

Background: Anthrax toxin is the primary cause of pathology from exposure to anthrax spores.
Results: Two linked single domain antibodies (VHHs), each neutralizing anthrax toxicity by different mechanisms, potently protect mice from anthrax spore challenge.
Conclusion: Linked, toxin-neutralizing VHHs (VNAs) are highly effective anthrax antitoxin agents.
Significance: VNAs offer excellent versatility in developing novel therapeutics for many toxin-mediated diseases.
Keywords: Anthrax Toxin, Antibody, Antibody Engineering, Receptor, Toxin
Abstract
Anthrax disease is caused by a toxin consisting of protective antigen (PA), lethal factor, and edema factor. Antibodies against PA have been shown to be protective against the disease. Variable domains of camelid heavy chain-only antibodies (VHHs) with affinity for PA were obtained from immunized alpacas and screened for anthrax neutralizing activity in macrophage toxicity assays. Two classes of neutralizing VHHs were identified recognizing distinct, non-overlapping epitopes. One class recognizes domain 4 of PA at a well characterized neutralizing site through which PA binds to its cellular receptor. A second neutralizing VHH (JKH-C7) recognizes a novel epitope. This antibody inhibits conversion of the PA oligomer from “pre-pore” to its SDS and heat-resistant “pore” conformation while not preventing cleavage of full-length 83-kDa PA (PA83) by cell surface proteases to its oligomer-competent 63-kDa form (PA63). The antibody prevents endocytosis of the cell surface-generated PA63 subunit but not preformed PA63 oligomers formed in solution. JKH-C7 and the receptor-blocking VHH class (JIK-B8) were expressed as a heterodimeric VHH-based neutralizing agent (VNA2-PA). This VNA displayed improved neutralizing potency in cell assays and protected mice from anthrax toxin challenge with much better efficacy than the separate component VHHs. The VNA protected virtually all mice when separately administered at a 1:1 ratio to toxin and protected mice against Bacillus anthracis spore infection. Thus, our studies show the potential of VNAs as anthrax therapeutics. Due to their simple and stable nature, VNAs should be amenable to genetic delivery or administration via respiratory routes.
Introduction
Anthrax, the disease caused by the Gram-positive bacterium Bacillus anthracis, is a major bioterror concern. After introduction in spore form and germination, the bacterium divides and manifests disease and lethality primarily through the action of two toxins, lethal toxin (LT)4 and edema toxin. These toxins have a common receptor binding component, protective antigen (PA) that is responsible for transport of the lethal factor metalloprotease (LF) or edema factor adenylate cyclase (EF) into the host cell cytosol. The injection of the toxins into animals can replicate symptoms of anthrax disease (for review see Refs. 1 and 2).
PA acts as the “gateway” that allows the translocation and action of both toxins. Full-length PA is an 83-kDa polypeptide (PA83) that is rapidly cleaved by cell surface proteases such as furin to a 63-kDa form (PA63). Only the PA63 form oligomerizes as heptamers or octamers that provide the binding sites for LF or EF. The oligomer bound to one or more molecules of LF/EF is then rapidly translocated into cells. When recombinant PA83 is intentionally cleaved before exposure to cells or purified as the PA63 polypeptide, it rapidly oligomerizes in solution, and the preformed oligomer can also bind and transport LF/EF into cells. The PA63 oligomer undergoes a conformational change in acidic endosomes to a heat and SDS-stable form that allows the translocation of LF and EF through a central pore into the cytosol. LF and EF can then act on their substrates and manifest toxic effects (for review, see Refs. 1 and 2).
During anthrax infection the accumulation of anthrax toxins in the blood leads to lethality. Because both toxins require PA for their action, this protein has been the primary target of therapeutics, including antibodies developed for treatment of anthrax (3). The efficacy of the currently licensed anthrax vaccine depends on its induction of antibodies to PA (4). The majority of neutralizing antibodies developed against PA act on the receptor binding domain (domain 4) to inhibit interaction of the toxin with cells. A few antibodies have also been identified that neutralize PA by other mechanisms (for review see Ref. 3).
Camelid animals produce a heavy chain-only antibody from which the 14-kDa variable domains (called VHHs) are well expressed in bacteria as recombinant proteins that are unusually stable to pH and elevated temperatures (5, 6). VHHs often target active sites that may be inaccessible to larger conventional antibodies (7, 8) and have proven to be effective as toxin neutralizing agents (9–16). We have found that linking two or more neutralizing VHHs recognizing non-overlapping epitopes into heteromultimers (VHH-based neutralizing agents (VNAs)) often provides major improvements in the in vivo protection from toxin exposure as compared with the unlinked component VHHs (11–13, 16).
In this paper we report the identification of a panel of VHHs that recognize PA. Potent toxin-neutralizing VHHs were identified that recognize two non-overlapping epitopes. Characterization of the mechanisms by which these VHHs neutralize anthrax toxin reveals that one VHH class (represented by JIK-B8) binds to the well characterized neutralizing epitope through which PA (both PA83 and PA63 forms) binds to its receptor. A second neutralizing and unique VHH, JKH-C7, inhibits transition of the cell surface-generated PA63 oligomer from pre-pore to the acid and SDS-stable pore-forming conformation in endosomes by blocking endocytosis of cell surface-generated PA63. Linking JIK-B8 and JKH-C7 VHHs into a heterodimeric VNA resulted in an agent with an IC50 of ∼100 pm that proved highly effective in protecting mice from anthrax toxin or spore challenges. This VNA should offer new and cost-effective therapeutic options for treating anthrax exposures.
EXPERIMENTAL PROCEDURES
Ethics Statement
All studies followed protocols approved by the Tufts University and NIAID Animal Care and Use Committees. Work with alpacas was done at Tufts under approved protocol Tufts University School of Veterinary Medicine IACUC #G2011-08. All mouse studies were performed at NIAID under approved protocols LPD8E and LPD9E.
Toxins and Spores
Endotoxin-free mutant PA proteins, wild type PA83, PA63, and LF were purified from B. anthracis as previously described (17). The PAΔΔ is a mutant in which amino acids 162–167 and 304–317 are deleted such that it cannot be cleaved by furin and accumulates on the cell surface (reported here). PAdFF is a mutant in which phenylalanine residues 313 and 314 have been deleted, making it unable to translocate LF and EF (18). Concentrations of LT correspond to the concentration of each toxin component (i.e. 1 μg/ml LT is 1 μg/ml PA + 1 μg/ml LF). Spores of the non-encapsulated, toxigenic Sterne-like strain A35 (19) used to infect mice were prepared as previously described (20).
Reagents
Rabbit anti-PA83 polyclonal serum #5308 was made in one of our laboratories as was the neutralizing anti-PA mouse monoclonal antibody (mAb) 14B7, which blocks binding of PA (both PA83 and PA63) to its cellular receptors (21). Antibodies against the N terminus of MEK1 (Calbiochem), horse radish peroxidase (HRP)-conjugated and non-conjugated anti-E-tag polyclonal antibodies (Bethyl Laboratories, Montgomery, TX), and various IR-dye tagged secondary antibodies (Rockland Immunochemicals, Pottstown, PA) were purchased. The dye 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma.
VHH-display Library Preparation from Immunized Alpacas
Three alpacas were immunized with PA83 (100 μg) by five successive multi-site subcutaneous injections at 3-week intervals. For the first immunization, the adjuvant was alum/CpG, whereas subsequent immunizations used only alum. All alpacas achieved ELISA anti-PA titers of 1:1,000,000. Blood was obtained for lymphocyte preparation 7 days after the fifth immunization, and RNA was prepared using the RNeasy kit (Qiagen, Valencia, CA). Two VHH-display phage libraries were prepared as described previously (22, 23) except that the forward and reverse primers used to amplify the VHH coding region repertoire contained Not1 and Asc1 sites, which were used for ligation into the JSC vector for gene III phage display. The first library (JIG-2) was from peripheral blood lymphocytes of one immunized alpaca and contained ∼1 × 107 independent clones, whereas the second library (JKF-1) was generated from a pool of peripheral blood lymphocytes of the other two alpacas and contained ∼3 × 107 independent clones.
ELISAs
Purified VHH agents were serially diluted onto ELISA plates coated with 1 μg/ml different PA proteins, incubated for 1 h at room temperature, washed, and then incubated 1 h with HRP-anti-E-tag. Bound HRP was detected using 3,3′,5,5′-tetramethylbenzidine (Sigma), and values were plotted as a function of the input VHH concentration. EC50 values were calculated as the VHH concentration that produced a signal equal to 50% of the maximum signal.
Anti-PA VHH Identification and Preparation
Phage library panning, phage recovery, and clone fingerprinting were performed as previously described (11, 22, 23) with the following variations. The first panning process utilized the JIG-2 VHH-display library and employed purified PA83 or PA63 coated onto Nunc Immunotubes at 10 μg/ml for the first low stringency pan and then 1 μg/ml for the second high stringency pan. After two panning cycles, 70% of random clones selected on each target produced a signal >2× background, and clones producing the strongest “bug supernatant” ELISA (12) signals on plates coated with 0.5 μg/ml PA83 were fingerprinted. All VHHs panned on PA83 or PA63 recognized both PA83 and PA63. VHH coding sequences were determined for 24 clones displaying clearly unique fingerprints (12). Sequence alignments revealed 11 distinct homology groups. Amino acid sequences of clones representing each group are shown in Fig. 1A. A second panning process using the JKF-1 library was performed as for the first panning but using only PA83 as the target. ∼300 colonies were picked randomly and screened by bug supernatant ELISA on replica plates coated with either 1) 0.5 μg/ml PA or 2) 3 μg/ml 14B7 mAb (24) followed by 1 μg/ml PA83. Screening on 14B7-captured PA83 was performed to block binding of VHHs recognizing the dominant epitope (C-group 1, Fig. 1B) that was identified after the screening of the first library. ∼70% of clones recognized PA83, whereas only ∼20% recognized 14B7-captured PA83. Based on ELISA data and DNA fingerprinting, ∼70 different VHH coding sequences were obtained. Their alignment permitted the identification of eight new homology groups not previously identified in the first screen (bottom 8 in Fig. 1, A and B). At least one VHH from each homology group was selected for protein expression. Expression and purification of VHHs in Escherichia coli as recombinant thioredoxin fusion proteins containing hexahistidine were performed as previously described (23). VHH heterodimers were engineered to be linked by a 15-amino acid flexible spacer ((GGGGS)3). All VHHs were expressed with a C-terminal E-tag epitope. Competition ELISA analysis was performed as previously described with minor modifications (11).
FIGURE 1.

PA binding VHHs identified in this work. A, amino acid sequences of unique VHHs selected for binding to anthrax PA are shown. Sequences shown begin within framework 1 at the site of the primer binding employed in coding sequence DNA amplification from the immune alpaca cDNA (43) and continue through the end of framework 4. The parentheses at the end indicate whether the VHH contains a long hinge (lh) or a short hinge (sh). The three complementarity determining regions (CDR) are indicated at the top. B, table of the VHH names and binding properties. The first 11 VHHs were obtained by panning on PA83 bound to plastic, and the KD values for these VHHs were assessed by SPR-Proteon. The second group of nine VHHs were obtained by panning on 14B7-bound PA83 and the KD values assessed by SPR-Biacore. The KD for JIK-B8, obtained as an internal reference for SPR-Biacore group, was 1 ± 0.7. EC50 values were assessed by dilution ELISAs. IC50 values were assessed by toxin neutralization assays on macrophages and competition groups (C-groups) by competition ELISA. N/A refers to antibodies that did not neutralize toxin and thus have no measurable IC50.
Affinity Analyses
Studies to assess the kinetic parameters of the VHHs were performed by surface plasmon resonance using either a ProteOn XPR36 Protein Interaction Array System (Bio-Rad; upper antibody set in Fig. 1B) or a Biacore 3000 (GE Healthcare; lower antibody set in Fig. 1B). In each case the VHH was immobilized to the chip (GLH for ProteOn, CM5 for Biacore) by amine coupling chemistry, involving sequential, 1) activation of the chip surface with a mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and sulfo-N-hydoxysuccinimide (sulfo-NHS), 2) injection of PA83 at pH 5 (sodium acetate buffer), and 3) deactivation with an ethanolamine injection.
For the ProteOn data set, a range of PA concentrations was passed over the chip surface at 100 μl/min for 60 s, and dissociation was recorded for 600 or 1200 s. Running buffer for these studies was 10 mm Hepes, pH 7.4, 150 mm NaCl, 0.005% Tween 20. The surface was regenerated between runs with a 30-s injection of 50 mm HCl at 50 μl/min. Data were evaluated with ProteOn Manager software (Version 3.1.0.6) using the Langmuir interaction model to obtain KD values. Reported values are the mean of at least four runs.
For the Biacore data set, VHHs were passed over the PA immobilized on the chip surface at 100 nm and 100 μl/min for 60 s, and dissociation was recorded for 600 or 1200 s. Running buffer for these studies was 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% Tween 20. The surface was regenerated between runs with a 30-s injection of 10 mm glycine, pH 3, at 50 μl/min. Dissociation and association phases of each curve were fit separately using BIAevaluation software (GE Healthcare) using the 1:1 Langmuir model to obtain KD values. Reported values are the means of three runs. A series of four runs at 100 nm through 2 μm JKO-B8 resulted in comparable KD values at each concentration. A negative control VHH (anti-EF) did not exhibit any binding to the PA-coated chip. JIK-B8 was run at the beginning and end of the series to provide a point of comparison to the ProteOn data set.
Toxicity and Neutralization Assays
RAW264.7 mouse macrophages were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 10 mm HEPES, and 50 μg/ml gentamicin (all purchased from Invitrogen). For neutralization assays PA83 and LF (250 ng/ml) in serum-free Dulbecco's modified Eagle's medium) were incubated with various dilutions of antibody in 96-well plates for 1 h before the addition to RAW264.7 macrophages. Viability was assessed by MTT staining as previously described (25), when >90% of toxin-treated controls were lysed as assessed by light microscopy. In certain experiments PA83 or PA63 (1 μg/ml) were prebound to antibodies or were added to cells at 37 °C or 4 °C followed after 1 h by washing with serum-free DMEM at the same temperature and the addition of medium containing LF or antibodies prepared in LF (1 μg/ml). Cells were then incubated at 37 °C for 12–16 h. Viability was assessed after this period by MTT staining relative to untreated cell controls.
Cell Binding and Uptake Studies
Analyses of PA binding and oligomer formation were performed using CHO-WTP4 cells. Toxin or antibody prebound toxin were prepared in serum-free DMEM and bound to cells for 1 h before washing and lysis in radioimmune precipitation assay buffer as previously described (26). In parallel experiments, trypsin (0.25% trypsin-EDTA, Invitrogen) was used to detach cells and strip cell surface proteins. Trypsin was inactivated by the addition of 50% fetal calf serum, cells were centrifuged (1000 rpm, 10 min), washed twice with PBS, and lysed in radioimmune precipitation assay buffer. Western blot analyses were performed as previously described with loading of equal amounts of cell lysate protein in each lane (26).
Mouse Studies
For toxin challenge, Balb/cJ mice (female, 8 weeks old, The Jackson Laboratory, Bar Harbor, ME) were treated with antibody agents by the intravenous route at the doses (molar ratios relative to PA) and times indicated in figure legends. Mice were challenged with LT (45 μg, intravenous) and monitored for 10 days for survival. For spore challenges, C57BL/6J mice (8 weeks old, female, The Jackson Laboratory) were challenged with the lethal dose of 2 × 107 spores (subcutaneously, 200 μl) before or after antibody administration (subcutaneously) at various doses or times as noted in figure legends.
RESULTS
Anthrax PA Binding VHHs
VHH-display phage libraries were prepared from three alpacas immunized with purified anthrax PA83. Two separate libraries were selected for clones binding to PA83 or, alternatively, to PA83 immobilized on mAb 14B7. The mAb 14B7 is a well characterized neutralizing mAb that binds to an immunodominant epitope through which PA binds to its receptor. A total of 19 “unique” VHHs (with apparently unrelated coding sequences) were identified (Fig. 1A). Competition assays between the various VHHs and with 14B7 and 2D3 mAbs (which bind distinct regions of PA83 (24)) showed the identified VHHs fall into four distinct competition groups (C-groups designated in Fig. 1B). Thus these antibodies likely bind to four non-overlapping epitopes on PA. Not surprisingly, 10 of 11 VHHs selected by binding to PA83-coated tubes (Fig. 1B, top half) competed with 14B7 (Fig. 2A and C-group 1 in Fig. 1B). Eight unique PA binding VHHs, including six new VHHs that bind PA83 at sites different than 14B7 (Fig. 2A and C-groups 2, 3, and 4 in Fig. 1B) were subsequently selected by binding to 14B7-immobilized (and thereby blocked) PA (Fig. 1B, second half). Binding of one VHH (JIJ-B8) was not blocked by either 14B7 or 2D3 (data not shown), and binding of another (JKO-H2) was inhibited by both (Fig. 2, A and B). All VHHs were characterized for PA affinity by dilution ELISA (for EC50) and by surface plasmon resonance (for KD) (Fig. 1B). A selected VHH representative of each of the four epitope competition groups is shaded in Fig. 1B. These are JIK-B8 (C-group 1), JKH-D12 (C-group 2), JKH-C7 (C-group 3), and JKO-H2 (C-group 4).
FIGURE 2.
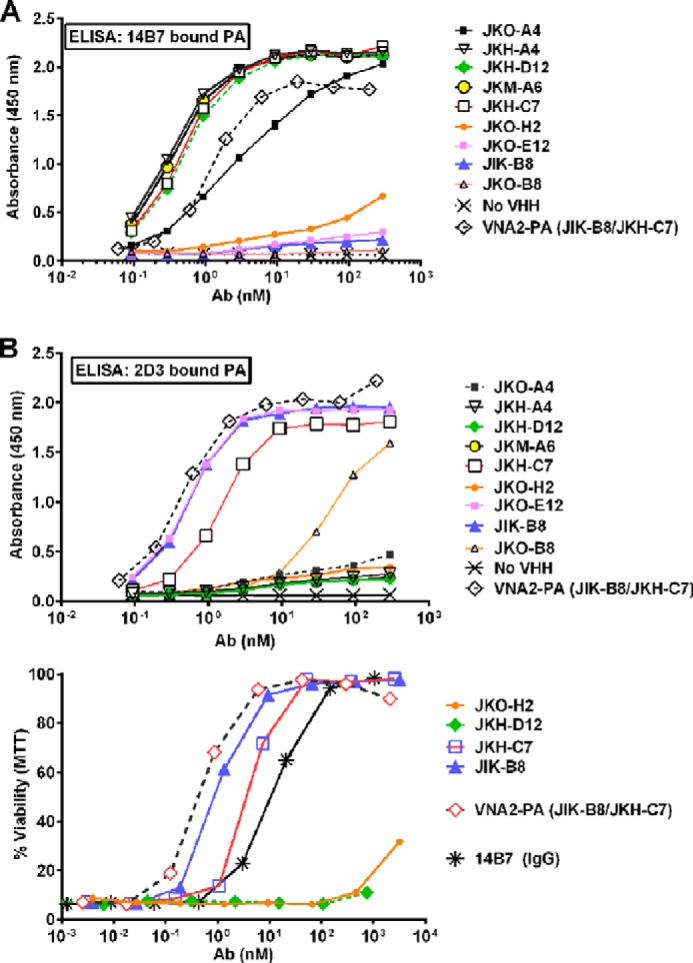
Competition ELISAs and neutralization assays. A and B, 14B7- or 2D3-captured PA was bound to plates, and varying concentration of each VHH were then added and their binding assessed with an HRP-conjugated anti-His6 or E-tag antibody (Ab) using standard ELISA protocols. C, representative neutralization assays for the four VHHs representing the four competition groups, a heteromultimer of two neutralizing VHHs, and mAb 14B7 are shown. All assays are representative of at least two separate experiments.
Anthrax Toxin Neutralization
Cell-based anthrax toxin neutralization assays performed on all 19 unique VHHs showed potencies ranging from IC50 of ∼200 pm to no activity in an assay using PA at 1.25 nm (Fig. 1B; representative assay with antibodies from each competition group shown in Fig. 2C). VHHs recognizing the immunodominant PA domain (C-group 1) differed widely in their ability to neutralize the toxin, with 4 of 12 showing no neutralizing ability. This was not surprising as our mutagenesis studies (21) previously identified single amino acid substitutions in this region that substantially altered binding to the anthrax receptor. VHHs JIK-B8 and JKO-E12 of the C-group 1 class displayed the highest affinity and lowest IC50 values, whereas only one VHH recognizing a second epitope (JKH-C7, C-group 3, Fig. 1B) showed potent anthrax neutralizing activity (Fig. 2C). VHHs recognizing C-groups 2 and 4 showed weak or undetectable toxin neutralizing activity (Fig. 2C). VHH JKO-H2 (C-group 4) displayed no recognition of PA63, suggesting that furin cleavage either removes the epitope or alters it in a manner that it cannot be recognized.
Mechanisms of VHH-mediated Neutralization
To decipher the mechanism of neutralization by VHH JIK-B8 and JKH-C7, we first determined whether the antibodies, as expected, prevented LF entry into the cytoplasm. This was assessed by monitoring cleavage of the toxin MEK1 substrate in RAW264.7 macrophages. Both VHHs prevented cleavage of MEK1 (Fig. 3). We next tested the effect of these VHHs on binding of PA83 to cells, cleavage of PA83 to PA63 by cell surface proteases, and formation of the SDS- and heat-stable PA63 oligomer. The stable oligomer forms only when PA63 is endocytosed through the receptor-mediated pathway and reaches an acidic intracellular endosome compartment (27, 28). PA83 is not endocytosis-competent, whereas PA63 is almost instantly recruited to lipid rafts and endocytosed (29). JIK-B8, previously shown to bind the same epitope as mAb 14B7, blocked PA from cell binding (Fig. 4A) as expected.
FIGURE 3.
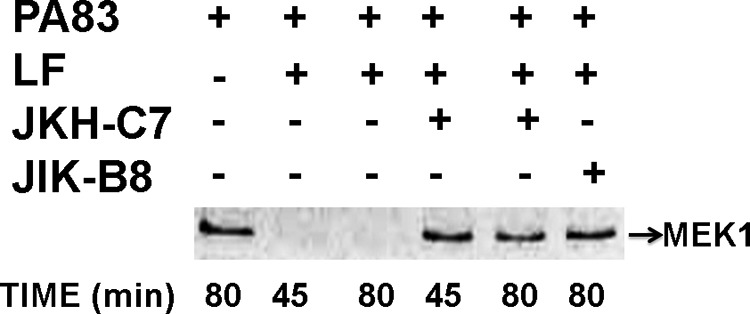
JKH-C7 and JIK-B8 inhibit LF-mediated MEK-1 cleavage in macrophages. RAW264.7 cells were treated with PA83 + LF (200 ng/ml each) for the shown amounts of time or were first preincubated with 1 μg/ml VHHs for 30 min before the addition to cells for the indicated times. Cell lysates were subjected to Western blotting with an antibody that reacts to the MEK-1 N-terminal epitope that is lost after cleavage by LF. Western blots are representative of at least two separate experiments.
FIGURE 4.

JKH-C7 does not prevent PA binding or cleavage and inhibits oligomer endocytosis. Various PA83, mutant PA proteins, or PA63 (1 μg/ml) were preincubated with antibodies (VHH:toxin, 4:1) in serum-free DMEM for 30 min to 1 h before the addition to CHO WTP4 cells for 60 min. Cells were either lysed directly in radioimmune precipitation assay buffer without trypsinization or trypsinized as described under “Experimental Procedures” to remove all cell surface proteins before lysis. Western blotting was performed with anti-PA polyclonal (1:5000), and in B–E, the blots were re-probed with goat anti-E-tag antibody (1:1000) to detect the JKH-C7 VHH in or on cells. IR-dye-conjugated secondary antibodies of different wavelengths were used to detect the PA and anti-E-tag antibodies in panels B and D where the re-probed section of the blot is shown as a separate panel. In panels C and E a single wavelength IR-dye secondary was used for detection of both primary antibodies, and the whole re-probed blot is shown. In panel A both a color and black and white image for the same blot probed with a single IR-dye secondary is shown for better identification of oligomer and lanes. CR stands for “cross-reactive” band, which serves as a nonspecific equal loading control. Western blots are representative of at least two separate experiments.
JKH-C7 appeared to have a novel neutralizing mechanism. It did not prevent cleavage of PA83 to PA63 after binding of the toxin to cellular receptors, but no SDS-stable oligomer formed when PA83 was prebound to this VHH (Fig. 4A). This result suggested that the antibody either prevents formation of the PA63 oligomer at the cell surface, inhibits endocytosis of the oligomer, or prevents endosomal conversion of the pre-pore to the SDS-stable pore.
To test if PA63 was trapped on the cell surface by the VHH, we performed cell surface proteolysis experiments. PA83 was bound and internalized for 60 min, and then cells were treated with trypsin in a manner that strips all PA63 and PA83 from the cell surface. Lysates of cells treated in this way contained only the internalized SDS-resistant oligomer (lane 4, Fig. 4B). Under these conditions, the JKH-C7 antibody did not protect the PA63 from trypsin (lane 5, Fig. 4B), indicating that it was at the cell surface. Furthermore, the toxin-bound VHH could be detected in cell lysates using an anti-E-tag antibody, and it was also fully susceptible to proteolysis. This indicated the PA63-VHH complex was not endocytosed. Interestingly, a PA mutant PAΔΔ (deleted for two loops: one containing the furin cleavage site in PA83 and the other a flexible loop required for translocation-competent oligomer formation) was also able to localize the VHH to cells, as indicated by its detection in lysates (lane 7, Fig. 4B). This showed that these two loops were not part of the epitope recognized by this VHH. Surprisingly, we consistently found that the uncleaved mutant PAΔΔ toxin was partially protected from cell surface trypsin treatment, suggesting that this mutant has a trypsin-resistant conformation or, alternatively, could be endocytosed as a monomer. However, the PAΔΔ-associated VHH was susceptible to trypsin treatment (lane 9, Fig. 4B), arguing against endocytosis of the complex. We concluded that JKH-C7 VHH, although incapable of impacting PA83 binding to cellular receptors or its cleavage to PA63, could prevent endocytosis of the cell surface-generated PA63.
PA63 produced in vitro by cleavage with trypsin or furin and purified by anion-exchange chromatography is strictly oligomeric. Incubation of this purified PA63 with the JKH-C7 antibody before binding to cells did not alter its uptake and conversion to SDS and heat-stable oligomers (Fig. 4C). This suggested that the antibody recognizes full-length PA83 but not the preformed PA63 oligomer. Furthermore, even if bound to the oligomer, JKH-C7 is unable to prevent its endocytosis. This VHH bound equally well to a PA mutant, PAdFF, in which two phenylalanine residues required for oligomer formation are deleted (Fig. 4C), but bound poorly to purified PA63 (Fig. 4, C and D, E-tag reactive band). When purified PA63 was prebound to the VHH before exposure to cells and cell surface proteolysis experiments were performed, a substantial portion of the PA63 was protected from proteolysis. At the same time the small amount of JKH-C7 at the cell surface was completely trypsinized (Fig. 4E). Therefore, a larger portion of the purified PA63 monomers were endocytosed to a protected compartment in a manner not impacted by JKH-C7. We conclude that this VHH is a novel PA neutralizing antibody that binds to PA83 at a site independent of the furin cleavage site and inhibits cell surface-generated PA63 from endocytosis by the normal pathway that allows formation of stable oligomers. The antibody is less able, however, to prevent endocytosis of preformed PA63 oligomers.
We next wanted to test the functional ability of the antibody to block neutralization when PA83 or PA63 was prebound to cells before exposure to the antibody. When JKH-C7 was prebound to PA83 and then added to cells in the presence of LF, it was fully protective to the same level as the receptor blocker, JIK-B8. When PA83 was prebound to cells at 4 °C (without processing to PA63) and the VHH was added followed by a shift to 37 °C in media containing LF, there was only a 20–30% increase in cell viability (relative to controls in the same experiments that were not exposed to the antibody). This indicated that JKH-C7 was not as effective in binding to its epitope and targeting toxin endocytosis/oligomerization for receptor-bound PA83 compared with as yet unbound PA83 (Fig. 5A, left bars). Furthermore, when PA63 was utilized in these neutralization assays, the JKH-C7 was ineffective in protection both if the PA63 was prebound to cells at 4 °C or if the PA63 was exposed to antibody before cells (Fig. 5A, right bars). This indicated that PA63 (or its oligomer) does not have the epitope for this antibody present. Not surprisingly, JIK-B8 was also unable to provide protection if PA83 or PA63 was prebound to cells, although a slight (20%) protective effect was consistently seen with PA83.
FIGURE 5.
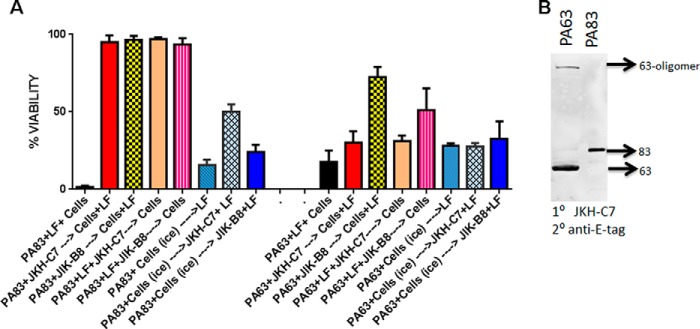
JKH-C7 does not inhibit preformed PA63 oligomer function. A, in columns 1–5 and 9–13, PA83 or PA63 (1 μg/ml) with or without LF (1 μg/ml) were incubated with VHH (4:1 VHH:toxin) or with medium (columns 1 and 9) for 1 h before the addition to cells. If LF was not present initially, it was present on cells (1 μg/ml) when the antibody-bound PA83 of PA63 was added. Cells were washed after 1 h of toxin binding and incubated overnight in complete medium before viability testing. In parallel plates, the PA83 or PA63 (1 μg/ml) was first bound to cells on ice for 1 h and washed with cold medium before the addition of the first VHH, transfer to 37 °C, and then LF in same amounts described above. Cells stayed at 37 °C for overnight incubation followed by viability assessment. The viability was calculated relative to untreated controls. Data presented in this graph are a representative experiment in which each column is the average of three replicate wells. B, purified PA63 or PA83 were boiled in SDS-loading buffer before SDS-PAGE and Western blotting in a triple sandwich using JKH-C7 as primary antibody, the goat anti-E-tag antibody as a secondary antibody and an IRDye-conjugated anti-goat antibody to detect the E-tag antibody. Western blots are representative of at least two separate experiments.
These results suggested that the epitope for JKH-C7 likely required full-length folded PA83 and was somehow eliminated in cell-bound PA63 or blocked in the preformed PA63 oligomers. Early observations that showed poor binding of JKH-C7 to plastic-immobilized PA83 suggested recognition of a conformation-dependent epitope. However, when used as a primary antibody in denaturing Westerns, JKH-C7 detected purified SDS-denatured PA83 and PA63 and was weakly reactive toward the PA63 oligomer (Fig. 5B). This suggests that the epitope, if dependent on conformation, is one that can renature after SDS treatment.
Heterodimeric VNAs Protect against Anthrax Toxin and Spore Infection in Mice
Linking toxin-neutralizing VHHs into heteromultimeric VNAs has been found to improve toxin affinity and, more importantly, to substantially improve in vivo antitoxin efficacy (11–13, 16). A heterodimeric VNA (VNA2-PA) consisting of the two potent neutralizing VHHs, JIK-B8 and JKH-C7, separated by a short unstructured peptide, was expressed and purified (amino acid sequence shown in Fig. 6A) and found to be potent in neutralizing toxin activity (Fig. 2C). VNA2-PA was then compared with monomeric VHHs for the ability to protect mice from anthrax toxin. The toxin dose administered by the intravenous route was between 1 and 2 LD100 (lethal dose 100%) (45 μg of LT) for the Balb/cJ strain. Treatment doses were selected so as to test efficacy at various molar ratios of agent to toxin. Heterodimeric VNAs each bind at two separate sites on each toxin, so a dose that can fully occupy both binding sites must be a 2:1 molar ratio agent:toxin. Neither monomer VHH was able to protect mice or provide any beneficial effect at a 1:1 molar ratio to toxin, whereas VNA2-PA was highly protective when used at this ratio (Fig. 6B). Thus VNA2-PA was able to shift the time to death significantly even at submolar ratios to toxin. Importantly, the heterodimer also performed better than a pool of the two VHHs used with toxin in a 1:1:1 ratio with toxin, providing evidence of the improved in vivo efficacy of the heterodimer form (Fig. 6B). Surprisingly, VNA2-PA treatment 2 h post-toxin administration was also highly protective (Fig. 6B). This finding was unexpected in light of the fact that the bulk of PA has been shown to be cleaved to PA63 and removed from circulation by 2 h after a similar bolus administration (30). This suggests that a significant amount of active PA not measurable in circulation (plasma) may remain accessible to antibody at crucial tissue sites. A second VHH heterodimer (VNA1-PA), incorporating the neutralizing JIK-B8 VHH with a non-neutralizing VHH (JIJ-B8), failed to provide any protection when administered 1:1 but was fully protective in this assay when provided at a 2-fold molar excess (Fig. 6, C and D).
FIGURE 6.

VNA amino acid sequence and antitoxin protection in animal models. A, VNA2-PA translation product sequence. Shown is the protein sequence of the entire VNA2-PA translation product expressed in E. coli. The VNA contains an N-terminal thioredoxin fusion partner and hexahistidine encoded by the pET32b expression vector. The VHH sequences are flanked by two E-tag peptides (underlined) and separated by an unstructured spacer ((GGGGS)3). A 14-amino acid albumin binding peptide (ABP), DICLPRWGCLEWED (36), is at the carboxyl end, separated from the second E-tag by a GGGGS spacer. For clarity, the eight defined protein segments are separated by slashed (///). B, Balb/cJ mice (n = 5/group, except PBS control group, n = 10) were injected intravenous with antibody at the indicated molar ratios (Ab:toxin) 10 min before injection with LT (45 μg for each toxin component, intravenous) except a single group marked (POST) that received antibody 2 h after toxin was administered. Control groups received PBS instead of antibody. Animals were monitored for 10 days post-intoxication for signs of malaise and survival. C, protein sequence of the entire VNA1-PA translation product with same specifications described in panel A. This VNA is a heterodimer of the neutralizing VHH JIK-B8 and the high affinity non-neutralizing antibody JIJ-B8. D, Balb/cJ mice were injected intravenously with antibody at the indicated molar ratios (Ab:toxin) 10 min before injection with LT (45 μg for each toxin component, intravenous). Control groups received PBS instead of antibody. Animals were monitored for 10 days post infection for signs of malaise and survival. E, C57BL/6J mice (n = 5/group, except PBS controls, n = 15) were treated with heterodimeric VNA2-PA (subcutaneously) at the indicated times and doses before or after spore infection (2 × 107 spores, also subcutaneously at the distal site). Control mice were treated with PBS at 15 min, 1 h, and 4 h post infection (n = 5) or at 5 min (n = 5) and 8 h (n = 5) post infection. Neutralizing mAb 14B7 was used as a positive control in these studies. Mice were monitored for survival and signs of malaise for 10 days.
VNA2-PA was also tested alongside 14B7 in protection of C57BL/6J mice against infection with a one LD100 (lethal dose 100%) dose of the A35 Sterne-like toxigenic B. anthracis strain. Antibody provided 15 min before subcutaneous spore infection or 3 times at 15, 60, and 240 min post-infection was also fully protective (Fig. 6E). No protection was provided when antibody was administered 20–24 h post-toxin (data not shown). The latter result was expected as our previous studies have shown that the survival of the un-encapsulated A35 strain in subcutaneous administrations depends on the disabling of the myeloid cells of the host innate immune system through production of toxins early in infection (31).
A single administration of the VNA2-PA antibody at the lower dose of 30 μg at 4 h post infection resulted in survival of 2/5 mice. Mice treated with this dose of 14B7 did not survive, likely because only one-third the number of antibody molecules was present compared with VNA2-PA. Increasing the time gap between spore infection and antibody administration to 8 h resulted in a complete loss of protection unless antibody was increased to a much higher dose of 250 μg, where a surprising full protection was observed (Fig. 6E).
DISCUSSION
Anthrax is a toxigenic disease that can rapidly progress to result in lethality for the host if left untreated. The bioterrorist attacks utilizing B. anthracis spores highlighted the need for cost-effective treatments that could be produced on a large scale if necessary. Almost all the therapeutics developed against the disease focus on the anthrax toxins, which have been demonstrated to be the primary virulence determinants. In this work we hypothesized that a novel recombinant antitoxin agent consisting of a heterodimer of two distinct toxin-neutralizing, camelid anti-anthrax PA heavy chain VHH binding domains would serve as an effective therapeutic for anthrax exposure. This hypothesis was based on our earlier findings of unusually high in vivo antitoxin potency achieved for Clostridium botulinum neurotoxins (11), ricin (13), Shiga toxins (12), and Clostridium difficile (16) toxins using heteromultimers of toxin-neutralizing VHHs called VNAs.
A number of conventional mAbs have been produced against the PA receptor binding component of the tripartite toxin that neutralizes anthrax in cell-based assays and is shown to provide protection in animal models (for review, see Ref. 3). Most antibodies target the same receptor binding epitope of the toxin and differ only in their affinities and clearance rates. The novel VHH-based VNA reported here consists of two anti-toxin VHHs targeting independent epitopes of PA and inhibits toxin functions at two different steps. The agent proved as effective in protecting against anthrax toxin and spore challenges in a mouse model as the well characterized neutralizing 14B7 mAb. 14B7 acts at the same epitope as the approved human anti-PA mAb, raxibacumab (Abthrax).
VHHs, the components of VNAs, have numerous potential commercial and therapeutic advantages over mAb products (for review, see Refs. 32 and 6). We have found that VHH heteromultimer VNAs possess additional potential therapeutic advantages over conventional mAb products such as 1) the option to co-administer an anti-tag effector antibody that binds multiple tags on the VNAs (11, 13) to promote antibody Fc effector functions such as serum clearance (33), 2) the ability to target multiple toxins with one biomolecule (12, 16), and 3) the use of VNA gene therapy as an effective means to provide prolonged antitoxin protection (34, 35). Simple addition of an albumin binding peptide (34, 36) can dramatically improve the serum stability of VNAs (34). Because VHHs can be obtained from preexisting libraries (37) and then affinity-matured for improved properties (38, 39), new antitoxins can be rapidly developed as treatments for emerging threat agents. VHHs are known to be poorly immunogenic (5), and immunogenicity can by reduced further by introducing targeted mutations (40).
The novel neutralization mechanism for one of the VHHs in the VNA2-PA, JKH-C7, has some similarities to anti-PA antibodies AVP-21D9 and AVP22G12, which inhibit PA oligomer formation (41). The difference is that these antibodies prevent PA63 oligomer formation in solution, whereas the VHH primarily acts when bound to PA83 and does not disrupt preformed PA63 oligomers. Similar to those antibodies, the JKH-C7 VHH does not prevent binding of PA83 to its cellular receptor and does not impact the cleavage of PA83 to PA63 by cellular furin, but it does prevent endocytosis of the cell-bound PA63. It binds equally well to a toxin mutant in which the furin loop has been deleted, indicating an epitope independent of the receptor binding domain or cleavage domain. The antibody is not effective against preformed PA63 oligomers or cell-bound PA63, suggesting an epitope that requires full-length PA83 for neutralizing efficacy or an epitope on a PA63 surface involved in monomer-monomer association.
The antitoxin strategy reported here, which uses VNAs consisting of two or more linked, toxin-neutralizing VHHs recognizing non-overlapping epitopes on PA, is a novel approach to treating toxin exposures. Although linking VHHs together increases the total contact area with toxin target and thus binding avidity, we find that VNAs with two or more toxin-neutralizing VHHs, such as VNA2-PA, often increase in vivo antitoxin efficacy beyond that which can be explained by improved avidity (11,13).5 This is highlighted by the comparison with VNA1-PA (Fig. 6, C and D) in which only one of the two VHHs is toxin-neutralizing. This VNA displays very similar EC50 and IC50 properties but is significantly less effective in mouse anthrax challenge models. A likely explanation is that VNA2-PA targets two different steps in the toxin action on cells. First, one arm of the VNA prevents binding of PA to cellular receptors (Fig. 7). Although this is typically sufficient to neutralize toxin, the preformed PA63 oligomer requires binding of multiple antibodies to efficiently prevent cell binding, as evidenced by the inability of JIK-B8 to completely stop the oligomer's association with cells (Fig. 4E). The second arm of the VNA prevents oligomerization and endocytosis that converts the pre-pore to SDS-stable pore (Fig. 7).
FIGURE 7.

Model for mechanism of action of two VHHs that make up neutralizing VNA2-PA: PA83 normally binds to cellular receptors and is cleaved by cellular furin to form PA63. PA63 monomers immediately oligomerize to form the binding sites for LF. The oligomer complex is endocytosed (with or without LF cargo) and upon arrival in the acidic endosomal environment alters in conformation to form a pore through which LF can be translocated to the cytosol where it cleaves its substrates. VNA2-PA, which is a heterodimer of JIK-B8 and JKH-C7, inhibits this process through two mechanisms. The JIK-B8 arm binds to the receptor binding domain of PA83 and inhibits binding of the toxin to its receptor. The JKH-C7 arm binds to a conformational epitope on PA83 that does not prevent cleavage of the toxin to PA63 form but prevents oligomerization of PA63 at the cell surface or its endocytosis into the cell.
Another benefit of VNAs, which target two distinct sites on the toxin, is the reduced likelihood of toxin mutants arising that can escape the antitoxin activity. Escape mutants may be even less likely in the case of VNA2-PA due to the JKH-C7 VHH component that neutralizes anthrax via an apparently complex mechanism clearly distinct from simple inhibition of receptor binding. The bulk of anti-PA neutralizing antibodies target the same receptor binding epitope that the JIK-B8 arm of VNA2-PA targets, and the receptor binding epitope can be destroyed by genetic manipulation of the PA antigen to eliminate reactivity with these neutralizing antibodies (21). The VNA2-PA antibody described in this work offers an alternative therapeutic in which the epitope for both arms is unlikely to be disrupted without impact on PA function.
VNA2-PA or similar VNAs should permit a variety of new options for prevention and treatment of anthrax intoxication. Studies presented here demonstrate that this VNA protects mice from toxin or anthrax spore challenge with similar or improved efficacy compared with a conventional mAb antitoxin product. Future directions will include testing the use of gene therapy to promote long term serum expression of antitoxin VNA to prophylactically prevent intoxication of individuals at high risk of exposure using an approach successfully applied in mice for botulism (34) and in pigs for Shiga toxin-producing E. coli infection (35). Gene therapy could include co-expression of VNA and effector antibody for enhanced potency (11–13), and potentially the gene delivery vehicle could selectively target the pulmonary system (42) for improved efficacy from aerosol exposures. Because VHHs are highly stable proteins, it should be possible to administer VNAs by aerosol or nasal routes of administration for more rapid and targeted treatment of anthrax exposures.
Acknowledgments
We thank Drs. Daniela Bedenice and Jean Mukherjee and members of the Mukherjee laboratory for excellent assistance in handling the alpacas, performing the immunizations, and obtaining serum and peripheral blood lymphocytes used in VHH-display library construction.
This work was supported, in whole or in part, by National Institutes of Health Grant U54 AI057159 (NIAID) and by the Intramural Research Program of the National Institutes of Health (NIAID).
M. Moayeri, C. E. Leysath, J. M. Tremblay, C. Vrentas, D. Crown, S. H. Leppla, and C. B. Shoemaker, unpublished information.
- LT
- lethal toxin
- VHH
- variable domain of a camelid heavy chain-only antibody
- VNA
- VHH-based neutralizing agent, a heteromultimer of neutralizing VHHs
- PA
- protective antigen
- EF
- edema factor
- LF
- lethal factor
- MTT
- 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide.
REFERENCES
- 1. Liu S., Moayeri M., Leppla S. H. (2014) Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 22, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moayeri M., Leppla S. H. (2009) Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 30, 439–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Z., Moayeri M., Purcell R. (2011) Monoclonal antibody therapies against anthrax. Toxins 3, 1004–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaur M., Singh S., Bhatnagar R. (2013) Anthrax vaccines: present status and future prospects. Expert Rev. Vaccines 12, 955–970 [DOI] [PubMed] [Google Scholar]
- 5. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 [DOI] [PubMed] [Google Scholar]
- 6. Hassanzadeh-Ghassabeh G., Devoogdt N., De Pauw P., Vincke C., Muyldermans S. (2013) Nanobodies and their potential applications. Nanomedicine 8, 1013–1026 [DOI] [PubMed] [Google Scholar]
- 7. Lauwereys M., Arbabi Ghahroudi M., Desmyter A., Kinne J., Hölzer W., De Genst E., Wyns L., Muyldermans S. (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17, 3512–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abderrazek R. B., Hmila I., Vincke C., Benlasfar Z., Pellis M., Dabbek H., Saerens D., El Ayeb M., Muyldermans S., Bouhaouala-Zahar B. (2009) Identification of potent nanobodies to neutralize the most poisonous polypeptide from scorpion venom. Biochem. J. 424, 263–272 [DOI] [PubMed] [Google Scholar]
- 10. Hmila I., Abdallah R., B. A., Saerens D., Benlasfar Z., Conrath K., Ayeb M. E., Muyldermans S., Bouhaouala-Zahar B. (2008) VHH, bivalent domains and chimeric heavy chain-only antibodies with high neutralizing efficacy for scorpion toxin AahI'. Mol. Immunol. 45, 3847–3856 [DOI] [PubMed] [Google Scholar]
- 11. Mukherjee J., Tremblay J. M., Leysath C. E., Ofori K., Baldwin K., Feng X., Bedenice D., Webb R. P., Wright P. M., Smith L. A., Tzipori S., Shoemaker C. B. (2012) A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PloS ONE 7, e29941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tremblay J. M., Mukherjee J., Leysath C. E., Debatis M., Ofori K., Baldwin K., Boucher C., Peters R., Beamer G., Sheoran A., Bedenice D., Tzipori S., Shoemaker C. B. (2013) A single VHH-based toxin-neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect Immun. 81, 4592–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vance D. J., Tremblay J. M., Mantis N. J., Shoemaker C. B. (2013) Stepwise engineering of heterodimeric single domain camelid VHH antibodies that passively protect mice from ricin toxin. J. Biol. Chem. 288, 36538–36547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richard G., Meyers A. J., McLean M. D., Arbabi-Ghahroudi M., MacKenzie R., Hall J. C. (2013) In vivo neutralization of α-cobratoxin with high-affinity llama single-domain antibodies (VHHs) and a VHH-Fc antibody. PloS ONE 8, e69495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bakherad H., Mousavi Gargari S. L., Rasooli I., Rajabibazl M., Mohammadi M., Ebrahimizadeh W., Safaee Ardakani L., Zare H. (2013) In vivo neutralization of botulinum neurotoxins serotype E with heavy-chain camelid antibodies (VHH). Mol. Biotechnol. 55, 159–167 [DOI] [PubMed] [Google Scholar]
- 16. Yang Z., Schmidt D., Liu W., Li S., Shi L., Sheng J., Chen K., Yu H., Tremblay J. M., Chen X., Piepenbrink K. H., Sundberg E. J., Kelly C. P., Bai G., Shoemaker C. B., Feng H. (2014) A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect Dis. 210, 964–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S., Leppla S. H. (2000) Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr. Purif. 18, 293–302 [DOI] [PubMed] [Google Scholar]
- 18. Singh Y., Klimpel K. R., Arora N., Sharma M., Leppla S. H. (1994) The chymotrypsin-sensitive site, FFD315, in anthrax toxin protective antigen is required for translocation of lethal factor. J. Biol. Chem. 269, 29039–29046 [PubMed] [Google Scholar]
- 19. Pomerantsev A. P., Sitaraman R., Galloway C. R., Kivovich V., Leppla S. H. (2006) Genome engineering in Bacillus anthracis using Cre recombinase. Infect Immun. 74, 682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moayeri M., Crown D., Newman Z. L., Okugawa S., Eckhaus M., Cataisson C., Liu S., Sastalla I., Leppla S. H. (2010) Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling, and neutrophil recruitment. PLoS Pathog. 6, e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosovitz M. J., Schuck P., Varughese M., Chopra A. P., Mehra V., Singh Y., McGinnis L. M., Leppla S. H. (2003) Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J. Biol. Chem. 278, 30936–30944 [DOI] [PubMed] [Google Scholar]
- 22. Maass D. R., Harrison G. B., Grant W. N., Shoemaker C. B. (2007) Three surface antigens dominate the mucosal antibody response to gastrointestinal L3-stage strongylid nematodes in field immune sheep. Int. J. Parasitol. 37, 953–962 [DOI] [PubMed] [Google Scholar]
- 23. Tremblay J. M., Kuo C. L., Abeijon C., Sepulveda J., Oyler G., Hu X., Jin M. M., Shoemaker C. B. (2010) Camelid single domain antibodies (VHHs) as neuronal cell intrabody binding agents and inhibitors of Clostridium botulinum neurotoxin (BoNT) proteases. Toxicon 56, 990–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Little S. F., Leppla S. H., Cora E. (1988) Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect Immun. 56, 1807–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z., Moayeri M., Crown D., Emerson S., Gorshkova I., Schuck P., Leppla S. H., Purcell R. H. (2009) Novel chimpanzee/human monoclonal antibodies that neutralize anthrax lethal factor, and evidence for possible synergy with anti-protective antigen antibody. Infect Immun. 77, 3902–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moayeri M., Wickliffe K. E., Wiggins J. F., Leppla S. H. (2006) Oxidized ATP protection against anthrax lethal toxin. Infect. Immun. 74, 3707–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Goot G., Young J. A. (2009) Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 30, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller C. J., Elliott J. L., Collier R. J. (1999) Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 38, 10432–10441 [DOI] [PubMed] [Google Scholar]
- 29. Abrami L., Liu S., Cosson P., Leppla S. H., van der Goot F. G. (2003) Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 160, 321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moayeri M., Wiggins J. F., Leppla S. H. (2007) Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun. 75, 5175–5184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu S., Miller-Randolph S., Crown D., Moayeri M., Sastalla I., Okugawa S., Leppla S. H. (2010) Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host. Microbe. 8, 455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muyldermans S., Atarhouch T., Saldanha J., Barbosa J. A., Hamers R. (1994) Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 7, 1129–1135 [DOI] [PubMed] [Google Scholar]
- 33. Sepulveda J., Mukherjee J., Tzipori S., Simpson L. L., Shoemaker C. B. (2010) Efficient serum clearance of botulinum neurotoxin achieved using a pool of small antitoxin binding agents. Infect. Immun. 78, 756–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mukherjee J., Dmitriev I., Debatis M., Tremblay J. M., Beamer G., Kashentseva E. A., Curiel D. T., Shoemaker C. B. (2014) Prolonged prophylactic protection from botulism with a single adenovirus treatment promoting serum expression of a VHH-based antitoxin protein. PloS ONE 9, e106422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheoran A. S., Dmitriev I. P., Kashentseva E. A., Cohen O., Mukherjee J., Debatis M., Shearer J., Tremblay J. M., Beamer G., Curiel D. T., Shoemaker C. B., Tzipori S. (2015) Adenovirus vector expressing Stx1/2-neutralizing agent protects piglets infected with E. coli O157:H7 against fatal systemic intoxication. Infect. Immun. 83, 286–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen A., Reyes A. E., 2nd, Zhang M., McDonald P., Wong W. L., Damico L. A., Dennis M. S. (2006) The pharmacokinetics of an albumin-binding Fab (AB. Fab) can be modulated as a function of affinity for albumin. PEDS 19, 291–297 [DOI] [PubMed] [Google Scholar]
- 37. Verheesen P., Roussis A., de Haard H. J., Groot A. J., Stam J. C., den Dunnen J. T., Frants R. R., Verkleij A. J., Theo Verrips C., van der Maarel S. M. (2006) Reliable and controllable antibody fragment selections from Camelid non-immune libraries for target validation. Biochim. Biophys. Acta 1764, 1307–1319 [DOI] [PubMed] [Google Scholar]
- 38. Boder E. T., Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 [DOI] [PubMed] [Google Scholar]
- 39. Razai A., Garcia-Rodriguez C., Lou J., Geren I. N., Forsyth C. M., Robles Y., Tsai R., Smith T. J., Smith L. A., Siegel R. W., Feldhaus M., Marks J. D. (2005) Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J. Mol. Biol. 351, 158–169 [DOI] [PubMed] [Google Scholar]
- 40. Vincke C., Loris R., Saerens D., Martinez-Rodriguez S., Muyldermans S., Conrath K. (2009) General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 284, 3273–3284 [DOI] [PubMed] [Google Scholar]
- 41. Wang F., Ruther P., Jiang I., Sawada-Hirai R., Sun S. M., Nedellec R., Morrow P. R., Kang A. S. (2004) Human monoclonal antibodies that neutralize anthrax toxin by inhibiting heptamer assembly. Hum. Antibodies 13, 105–110 [PubMed] [Google Scholar]
- 42. Alberti M. O., Deshane J. S., Chaplin D. D., Pereboeva L., Curiel D. T., Roth J. C. (2013) A myeloid cell-binding adenovirus efficiently targets gene transfer to the lung and escapes liver tropism. Gene. Ther. 20, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maass D. R., Sepulveda J., Pernthaner A., Shoemaker C. B. (2007) Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs). J. Immunol. Methods 324, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]