

Background: ADAMDEC1 is an ADAM-like metalloprotease with a rare active site affecting the proteolytic activity.
Results: Reconstruction of the ADAMDEC1 active site, based on the ADAM family consensus, increases proteolytic activity and susceptibility for inhibition.
Conclusion: Specific structural features may protect ADAMDEC1 from endogenous metalloprotease inhibitors.
Significance: ADAMDEC1 has evolved features resulting in narrow substrate specificity and restricted reactivity with endogenous protease inhibitors.
Keywords: ADAM, ADAMTS, Metalloprotease, Substrate Specificity, Tissue Inhibitor of Metalloproteinase (TIMP), Metzincin
Abstract
ADAMDEC1 is a proteolytically active metzincin metalloprotease displaying rare active site architecture with a zinc-binding Asp residue (Asp-362). We previously demonstrated that substitution of Asp-362 for a His residue, thereby reconstituting the canonical metzincin zinc-binding environment with three His zinc ligands, increases the proteolytic activity. The protease also has an atypically short domain structure with an odd number of Cys residues in the metalloprotease domain. Here, we investigated how these rare structural features in the ADAMDEC1 metalloprotease domain impact the proteolytic activity, the substrate specificity, and the effect of inhibitors. We identified carboxymethylated transferrin (Cm-Tf) as a new ADAMDEC1 substrate and determined the primary and secondary cleavage sites, which suggests a strong preference for Leu in the P1′ position. Cys392, present in humans but only partially conserved within sequenced ADAMDEC1 orthologs, was found to be unpaired, and substitution of Cys392 for a Ser increased the reactivity with α2-macroglobulin but not with casein or Cm-Tf. Substitution of Asp362 for His resulted in a general increase in proteolytic activity and a change in substrate specificity was observed with Cm-Tf. ADAMDEC1 was inhibited by the small molecule inhibitor batimastat but not by tissue inhibitor of metalloproteases (TIMP)-1, TIMP-2, or the N-terminal inhibitory domain of TIMP-3 (N-TIMP-3). However, N-TIMP-3 displayed profound inhibitory activity against the D362H variants with a reconstituted consensus metzincin zinc-binding environment. We hypothesize that these unique features of ADAMDEC1 may have evolved to escape from inhibition by endogenous metalloprotease inhibitors.
Introduction
ADAMDEC1 (decysin-1) is a novel “a disintegrin and metalloprotease” (ADAM)4-like protease bearing closest resemblance to ADAM-28 and ADAM-7, with sequence identities of 47 and 36%, respectively. The ADAM family comprises type 1 transmembrane metzincin metalloproteases, which, in addition to the metzincin metalloprotease domain, have multiple C-terminal ancillary domains, including a disintegrin-like domain, a Cys-rich domain, an EGF-like domain, a transmembrane domain, and a cytoplasmic tail (1, 2). In proteolytically active ADAM family members, the metalloprotease domain contains an active site consisting of an elongated zinc-binding motif, HEXXHXXGXXH, with three underlined zinc-coordinating ligands (2, 3).
Like other ADAMs, ADAMDEC1 mRNA encodes an N-terminal signal peptide directing secretion and a relatively large (173 residue) prodomain, which is processed during maturation, presumably by a member of the proprotein convertase family (4, 5). The mature protein comprises a metzincin metalloprotease domain and a short disintegrin-like domain. Consequently, ADAMDEC1 lacks most of the ancillary domains otherwise present in the ADAMs and is secreted as a soluble protein (5–7). The metalloprotease domain harbors a rare metzincin-type active site consensus sequence (HEXXHXXGXXD), where the third zinc-binding ligand is an Asp residue, otherwise only found in a few bacterial metzincins like snapalysin (2). ADAMDEC1 is suggested to be the first (and only) member of a novel subgroup of mammalian ADAMs based on the differences in primary structure (6).
ADAMDEC1 mRNA has been detected in monocytes, and the level increases during differentiation into mature macrophages (8). ADAMDEC1 mRNA is undetectable in immature dendritic cells, but expression is strongly induced during maturation (6, 8). In addition, ADAMDEC1 expression has been found to be differentially regulated in a number of pathological conditions (9–15). However, the physiological roles of ADAMDEC1 remain elusive.
Here we show that ADAMDEC1 cleaves carboxymethylated transferrin (Cm-Tf) and determine the cleavage sites. We also demonstrate that special features of ADAMDEC1, including an unpaired Cys residue (Cys392) near the active site in the metalloprotease domain and the active site Zn2+-ligating Asp residue, have functional impacts on proteolytic activity, specificity, and effect of inhibitors. Finally, we present a homology-based model of the tertiary structure of mature ADAMDEC1.
EXPERIMENTAL PROCEDURES
Materials
The FreeStyle 293 Expression System, including all additives, SDS-PAGE, zymography, and Western blot materials (Novex/iBlot System), was from Invitrogen. Azocasein and batimastat (BB-94) were from Sigma-Aldrich. Plasma-derived human α2-macroglobulin (α2M) was a kind gift from Prof. Lars Sottrup-Jensen (Aarhus University) and Henrik Østergaard (Novo Nordisk A/S). TIMP-1, TIMP-2, N-TIMP-3, and Cm-Tf were prepared as described previously (16–18).
Cloning, Site-directed Mutagenesis, and Protein Expression
An expression system for human ADAMDEC1 variants was described previously (5). A similar expression vector was generated for expression of the murine ADAMDEC1. cDNA encoding murine ADAMDEC1 was purchased from ImaGenes (Berlin, Germany) and cloned into a pCI-neo vector (Promega) using a forward (tacgactcactataggctagcatgctgcctgggact) and reverse (cctcactaaagggaagcggccgctcattctgtgatgtgg) primer. An HPC4 affinity tag inserted 2 amino acids downstream of the furin recognition site (RTSR203 (4)) was created by overlap extension PCR using two internal primers: cttcatttgggttttttttgccatcaatcagacgcggatccacc and gaacttccaggtcactcgaagatcaggtggatccgcgtctgattg. Site-directed mutagenesis of C392S in hADAMDEC1 was carried out using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies) with mutagenesis primers (ggacttcagcacaagctccagagcccacttcgag and the reverse complementary primer). All primers were purchased from MWG Eurofins (Ebersberg, Germany). For protein expression in HEK293-6E, cells were grown at 37 °C and 8% CO2 in FreeStyle 293 expression medium supplemented with 25 μg/ml G-418 Geneticin and 0.1% (v/v) Pluronic F-68. pCI-neo-based expression plasmid encoding ADAMDEC1 variants were used for transfection according to the FreeStyle 293 protocol (Invitrogen). As a control, the supernatant from non-transfected cells (mock transfection) was collected. When applicable, the HEK293-6E conditioned medium containing ADAMDEC1 variants was concentrated using Amicon Ultra 10,000 molecular weight cut-off centrifugal filters (Millipore).
Determination of Disulfide Pattern
Protein bands were excised from a Coomassie Brilliant Blue-stained gel and split into two parts, to one of which was added reducing agent (40 mm DTT final concentration) and to the other buffer. Both samples were alkylated using iodoacetic acid and subjected to in-gel digestion (19). The peptides were purified using C18 StageTips (20) and eluted onto an anchor chip target plate (Bruker) using 2,5-dihydroxybenzoic acid matrix in 0.1% (w/v) trifluoroacetic acid and 70% (v/v) acetonitrile. Samples were analyzed on a Bruker Reflex III MALDI-TOF mass spectrometer (Bruker Daltonik GmbH (Bremen, Germany), Basis-REFLEX SCOUT 384) in reflector mode. External calibration of the mass spectrometer was performed immediately before data acquisition using angiotensin II (Sigma-Aldrich), ACTH clip 18-39 (Sigma-Aldrich), bombesin (Fluka), and somatostatin (Sigma-Aldrich).
Proteolytic Activity Assays
The relative concentrations of ADAMDEC1 variants were evaluated by densitometric analysis of a Coomassie Brilliant Blue-stained SDS-polyacrylamide gel using the Phoretix 1D software (TotalLab). Proteolysis of human plasma-derived α2M was carried out by incubating 25 nm ADAMDEC1 variant protein with 800 nm α2M in reaction buffer (50 mm Hepes (pH 7.5), 100 mm NaCl, 5 mm CaCl2, 5 μm ZnCl2) for ∼20 h at 37 °C. Cross-linking of ADAMDEC1 to α2M was visualized by reducing SDS-PAGE followed by Western blot analysis using 1.3 μg/ml anti-HPC4 tag antibody and 1.0 μg/ml HRP-labeled goat anti-human IgG (PerkinElmer Life Sciences) for detection or 1.0 μg/ml anti-hADAMDEC1 (Abcam ab57224) with 1.0 μg/ml HRP-labeled rabbit anti-mouse IgG (Dako) for detection. Carboxymethylated transferrin (3.75 mg/ml) was used as a substrate for hADAMDEC1 variants (100–500 nm) in reaction buffer. Samples were analyzed by reducing SDS-PAGE, and densitometry analysis was carried out using Phoretix 1D software (TotalLab). Proteolytic activity against azocasein was measured by incubating 7.8 μm ADAMDEC1 with 94 μm substrate at 37 °C for 72 h. The proteolysis was terminated by the addition of 6% (w/v) trichloroacetic acid (TCA) followed by a 30-min incubation on ice and removal of undigested protein by centrifugation at 10,000 × g and 4 °C for 10 min. 0.26 mm NaOH was added to the supernatants, and the absorbance was measured at 440 nm using a SpectraMax 190 microplate reader (Molecular Devices). From the absorption data was subtracted the reaction buffer contribution, and the data were compared with the data obtained with supernatant from mock-transfected cells using one-way analysis of variance with Bonferroni's adjustment. Individual sample pairs were additionally compared using an unpaired, two-tailed Student's t test. For casein zymography, 4–16% zymogram blue casein gel and buffers (Invitrogen) were used according to the manufacturer's protocol. For inhibition of ADAMDEC1 with batimastat (BB-94), the ADAMDEC1 variants were preincubated with a 10–100-fold molar excess of BB-94 for 1 h at 37 °C before the addition of substrate. Characterization of TIMP inhibition of ADAMDEC1 was performed by preincubation of ADAMDEC1 with TIMP-1 (5–10-fold molar excess), TIMP-2 (5–10-fold molar excess), or N-TIMP-3 (up to 3-fold molar excess), respectively, for 4 h at 37 °C prior to the addition of substrate.
N-terminal Sequence Analysis
Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and stained using Coomassie Brilliant Blue. Individual bands were excised and subjected to Edman amino acid sequence analysis using an Applied Biosystems Procise HT protein sequencer with on-line identification of phenylthiohydantoin derivatives.
Homology Modeling
Amino acid sequences were obtained from the UniProt Knowledgebase and aligned using MUSCLE (21). In order to generate a structure model of hADAMDEC1, a homology modeling approach was pursued as implemented in the computer program Modeler (22). Based on an input sequence alignment between the mature hADAMDEC1 amino acid sequence and template proteins with known structures, a three-dimensional model was generated. Loop regions not resolved in the template protein were predicted by a limited functionality for ab initio structure prediction. In the present study, the template protein was ADAM-8 (PDB entry 4DD8, resolution 2.1 Å (23)) for the metalloprotease domain (residues 204–410) and ADAM-22 (PDB entry 3G5C, resolution 2.36 Å (24)) for the disintegrin-like domain structure (residues 411–470) and the relative domain orientation. The resulting model was processed by CHARMM (25). The active site region of hADAMDEC1 WT was modeled after snapalysin (PDB entry 1C7K, resolution 1 Å (26)) and relaxed by energy minimization, resulting in close coordination of active site residues to the catalytic zinc ion. Furthermore, N-TIMP-3 was docked in the model based on the structure of the ADAM-17·N-TIMP-3 complex (PDB entry 3CKI, resolution 2.30 Å (27)), and a substrate model addressing both the non-prime and prime substrate binding sites was generated inspired by the crystal structure of MMP-9 with bound active site probe (PDB entry 4JQG, resolution 1.85 Å (28)).
RESULTS
Human ADAMDEC1 Has an Unpaired, Reactive Cys in the Metalloprotease Domain
Comparing the amino acid sequences of human and murine ADAMDEC1 with that of the related hADAM-28 and hADAM-8 illustrates a modified Cys pattern of the ADAMDEC1 metalloprotease domain (Fig. 1A). In the majority of the mammalian ADAMs, the metzincin metalloprotease domain contains six Cys residues forming three intrachain disulfide bonds in a C1–C6, C2–C5, C3–C4 pattern (29, 30). However, the hADAMDEC1 metalloprotease domain contains only five Cys residues. Cys328, Cys369, Cys374, and Cys407 of hADAMDEC1 align with Cys residues in the related ADAMs, presumably forming the two disulfide bonds corresponding to C1–C6 (Cys328–Cys407) and C3–C4 (Cys369–Cys374) (Fig. 1A). Both human and murine ADAMDEC1 lack Cys residues corresponding to the C2–C5 disulfide bond. However, hADAMDEC1 contains a fifth Cys residue in the metalloprotease domain (Cys392), which we hypothesized to be unpaired. Indeed, hADAMDEC1 wild type (hWT) formed conjugates with thiol-reactive 5-kDa PEG-maleimide, resulting in a decrease in electrophoretic mobility. Furthermore, substitution of Cys392 for a Ser (hC392S) blocked the reaction with the PEG-maleimide (Fig. 1B), strongly suggesting that Cys392 exists as an unpaired and reactive Cys residue. The status of Cys392 was further probed by tryptic peptide mass fingerprinting analyzed by MALDI-TOF mass spectrometry (Fig. 1, C and D). hADAMDEC1 was excised from a non-reducing SDS-PAGE gel, alkylated under either reducing or non-reducing conditions, and digested with trypsin (Fig. 1C). In the non-reduced alkylated sample, a peptide with a mass corresponding to hADAMDEC1 residues 387–398, containing Cys392 in an alkylated state (MH+ 1513.2/1512.6 (observed/expected)), was observed, demonstrating that Cys392 is not bound to any other Cys residue (Fig. 1D). In addition, we observed a fragment mass in the non-reduced sample consistent with the expected tryptic peptide CPSGSCVMNQYLSSK containing Cys369 and Cys374 engaged in an internal disulfide bond (MH+ 1601.2/1601.7) (Fig. 1D). A peptide equivalent to the same fragment modified by oxidation (ox), presumably affecting the Met residue in the peptide, was also observed. Both of these fragments were absent in the reduced sample, and a new peak appeared, corresponding to the same peptide modified with two carboxymethyl groups (MH+ 1719.3/1717.69) (Fig. 1D). Together, this demonstrates that Cys369 and Cys374 form a disulfide bond corresponding to C3–C4 in other ADAMs. The mass spectrometry analysis was consistent with the presence of a Cys328–Cys407 disulfide bond; however, due to the close proximity of the disintegrin domain Cys residues with no Lys or Arg residues in between, the disulfide bond pattern of this peptide cluster cannot be fully resolved by the method employed (data not shown).
FIGURE 1.

ADAMDEC1 contains a unique Cys pattern. A, multiple sequence alignment comparing the full sequence of mature hADAMDEC1 and mADAMDEC1 with the N-terminal part of mature hADAM-28 and -8. Disulfide bonds in the metalloprotease domain of hADAM-8 are shown by black lines connecting Cys residues (gray boxes) and are based on the crystal structure (23). The metzincin active site, the conserved Met residue in the hallmark Met-turn (Met376), and a conserved structurally important hydrophobic Phe anchor (Phe388) are all marked by black boxes. The identified proprotein convertase-processing sites of human and murine ADAMDEC1 are marked by a separate black box (4, 5). The numbering above selected key residues refers to the hADAMDEC1 full-length sequence. B, thiol-specific PEGylation of hWT and hC392S analyzed by SDS-PAGE. C, experimental setup for tryptic peptide mass fingerprinting using MALDI-TOF mass spectrometry. D, tryptic peptide mass fingerprinting of non-reduced (blue) and reduced (red) human pro-ADAMDEC1 (hADAMDEC1 R56A/R200K/R203A (5)). The mass spectrum of m/z 1500–1730 is shown. Two additional expected ADAMDEC1 tryptic peptides, VVPSASTTFDNFLR (*, residues 279–292) and QTPELTLHEIVCmPK (**, residues 35–48), where Cm denotes a carboxymethylated Cys residue, are also visible in the depicted mass range.
Cys392 Affects hADAMDEC1 Activity in a Substrate-specific Manner
Where hADAMDEC1 has an unpaired Cys at position 392, mADAMDEC1 has a Ser residue instead (Fig. 1A). To investigate the possible significance of Cys392 for the proteolytic activity of hADAMDEC1, it was substituted for a Ser residue (hC392S). The reactivity with human α2M was analyzed using an α2M-cross-linking assay, demonstrating a small increase in the activity of hC392S relative to hADAMDEC1 (Fig. 2A). Thus, a Ser residue at position 392 changes the reactivity of hADAMDEC1 toward human α2M. Interestingly, murine ADAMDEC1 (mWT), also with a Ser residue at position 392, exhibits significantly higher reactivity toward human α2M than both hADAMDEC1 and hC392S (Fig. 2A). In contrast to the observation in the α2M-cross-linking assay, the C392S substitution does not impact the caseinolytic activity of hADAMDEC1, measured by the release of TCA-soluble azo-labeled peptides (Fig. 2B). In addition to the two known substrates of ADAMDEC1, Cm-Tf is cleaved into two major products, with apparent molecular masses of 76 and 20 kDa, respectively, when incubated with hADAMDEC1. These products are not present when incubating with supernatant from mock-transfected cells, demonstrating hADAMDEC1-specific cleavage of Cm-Tf at one primary site (Fig. 2C). As with casein, the C392S-substitution does not change the activity toward Cm-Tf. Interestingly, mADAMDEC1 does not display any measurable caseinolytic activity or any observable cleavage of Cm-Tf (data not shown). Substitution of the catalytic Glu353 for an Ala in hADAMDEC1 (hE353A) completely abrogated the catalytic activity against all substrates (Fig. 2, B and C; shown previously (5)).
FIGURE 2.
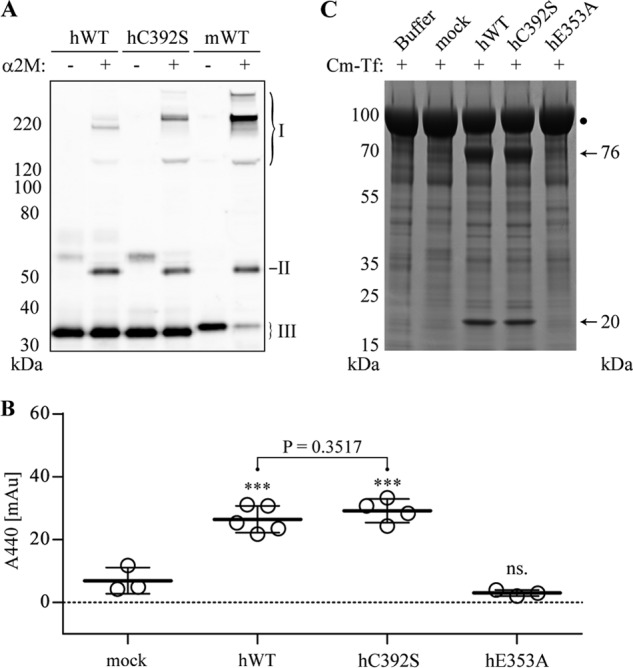
Influence of Cys392 on the proteolytic activity of hADAMDEC1. A, α2M-cross-linking assay with hADAMDEC1 variants and mWT visualized by Western blot analysis using an anti-HPC4-tag mAb against a N-terminal HPC4 tag on the ADAMDEC1 variants. The different α2M·ADAMDEC1 complexes (I) are observed at the top of the blots, whereas the added protease (III) is seen at the bottom. Also, cross-reactivity of antibodies with an impurity in the α2M preparation is observed (II). B, proteolytic activity against soluble, azo-labeled casein. The sample means are shown by thick horizontal lines. For one-way analysis of variance: ***, p < 0.0001; ns, not significant relative to mock. Student's t test comparing hWT and hC392S is shown with a horizontal bracket. C, proteolytic assay using Cm-Tf (marked by a black dot) as substrate for the indicated hADAMDEC1 variants (500 nm). Samples were analyzed by reducing SDS-PAGE, and primary product bands are marked by black arrows.
The Activity Enhancement of the D362H Active Site Reconstruction Is Augmented by the C392S Substitution
We previously demonstrated that reconstruction of the hADAMDEC1 active site by substituting the zinc-binding Asp362 for a His residue (hD362H) increases the proteolytic activity of hADAMDEC1 toward α2M and azocasein (5). Interestingly, when combining the D362H and C392S substitutions (hD362H/C392S), we observe a synergistic effect on the proteolytic activity toward human α2M relative to hADAMDEC1, hC392S, and hD362H (Fig. 3A). Also, both hD362H and hD362H/C392S demonstrated significantly increased proteolysis of Cm-Tf compared with hADAMDEC1 and hC392S, again with hD362H/C392S being the most active (Fig. 3B). Whereas digestion of Cm-Tf by hADAMDEC1 primarily generated two fragments, hD362H and hD362H/C392S produced a range of fragments under the conditions employed (Fig. 3B). Finally, hD362H/C392S also exhibited significantly increased caseinolytic activity, shown by the proteolysis of azo-labeled casein (Fig. 3C) and further demonstrated by casein zymography (Fig. 3D). In fact, hD362H/C392S is the only variant displaying visible activity in the casein zymogram.
FIGURE 3.

Influence of the C392S substitution on hADAMDEC1 D362H. A, α2M-cross-linking assay with hADAMDEC1 variants visualized by anti-hADAMDEC1 Western blot analysis. The different α2M·ADAMDEC1 complexes (I) are observed at the top of the blots, whereas the added protease (III) is seen at the bottom. B, digestion of Cm-Tf (marked by a black dot) by a 500 nm concentration of the indicated hADAMDEC1 variants analyzed by reducing SDS-PAGE. C, proteolytic activity against soluble, azo-labeled casein. The sample means are shown by thick horizontal lines. For one-way analysis of variance: ***, p < 0.0001; ns, not significant relative to mock. Student's t tests comparing individual sets of data are shown with a horizontal bracket. D, casein zymogram of hADAMDEC1 variants. Caseinolytic activity is observed by the appearance of white bands (indicated by arrows). A distinct band of ∼32 kDa, consistent with the molecular mass of hADAMDEC1 D362H/C392S, is observed.
Identification of hADAMDEC1 Cleavage Sites in Cm-Tf
As described in the legend to Fig. 2B, proteolysis of Cm-Tf by hC392S generated a cleavage pattern similar to that of hADAMDEC1. The combined molecular mass of the two primary bands (76 and 20 kDa) is in good agreement with the observed mass of the undigested Cm-Tf (95 kDa; marked by a black dot). Thus, the fragment pattern is consistent with a single cleavage event giving rise to the two primary proteolytic fragments. In comparison, hD362H and hD362H/C392S generated multiple cleavage products, indicating not only an increased proteolytic activity but also a change in substrate specificity compared with hADAMDEC1 and hC392S (Fig. 3B). The most intense fragments were sequenced by N-terminal Edman degradation, and four cleavage sites were identified (Fig. 4A). The 76-kDa fragment stems from cleavage of the Cys(Cm)194–Leu195 peptide bond, identifying the major cleavage site of hADAMDEC1 in Cm-Tf. The N-terminal sequence of the 20-kDa fragment is identical to the N terminus of mature transferrin, suggesting that the 76- and 20-kDa fragments together make up mature Cm-Tf (Fig. 4B). Three additional main cleavage sites between Ala346 and Leu347, Gly465 and Leu466, and Cys(Cm)523 and Leu524 in Cm-Tf were identified when incubated with hD362H and hD362H/C392S. Our previous studies identified a cis-autocleavage of hADAMDEC1 between Pro161 and Leu162 in the prodomain (5). The sequences flanking these sites are remarkably similar to each other, and all identified sites have a Leu residue in the P1′ position (Fig. 4C). In addition, the identified cleavage sites may suggest a preference for a charged group in P2 and a hydrophobic residue in P3. Finally, both scissile bonds identified as cleaved by hADAMDEC1 wild type contain a Lys at P2′, whereas variability at this position is observed in D362H-based variants (Fig. 4C).
FIGURE 4.
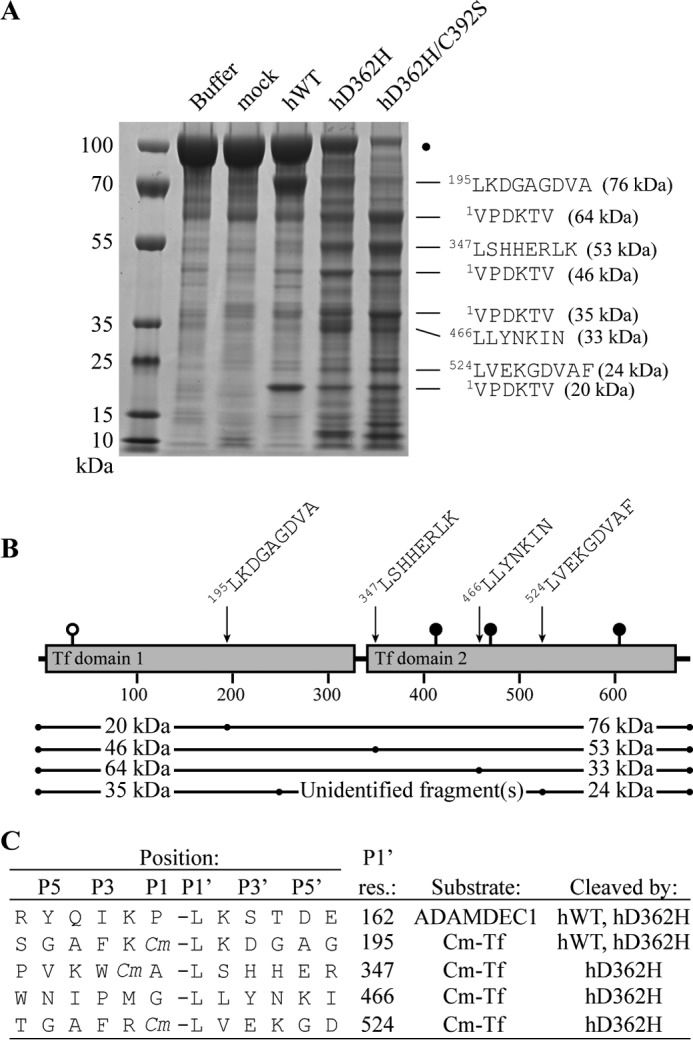
Substrate specificity of hADAMDEC1 variants. A, N-terminal Edman sequencing of the dominant proteolytic bands of Cm-Tf by hADAMDEC1, hD362H, and hD362H/C392S. Mature numbering is used for Cm-Tf. The SDS-polyacrylamide gel is identical to that of Fig. 3B and serves only to illustrate sequenced bands. B, the four identified cleavage sites at residues 195, 347, 466, and 524 giving rise to 76, 53, 33, and 24 kDa product bands, respectively, are mapped on the schematic domain structure of mature transferrin. Suggested fragment pairs making up mature Cm-Tf are shown below. Transferrin is modified by one O-linked and three N-linked glycosylations illustrated by white and black lollipops, respectively. C, sequence alignment of the identified cleavage sites in Cm-Tf and in the ADAMDEC1 prodomain (5). Cm, carboxymethyl-modified Cys residue.
ADAMDEC1 Is Inhibited by Batimastat
To investigate whether the ADAMDEC1-specific features influence the conformation of the active site, inhibition by the metzincin active site inhibitor batimastat (BB-94) was evaluated using the α2M-cross-linking and Cm-Tf proteolysis assays (Fig. 5). BB-94 inhibited the α2M cross-linking of both human and mouse ADAMDEC1 wild type and the hC392S, hD362H, and hD362H/C392S variants (Fig. 5, A–C). BB-94 also inhibited the proteolysis of Cm-Tf by all tested variants, and neither of the substitutions has an apparent effect on the inhibition (Fig. 5D). Because BB-94 primarily probes the S1′ pocket, inhibition by BB-94 indicates that neither the C392S nor the D362H substitution imposes dramatic changes to the conformation of the S1′ pocket, also illustrated by the retained preference for a P1′ Leu residue in the identified cleavage sites.
FIGURE 5.
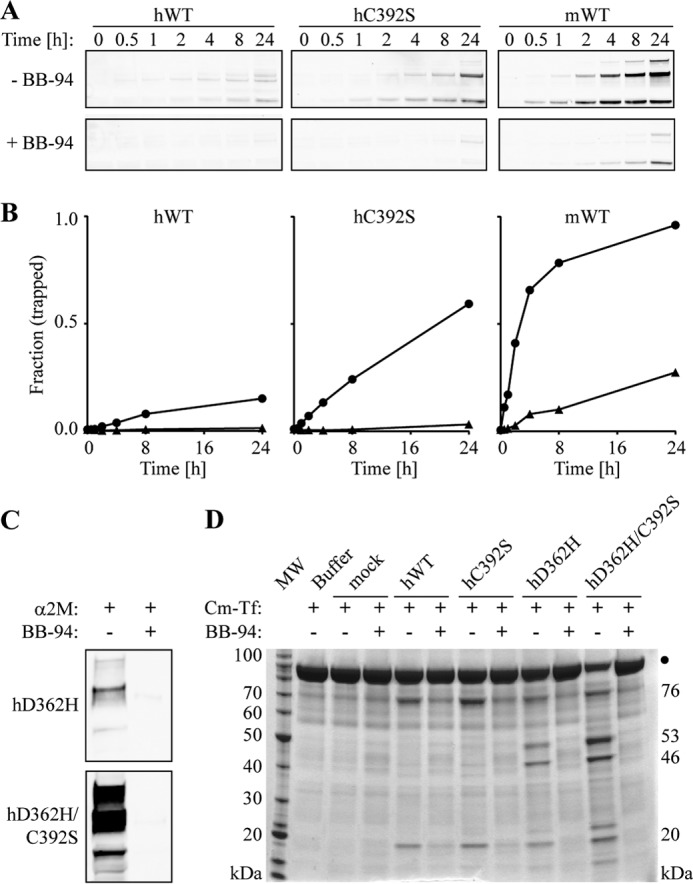
Effects of batimastat (BB-94) on the proteolytic activity of ADAMDEC1 variants. A, time-course of α2M cross-linking in the absence and presence of a 100-fold molar excess of BB-94 visualized by anti-HPC4 tag Western blot analysis. Only the α2M·ADAMDEC1 complexes are shown. B, densitometry analysis showing the fraction of hADAMDEC1, hC392S, and mADAMDEC1 cross-linked by α2M in the absence (circles) and presence (triangles) of BB-94. C, α2M cross-linking of hD362H and hD362H/C392S ADAMDEC1 variants in the absence and presence of a 10-fold molar excess of BB-94. D, proteolysis of Cm-Tf by ADAMDEC1 variants in the absence and presence of 3.5 μm BB-94 (∼10-fold molar excess). The primary product bands of hADAMDEC1 and hC392S (76 and 20 kDa) as well as those of hD362H and hD362H/C392S (46 and 53 kDa) are indicated on the right.
TIMPs Do Not Inhibit ADAMDEC1, but N-TIMP-3 Inhibits ADAMDEC1 with a Reconstituted ADAM Active Site
The TIMPs regulate matrix metalloproteinase activities in vivo, and especially TIMP-3 has been shown to have a broad specificity toward the ADAMs and ADAMTSs (17, 31–33). To investigate whether members of this endogenous inhibitor family affect hADAMDEC1 proteolytic activity, mature TIMP-1 and TIMP-2 as well as N-TIMP-3 were incubated with hADAMDEC1 and variants before the addition of Cm-Tf. TIMP-1 and TIMP-2 did not inhibit the proteolysis of Cm-Tf by any of the investigated hADAMDEC1 variants (data not shown). Similarly, N-TIMP-3 in up to a 3-fold molar excess did not show any inhibition of Cm-Tf digestion by hADAMDEC1 or hC392S (Fig. 6, A and B). However, the hD362H variant with enhanced proteolytic activity was efficiently inhibited by a 3-fold molar excess of N-TIMP-3 (Fig. 6C). In addition, equimolar concentration of N-TIMP-3 demonstrated efficient inhibition of the superactive hD362H/C392S variant (Fig. 6D). To account for the higher proteolytic activity of hD362H and hD362H/C392S compared with that of hWT and hC392S, D362H-based variants were incubated half as long with Cm-Tf. Thus, the D362H-substitution, reconstituting the consensus three-His Zn2+-binding site appears to be needed for ADAMDEC1 susceptibility to inhibition by N-TIMP-3.
FIGURE 6.
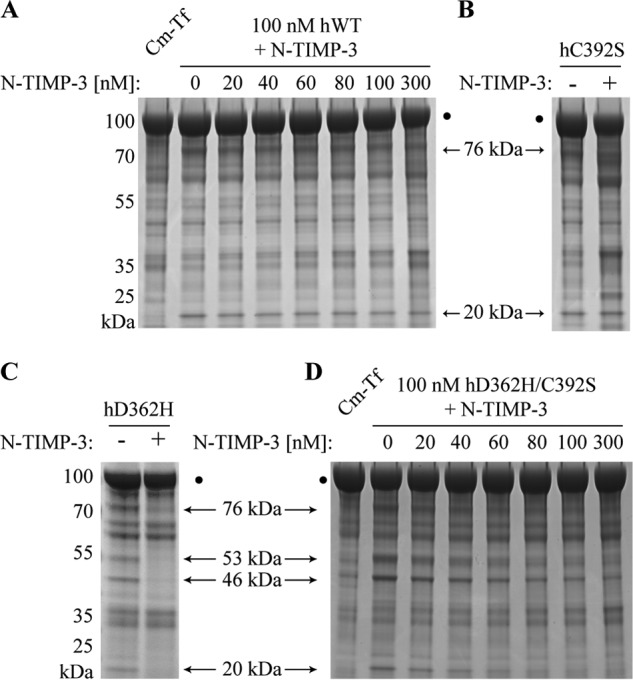
Inhibition by N-TIMP-3. A, inhibitory effects of N-TIMP-3 on Cm-Tf digestion by 100 nm hWT (A) and 100 nm hD362H/C392S (D). The molar concentrations of N-TIMP-3 are stated above each lane in the gel. The inhibitory effect of a 3-fold molar excess of N-TIMP-3 on Cm-Tf digestion by hC392S (B) and hD362H (C) variants is shown for comparison with the effect on hWT and hD362H/C392S activity. Cm-Tf digestion is visualized by reducing SDS-PAGE analysis showing the 76- and 20-kDa product bands for hADAMDEC1 and hC392S as well as the 76-, 53-, 46-, and 20-kDa product bands for hD362H and hD362H/C392S.
Homology Modeling of the hADAMEC1 Structure
To gain insight into how the rare and unique features of the ADAMDEC1 amino acid sequence may impact the structure of ADAMDEC1, a homology model was created using the crystal structures of hADAM-8, hADAM-22, and snapalysin as templates (23, 24, 26). The metalloprotease domain was modeled after the hADAM-8 structure; the active site was based on snapalysin and refined by energy minimization. The disintegrin-like domain and its relative positioning were based on that of hADAM-22 (Fig. 7). The homology model of hADAMDEC1 contains the hallmarks of a metzincin metalloprotease, including a central highly twisted five-stranded β-sheet (I–V) and three conserved α-helices (A–C) as well as two additional α-helices specific for the ADAMs (Fig. 7, A and B) (1, 34). Contrary to the metalloprotease domain, the disintegrin-like domain consists mainly of loops and turns (Fig. 7B). The model suggests the disulfide bond pattern of the mature ADAMDEC1 protein from the template crystal structures. These are Cys328–Cys407, Cys369–Cys374, Cys423–Cys452, Cys434–Cys447, Cys436–Cys442, and Cys446–Cys460, where the latter four appear to hold the disintegrin-like domain together. In hADAM-22, the Cys residues aligning to ADAMDEC1 Cys446 and Cys460 are not connected to each other but form disulfide bonds with Cys residues absent in ADAMDEC1. However, the model orients Cys446 and Cys460 within disulfide bonding distance, suggesting that these could be connected. In the metalloprotease domain, a putative calcium binding site is formed by the side chains of Asp221, Asp304, and Gln410, the backbone carbonyl oxygen of Cys407, and two water molecules (Fig. 7B). Another putative calcium binding site in the disintegrin-like domain is made up by the side chains of Asn425, Glu429, Glu432, and Asp435 in addition to the backbone carbonyls of Val422 and Leu427. To accommodate Zn2+ coordination, Asp362 is predicted to influence the active site architecture, as illustrated by a superposition of the modeled active sites of hWT and hD362H (Fig. 8A) showing a 1.9-Å offset of the Cα of hWT Asp362 compared with hD362H His362. The active site architecture of ADAMDEC1 is suggested to be similar to that of snapalysin, and substitution of Asp362 for His is predicted to adopt an active site like hADAM-8. The model places the unpaired Cys392 in the N-terminal of α-helix C below the active site and suggests that Ser392 in the hC392S variant stabilizes α-helix C by forming an N-cap hydrogen bond to Ser389 (Fig. 8A). The modeled active site readily accommodates the AFKCm↓LKDG peptide representing the dominant cleavage site of hADAMDEC1 in Cm-Tf (Fig. 8, B and C). The modeled peptide docking suggests that the conserved P1′ Leu residue fits easily into the deep and hydrophobic S1′ cavity and that also the preferred hydrophobic residue in the P3 position is bound in a hydrophobic pocket formed by Phe287, Leu321, and Ala323. The model further predicts favorable electrostatic interactions between the negatively charged carboxymethylated Cys in the P1 position and Arg318 as well as between the P3′ Asp and Lys339/Lys340 of hADAMDEC1 (Fig. 8B). The crystal structure of N-TIMP-3 docks into the hADAMDEC1 model with the N-terminal ridge of the N-TIMP-3 binding the catalytic Zn2+ through the backbone of Cys1 (Fig. 8D). In addition, residues flanking Cys1 and Cys68 are predicted to interact with the substrate binding pockets. Whereas hADAMDEC1 is not inhibited by N-TIMP-3, the proposed backbone rearrangement in the active site of the hD362H variant is predicted to accommodate interactions with N-TIMP-3 Glu65, thereby enabling inhibition of the metalloprotease (Fig. 8E).
FIGURE 7.

Structural modeling of human ADAMDEC1. A, standard orientation of mature hADAMDEC1 homology model based on the crystal structure of hADAM-8, snapalysin, and hADAM-22 (PDB entries 4DD8, 1C7K, and 3G5C) (23, 24, 26). The amino acid backbone of the metalloprotease domain and the disintegrin-like domain are shown in light and dark gray, respectively. The three α-helices (A–C) and five β-sheets (I–V), characteristic for the catalytic domain of metzincin metalloproteases, are labeled accordingly, and the sheets are colored blue. The catalytic zinc ion (dark gray) and the two putative calcium (green) ions are shown as spheres. B, 90º rotation of A, showing the disintegrin-like domain and the two putative calcium binding sites.
FIGURE 8.

Proposed structural effects of hADAMDEC1 mutations and basis of substrate/inhibitor interactions. A, superposition of the modeled Zn2+-binding active sites of hWT (gray), hD362H (green), and hC392S (cyan). Catalytic water molecules are marked by a light red cross. The identified disulfide bond connecting Cys369 and Cys374 is visible on the right. B and C, proposed docking of a peptide substrate (dark gray sticks) in an electrostatic surface model of hADAMDEC1. The substrate, AFKCm↓LKDG, represents the dominant hADAMDEC1-mediated cleavage of Cm-Tf. D, docking of N-TIMP-3 into the structural model of ADAMDEC1 based on the crystal structure of N-TIMP-3 in complex with ADAM-17 (PDB entry 3CKI (27)). N-TIMP-3 interacts directly with the catalytic Zn2+ through the backbone amino and carbonyl groups of Cys1 and with the substrate binding pockets through multiple amino acid side chains (as indicated in the figure). E, the interaction of N-TIMP-3 Glu65 with the backbone of Asp362 in hWT (gray) and His362 of the hD362H variant (green).
DISCUSSION
ADAMDEC1 is an unusual metzincin metalloprotease with an unknown biological function. It has a rare active site with a zinc-coordinating Asp residue and a short domain structure terminating in the disintegrin-like domain. Here we have demonstrated that ADAMDEC1 also contains a different Cys pattern of the metalloprotease domain, resulting in an unpaired Cys residue, Cys392, in the human ortholog. We show that Cys392 of hADAMDEC1 can react with a maleimide group coupled to a 5-kDa polyethylene glycol moiety, indicating that Cys392 is reactive and at least partially surface-exposed. Cys392 appears to modulate the ability of ADAMDEC1 to be cross-linked by α2M because its relatively subtle substitution for Ser increases α2M-mediated trapping of the protease. Substituting the Cys392 for a Ser is predicted by structural modeling to stabilize α-helix C by forming an N-cap hydrogen bond to Ser389. Interestingly, the C392S substitution does not change the activity against the other identified ADAMDEC1 substrates; thus, Cys392 may have evolved in the human ortholog to limit the risk of inhibitory trapping by the α2M protease inhibitor. We previously showed that the zinc-coordinating Asp dampened the proteolytic activity of ADAMDEC1, in that reconstruction of the canonical metzincin active site (hD362H) increases the activity (5). Surprisingly, the combination of the D362H and C392S substitutions (hD362H/C392S) has a synergistic effect, resulting in a significant enhancement of the proteolytic activity against all identified hADAMDEC1 substrates.
Human Cm-Tf is a previously used metalloprotease substrate that contains two homologous transferrin domains. hADAMDEC1 cleaves Cm-Tf at one primary site, between Cys(Cm)194 and Leu195 in the transferrin domain 1, generating two major Cm-Tf fragments. The D362H and D362H/C392S variants not only displayed enhanced proteolytic activity but also exhibited altered specificity, identifying the Ala346–Leu347 bond as an additional preferred scissile bond. In addition, we identified secondary cleavage sites at Gly465↓Leu466, Cys(Cm)523↓Leu524, and the site preferred by wild-type hADAMDEC1, Cys(Cm)194↓Leu195. The data demonstrate a clear change in the substrate specificity of the D362H and D362H/C392S variants, yet all identified sites have a Leu residue at the P1′ position. We previously determined an autocatalytic processing site in the prodomain of hADAMDEC1 (RYQIKPLKSTDE (5)), which also contains a Leu residue at the P1′ position as well as a hydrophobic residue in the P3 position. Interestingly, the main cleavage site of hADAMDEC1 at Cys(Cm)194-Leu195 and the secondary cleavage site of hD362H and hD362H/C392S at Cys(Cm)523-Leu524 have both been found to be proteolyzed by ADAMTS-4 and -5 (35, 36). In contrast, the primary cleavage site of D362H-substituted variants at Ala346↓Leu347 has been shown to be cleaved by ADAM-12 (37). To our knowledge, proteolysis of the Gly465–Leu466 peptide bond has not previously been reported. Comparison of the two identified cleavage sites for hADAMDEC1 and the three sites specific for the D362H substitution suggests that a positively charged Lys residue in the P2′ position may be preferred or needed for hADAMDEC1 (Fig. 4C). Because a single change of the third Zn2+ binding residue from the negatively charged Asp362 to the uncharged His seems to alleviate the need for a positively charged P2′ residue in the substrate, we suggest that the P2′ may interact directly with Asp362 or through a hydrogen bond network to reduce the increased electron density or negative charge of the Zn2+ ligand. The P2′ Lys may thus be part of creating a Zn2+ environment for ADAMDEC1, which may be closer to that of the three-His-coordinated ion.
hADAMDEC1 is not inhibited by TIMP1 to -3 under the investigated conditions, and substitution of Cys392 for Ser has no effect on TIMP inhibition. However, substitution of the zinc-binding Asp362 for a His increases the susceptibility to N-TIMP-3 inhibition. The crystal structure of hADAM-17 in complex with N-TIMP-3 reveals that Glu65 of N-TIMP-3 interacts with the backbone nitrogen of His415, the third zinc ligand of hADAM-17 (27). Because the amino acid side chain of Asp is shorter than that of His, the backbone of the active site Asp362 in ADAMDEC1 is predicted to be moved closer to the Zn2+ in order to accommodate binding (Fig. 8A). Hence, the D362H substitution is predicted to push the backbone down 1.9 Å, allowing the substituting His362 to coordinate the active site zinc ion while at the same time allowing Glu65 of N-TIMP-3 to interact with the backbone nitrogen of His362. We suggest that this may in part account for the increased inhibition by N-TIMP-3 when substituting Asp362 for a His residue, in addition to the changed chemical environment of the catalytic Zn2+ ion. Although TIMP-1, -2, and -3 are structurally similar, only N-TIMP-3 inhibits hD362H. This selectivity may in part arise from a favorable interaction of N-TIMP-3 Glu62 with Arg318 of ADAMDEC1 (data not shown). In contrast, TIMP-1 has a Pro and TIMP-2 has an Ala at the corresponding position, and they are thus incapable of forming a hydrogen bond or a salt bridge to Arg318. Additionally, TIMP-1 Thr97 and TIMP-2 Thr99 are predicted to interfere with binding of ADAMDEC1 by coming into too close proximity of Arg318. TIMP-3, on the other hand, has Gly93 on the corresponding position, which is not predicted to influence the binding of N-TIMP-3 to hD362H.
All investigated hADAMDEC1 variants are inhibited by the synthetic active site inhibitor batimastat (BB-94), suggesting that neither of the substitutions change the S1′ binding pocket. This is in accordance with the finding that all tested variants proteolyze Cm-Tf N-terminal to a Leu residue.
Taken together, the ADAMDEC1-specific features result in narrower substrate specificity but also a dampening of the proteolytic activity. The observed effects lead us to hypothesize that hADAMDEC1 has evolved with the rare active site and the unpaired Cys392 allowing escape from inhibition by TIMP-3 and α2M.
This work was supported, in whole or in part, by National Institutes of Health, NIAMS, Grant AR40994 (to H. N.).
- ADAM
- a disintegrin and metalloprotease
- TIMP
- tissue inhibitor of metalloproteases
- N-TIMP-3
- N-terminal inhibitory domain of TIMP-3
- α2M
- α2-macroglobulin
- Cm-Tf
- carboxymethylated transferrin
- hWT and mWT
- human and mouse ADAMDEC1 wild type, respectively
- hADAM
- human ADAM
- hADAMDEC1 and mADAMDEC1
- human and mouse ADAMDEC1, respectively
- hD362H
- hC392S, and hD362H/C392H, human D362H, C392S, and D362H/C392H ADAMDEC1 active site mutations, respectively
- PDB
- Protein Data Bank.
REFERENCES
- 1. Bode W., Gomis-Rüth F. X., Stöckler W. (1993) Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the “metzincins”. FEBS Lett. 331, 134–140 [DOI] [PubMed] [Google Scholar]
- 2. Gomis-Rüth F. X. (2003) Structural aspects of the metzincin clan of metalloendopeptidases. Mol. Biotechnol. 24, 157–202 [DOI] [PubMed] [Google Scholar]
- 3. Gomis-Rüth F. X. (2009) Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem. 284, 15353–15357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mueller C. G. F., Cremer I., Paulet P. E., Niida S., Maeda N., Lebeque S., Fridman W. H., Sautès-Fridman C. (2001) Mannose receptor ligand-positive cells express the metalloprotease decysin in the B cell follicle. J. Immunol. 167, 5052–5060 [DOI] [PubMed] [Google Scholar]
- 5. Lund J., Olsen O. H., Sørensen E. S., Stennicke H. R., Petersen H. H., Overgaard M. T. (2013) ADAMDEC1 is a metzincin metalloprotease with dampened proteolytic activity. J. Biol. Chem. 288, 21367–21375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mueller C. G. F., Rissoan M. C., Salinas B., Ait-Yahia S., Ravel O., Bridon J. M., Briere F., Lebecque S., Liu Y. J. (1997) Polymerase chain reaction selects a novel disintegrin proteinase from CD40-activated germinal center dendritic cells. J. Exp. Med. 186, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bates E. E., Fridman W. H., Mueller C. G. (2002) The ADAMDEC1 (decysin) gene structure: evolution by duplication in a metalloprotease gene cluster on chromosome 8p12. Immunogenetics 54, 96–105 [DOI] [PubMed] [Google Scholar]
- 8. Fritsche J., Müller A., Hausmann M., Rogler G., Andreesen R., Kreutz M. (2003) Inverse regulation of the ADAM-family members, decysin and MADDAM/ADAM19 during monocyte differentiation. Immunology 110, 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Papaspyridonos M., Smith A., Burnand K. G., Taylor P., Padayachee S., Suckling K. E., James C. H., Greaves D. R., Patel L. (2006) Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 26, 1837–1844 [DOI] [PubMed] [Google Scholar]
- 10. Crouser E. D., Culver D. A., Knox K. S., Julian M. W., Shao G., Abraham S., Liyanarachchi S., Macre J. E., Wewers M. D., Gavrilin M. A., Ross P., Abbas A., Eng C. (2009) Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 179, 929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dommels Y. E., Butts C. A., Zhu S., Davy M., Martell S., Hedderley D., Barnett M. P., McNabb W. C., Roy N. C. (2007) Characterization of intestinal inflammation and identification of related gene expression changes in mdr1a(−/−) mice. Genes Nutr. 2, 209–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu J., Liu L., Zheng X., You C., Li Q. (2012) Expression and inhibition of ADAMDEC1 in craniopharyngioma cells. Neurol. Res. 34, 701–706 [DOI] [PubMed] [Google Scholar]
- 13. Lucchesi W., Brady G., Dittrich-Breiholz O., Kracht M., Russ R., Farrell P. J. (2008) Differential gene regulation by Epstein-Barr virus type 1 and type 2 EBNA2. J. Virol. 82, 7456–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macartney-Coxson D. P., Hood K. A., Shi H. J., Ward T., Wiles A., O'Connor R., Hall D. A., Lea R. A., Royds J. A., Stubbs R. S., Rooker S. (2008) Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer 8, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McClellan M. J., Khasnis S., Wood C. D., Palermo R. D., Schlick S. N., Kanhere A. S., Jenner R. G., West M. J. (2012) Downregulation of integrin receptor-signaling genes by Epstein-Barr virus EBNA 3C via promoter-proximal and -distal binding elements. J. Virol. 86, 5165–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Troeberg L., Tanaka M., Wait R., Shi Y. E., Brew K., Nagase H. (2002) E. coli expression of TIMP-4 and comparative kinetic studies with TIMP-1 and TIMP-2: insights into the interactions of TIMPs and matrix metalloproteinase 2 (gelatinase A). Biochemistry 41, 15025–15035 [DOI] [PubMed] [Google Scholar]
- 17. Kashiwagi M., Tortorella M., Nagase H., Brew K. (2001) TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 276, 12501–12504 [DOI] [PubMed] [Google Scholar]
- 18. Nagase H. (1995) Human stromelysins 1 and 2. Methods Enzymol. 248, 449–470 [DOI] [PubMed] [Google Scholar]
- 19. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 20. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 21. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fiser A., Sali A. (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 [DOI] [PubMed] [Google Scholar]
- 23. Hall T., Shieh H. S., Day J. E., Caspers N., Chrencik J. E., Williams J. M., Pegg L. E., Pauley A. M., Moon A. F., Krahn J. M., Fischer D. H., Kiefer J. R., Tomasselli A. G., Zack M. D. (2012) Structure of human ADAM-8 catalytic domain complexed with batimastat. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 616–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu H., Shim A. H. R., He X. (2009) Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase. J. Biol. Chem. 284, 29077–29086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 26. Kurisu G., Kai Y., Harada S. (2000) Structure of the zinc-binding site in the crystal structure of a zinc endoprotease from Streptomyces caespitosus at 1 Å resolution. J. Inorg. Biochem. 82, 225–228 [DOI] [PubMed] [Google Scholar]
- 27. Wisniewska M., Goettig P., Maskos K., Belouski E., Winters D., Hecht R., Black R., Bode W. (2008) Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 381, 1307–1319 [DOI] [PubMed] [Google Scholar]
- 28. Tranchant I., Vera L., Czarny B., Amoura M., Cassar E., Beau F., Stura E. A., Dive V. (2014) Halogen bonding controls selectivity of FRET substrate probes for MMP-9. Chem. Biol. 21, 408–413 [DOI] [PubMed] [Google Scholar]
- 29. Gomis-Rüth F. X., Kress L. F., Bode W. (1993) First structure of a snake venom metalloproteinase: a prototype for matrix metalloproteinases/collagenases. EMBO J. 12, 4151–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cerdà-Costa N., Gomis-Rüth F. X. (2014) Architecture and function of metallopeptidase catalytic domains. Protein Sci. 23, 123–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murphy G. (2011) Tissue inhibitors of metalloproteinases. Genome Biol. 12, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Amour A., Slocombe P. M., Webster A., Butler M., Knight C. G., Smith B. J., Stephens P. E., Shelley C., Hutton M., Knäuper V., Docherty A. J. P., Murphy G. (1998) TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39–44 [DOI] [PubMed] [Google Scholar]
- 33. Amour A., Knight C. G., Webster A., Slocombe P. M., Stephens P. E., Knäuper V., Docherty A. J. P., Murphy G. (2000) The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 473, 275–279 [DOI] [PubMed] [Google Scholar]
- 34. Stöcker W., Grams F., Baumann U., Reinemer P., Gomis-Rüth F. X., McKay D. B., Bode W. (1995) The metzincins: topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. 4, 823–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kashiwagi M., Enghild J. J., Gendron C., Hughes C., Caterson B., Itoh Y., Nagase H. (2004) Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J. Biol. Chem. 279, 10109–10119 [DOI] [PubMed] [Google Scholar]
- 36. Gendron C., Kashiwagi M., Lim N. H., Enghild J. J., Thøgersen I. B., Hughes C., Caterson B., Nagase H. (2007) Proteolytic activities of human ADAMTS-5. J. Biol. Chem. 282, 18294–18306 [DOI] [PubMed] [Google Scholar]
- 37. Jacobsen J., Visse R., Sørensen H. P., Enghild J. J., Brew K., Wewer U. M., Nagase H. (2008) Catalytic properties of ADAM12 and its domain deletion mutants. Biochemistry 47, 537–547 [DOI] [PubMed] [Google Scholar]