane have been extensively studied (1). However, up to now, the molecular mechanisms of cytoskeletal organization at intracellular, vesicular membranes are still poorly characterized. Proteins of the Spir family are cytoskeletal organizers at the crossroad of actin and vesicle processes (2). Mouse genetics and cell biological experiments point toward a function of the Spir·FMN3 complex in oocyte maturation and emotional learning (3–5). In mouse oocytes, Spir-1 and Spir-2 proteins cooperate with formin-2 (Fmn-2; FMN subgroup of formins) in the generation of a dynamic actin meshwork, which is thought to originate from the surface of vesicles (3, 6). The Spir/Fmn-2 nucleated actin filaments serve as tracks for myosin Vb actin motor protein-dependent long range transport of Rab11 vesicles in oocytes (6).
Besides a function in actin-dependent long range vesicle transport in oocytes, a role of Spir proteins in exocytic vesicle transport and in transport processes beyond early endosomes was described in somatic cells (7, 8). In all cell types characterized so far, Spir proteins highly colocalize with Rab11, indicating a close functional relationship of the actin organizer and the small G protein of the Ras superfamily. Rab11 is localized at the trans-Golgi network, post-Golgi vesicles, and recycling endosome and is thought to be a master regulator in the orchestration of cell surface proteins such as growth factor receptors, ion channels, and cell adhesion proteins (9).
On a molecular level, Spir proteins were shown to interact with FMN-type formins and regulate each other in a mutual fashion (10). In vitro, Spir proteins alone cannot nucleate the polymerization of profilin-bound actin, which is thought to be the major source for actin filament polymerization in cells (1, 11, 12). However, the Spir·FMN complex is a very potent nucleator of profilin/actin (12). The in vitro data are confirmed by findings in oocytes showing that the vesicle-originated cytoplasmic actin meshwork cannot be formed by one of the nucleators alone but requires the cooperative action of both Spir and Fmn-2 (3, 6). The cooperation is conserved between mammals and flies as the Drosophila Spir protein and Drosophila FMN-family protein Cappuccino also cooperate in building an oocytic actin meshwork during fly oogenesis (13, 14).
The physical interaction of Spir and FMN proteins is mediated by the evolutionarily conserved formin/Spir interaction (FSI) sequence at the C terminus of FMN-type formins and involves the N-terminal kinase non-catalytic C-lobe domain (KIND) of Spir (15). The KIND domain evolved from the catalytic protein kinase fold into a protein interaction module (10, 16–19). The complex crystal structure disclosed a large interface of positively charged residues of the formin-2 FSI sequence mediating contacts to an acidic groove at the surface of the Spir-1-KIND domain (18, 19).
The introduction of a positive charge into the acidic groove of the KIND domain (human Spir-1 Y134K and Drosophila Spir Y232K mutations) strongly impaired the Spir/formin interaction in vitro (18, 19). Transgenic expression of Drosophila Spir Y232K mutant protein in flies having a spir mutant background failed to rescue the oocytic actin meshwork (14). Direct physical interaction of Spir and the formin Cappuccino is therefore required to build up the oocytic actin mesh during fly oogenesis (14).
Targeting of Spir proteins toward vesicle membranes and the assembly of an active membrane-bound Spir·FMN actin nucleator complex can be regarded as essential steps in the regulation of Spir functions. The localization of Spir proteins at intracellular membranes is mediated by a C-terminal FYVE-type zinc finger membrane interaction module, a structural domain that is typically found in proteins involved in membrane trafficking (7). FYVE zinc fingers comprise eight cysteine residues, which bind two zinc ions (20, 21). The structure forms a hydrophobic “turret loop,” which penetrates the membrane (22, 23). A second hallmark of canonical FYVE domain proteins such as EEA1 or the yeast VPS27 proteins is its specific interaction with phosphatidylinositol 3-phosphate, a key component in the integration of cell signaling cascades and vesicle transport processes (24, 25). The Spir FYVE-type domain is different from the canonical FYVE domain in that it lacks the conserved phosphoinositide-binding motifs and has an insertion loop between cysteines 6 and 7 of the FYVE consensus sequence (7, 26) (see Fig. 1A). Other FYVE-type zinc finger proteins have been identified that also share with the canonical FYVE domains the two zinc ion complexing centers and the hydrophobic turret loop but lack the consensus sequences forming the phosphatidylinositol 3-phosphate binding pocket. Members of this group are the Rab3 small G protein effectors Rim1, Rim2, Rabphilin-3A, and Noc2 and the Rab11 effector protrudin (27, 28) (see Fig. 1A). Lipid interaction studies of the protrudin FYVE-type domain revealed that the membrane binding domain preferentially binds phosphatidylinositol bis- and trisphosphates but not monophosphates as the canonical FYVE domains do (29). Until now, membrane binding characteristics of the Spir FYVE-type domains have not been addressed.
FIGURE 1.

Organization of Spir and formin proteins. A, sequence alignment of FYVE-type zinc finger domains of vertebrate Spir and proteins containing a canonical FYVE domain as a reference (conserved sequences, gray/yellow). Within the consensus sequence, eight cysteine residues define two coordination centers for zinc ions (C1–C8; teal). The hydrophobic amino acids at the tip of the turret loop are highlighted (red). Canonical FYVE domains (EEA1, SARA, Vps27, and Hrs) contain the WXXD, R+HHCR, and RVC sequence motifs, which form a phosphatidylinositol 3-phosphate binding pocket (green). Accession numbers and aa are as follows: human Spir-1, aa 582–711 (NM_001128626); human Spir-2, aa 574–692 (AJ422077); mouse Spir-2, aa 564–682 (AJ459115); chicken Spir-2, aa 575–702 (XM_004944321); human Rabphillin-3A, aa 87–149 (NM_001143854); human protrudin, aa 344–416 (NM_001002261); human EEA1, aa 1346–1411 (NM_003566); human SARA, aa 693–759 (NM_004799); yeast Vps27, aa 167–231 (NM_001183183); and human Hrs, aa 157–221 (NM_004712). B, domain structure of wild-type human Spir-2 and murine Fmn-2 (NM_019445) and fragments thereof used in this study. *, wild-type protein and two mutants in which amino acids were replaced: removal of four zinc binding cysteines (C1,2,7,8S) and exchange of the two hydrophobic residues (LAFA) in the turret loop; **, wild-type sequence and the point mutation Y120K.
In our aim to understand how the Spir/FMN actin nucleators assemble site-specifically into a functional complex, we investigated the membrane and protein interactions of the Spir-2 FYVE-type domain. Experiments using biomimetic membrane systems showed that the Spir-2 FYVE-type domain promiscuously interacts with negatively charged lipids. Intriguingly, we discovered an interaction of the Spir-2 FYVE-type zinc finger with the N-terminal Spir-2-KIND domain, which could not be detected at membranes. Based on data showing that the full-length Spir-2 protein is a monomer in the cytosol and only weakly interacts with Fmn-2 when released from membranes, we propose a model according to which Spir-2 exists in two different states, a cytosolic backfolded monomer and a membrane-bound state in which the KIND domain is released and accessible for subsequent formin-2 recruitment.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
Prokaryotic and eukaryotic expression vectors were generated by standard cloning techniques using Pfu polymerase (Fermentas), restriction endonucleases (New England Biolabs), and T4 DNA ligase (New England Biolabs). Point mutants were generated with the QuikChange site-directed mutagenesis kit (Agilent, Stratagene). The sequence of the constructs was verified by DNA sequencing (MWG-Biotech/Eurofins). A description of all constructs used in this study is given in Table 1. Prokaryotic glutathione S-transferase (GST)- and His6-AcGFP-tagged constructs were expressed in Escherichia coli Rosetta(DE3) pLysS/BL21(DE3) pLysS. Bacteria were grown in LB (100 mg/liter ampicillin and 30 mg/liter chloramphenicol) at 37 °C up to an A600 of 0.6–0.8. Recombinant protein expression was induced by 100–300 μm isopropyl 1-thio-β-d-galactopyranoside and continued at 18–20 °C for 20 h. Bacteria were harvested and lysed by ultrasonication, and soluble proteins were purified using an ÄKTA purifier system (GE Healthcare) using GSH-Sepharose HP-, Ni-NTA FF, Ni-NTA-agarose beads (Qiagen), GSH-Sepharose 4B beads (GE Healthcare), and size exclusion chromatography (Superdex G200 16/60, GE Healthcare). GST tags were cleaved where indicated by tobacco etch virus protease. Protein concentration was increased by ultrafiltration using Amicon Ultra-4 ultracentrifugation devices with respective molecular weight cutoffs (Millipore). Protein purity was estimated by SDS-PAGE and Coomassie staining. For all bacterially expressed recombinant proteins used in this study, the purity was >90% except for the GFP-Spir-2-ΔKWL-C1,2,7,8S protein, which was very unstable and had an estimated purity of 60–70%.
TABLE 1.
Compilation of plasmid DNA expression vectors used in this study
Seph., Sepharose; TEV, tobacco etch virus.
| Construct | Plasmid | Description | Fragment boundaries (restriction sites) | Purification | Purpose |
|---|---|---|---|---|---|
| His-Spir2-KIND | pProExHTb-Spir2-KIND | His-Spir2-KIND | aa 18–207 (BamHI/XhoI) | Ni-NTA, Superdex G200 | GUV binding/FCCS in vitro |
| GFP | pProExHTb-AcGFP | His-AcGFP | aa 1–238 (BamHI/KpnI) | Ni-NTA FF, Superdex G200 | GUV binding/FCCS in vitro |
| GFP-Spir2-ΔKWL | pProExHTb-AcGFP-Spir2-ΔKWL | His-AcGFP-Spir2-ΔKWL | aa 502–728 (BamHI/EcoRI/HindIII) | Ni-NTA, Superdex G200 | GUV binding/FCCS in vitro |
| GFP-Spir2-ΔKWL-LAFA | pProExHTb-AcGFP-Spir2-ΔKWL-LAFA | His-AcGFP-Spir2-ΔKWL-L595A,F596A | aa 502–728 (BamHI/EcoRI/HindIII) | Ni-NTA FF, Superdex G200 | GUV binding |
| GFP-Spir2-ΔKWL-C1,2,7,8S | pProExHTb-AcGFP-Spir2-ΔKWL-C1,2,7,8S | His-AcGFP-Spir2-ΔKWL-C1,2,7,8S C586S,C589S,C683S,C686S | aa 502–728 (BamHI/EcoRI/HindIII) | Ni-NTA FF, Superdex G200 | GUV binding |
| Fmn2-eFSI | pGEX4T1-NTEV-Fmn2-eFSI | GST-Fmn2-eFSI/Fmn2-eFSI | aa 1523–1578 (EcoRI/SalI) | GSH-Seph. FF, TEV, SP-Sepharose XL | FCCS in vitro/GST pulldown |
| GST-Spir2-KIND | pGEX4T1-NTEV-Spir2-KIND | GST-Spir2-KIND | aa 18–207 (BamHI/XhoI) | GSH-Seph. 4B | GST pulldown |
| GST-Spir2-KIND-Y120K | pGEX4T1-NTEV-Spir2-KIND-Y120K | GST-Spir2-KIND-Y120K | aa 18–207 (BamHI/XhoI) | GSH-Seph. 4B | GST pulldown |
| GFP-Spir2 | pEGFP-C1-Spir2 | eGFP-Spir2 | aa 1–728 (BamHI/NotI) | FCCS in HeLa/transient expression in HeLa | |
| GFP-Spir2-LAFA | pEGFP-C1-Spir2-LAFA | eGFP-Spir2-L595A,F596A | aa 1–728 (BamHI/NotI) | Transient expression in HeLa | |
| GFP-Spir2-C1,2,7,8S | pEGFP-C1-Spir2-C1,2,7,8S | eGFP-Spir2-C1,2,7,8S | aa 1–728 (BamHI/NotI) | Transient expression in HeLa | |
| GFP-Spir2-KIND | pEGFP-C1-Spir2-KIND | eGFP-Spir2-KIND | aa 18–207 (XhoI/BamHI) | FCCS in HeLa | |
| GFP-Spir2-FYVE | pEGFP-C1-Spir2-FYVE | eGFP-Spir2-FYVE | aa 577–696 (XhoI/BamHI) | GST pulldown | |
| GFP-Spir2-ΔKW | pEGFP-C1-Spir2-ΔKW | eGFP-Spir2-ΔKW | aa 375–728 (EcoRI/XbaI) | GST pulldown/transient expression in HeLa | |
| GFP-Spir2-ΔKW-LAFA | pEGFP-C1-Spir2-ΔKW-LAFA | eGFP-Spir2-ΔKW-L595A,F596A | aa 375–728 (EcoRI/XbaI) | GST pulldown/transient expression in HeLa | |
| GFP-Spir2-ΔKW-C1,2,7,8S | pEGFP-C1-Spir2-ΔKW-C1,2,7,8S | eGFP-Spir2-ΔKW-C1,2,7,8S C586S,C589S,C683S,C686S | aa 375–728 (EcoRI/XbaI) | GST pulldown/transient expression in HeLa | |
| 1xGFP | pCMSV-1xEGFP | eGFP | aa 1–238 (BsrGI) | FCS/FCCS in HeLa | |
| 2xGFP | pCMSV-2xEGFP | eGFP-Glu-Gly-Ala-Gly-Ala-eGFP | aa 1–238 (BsrGI) | FCS in HeLa | |
| 3xGFP | pCMSV-3xEGFP | eGFP-Glu-Gly-Ala-Gly-Ala-eGFP-Glu-Gly-Ala-Gla-Ala-eGFP | aa 1–238 (BsrGI) | FCS in HeLa | |
| GFP-linker-mStrawberry | pEGFP-C1-linker-mStrawberry | eGFP-linker(Ala-Ser-Gly-Ala-Gly)-mStrawberry | aa 1–236 (BspEI/BglII) | FCCS in HeLa | |
| mStrawberry-Spir2-KIND | pmStrawberry-C2-hs-Spir2-KIND | mStrawberry-hs-Spir2-KIND | aa 18–207 (BamHI/EcoRI/XbaI) | Transient expression in HeLa | |
| mStrawberry-Fmn2-FH2-FSI | pmStrawberry-C1-mm-Fmn2-FH2-FSI | mStrawberry-mm-Fmn2-FH2-FSI | aa 1134–1578 (KpnI/XbaI) | FCCS in HeLa | |
| Fmn2-eFSI-K1571A | pGEX4T1-NTEV-Fmn2-eFSI-K1571A | GST-Fmn2-eFSI-K1571A/Fmn2-eFSI-K1571A | aa 1523–1578 (BamHI/XhoI) | GSH-Seph. 4B, Superdex G200, TEV, Superdex G200 | GST pulldown |
| GFP-EEA1-FYVE | pProExHTb-AcGFP-EEA1-FYVE | His-AcGFP-EEA1-FYVE | aa 1277–1411 (EcoRI/KpnI) | Ni-NTA, Superdex G200 | GUV binding |
| GFP-2xHrs-FYVE | pEGFP-C2–2xHrs-FYVE (from Harald Stenmark) | eGFP-2xHrs-FYVE | aa 147–223 duplicate, separated by linker QGQGS (EcoRI/SalI) | Transient expression in HeLa |
Fluorophore Protein Labeling
Purified proteins were dialyzed extensively against PBS with Slide-A-Lyzers (cutoff, 10 kDa). The proteins were diluted to 100 μm, and the dye (Alexa Fluor 647 C2-maleimide (Invitrogen)) was solved in DMSO and then diluted in PBS. An excess of 5–7 times of the dye was used for the labeling reaction. The reaction took place for 2 h at room temperature and then overnight at 4 °C on a rotating wheel. The excess dye was quenched with 2 mm DTT for 1 h at room temperature. The non-reacted dye was removed by extensive dialysis (cutoff, 10 kDa) against PBS. The efficiency was determined by absorption measurements and was in the range of 85–98%.
GST Pulldown Assay
HEK 293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (FCSIII, HyClone), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2. HEK 293 cells were transfected with expression vectors encoding eGFP-tagged constructs by use of LipofectamineTM/Lipofectamine2000TM (Invitrogen). 24 h post-transfection cells were lysed for 40 min in lysis buffer (25 mm Tris, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 10% (v/v) glycerol, and 0.1% (v/v) Nonidet P-40) supplemented with protease inhibitor mixture (Complete Mini, EDTA-free, Roche Applied Science) and 1 mm phenylmethylsulfonyl fluoride (PMSF) and centrifuged at 14,000 rpm at 4 °C for 20 min to remove insoluble debris. Supernatant was incubated with 50 μg of GST fusion protein coupled to 40 μl of GSH-Sepharose 4B beads (1:1 suspension) for 2 h at 4 °C on a rotating wheel. Beads were washed six times with wash buffer (25 mm Tris, pH 7.4, 300 mm NaCl, 5 mm MgCl2, 10% (v/v) glycerol, 0.1% (v/v) Nonidet P-40, and 2 mm dithioerythritol). Bound proteins were eluted by 1× SDS sample buffer and denatured for 10 min at 95 °C. For competition experiments, 5 μm purified Fmn-2-eFSI peptide and Fmn-2-eFSI-K1571A mutant peptide, respectively, was preincubated with GST-Spir-2-KIND coupled to GSH beads for 1 h at 4 °C on a rotating wheel. Subsequent pulldowns were performed as described above. Proteins were separated by SDS-PAGE and analyzed by Western blotting using an anti-eGFP antibody (Aequorea victoria Living Colors®, rabbit polyclonal, 1 μg/ml; Clontech) and horseradish peroxidase-coupled anti-rabbit secondary antibody (1:5000; GE Healthcare) using the enhanced chemiluminescence method (ECL, GE Healthcare). The ECL signal was recorded with an ImageQuant LAS4000 system (GE Healthcare).
Giant Unilamellar Vesicle (GUV) Binding Assay
GUVs were prepared by the electroformation method (30). Lipids were from Avanti Polar Lipids or Sirius Fine Chemicals SiChem and dissolved in chloroform, but phosphoinositides were dissolved in 1:2:0.8 CHCl3:MeOH:H2O. In brief, 5 μl of lipid mixture (1 mg/ml) was spread onto two platinum wire electrodes. After drying the lipid film, the electrodes with the lipid films were placed into a chamber containing 300 mm sucrose solution. Platinum wires were connected to a power generator. Electroformation was performed at 10 Hz and 2.3 V for 1 h at room temperature. The GUVs were released from the electrodes by changing the frequency to 2 Hz for 30 min. 50 μl of GUVs were sedimented in 150 μl of TBS buffer containing up to 200 nm His6-AcGFP-Spir-2-ΔKWL or His6-AcGFP. After 5 min of incubation at room temperature, protein binding to the GUVs was analyzed by confocal microscopy.
Large Unilamellar Vesicle (LUV) Flotation Assay
The LUV flotation assay was performed according to Höfer et al. (31). Solutions of 25 and 75% sucrose were prepared in 1× TBS buffer. For the preparation of LUVs, a lipid suspension in 1× TBS was subjected to eight freeze-thaw cycles and extruded 19 times through a 100-nm-diameter polycarbonate filter (Avanti Polar Lipids) using a Mini-Extruder (Avanti Polar Lipids). 40 μl of protein (2 μm) was mixed with 80 μl of LUVs (2 mg/ml) in a total volume of 150 μl in 1.5-ml polyallomer microfuge tubes (Beckman Instruments Inc.) and incubated for 10 min. The protein/liposome solution was then mixed with 100 μl of the prepared 75% sucrose solution, giving a final sucrose solution of 30% at the bottom. This fraction was overlayered with 200 μl of 25% sucrose and subsequently 100 μl of 1× TBS buffer, giving three layers of different sucrose concentrations (0, 25, and 30% sucrose). The tubes were centrifuged for 2 h at 50,000 rpm at 4 °C using an ultracentrifuge (OptimaTM Max, Beckman Instruments Inc.) and a TLA-55 rotor (Beckman Instruments Inc.). After centrifugation, LUVs and the bound protein floated on the top fraction in 0% sucrose. The fractions were gently collected and analyzed by SDS-PAGE and Western blotting.
Fluorescence Microscopy
HeLa cells were transfected with eukaryotic expression plasmids encoding eGFP-tagged Spir-2 constructs and mStrawberry-tagged Spir-2-KIND using Turbofect (Invitrogen). 24 h after transfection, cells were washed two times with air buffer (150 mm NaCl, 20 mm HEPES, pH 7.4, 15 mm glucose, 150 μg/ml BSA, 20 mm trehalose, 5.4 mm KCl, 0.85 mm MgSO4, 0.6 mm CaCl2). The cells were analyzed by confocal microscopy (Zeiss LSM780 microscope equipped with a 40× numerical aperture 1.2W UV-visible IR C-Apochromat water immersion objective). The sample was excited either by a 488 (GFP), 561 (mStrawberry), or a 633 nm (Alexa647) laser. The fluorescence was then collected in the detection range of 490–560 or 490–604 (GFP), 568–691 (mStrawberry), and 638–758 nm (Alexa647).
Electrochemiluminescence Immunoassay (EIA)
EIA was used to measure protein/protein interactions and protein/lipid interactions (32, 33). Protein (2 μl of 25 ng/μl Spir-2-KIND) or liposomes (2 μl of 2 mg/ml LUVs of different compositions) were passively adsorbed on the electrode surface of 384-well high bind plates (Meso Scale Discovery, Rockville, MD) for 1 h at 23 °C. The surface was blocked with 0.5% porcine gelatin in 1× TBS buffer for 1 h at 23 °C. Defined concentrations of protein (His6-AcGFP-Spir-2-ΔKWL) were added to each well, and binding was carried out for 2 h at 23 °C. Afterward, primary antibodies against GFP (Living Colors full-length GFP polyclonal antibody, Clontech) were applied for 1 h at 23 °C followed by secondary antibodies (goat anti-rabbit SULFO-TAG from Meso Scale Discovery; 1.25 μg/ml; 23 °C for 1 h). Each of the above steps was followed by washing three times with 1× TBS. The signal readout from the bound fraction was performed on the SECTOR Imager 6000 (Meso Scale Discovery) using surfactant-free read buffer provided by the manufacturer. As a background, both primary and secondary antibody binding to gelatin-blocked electrodes was determined and subtracted. The data were analyzed in SigmaPlot 12.5 software (Systat Software Inc.) assuming a single binding site with maximum signal change upon binding amplitude (Bmax) and equilibrium dissociation constant (Kd).
 |
Fluorescence Cross-correlation Spectroscopy (FCCS)
Position-dependent FCS/FCCS was measured with a laser scanning microscope (LSM780, Zeiss) equipped with a Confocor3 unit providing single molecule sensitivity of avalanche photodiodes. All measurements were performed at 25 °C. The samples were excited by using the 488-nm line of an argon ion laser, the 561 and 633 nm HeNe laser with a power of 2–2.5 microwatts. A dichroic beam splitter (488/561 or 488/633 nm) was used for simultaneous excitation of GFP variants and mStrawberry or GFP and Alexa647, respectively. Detection channels were split by dichroic beam splitters (545 or 635 nm) and spectrally confined to GFP emission (505–540 or 505–610 nm), mStrawberry emission (580 nm long pass), and Alexa647 emission (655-nm long pass filter). To derive concentrations, the focal volumes were calibrated by Alexa Fluor 488 (D = 435 μm2/s (34)), Alexa Fluor 568 (D = 332 μm2/s (35)), and Alexa647 (D = 330 μm2/s (36)) for each individual Lab-Tek slide. To remove dominant after-pulsing, dye calibrations were determined by cross-correlating the signal between two identical detection channels. For dye calibrations, we applied a one-component three-dimensional diffusion model with triplet states (T+3D). FCS curves were evaluated with Zeiss Zen 2010 or open source software (PyCorrFit (37)). Statistical analysis was performed using SigmaPlot 12.5 software. For single comparisons, Student's t tests were performed if the data passed normality assumptions. If the data did not pass normality assumptions, the Mann-Whitney U test was performed. Significance levels are marked accordingly: *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant, p > 0.05.
FCCS in Solution
Binding between Spir-2-KIND*Alexa647 and His6-AcGFP-Spir-2-ΔKWL was determined by single point FCCS in BSA-coated 8-well Lab-Tek chambers (Nunc). 12–24 recordings (10 s) were measured in each reaction at a distance of 100 μm above the coverslip. Intensity traces showing significant variations due to aggregates were discarded. The number of complexes (Ngr) was calculated from the ratio of background-corrected cross- and autocorrelation amplitudes (38). Bleed-through from the GFP into the Alexa647 channel was negligible (below 1%). Correlation curves of His6-AcGFP-Spir-2-ΔKWL were evaluated by applying a one-component three-dimensional diffusion model with a blinking term (T+3D). Residual non-conjugated Alexa647 was resolved by a two-component diffusion model (T+3D+3D) in which the diffusion time of the dye was fixed to 30 μs. For cross-correlation analysis, the fraction of free Alexa647 dye (∼30%) was subtracted from the total number of red particles (Nr) in the Alexa647 channel.
To determine dissociation constants (Kd), increasing concentrations of (up to 1 μm) Spir-2-KIND*Alexa647 were titrated into 1× TBS buffer containing 25 nm His6-AcGFP-Spir-2-ΔKWL. Prior to measurement, the samples were incubated for 20 min at room temperature. The cross-correlation levels reflecting progressive binding were evaluated with a simple 1:1 interaction (39). In solutions containing GUVs, FCCS was measured at two positions, in the supernatant (free solution) and at the apical pole of the membrane. GUV measurements were evaluated by a mixed two-component diffusion model (T+3D+2D) to account for two-dimensional diffusion in the membrane plane. As a negative control, we used His6-AcGFP for which we obtained a significantly lower cross-correlation (2%) at both positions.
FCCS in Cells
Molecular interactions between mStrawberry-tagged Fmn-2 and GFP-tagged Spir-2 constructs were measured in HeLa cells plated in 8-well Lab-Tek (Nunc) and transiently transfected with 50–150 ng of each plasmid. Prior to measurement, cells were washed twice for 5 min with air buffer. To dissolve the vesicular pattern of full-length Spir constructs, the samples were treated with 5 μg/ml brefeldin A (BFA) in air buffer for at least 30 min. For positioning in cells, we adjusted the focal depth (z axis) to the equatorial plane of the nucleus and positioned the laser focus in a region with homogeneous fluorescence distribution in the cytoplasm. Excitation power was adjusted to minimize bleaching to less than 10% during measurements (15–20 s), resulting in a molecular brightness of 1–5 kHz. The cytoplasmic correlation curves were evaluated by applying a two-component T+3D+3D diffusion model. For fitting intracellular correlation data, time bins shorter than 10 μs were ignored; a distinct submillisecond blinking time was taken into account if significant. For molecular brightness analysis, the average counts per particle were calculated from the fluorescence intensity (kHz) of the trace and the number of particles as derived from the fluctuation amplitude (both background-corrected). The average counts per particle were finally normalized to monomeric GFP. The maximum cross-correlation levels were calculated individually for each set of auto- and cross-correlation amplitudes (38). Bleed-through from mStrawberry into the GFP channel was negligible; however, GFP produced 12% signal in the mStrawberry channel, which was back-corrected (40). The maximum of the corrected cross-correlation ratio (CCmax) of multiple runs were averaged for the particular cell. To compare the diffusion behavior, multiple autocorrelation curves (comprising a total of 4–6-min measurement time) for each construct were normalized (amplitude = 1) and averaged.
RESULTS
The FYVE-type Domain of Spir-2
FYVE domains share two zinc ion complexing centers (C1–C8) and a hydrophobic turret loop (conserved in human Spir-2: Leu-595 and Phe-596) that mediate lipid binding at the cytoplasmic face of intracellular vesicles (Fig. 1A). For canonical FYVE domains, it is well established that the specific phosphatidylinositol 3-phosphate (PI(3)P) binding is mediated by a pocket comprising the conserved WXXD, R(R/K)HHCR, and RVC sequence motifs of the FYVE consensus sequence (23) (Fig. 1A). However, the Spir FYVE-type domain is lacking these motifs. Instead, it has an insertion loop between C6 and C7 of the FYVE consensus sequence (7, 26) (Fig. 1A). Other FYVE-type zinc finger proteins such as Rabphilin-3A and protrudin also lack the canonical PI(3)P binding pocket and may therefore be classified, together with Spir-2, into a separate subfamily (Fig. 1A). However, the biochemical nature of these non-canonical FYVE-type-mediated membrane interactions is still unknown. To experimentally characterize the assembly and membrane binding properties of the Spir·FMN actin nucleator complex, we therefore generated a set of vectors encoding tagged Spir-2 and Fmn-2 proteins for their expression in human and bacterial cells (Fig. 1B and Table 1).
Membrane Interactions of Spir-2
Live cell imaging of transiently expressed full-length GFP-tagged Spir-2 or a truncated C-terminal fragment encoding the Spir box and the FYVE-type domain but lacking the N-terminal KIND and WH2 domains (GFP-Spir-2-ΔKW) showed the expected localization at endogenous vesicular compartments (Fig. 2A, first and second panels). In contrast, point mutants in the FYVE-type domain, cysteine replacements that destroy the zinc complexing centers (C1,2,7,8S), or mutations in the turret loop, L595A,F596A (LAFA), show a homogeneous fluorescence distribution, suggesting that these protein variants remain soluble in the cytoplasm (Fig. 2A, third and fourth panels).
FIGURE 2.

The Spir-2 FYVE-type zinc finger domain mediates membrane binding. A, confocal images showing the subcellular distribution of GFP-tagged Spir-2 constructs in HeLa cells. Wild-type Spir-2 and the deletion construct containing a functional FYVE-type domain, Spir-2-ΔKW, accumulate at intracellular vesicular membranes, whereas the mutants C1,2,7,8S and LAFA are evenly distributed in the cytoplasm. B–E, binding of recombinant GFP-tagged Spir-2 constructs at the membrane of GUVs composed of DOPC supplemented by the indicated differently charged lipid species (%). B, GFP-Spir-2-ΔKWL binds to negatively charged GUVs (DOPA, 1,2-dioleoyl-sn-glycero-3-phosphatidylglycerol (DOPG), or 1,2-dioleoyl-sn-glycero-3-phosphatidylserine (DOPS)) but not to neutral GUVs (DOPC and 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE)). C, membrane binding is reduced in the presence of 50 mm EDTA, which chelates the positively charged zinc ions. Disintegration of the FYVE-type domain (C1,2,7,8S) or its removal (free GFP) prevents membrane binding. D, the wild-type Spir-2 FYVE-type domain requires a lower content of negatively charged lipids to mediate binding than the less hydrophobic LAFA mutant. E, quantification of membrane-associated fluorescence with line profiles. Scale bars represent 10 μm. Error bars represent S.D.
To distinguish lipid binding from binding modes caused indirectly by protein/protein interactions, we established an assay using GUVs composed of defined lipid mixtures. Because of solubility issues, we did not succeed in purifying the full-length Spir-2 protein. However, we achieved sufficient yields for a C-terminal fragment containing the FYVE-type domain (GFP-Spir-2-ΔKWL) and the corresponding point mutants (GFP-Spir-2-ΔKWL-C1,2,7,8S and GFP-Spir-2-ΔKWL-LAFA; see above) (Fig. 1B). Lipid binding was studied by investigating the absence or presence of accumulated fluorescent protein at the lipid surface of GUVs in free solution by confocal imaging.
The FYVE-type domain-containing protein GFP-Spir-2-ΔKWL did not bind to neutral GUVs composed of either 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) or DOPC supplemented with 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE) (Fig. 2B, first and second panels). To induce significant binding, the GUVs had to be supplemented with negatively charged lipids, e.g. 20% 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), 20% 1,2-dioleoyl-sn-glycero-3-phosphatidylglycerol (DOPG), or 1,2-dioleoyl-sn-glycero-3-phosphatidylserine (DOPS) (Fig. 2B, third, fourth, and fifth panels). The interaction seems to depend on the structural integrity of the FYVE-type zinc finger because removal of the zinc ions by EDTA redistributed GFP-Spir-2-ΔKWL into bulk solution (Fig. 2C, first panel). Accordingly, the Spir-2 variant containing replacements of the zinc complexing cysteines (C1,2,7,8S) was binding-incompetent to the same degree as a recombinant GFP protein control (Fig. 2C, second and third panels).
To further dissect electrostatic from hydrophobic contributions mediated by the turret loop, we systematically varied the content of negatively charged lipids in GUVs and compared binding of the purified wild-type Spir-2 fragment with the corresponding LAFA mutant. We saw the onset of binding for GFP-Spir-2-ΔKWL at about 5% DOPA (Fig. 2D, upper panels), whereas the LAFA mutant required at least 8% DOPA (Fig. 2D, lower panels). The increased density for the LAFA mutant suggests that additional charges compensate for the missing hydrophobic contributions mediated by the tip of the turret loop, which is thought to intrude the lipid bilayer (Fig. 2E). In fact, the hydrophobic contribution might be more significant in cells where the LAFA mutant showed an entirely cytoplasmic distribution (Fig. 2A, fourth panel).
Thus, like the canonical FYVE domains, the FYVE-type domain of Spir-2 mediates vesicle binding mediated by electrostatic, polar, and hydrophobic contributions. The complexed zinc ions play a crucial role for membrane binding, although this may include indirect effects related to domain structure. However, we could not pinpoint a steric preference for particular lipid headgroups.
The Spir-2 FYVE-type Zinc Finger Binds Strongly but Nonspecifically to Phosphoinositides
Among negatively charged lipids, phosphoinositides are distinguished by their importance for regulating vesicular trafficking. PIPs serve as intracellular address tags that recruit canonical FYVE domains by a PI(3)P-specific interaction. We first tested vesicular membrane association in HeLa cells in the presence and absence of wortmannin, a fungal metabolite that acts as inhibitor of PI 3-kinase and reduces the intracellular PI(3)P levels (41, 42). In contrast to a classical, Hrs-derived FYVE domain (43, 44), wortmannin was incapable to release Spir-2 from the endogenous membranes (Fig. 3A). Thus, although Spir-2 exhibits affinity for negatively charged membranes, in cells, we did not see a PI(3)P dependence.
FIGURE 3.
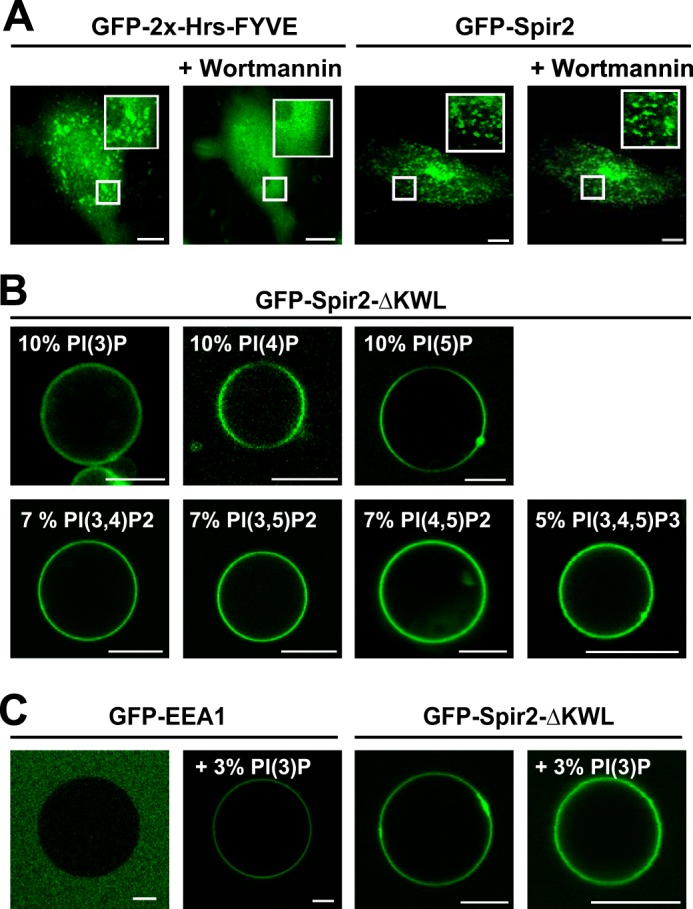
Promiscuous interaction of Spir-2 with phosphoinositides. A, confocal images showing the subcellular distribution of the canonical Hrs FYVE domain (GFP-2x-Hrs-FYVE; mouse Hrs, aa 147–223) and the Spir-2 FYVE-type domain (GFP-Spir-2) in HeLa cells in the presence and absence of 200 nm PI(3)P kinase inhibitor wortmannin. Under wortmannin, Spir-2 remains membrane-associated. B, nonspecific binding of GFP-Spir-2-ΔKWL to GUVs composed of DOPC and different PIP species. C, PI(3)P-specific binding of the classical FYVE domain GFP-EEA1 (human EEA1, aa 1277–1411) and nonspecific binding of GFP-Spir-2-ΔKWL to GUVs composed of 50% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 20% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 20% cholesterol, and 10% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS) ± 3% PI(3)P. Scale bars represent 10 μm.
Applying GFP-Spir-2-ΔKWL to GUVs composed of DOPC supplemented with all combinations of phosphorylated PIPs (Fig. 3B) showed the expected binding to negatively charged membranes albeit without any specificity among the different PIP species. Furthermore, under conditions where binding of a classical EEA1-derived FVYE domain shows PI(3)P-specific membrane association, the Spir-2 protein was constitutively bound (Fig. 3C).
Under steady state conditions, accumulation of fluorescent GFP-Spir-2-ΔKWL at the surface of GUVs does not per se allow a conclusion to be drawn on the binding strength of the protein/lipid interactions. Small amounts of bound protein cannot be detected due to the dominant signal from the supernatant; inhomogeneity in imaging and protein distributions exacerbate quantifications (45). Therefore we aimed to uncover a putative hidden structure of differential PIP binding patterns for the Spir-2 FYVE-type domain with more sensitive approaches.
First we set up a well established liposome flotation assay (46). LUVs were produced from defined lipid compositions of comparable net charge and mixed with the target protein GFP-Spir-2-ΔKWL. Ultracentrifugation was applied to separate the LUV-bound protein fraction from bulk solution with a sucrose gradient. Layers containing different content of sucrose were assayed by immunoblotting against GFP. Centrifugation conditions were such that the unbound protein sedimented into the sucrose cushion (30%), whereas the LUV fraction, filled with buffer of lower density, retained the bound protein in top layers of low sucrose (Fig. 4A, first panel). In agreement with GUV imaging, LUVs of pure 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) retained only trace amounts GFP-Spir-2-ΔKWL, whereas a content of 20% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) concentrated the protein almost quantitatively in the top layer of the gradient (Fig. 4A, second and third panels).
FIGURE 4.
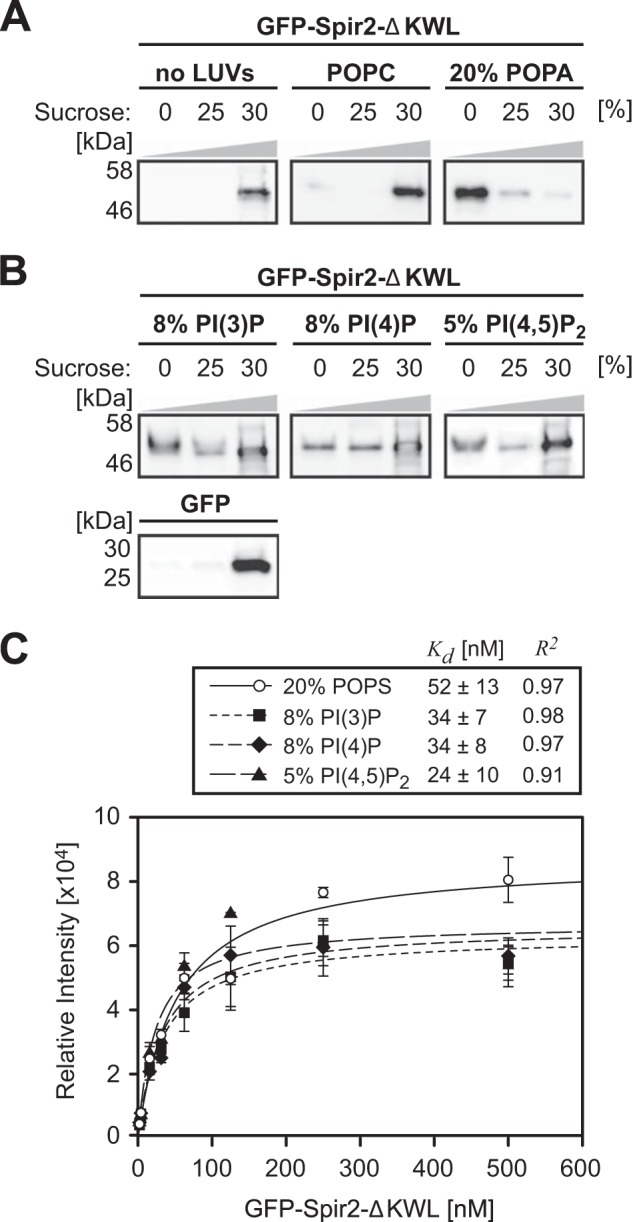
Binding of Spir-2 to PIP-containing liposomes. A, phosphoinositide binding assessed by LUV flotation assay. Mixtures of LUVs composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) and protein were resolved by ultracentrifugation in a sucrose gradient (0–30%). The indicated fractions were analyzed by SDS-PAGE followed by immunoblotting (anti-GFP). Proteins in bulk solution show complete sedimentation (30% fraction), whereas supplementation of negatively charged 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) restrains GFP-Spir-2-ΔKWL on top of the column (0%). B, in agreement with A, different phosphoinositides mediate similar GFP-Spir-2-ΔKWL retention. C, EIA quantifying surface binding of GFP-Spir-2-ΔKWL at the membrane of LUVs composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and the indicated amounts of negatively charged lipids. Saturation curves were analyzed with a single site binding model. The apparent dissociation constants indicate a rather uniform binding strength among phosphoinositides that were applied with similar surface charge density. Error bars represent S.D. (n = 3). POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine.
However, LUVs containing different PIP species appeared more evenly distributed in layers between 0 and 25%, indicating a weaker binding as for LUVs containing 20% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) (Fig. 4, compare B, first, second, and third panels with A, third panel). Binding to polyanionic PI(3)P, PI(4)P, or PI(4,5)P2 retained slightly smaller amounts of protein, suggesting that a single charge per molecule may be sufficient to recruit the FYVE-type domain. Quantifying band intensities showed no significant difference between the PIP species.
Next we measured saturating binging curves with a recently established EIA (32). With EIA, LUVs of different lipid composition were adsorbed at a sensor surface to provide immobilized binding sites for the target protein. Vesicle-bound GFP-Spir-2-ΔKWL protein was detected with an antibody sandwich coupled to an electrochemiluminescent enzyme; the signal therefore reflects the amount of surface-bound protein after subsequent washes. Binding curves were fitted with an equation assuming a non-cooperative 1:1 binding model with an arbitrary number of surface binding sites (Fig. 4C). Again, within error, the apparent Kd values were indistinguishable among the different PIP species (24–34 nm). The dissociation constants were of the same order of magnitude as that measured for LUVs supplemented with 20% negatively charged 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS). In conclusion, the Spir-2 FYVE-type domain mediates a nanomolar interaction to negatively charged lipids in a rather promiscuous fashion, suggesting that additional cues like protein/protein interactions may be required to establish targeting at specific subpopulations of endogenous vesicles.
The C-terminal Spir-2 FYVE-type Zinc Finger Interacts with the N-terminal Spir-2-KIND Domain
The strong interaction with negatively charged lipid headgroups suggested that the FYVE-type zinc finger exposes positively charged residues at the protein surface (21, 22). Indeed, the isoelectric point (pI) of the Spir-2 FYVE-type domain is basic (amino acids (aa) 577–696, pI = 9.1) and, strikingly, opposite to that of Spir-2 N-terminal KIND (human Spir-2, GenBankTM accession number AJ422077; aa 32–221, pI = 4.4). Because it is a common theme for kinases that intramolecular backfolding keeps the enzymes in a quiescent state (47) and considering the evolutionary kinase origin of the KIND domain (16), we therefore hypothesized that such intramolecular interactions also exist for Spir.
To test this, we performed pulldown assays using GST as an affinity tag. Purified GST fusion proteins were preloaded on glutathione-Sepharose beads. GFP constructs of potential interaction partners were transiently expressed in HeLa cells and pulled down from the cell lysate; GFP was used to check the cellular expression and for immunodetection. Indeed, for GST-Spir-2-KIND, we obtained a strong band when pulling down a C-terminal fragment containing the FYVE-type domain (GFP-Spir-2-ΔKW; Fig. 1B) but not for the GFP control (Fig. 5A). Furthermore, the interaction was almost completely suppressed by replacing a single amino acid (Y120K; Fig. 1B) at a position that is analogous to a recently described point mutation in the Spir-1-KIND domain (Y134K) (18). Interestingly, this mutation was described to impair the interaction with Fmn-2, already providing a hint that Spir-2 interactions in cis and in trans may be competitive.
FIGURE 5.

Spir-2 C-terminal FYVE-type domain strongly binds N-terminal KIND in solution. A and B, GST pulldown experiments using GST-tagged Spir-2-KIND as a bait to probe mutual interactions with GFP-tagged C-terminal Spir-2 proteins (Input) transiently expressed in HEK 293 cells. The cell lysates were analyzed by SDS-PAGE followed by immunoblotting (anti-GFP) and Ponceau S staining to document the protein content. A, the C-terminal fragment (GFP-Spir-2-ΔKW) shows strong interactions with the N-terminal fragment (GST-Spir-2-KIND) but not a dysfunctional KIND mutant (GST-Spir-2-KIND-Y120K) (experimental repeats, n = 4). B, a functional FYVE-type domain (GFP-Spir-2-ΔKW and GFP-Spir-2-FYVE) is sufficient for binding of GST-Spir-2-KIND. Mutations in the turret loop had little effect (LAFA mutant), whereas cysteine replacements impaired binding (n = 3). C and D, binding studies using purified recombinant Spir-2 fragments. C, titration of GFP-Spir-2-ΔKWL to quantify binding of non-tagged Spir-2-KIND analyzed by EIA. Progressive binding (average ECL signal) was evaluated by assuming a single binding site. D, complementary binding assay in homogenous solution. Titration of fluorescently labeled Spir-2-KIND*Alexa647 was carried out with constant amounts of GFP-Spir-2-ΔKWL (25 nm). The fraction that was ligand-bound was quantified by FCCS. Evaluation of the curve assumes a 1:1 binding stoichiometry (39). Error bars represent S.D. (n = 3). WB, Western blotting.
We further analyzed different constructs containing the FYVE-type domains for their potential to interact with GST-Spir-2-KIND (Fig. 5B) and detected significant interactions for both truncations, the GFP-Spir-2-ΔKW, as well as the isolated FYVE-type domain, which seems sufficient for binding (Fig. 1B). The isolated FYVE-type zinc finger interacted slightly stronger than the Spir-2-ΔKW protein, which in addition encodes the Spir box and a linker region (Fig. 5B). Quantification of three individual experimental repeats confirmed an increased band intensity of the pulled down FYVE-type domain by a factor of 1.3 that, however, was not significant (p value = 0.14). Point mutations in the turret loop (LAFA) had no effect, whereas removal of the complexed zinc ions (C1,2,7,8S) drastically reduced the band intensity. This suggests that electrostatic contributions and the structural integrity of the FYVE-type domain also play an important role for this intramolecular protein/protein interaction.
To quantify the strength of the KIND/FYVE interaction, we first used EIA. Different concentrations of purified GFP-Spir-2-ΔKWL were titrated to a constant amount of surface-adsorbed non-tagged Spir-2-KIND protein (Fig. 5C). The hyperbolic signal increase confirmed that both protein constructs interacted directly without any hidden cofactors that might be present in cell lysates. Fitting a 1:1 binding model returned an apparent dissociation constant (Kd) of 16 nm.
In a second attempt, we applied FCCS, which is a suitable technique to investigate molecular interactions in homogeneous solution (38). First we characterized the recipient protein GFP-Spir-2-ΔKWL with single color FCS. The molecular brightness (average counts per particle per transit through the detection volume) of GFP-Spir-2-ΔKWL was the same as that of purified monomeric GFP, indicating that at lower nanomolar concentrations FYVE-type domains do not mediate homotypic oligomerization. For dual color FCCS, purified KIND was labeled with an amino-reactive dye of orthogonal color (Alexa647). The degree of labeling was about one dye molecule per protein as determined by photometry and FCS. To avoid cross-talk-related false positive cross-correlation, we titrated in the red color channel (Spir-2-KIND*Alexa647), which produced a saturating binding curve. Evaluating a one-site binding model, we obtained a Kd value 50 ± 30 nm (Fig. 5D). A slightly reduced affinity in FCCS as compared with EIA may be attributed to partially inactivated Spir-2-KIND*Alexa647 due to chemical modification. However, within error, the affinities obtained by these two different techniques are in good agreement and represent sufficient strength to stabilize a backfolded conformation of Spir-2 in free solution.
Membrane Binding and the Intramolecular Spir-2 FYVE-type/KIND Interaction Are Mutually Exclusive
We next addressed the interplay between membrane binding and the intramolecular Spir-2 FYVE-type/KIND interaction. In HeLa cells, transient co-expression of the FYVE-type zinc finger (GFP-Spir-2-ΔKW) and KIND (mStrawberry-Spir-2-KIND) showed the expected vesicular distribution in the GFP channel. However, for all observed expression levels, KIND is neither capable to bind intracellular vesicles on its own (not shown) nor recruited by the C-terminal fragment GFP-Spir-2-ΔKW (Fig. 6A). This behavior could be reproduced with our GUV membrane model when applying a mixture of purified fluorescent constructs (GFP-Spir-2-ΔKWL and Spir-2-KIND*Alexa647). The FYVE-type zinc finger protein GFP-Spir-2-ΔKWL showed membrane binding at negatively charged GUVs but could not recruit the Alexa647-labeled KIND, which remained in bulk solution (Fig. 6B).
FIGURE 6.
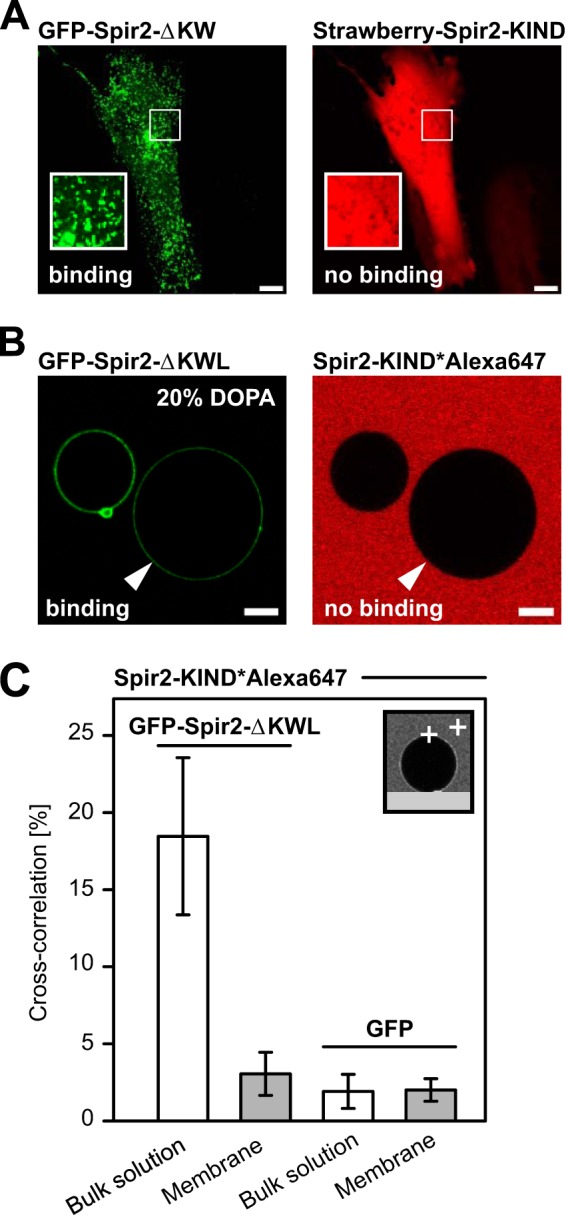
Membrane binding of the Spir-2 FYVE-type domain prevents interactions with Spir-2-KIND. A, confocal images of HeLa cells transiently co-expressing the FYVE-type domain-containing fragment GFP-Spir-2-ΔKW (green) and mStrawberry-Spir-2-KIND (red). The proteins do not colocalize at endogenous vesicular membranes. B, confocal cross-section of GUVs in a solution of purified GFP-Spir-2-ΔKWL and fluorescently labeled Spir-2-KIND*Alexa647. GFP-Spir-2-ΔKWL (green) accumulates at the surface of the negatively charged GUVs (see also Fig. 2), whereas Spir-2-KIND*Alexa647 (red) remains in bulk solution. C, binding analysis at the molecular level by FCCS in bulk solution and the membrane on top of GUVs (inset). Significant cross-correlation confirms the FYVE-type domain/KIND interaction in bulk solution and a reduced level on top of the GUVs that is comparable with a GFP control. Scale bars represent 10 μm. Error bars represent S.D. (n = 10–24).
Finally, to confirm this observation on a molecular level, we performed FCCS. In agreement with Fig. 5D, cross-correlation indicated co-diffusion of the two constructs in bulk solution (Fig. 6C). However, when positioning at the pole of GUVs, cross-correlation dropped to a level slightly above that of the His-tagged GFP control for which we enforced membrane binding by Ni-NTA lipids (DOPC, 20% DOPA, and 3% DOGS-Ni-NTA). Based on diffusion times, the remaining signal reflects contributions from soluble complexes because the three-dimensional detection volume encompasses both a cross-sectioning GUV membrane and some volume from the bulk. In conclusion, lipid binding and KIND interaction mediated by the Spir-2 FYVE-type domain seem to be mutually exclusive. Therefore Spir-2 seems to occur in two different conformations, a backfolded compact structure in free solution and a more extended conformation in which the FYVE/KIND interaction is released when bound to negatively charged lipid bilayers.
Fmn-2 Binding and the Intramolecular Spir-2 FYVE-type/KIND Interaction Are Mutually Exclusive
It was proposed that the Spir·FMN actin nucleator complex exerts its function at vesicular membranes (6). Therefore how formins are recruited into the complex and which of the two Spir conformations is engaged remain important questions. We recently showed that a short C-terminal sequence (eFSI; 56 amino acids; Fig. 1B) provides the binding epitope for both human Spir proteins (15). We used this peptide for competition experiments to validate its role in our intramolecular Spir-2 FYVE-type/KIND interaction. To our surprise, the competing activity of Fmn-2-eFSI was very efficient in the GST pulldown assay where the GFP-Spir-2-FYVE band completely disappeared in the presence of 5 μm Fmn-2-eFSI (Fig. 7A). In contrast, the same peptide carrying a point mutation (K1571A) that was described to strongly impair the Spir/formin interaction (18, 19) was inactive (Fig. 7B).
FIGURE 7.
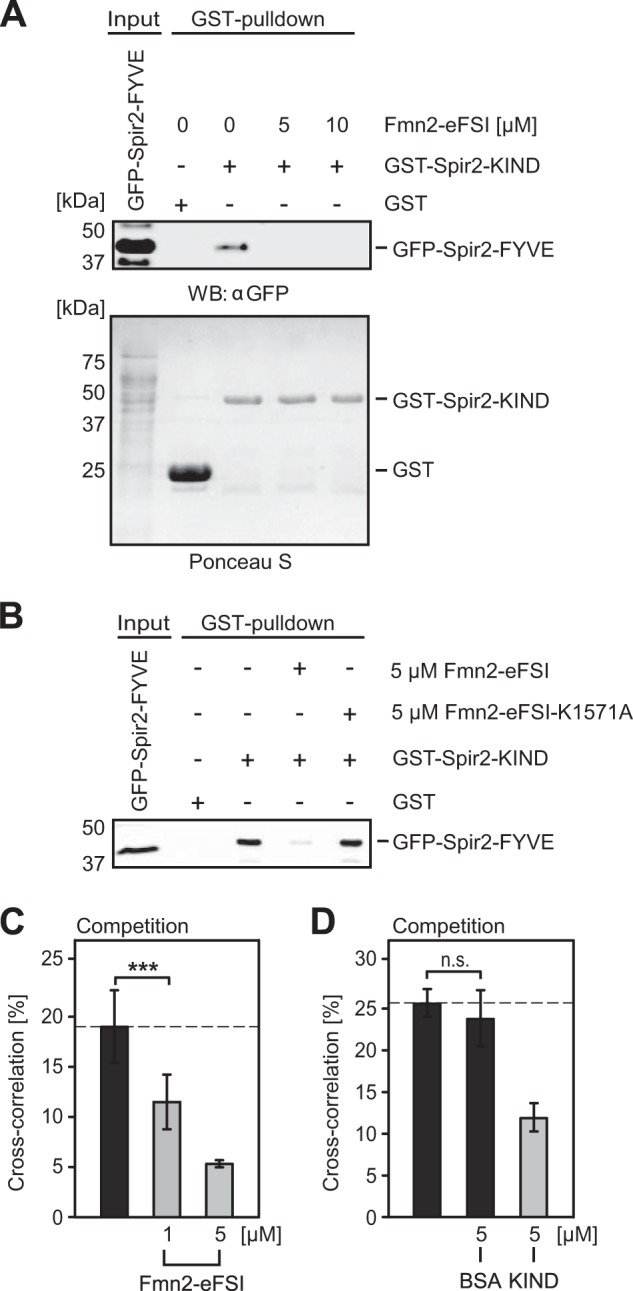
Spir-2-FYVE and Fmn-2-eFSI compete for overlapping binding sites at Spir-2-KIND. A, GST pulldown experiment of recombinant bacterially expressed GST-Spir-2-KIND and GFP-Spir-2-FYVE (Input) transiently expressed in HEK 293 cells. The pulldown experiment was performed in the presence of increasing amounts of purified Fmn-2-eFSI peptide, which fully abolished complex detection (experimental repeats, n = 4). B, same experiment as in A using a point mutated (K1571A), inactive eFSI peptide, which does not compete for binding (n = 2). C and D, FCCS binding assay in homogenous solution using purified GFP-Spir-2-ΔKWL and fluorescently labeled Spir-2-KIND*Alexa647. C, cross-correlation is reduced in the presence of unlabeled Fmn-2-eFSI in a concentration-dependent manner. D, complementary FCCS experiment with unlabeled Spir-2-KIND as competitor shows reduced cross-correlation, whereas BSA has no effect. Error bars represent S.D. (n = 4–8). ***, p < 0.001, n.s., not significant, p > 0.05. WB, Western blotting.
Competition was also observed with dual color FCCS in free solution containing purified GFP-Spir-2-ΔKWL and Alexa647-labeled KIND protein. The normalized cross-correlation amplitude decreased in the presence of Fmn-2-eFSI in a concentration-dependent manner (Fig. 7C). Control experiments with 5 μm BSA showed no effect, whereas addition of unlabeled KIND protein diluted the double labeled complexes and therefore reduced the cross-correlation (Fig. 7D). The latter confirms specificity of the observed interactions in the presence of fluorescent tags.
Intramolecular Backfolding of Monomeric Spir-2 Modulates Fmn-2 Binding in the Cytoplasm of HeLa Cells
A possible regulatory function of the intramolecular Spir-2 FYVE/KIND interaction could be the mechanistic coupling of membrane binding with the assembly of a functional Spir·FMN actin nucleator complex. Therefore we asked whether the accessibility of the Spir-2-KIND domain (in cis) affects formin binding (in trans). Because the full-length protein could not be purified, we turned to transient expression in HeLa cells where we aimed to characterize binding between fluorescent Fmn-2 and different Spir-2 constructs by dual color FCCS.
As shown previously, Spir-2, but not Fmn-2, localized at endogenous membranes when overexpressed (15) (Fig. 8, A and B). Membrane binding depends on the C-terminal half of the protein that contains the FYVE-type domain; thus the isolated KIND domain appears homogeneous (Fig. 8C). Vesicular localization is conveyed to Fmn-2 when co-expressing full-length Spir-2 (Fig. 8D) but is lost for the deletion mutant GFP-Spir-2-ΔKW that lacks the KIND domain and is therefore potentially unable to recruit Fmn-2 via eFSI-mediated interactions (Fig. 8E). These observations already suggested that recruitment of Fmn-2 and Spir-2 to these membranes is mechanistically linked presumably by direct molecular interactions. Unfortunately, the vesicular pattern of full-length Spir-2 seemed unsuitable for a molecular validation by intracellular FCCS where the binding partners should be observed in a diffusive state. To achieve this, we applied the fungal toxin BFA, an efficient inhibitor of the small GTP-binding protein Arf (48) that we and others have found to dissolve the vesicular pattern of Spir-2 by a yet unknown mechanism (49) (Fig. 8F). Note that, in agreement with our working hypothesis, BFA treatment concomitantly dissolved the vesicular Fmn-2 pattern.
FIGURE 8.
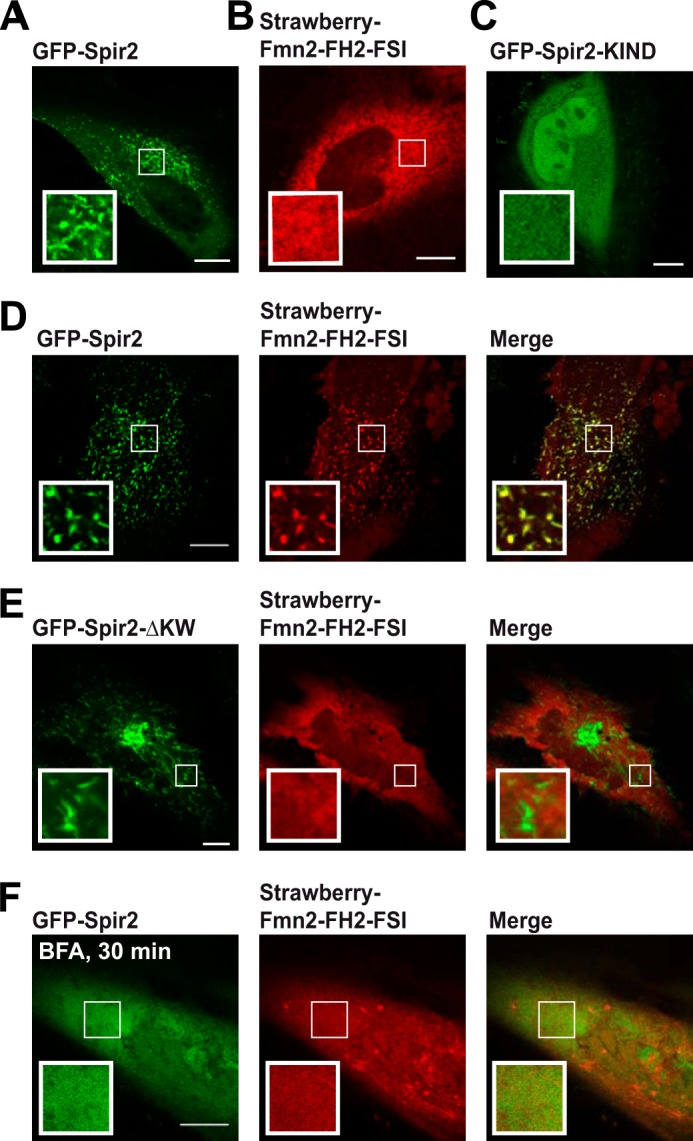
Confocal images showing subcellular distributions of fluorescently tagged Spir-2 and Fmn-2 constructs in HeLa cells. A–C, individual constructs. Full-length GFP-Spir-2 (A) shows a vesicular pattern but not mStrawberry-Fmn-2-FH2-FSI (B). C, the vesicular pattern is lost for GFP-Spir-2-KIND, a mutant that lacks the FYVE-type domain. D–F, co-expression of mStrawberry-Fmn-2-FH2-FSI with different Spir-2 constructs. Full-length GFP-Spir-2 (D) induces a vesicular pattern of Fmn-2, whereas GFP-Spir-2-ΔKW lacking the KIND domain (E) is not able to recruit Fmn-2 to membranes. F, both full-length GFP-Spir-2 and the Fmn-2 constructs redistribute into the cytoplasm in the presence of BFA. Scale bars represent 10 μm.
Under BFA, GFP-Spir-2 showed sufficient mobility in the cytoplasm to be characterized by FCS. For the autocorrelation analysis of individually expressed constructs, a three-dimensional two-component diffusion model for fitting provided sufficient degree of freedom to represent the correlation data (Fig. 9A). We determined in the cytoplasm a short correlation time of 2.4 ms (fraction, 76%) and a significantly longer correlation time of 75.7 ms (24%). In contrast, the deletion mutant GFP-Spir-2-KIND (47.8 kDa) diffused faster with almost a single component of 1.1 ms (94%). Using the same settings, we obtained for a 3-fold GFP tandem 1.2 ms (94%). Thus, in contrast to Spir-2-KIND and the GFP constructs, the decay of the full-length Spir-2 autocorrelation curve was pronounced biphasic. Because diffusion times scale roughly with the cubic root of the molecular mass, the very long correlation time of full-length Spir-2 cannot be justified by molecular size (106.6 kDa). Instead, such long tail correlations are indicative of large scale aggregations or transient binding events at large structures like membranes that may remain detectable even after BFA treatment.
FIGURE 9.

Characterization of the Spir-2 dynamics and interactions by FCCS. A, averaged normalized autocorrelation (AC) functions of the indicated fluorescent molecules reveal the progressively slower diffusion according to molecular size as well as a pronounced long tail correlation for full-length Spir-2 (arrow). B, oligomerization of cytoplasmic full-length GFP-Spir-2 probed by single color FCS in the cytoplasm of HeLa cells. The relative brightness was calibrated with multiple GFP tandem fusion constructs. Both GFP-Spir-2 in the presence of BFA and the LAFA mutant are monomeric (n = 5–13 cells; 1xGFP versus Spir-2, not significant, p > 0.1). C and D, FCCS measured with mStrawberry-Fmn-2-FH2-FSI co-expressed with either GFP-Spir-2 or GFP-Spir-2-KIND. Focal volume was positioned in the confocal cross-sections (white cross) where intensity traces and correlation curves were recorded for both color channels simultaneously. The resulting auto- and cross-correlation functions allow quantification of the cross-correlation ratio (CCmax) as a measure for the degree of binding. Error bars represent S.D.
To rule out aggregations, we first determined the molecular brightness of GFP-Spir-2 in comparison with tandems of multiple GFP domains (Fig. 9B). The GFP chain nicely reproduced a linear increase, whereas full-length GFP-Spir-2 in the presence of BFA as well as the homogeneously distributed LAFA mutant diffused clearly as monomers. Encouraged by the monomeric nature of Spir-2, we measured cross-correlation in cells co-expressing mStrawberry-Fmn-2-FH2-FSI with either GFP-Spir-2 or GFP-Spir-2-KIND (Fig. 9, C and D). We placed the laser focus in the cytoplasm or the nucleus (not shown) at positions that produced stable intensity traces. The amplitude of the autocorrelation curves varied due to different expression levels in the upper nanomolar range (green and red autocorrelation functions in Fig. 9, C and D). As shown in these two example cells, the cross-correlation amplitude (blue) was higher for GFP-Spir-2-KIND than for the full-length Spir-2 protein, indicating the increased abundance of double labeled complexes for GFP-Spir-2-KIND.
To assess this difference in a more systematic way, we determined average values across a small population of individual cells. When co-expressed, the different mobility of full-length GFP-Spir-2 and GFP-Spir-2-KIND was largely maintained (compare Fig. 10A with 9A), whereas the Fmn-2 construct diffused about the same as full-length Spir-2 under both conditions (Fig. 10A). Therefore the mobility pattern was not a suitable readout for binding. To evaluate cross-correlation amplitudes, we included controls for spectral cross-talk and chromatic mismatch between the color channels. The accessible cross-correlation range was about 50% as determined with a strictly co-diffusing GFP-linker-mStrawberry tandem (positive control; Fig. 10B) and separately co-expressed fluorescent proteins (negative control). Note that the baseline measured for negative control is close to zero only when correcting for 12% bleed-through from GFP into the red channel (38, 40). In agreement with our previous results, cross-correlation indicated significant co-diffusion and hence binding between the Fmn-2 fragment and the KIND protein in the presence and absence of BFA. However, the average cross-correlation was drastically reduced for full-length Spir-2 under BFA as well as the LAFA mutant that exhibits by itself a homogeneous distribution in the cytoplasm (Fig. 2A). Thus, full-length Spir-2 proteins show consistently lower interaction potential toward Fmn-2 as compared with the isolated KIND domain.
FIGURE 10.

Isolated KIND domain shows increased affinity toward Fmn-2. A, normalized autocorrelation (AC) curves demonstrate the faster diffusion of the smaller GFP-Spir-2-KIND construct (green, dashed) in comparison with full-length GFP-Spir-2 (green, solid). The autocorrelation curves of mStrawberry-Fmn-2-FH2-FSI (red) are indistinguishable between the experiments. B, cross-correlation ratio (CCmax) for different cell populations in the cytoplasm: a GFP-mStrawberry fusion construct (positive control (PC)), the individually co-expressed proteins (negative control (NC)), co-expressed mStrawberry-Fmn-2-FH2-FSI with GFP-Spir-2-KIND in the presence and absence of BFA, full-length GFP-Spir-2 in the presence of BFA, and GFP-Spir-2-LAFA. C, the cross-correlation ratio correlates with protein expression levels of the individual cells. D, same experiment as B except with positioning in the nucleus. Data points represent average values of individual cells. Box-and-whisker plots indicate second and third quartile (box), mean (square), median (horizontal line), and 1.5× interquartile range (whiskers). ***, p < 0.001.
Each data point in Fig. 10B represents the average value for a single cell, and the broad distribution of cross-correlation levels reflects variations within the cell population. In fact, cross-correlation and hence the degree of binding increased linearly with increased expression levels albeit with a smaller slope for full-length Spir-2 than for isolated KIND (Fig. 10C). A correlation between expression level and the degree of binding further supports the notion that the cross-correlation values reflect a dynamic binding equilibrium. Finally, we determined cross-correlation levels for nuclear fractions of the overexpressed proteins. Although expression levels were asymmetric, these nuclear cross-correlation levels fully recapitulate the behavior in the cytoplasm (Fig. 10, compare B and D). Because the nuclear compartment is devoid of endogenous membranes, we can faithfully interpret elevated cross-correlation as a true molecular interaction that does not depend on a joint residence at some sort of vesicle.
In summary, Spir-2·Fmn-2 complexes show a dynamic, KIND/FSI-mediated interaction in the submicromolar concentration range. The binding data corroborate our view that a backfolded conformation of full-length Spir-2 protein indeed exhibits a weaker affinity toward Fmn-2.
DISCUSSION
In this study, we characterize the molecular interactions that orchestrate the assembly of a Spir·FMN actin nucleation complex at membranes. A key player is the Spir FYVE-type domain. Consistent with the lack of the conserved amino acids for phosphatidylinositol 3-phosphate binding, the Spir-2 FYVE-type zinc finger domain did not show a preferential binding to different phosphoinositides; instead it binds rather nonspecifically to a broad variety of negatively charged lipids. Furthermore, the interaction is strengthened by additional hydrophobic contributions from the turret loop that, at least in cells, seem critical for vesicular localization. The promiscuous affinity for phosphoinositides differs markedly from that of protrudin, another FYVE-type domain-containing protein that is also closely associated with Rab11 (29, 50). Moreover, as both the electrostatic and hydrophobic contributions of Spir to membrane binding are rather unspecific, we argue that the targeting toward a specified subcellular compartment needs further regulatory steps, most likely the complex formation with other membrane-bound proteins.
In membrane targeting, interactions of cationic protein clusters with electronegatively charged membranes is a major mode of binding that was not always well acknowledged (51). Here we show that the Spir-2 protein has a high affinity for negatively charged membranes. Indeed, with a pI of 9.1, the Spir-2 FYVE-type domain is supposed to be highly positively charged at physiological pH.
Phosphatidylserine (PS) is the most abundant negatively charged phospholipid in eukaryotic cells (51). PS is synthesized at the inner leaflet of the endoplasmic reticulum. However, experiments using the PS-binding lactadherin C2 domain showed that the PS headgroup is flipped to the cytosolic side in or close to the trans-Golgi network (52). From there, PS is transported via the exocytic pathway to the plasma membrane. Thus, the subcellular PS distribution reflects largely that of the Spir protein: trans-Golgi network and post-Golgi vesicles (7).
In absolute quantities, subcellular PS concentrations are as yet poorly determined. Some studies report concentrations of about 5% PS in the Golgi membranes and an overall enrichment of PS in the recycling endosome (51, 53, 54). It is therefore likely that compartments associated with Spir already contain 5–10% PS at the cytosolic leaflet, which would be enough for nonspecific recruitment. Other negatively charged lipids like PI, which was also described to be concentrated in the Golgi membrane (7–12% (53)) may even add up to this effect. However, because we do not know their exact local concentrations, it remains to be shown in which way specific lipid compositions contribute to establish the characteristic Spir distribution under physiological conditions.
We show here that the FYVE-type domain is also a protein/protein interaction module. Studying Spir-2 fragments in free solution, we discovered a strong interaction between the C-terminal FYVE-type zinc finger and the N-terminal KIND domain. Importantly, when engaged by membranes, the FYVE-type domain was unable to recruit KIND domain from bulk solution. It seems that the positive charges associated with the FYVE-type domain are screened by the negatively charged KIND domain when released from the membrane. Affinities as determined by various methods are in the lower nanomolar range and thus in the same order of magnitude as the membrane interactions, already suggesting a dynamic equilibrium of two conformational states. Brightness analysis of fluorescently tagged full-length Spir-2 in the cellular cytoplasm showed that Spir-2 diffuses as a monomer and rules out a dimeric head-to-tail conformation. Such intramolecular interactions thus could stabilize a compact, backfolded conformation as it was described for several kinases (55, 56). Because backfolded conformations are associated with autoinhibitory functions and the KIND domain is evolutionarily related to kinase domains (16), it is intriguing to speculate that the KIND/FYVE interaction also has autoregulatory functions.
Similar to the FYVE/KIND interaction, recruitment of formins involves polar interactions. The crystal structure of the Spir-1-KIND·Fmn-2-FSI complex revealed an electrostatic nature of the Spir/FMN interaction (18, 19). The positively charged C-terminal FMN FSI motif binds a negatively charged groove on the surface of the KIND domain. Here we show biochemically that our novel, intramolecular Spir-2-KIND/FYVE interaction indeed strongly competes with recruitment of Fmn-2-FSI presumably because the FMN proteins and the FYVE-type zinc finger bind to overlapping sequences within the Spir-KIND domain. These observations combine into a sequence of polar interactions that involve both actin nucleators and the vesicle membrane (Fig. 11). We propose the following model for membrane targeting of the Spir·FMN complex. Spir proteins may exist in free solution as backfolded monomers in which the two oppositely charged protein domains stabilize a presumably inactive conformation. Only upon membrane binding does the FYVE-type domain release the KIND domain, which is now accessible to recruit FMN proteins via their FSI sequence. As a result, our model provides a causal link between membrane binding of Spir and the assembly of a functional actin nucleator complex.
FIGURE 11.
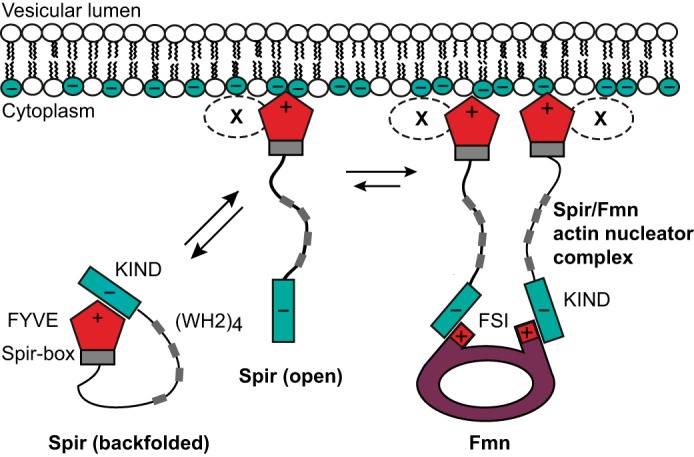
Sequential membrane targeting model of the Spir·formin actin nucleator complex. The scheme summarizes the different molecular states of the Spir/FMN interaction network. The high affinity of the FYVE/KIND interaction suggests that cytosolic Spir occurs largely as a backfolded monomer (closed conformation). Transient membrane binding mediated by the FYVE-type domain at negatively charged vesicles may release KIND (open conformation) for subsequent interaction with formins. Because the FYVE/KIND interaction involves strong contributions of charged residues, it is intriguing to speculate that the terminal Spir domains must bridge between a negatively charged vesicular membrane and the positively charged FMN FSI domain to initiate formation of a functional actin nucleator complex. Additional factors (X) may contribute in providing specificity for targeting the correct subpopulation of vesicles.
We were not yet able to characterize full-length Spir proteins in free solution. Therefore we studied transiently expressed Spir-2/Fmn-2 constructs in the cytoplasm of HeLa cells. We used dual color FCCS to measure the diffusion and interactions between differently labeled Spir-2 and Fmn-2 constructs. FCCS was measured under the continuous presence of BFA, a potent Arf inhibitor that was proposed to release the proteins from Rab11-positive vesicles in oocytes (49). Although BFA treatment produced homogeneous fluorescence distributions also in HeLa cells, autocorrelation analysis revealed that both proteins show a pronounced biphasic diffusion behavior with a second correlation time too long to represent free diffusion of protein monomers with the respective molecular size. The fact that the N-terminal Spir-2-KIND domain, which lacks an FYVE-type domain, showed much faster diffusion suggests that, even in the presence of BFA, Spir-2 and Fmn-2 show transient membrane interactions. The transient interactions of Fmn-2 may be indirectly mediated in complex with Spir-2. We detected significant interactions between Fmn-2 and the isolated KIND domain. However, in support of our model, interactions with full-length Spir-2 were much reduced, most likely reflecting intramolecular competition by the FYVE-type domain.
Our study has provided first molecular insights into the complex network of competing interactions that establish a vesicle-associated Spir·FMN complex. Such assembly is a necessary requirement for a site-specific activation of the actin machinery and the subsequent spatiotemporal regulation of actin-mediated vesicular transport processes.
Acknowledgments
We thank Christian Bökel (TU Dresden, Germany) for providing dimeric and trimeric eGFP expression vectors. We further thank Harald Stenmark (Oslo, Norway) for providing the eGFP-2x-Hrs-FYVE expression vector.
The work was supported by German Research Foundation (Deutsche Forschungsgemeinschaft (DFG), priority program SPP 1464) (to P. S. and E. K.) and the Bavarian Genome Research Network (BayGene) (to E. K.).
- FMN
- formin subgroup of formins
- KIND
- kinase non-catalytic C-lobe domain
- FSI
- formin/Spir interaction
- Fmn-2
- formin-2
- Alexa647
- Alexa Fluor 647
- AcGFP
- Aequorea coerulescens GFP
- GSH
- glutathione
- Ni-NTA
- nickel-nitrilotriacetic acid
- FF
- fast flow
- eGFP
- enhanced GFP
- eFSI
- extended FSI
- GUV
- giant unilamellar vesicle
- LUV
- large unilamellar vesicle
- EIA
- electrochemiluminescence immunoassay
- FCCS
- fluorescence cross-correlation spectroscopy
- FCS
- fluorescence correlation spectroscopy
- BFA
- brefeldin A
- LAFA
- L595A,F596A
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPA
- 1,2-dioleoyl-sn-glycero-3-phosphate
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- PIP
- phosphatidylinositol phosphate
- aa
- amino acids
- PS
- phosphatidylserine
- SARA
- SMAD anchor for receptor activation.
REFERENCES
- 1. Pollard T. D., Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dietrich S., Weiß S., Pleiser S., Kerkhoff E. (2013) Structural and functional insights into the Spir/formin actin nucleator complex. Biol. Chem. 394, 1649–1660 [DOI] [PubMed] [Google Scholar]
- 3. Pfender S., Kuznetsov V., Pleiser S., Kerkhoff E., Schuh M. (2011) Spire-type actin nucleators cooperate with Formin-2 to drive asymmetric oocyte division. Curr. Biol. 21, 955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R. C., Cota P., Wittnam J. L., Gogol-Doering A., Opitz L., Salinas-Riester G., Dettenhofer M., Kang H., Farinelli L., Chen W., Fischer A. (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756 [DOI] [PubMed] [Google Scholar]
- 5. Pleiser S., Banchaabouchi M. A., Samol-Wolf A., Farley D., Welz T., Wellbourne-Wood J., Gehring I., Linkner J., Faix J., Riemenschneider M. J., Dietrich S., Kerkhoff E. (2014) Enhanced fear expression in Spir-1 actin organizer mutant mice. Eur. J. Cell Biol. 93, 225–237 [DOI] [PubMed] [Google Scholar]
- 6. Schuh M. (2011) An actin-dependent mechanism for long-range vesicle transport. Nat. Cell Biol. 13, 1431–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kerkhoff E., Simpson J. C., Leberfinger C. B., Otto I. M., Doerks T., Bork P., Rapp U. R., Raabe T., Pepperkok R. (2001) The Spir actin organizers are involved in vesicle transport processes. Curr. Biol. 11, 1963–1968 [DOI] [PubMed] [Google Scholar]
- 8. Morel E., Parton R. G., Gruenberg J. (2009) Annexin A2-dependent polymerization of actin mediates endosome biogenesis. Dev. Cell 16, 445–457 [DOI] [PubMed] [Google Scholar]
- 9. Welz T., Wellbourne-Wood J., Kerkhoff E. (2014) Orchestration of cell surface proteins by Rab11. Trends Cell Biol. 24, 407–415 [DOI] [PubMed] [Google Scholar]
- 10. Quinlan M. E., Hilgert S., Bedrossian A., Mullins R. D., Kerkhoff E. (2007) Regulatory interactions between two actin nucleators, Spire and Cappuccino. J. Cell Biol. 179, 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quinlan M. E., Heuser J. E., Kerkhoff E., Mullins R. D. (2005) Drosophila Spire is an actin nucleation factor. Nature 433, 382–388 [DOI] [PubMed] [Google Scholar]
- 12. Montaville P., Jégou A., Pernier J., Compper C., Guichard B., Mogessie B., Schuh M., Romet-Lemonne G., Carlier M. F. (2014) Spire and Formin 2 synergize and antagonize in regulating actin assembly in meiosis by a ping-pong mechanism. PLoS Biol. 12, e1001795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dahlgaard K., Raposo A. A., Niccoli T., St Johnston D. (2007) Capu and Spire assemble a cytoplasmic actin mesh that maintains microtubule organization in the Drosophila oocyte. Dev Cell 13, 539–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quinlan M. E. (2013) Direct interaction between two actin nucleators is required in Drosophila oogenesis. Development 140, 4417–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pechlivanis M., Samol A., Kerkhoff E. (2009) Identification of a short Spir interaction sequence at the C-terminal end of formin subgroup proteins. J. Biol. Chem. 284, 25324–25333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ciccarelli F. D., Bork P., Kerkhoff E. (2003) The KIND module: a putative signalling domain evolved from the C lobe of the protein kinase fold. Trends Biochem. Sci. 28, 349–352 [DOI] [PubMed] [Google Scholar]
- 17. Huang J., Furuya A., Furuichi T. (2007) Very-KIND, a KIND domain containing RasGEF, controls dendrite growth by linking Ras small GTPases and MAP2. J. Cell Biol. 179, 539–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeth K., Pechlivanis M., Samol A., Pleiser S., Vonrhein C., Kerkhoff E. (2011) Molecular basis of actin nucleation factor cooperativity: crystal structure of the Spir-1 kinase non-catalytic C-lobe domain (KIND)*formin-2 formin SPIR interaction motif (FSI) complex. J. Biol. Chem. 286, 30732–30739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vizcarra C. L., Kreutz B., Rodal A. A., Toms A. V., Lu J., Zheng W., Quinlan M. E., Eck M. J. (2011) Structure and function of the interacting domains of Spire and Fmn-family formins. Proc. Natl. Acad. Sci. U.S.A. 108, 11884–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stenmark H., Aasland R., Toh B. H., D'Arrigo A. (1996) Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J. Biol. Chem. 271, 24048–24054 [DOI] [PubMed] [Google Scholar]
- 21. Kutateladze T. G. (2006) Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim. Biophys. Acta 1761, 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Misra S., Hurley J. H. (1999) Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE domain of Vps27p. Cell 97, 657–666 [DOI] [PubMed] [Google Scholar]
- 23. Dumas J. J., Merithew E., Sudharshan E., Rajamani D., Hayes S., Lawe D., Corvera S., Lambright D. G. (2001) Multivalent endosome targeting by homodimeric EEA1. Mol. Cell 8, 947–958 [DOI] [PubMed] [Google Scholar]
- 24. Gaullier J. M., Simonsen A., D'Arrigo A., Bremnes B., Stenmark H., Aasland R. (1998) FYVE fingers bind PtdIns(3)P. Nature 394, 432–433 [DOI] [PubMed] [Google Scholar]
- 25. Burd C. G., Emr S. D. (1998) Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol. Cell 2, 157–162 [DOI] [PubMed] [Google Scholar]
- 26. Otto I. M., Raabe T., Rennefahrt U. E., Bork P., Rapp U. R., Kerkhoff E. (2000) The p150-Spir protein provides a link between c-Jun N-terminal kinase function and actin reorganization. Curr. Biol. 10, 345–348 [DOI] [PubMed] [Google Scholar]
- 27. Ostermeier C., Brunger A. T. (1999) Structural basis of Rab effector specificity: crystal structure of the small G protein Rab3A complexed with the effector domain of rabphilin-3A. Cell 96, 363–374 [DOI] [PubMed] [Google Scholar]
- 28. Fukuda M. (2003) Distinct Rab binding specificity of Rim1, Rim2, rabphilin, and Noc2. Identification of a critical determinant of Rab3A/Rab27A recognition by Rim2. J. Biol. Chem. 278, 15373–15380 [DOI] [PubMed] [Google Scholar]
- 29. Gil J. E., Kim E., Kim I. S., Ku B., Park W. S., Oh B. H., Ryu S. H., Cho W., Heo W. D. (2012) Phosphoinositides differentially regulate protrudin localization through the FYVE domain. J. Biol. Chem. 287, 41268–41276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Angelova M. I., Dimitrov D. S. (1986) Liposome electroformation. Faraday Discuss. 81, 303 [Google Scholar]
- 31. Höfer C. T., Herrmann A., Müller P. (2010) Use of liposomes for studying interactions of soluble proteins with cellular membranes. Methods Mol. Biol. 606, 69–82 [DOI] [PubMed] [Google Scholar]
- 32. Wolny M., Grzybek M., Bok E., Chorzalska A., Lenoir M., Czogalla A., Adamczyk K., Kolondra A., Diakowski W., Overduin M., Sikorski A. F. (2011) Key amino acid residues of ankyrin-sensitive phosphatidylethanolamine/phosphatidylcholine-lipid binding site of βI-spectrin. PLoS One 6, e21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kolondra A., Lenoir M., Wolny M., Czogalla A., Overduin M., Sikorski A. F., Grzybek M. (2010) The role of hydrophobic interactions in ankyrin-spectrin complex formation. Biochim. Biophys. Acta 1798, 2084–2089 [DOI] [PubMed] [Google Scholar]
- 34. Petrásek Z., Schwille P. (2008) Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophys. J. 94, 1437–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montis C., Baglioni P., Berti D. (2014) Monitoring the interaction of nucleolipoplexes with model membranes. Soft Matter 10, 39–43 [DOI] [PubMed] [Google Scholar]
- 36. Kapusta P. (2010) Absolute diffusion coefficients: compilation of reference data for FCS calibration, in Application Note, PicoQuant GmbH, Berlin [Google Scholar]
- 37. Müller P., Schwille P., Weidemann T. (2014) PyCorrFit—generic data evaluation for fluorescence correlation spectroscopy. Bioinformatics 30, 2532–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weidemann T., Schwille P. (2013) Dual-color fluorescence cross-correlation spectroscopy with continuous laser excitation in a confocal setup. Methods Enzymol. 518, 43–70 [DOI] [PubMed] [Google Scholar]
- 39. Weidemann T., Worch R., Kurgonaite K., Hintersteiner M., Bökel C., Schwille P. (2011) Single cell analysis of ligand binding and complex formation of interleukin-4 receptor subunits. Biophys. J. 101, 2360–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bacia K., Petrášek Z., Schwille P. (2012) Correcting for spectral cross-talk in dual-color fluorescence cross-correlation spectroscopy. Chemphyschem 13, 1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ui M., Okada T., Hazeki K., Hazeki O. (1995) Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 20, 303–307 [DOI] [PubMed] [Google Scholar]
- 42. Shpetner H., Joly M., Hartley D., Corvera S. (1996) Potential sites of PI-3 kinase function in the endocytic pathway revealed by the PI-3 kinase inhibitor, wortmannin. J. Cell Biol. 132, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Komada M., Soriano P. (1999) Hrs, a FYVE finger protein localized to early endosomes, is implicated in vesicular traffic and required for ventral folding morphogenesis. Genes Dev. 13, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillooly D. J., Morrow I. C., Lindsay M., Gould R., Bryant N. J., Gaullier J. M., Parton R. G., Stenmark H. (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Czogalla A., Grzybek M., Jones W., Coskun U. (2014) Validity and applicability of membrane model systems for studying interactions of peripheral membrane proteins with lipids. Biochim. Biophys. Acta 1841, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 46. Busse R. A., Scacioc A., Hernandez J. M., Krick R., Stephan M., Janshoff A., Thumm M., Kühnel K. (2013) Qualitative and quantitative characterization of protein-phosphoinositide interactions with liposome-based methods. Autophagy 9, 770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lei M., Lu W., Meng W., Parrini M. C., Eck M. J., Mayer B. J., Harrison S. C. (2000) Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102, 387–397 [DOI] [PubMed] [Google Scholar]
- 48. Renault L., Guibert B., Cherfils J. (2003) Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 426, 525–530 [DOI] [PubMed] [Google Scholar]
- 49. Holubcová Z., Howard G., Schuh M. (2013) Vesicles modulate an actin network for asymmetric spindle positioning. Nat. Cell Biol. 15, 937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shirane M., Nakayama K. I. (2006) Protrudin induces neurite formation by directional membrane trafficking. Science 314, 818–821 [DOI] [PubMed] [Google Scholar]
- 51. Leventis P. A., Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427 [DOI] [PubMed] [Google Scholar]
- 52. Fairn G. D., Schieber N. L., Ariotti N., Murphy S., Kuerschner L., Webb R. I., Grinstein S., Parton R. G. (2011) High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 194, 257–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zambrano F., Fleischer S., Fleischer B. (1975) Lipid composition of the Golgi apparatus of rat kidney and liver in comparison with other subcellular organelles. Biochim. Biophys. Acta 380, 357–369 [DOI] [PubMed] [Google Scholar]
- 54. Gagescu R., Demaurex N., Parton R. G., Hunziker W., Huber L. A., Gruenberg J. (2000) The recycling endosome of Madin-Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol. Biol. Cell 11, 2775–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pufall M. A., Graves B. J. (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18, 421–462 [DOI] [PubMed] [Google Scholar]
- 56. Boggon T. J., Eck M. J. (2004) Structure and regulation of Src family kinases. Oncogene 23, 7918–7927 [DOI] [PubMed] [Google Scholar]