

Background: Super-low-dose endotoxemia contributes to cell stress and low-grade inflammation.
Results: Super-low-dose LPS removed Tollip from late endosomes/lysosomes and blocked lysosome fusion with endosomes or autophagosomes. Tollip knock-out mice had impaired wound healing.
Conclusion: Super-low-dose LPS leads to cell stress through clearing Tollip and blocking lysosome fusion.
Significance: Our data reveal molecular dynamics of innate immunity regulation.
Keywords: Cell Signaling, Innate Immunity, Lipopolysaccharide (LPS), Lysosome, Macrophage, Endosome-Lysosome Fusion, Low-grade Inflammation, Macrophages, Super-low-dose Endotoxin
Abstract
Subclinical super-low-dose endotoxin LPS is a risk factor for the establishment of low-grade inflammation during the pathogenesis and progression of chronic diseases. However, the underlying mechanisms are not well understood. At the cellular level, a disruption of lysosome fusion with endosomes or autophagosomes may contribute to the potentiation of low-grade inflammation. In this study, we identified that subclinical super-low-dose endotoxin LPS can potently inhibit the process of endosome acidification and lysosome fusion with endosomes or autophagosomes in primary macrophages. Super-low-dose LPS induced the inhibitory phosphorylation of VPS34, thus leading to the disruption of endosome-lysosome fusion. This effect may depend upon the clearance and relocation of Tollip in macrophages by super-low-dose LPS. Consistent with this notion, Tollip-deficient macrophages had constitutively elevated levels of VPS34 inhibitory phosphorylation and constitutive disruption of endosome-lysosome fusion. By employing a skin excision wound-healing model, we observed that Tollip-deficient mice had significantly elevated levels of cell stress and reduced wound repair. This study reveals a novel mechanism responsible for the modulation of endosome-lysosome fusion and low-grade inflammation in innate macrophages.
Introduction
Chronic low-grade inflammation is a common feature of numerous debilitating diseases and disorders (1–8). Subclinical super-low-dose endotoxin in the blood circulation is one of the emerging risk factors that can cause and sustain chronic low-grade inflammation in humans and animals (9–11). Super-low-dose endotoxin selectively induces mild inflammatory responses in innate leukocytes without triggering compensatory anti-inflammatory responses (12). As a consequence, this may explain the nonresolving nature of low-grade inflammation that often accompanies low-grade endotoxemia.
At the cellular level, alterations of intracellular trafficking processes, such as endosome acidification and autophagy initiation and completion, as well as fusion of lysosomes with endosomes or autophagosomes, are critically involved in the propagation of low-grade inflammation (6, 7, 13, 14). Of particular interest, timely and orderly fusion of lysosomes with endosomes or autophagosomes would enable efficient clearance of cell debris and resolution of inflammation (15). Limited studies suggest that the disruption of endosome acidification and/or endosome-lysosome fusion may be correlated with the modulation of inflammatory phenotypes in monocytes and macrophages (16, 17).
At the biochemical level, several distinct gate-keeping enzymes are responsible for the initiation of autophagy and the fusion of lysosomes with autophagosomes and/or endosomes. The cell stress-sensing protein kinase adenosine monophosphate-activated protein kinase (AMPK)3 is responsible for autophagy initiation through the activation of ULK1 (unc-51-like autophagy-activating kinase 1) (18). Upon phosphorylation, AMPK becomes activated and is able to phosphorylate and thus activate ULK1, triggering the autophagic cascade (18–20). Activated ULK1 is recruited to the phagophore, where it binds to regulatory proteins and begins the assembly process of the autophagosome.
Activation of the class III PI3K VPS34, also known as PI3KC3, plays a key role during the completion of autophagy and fusion of lysosomes with endosomes, phagosomes, or autophagosomes. VPS34 is responsible for the phosphorylation of phosphatidylinositol to generate phosphatidylinositol 3-phosphate (PI(3)P), an essential signaling lipid in the regulation of lysosome fusion with endosomes/autophagosomes (21–24). VPS34 is negatively regulated via phosphorylation by various kinases, including cyclin-dependent kinase (25, 26). The phosphorylation of VPS34 inhibits the proper fusion of endosomes and/or autophagosomes with lysosomes (27).
Although subclinical low-grade endotoxemia has been associated with nonresolving low-grade inflammation in humans and experimental animals, limited studies are available to clarify the underlying mechanisms. Previous studies suggest that Tollip (Toll-interacting protein), one of the key signaling molecules downstream of the TLR4 (Toll-like receptor 4) pathway, can interact with Tom1 and may facilitate lysosome fusion with lysosomes (28). However, the role of Tollip in the context of innate immunity is not well understood and may be highly context-dependent. High-dose LPS induces the expression of Tollip and may facilitate the resolution of inflammation (29, 30). Tollip may also be subjected to ubiquitination and degradation (31). In contrast, we previously reported that super-low-dose LPS effectively relocates Tollip away from late endosomes/lysosomes to mitochondria (see Fig. 4) (12). As a consequence, super-low-dose LPS induces mitochondrial reactive oxygen species and low-grade inflammation (12).
FIGURE 4.
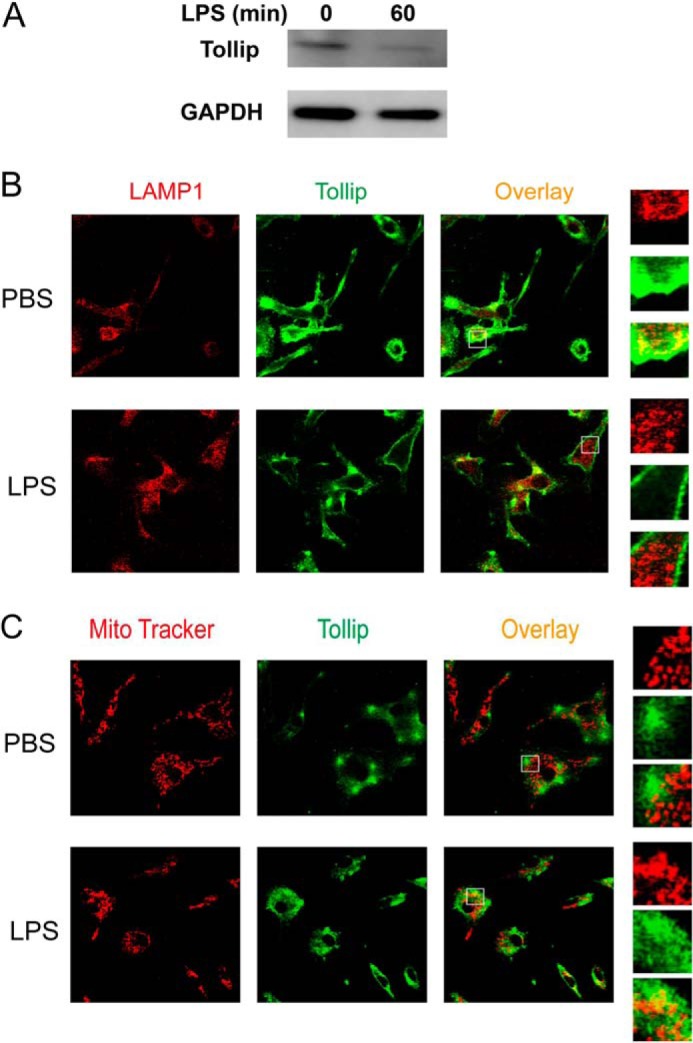
Clearance and removal of Tollip from late endosomes/lysosomes by super-low-dose LPS. A, WT macrophages were treated with PBS or 50 pg/ml LPS for 1 h. Total cell lysates were harvested, and equal amounts of protein lysates were resolved by SDS-PAGE. The levels of Tollip and control GAPDH were visualized by Western blotting. WT macrophages were treated with 50 pg/ml LPS for either 1 h (B) or 24 h (C). Cells were stained with goat anti-mouse Tollip antibody, followed by Alexa Fluor 488-labeled rabbit anti-goat IgG together with either Cy3-conjugated LAMP1-specific antibody (B) or MitoTracker Red (C). The intracellular distribution of Tollip in late endosomes/lysosomes or mitochondrial compartments was visualized by confocal fluorescence microscopy.
In this study, we further tested the hypothesis that the clearance and relocation of Tollip by super-low-dose LPS may be responsible for the disruption of lysosome fusion with endosomes/autophagosomes and cell stress in innate leukocytes. Using macrophages harvested from WT and Tollip-deficient mice, we examined the role of Tollip during the process of lysosome fusion. We determined that super-low-dose LPS caused cell stress in macrophages by reducing the total levels of Tollip and by clearing Tollip away from late endosomes/lysosomes and thus hindering lysosome fusion with endosomes/autophagosomes. We further examined the pathological relevance by using a skin excision wound repair model in WT and Tollip-deficient mice. Collectively, our results define a novel mechanism responsible for the potentiation of low-grade inflammation through the disruption of endosome-lysosome fusion.
EXPERIMENTAL PROCEDURES
Reagents
LPS (Escherichia coli O111:B4) was obtained from Sigma-Aldrich. Anti-GAPDH antibody was obtained from Santa Cruz Biotechnology. Antibodies against phospho-JNK, total JNK, phospho-ERK, total ERK, phospho-p38, total p38, SQSTM1/p62, phospho-AMPK, and total AMPK were obtained from Santa Cruz Biotechnology and Cell Signaling Technology. Anti-PI3KC3/VPS34 antibody was obtained from Aviva Systems Biology. Alexa Fluor 546-, Alexa Fluor 647-, FITC-, and tetramethylrhodamine-labeled dextran and zymosan A BioParticles were obtained from Invitrogen.
Mice and Cell Culture
WT C57BL/6 mice were purchased from Charles River Laboratories. Tollip−/− mice from a C57BL/6 background were provided by Dr. Jürg Tschopp (University of Lausanne, Lausanne, Switzerland). All mice were bred and housed at the Virginia Tech animal facility in compliance with approved Virginia Tech Animal Care and Use Committee protocols. Bone marrow-derived macrophages (BMDMs) were isolated from the tibias and femurs of WT and Tollip−/− mice and cultured in DMEM containing 30% L929 cell supernatant in untreated tissue culture dishes for 3 days. The cells were fed an additional 20 ml of fresh medium and cultured for an additional 3 days. Cells were harvested with PBS, resuspended in DMEM supplemented with 1% fetal bovine serum, and allowed to rest overnight before additional treatment. WT and Tollip-deficient murine embryonic fibroblast (MEF) cells and Tollip mutant MEFs harboring the CUE domain M240A/F241A mutation were cultured in complete DMEM.
Analysis of Lysosome Fusion with Endosomes or Autophagosomes
BMDMs harvested from WT and Tollip-deficient mice were incubated with Alexa Fluor 546-tagged dextran (1 mg/ml) for 1 h. Cells were then washed several times and reincubated in fresh medium for 3 h to allow lysosome translocation of dextran. Cells were subsequently incubated with FITC-conjugated zymosan for 1 h, followed by washing and fixation with 4% paraformaldehyde in PBS for 15 min at room temperature. Data were collected using a Zeiss LSM 510 laser-scanning confocal microscope. Experiments were conducted at 37 °C. Phagosomes (intracellular zymosan, green) fused with lysosomes (red) appeared yellow.
To determine whether the Tollip mutation affects lysosome fusion, we employed the Tollip mutant MEFs stably expressing the Tollip M240A/F241A mutant in a Tollip-deficient MEF background. These MEF cell lines were kindly provided by Drs. Kimberly Burns (University of Lausanne, Lausanne, Switzerland) and Jürg Tschopp. Unlike macrophages, which are efficient phagocytes and proficient in endosome acidification, MEFs are not efficient in uptake of zymosan and phagosomal/endosomal trafficking. Thus, we specifically examined the fusion of lysosomes with intracellular autophagosomes, as represented by the co-localization of LC3 and LAMP1 by confocal microscopy.
Electron Microscopy
WT and Tollip-deficient BMDMs were harvested after the specified treatments and washed with PBS. The cells were pelleted in a microcentrifuge tube, and 1 ml of 2% glutaraldehyde with 0.1 m cacodylate buffer (pH 7.4) fixative was placed on top of the cells without causing resuspension. Samples were sectioned and prepared on grids to be visualized under a JEOL JEM-1400 transmission electron microscope.
Immunoblotting
Cells were washed with PBS, harvested in SDS lysis buffer containing protease inhibitor mixture, and subjected to SDS-PAGE. The protein bands were transferred to an Immun-Blot PVDF membrane (Bio-Rad). Western blot analyses were performed with the specified antibodies as described previously (12).
Measuring Endosome Acidification
The effect of LPS on endosome acidification was measured using pH-sensitive dextran double-labeled with FITC and tetramethylrhodamine. Cells treated with LPS for the indicated times were then pulsed with dextran (1 mg/ml) for 10 min at 37 °C. Cells were extensively washed with cold PBS and immediately analyzed by FACS. The ratio of the mean fluorescence intensity emission between both dyes was determined. An increase in mean fluorescence intensity represents a decrease in acidification.
Confocal Microscopy
BMDMs harvested from WT mice were seeded on coverslips and treated with or without super-low-dose LPS (50 pg/ml) for 1 or 24 h. Some samples were labeled with MitoTracker Red (200 nm; Invitrogen) for 20 min. After fixation with 4% paraformaldehyde and permeabilization with 0.2% saponin, the cells were stained with goat anti-mouse Tollip antibody (Santa Cruz Biotechnology), followed by Alexa Fluor 488-labeled rabbit anti-goat IgG (Invitrogen). In some experiments, cells were also co-stained with Cy3-conjugated rabbit anti-mouse LAMP1 antibody (Abcam). WT and Tollip M240A/F241A mutant MEF cells were seeded on coverslips and treated with or without super-low-dose LPS (50 pg/ml). After 4 h, cells were starved with 1% FBS for 20 h to induce autophagy with or without LPS treatment. After fixation with 4% paraformaldehyde and permeabilization with methanol, the cells were stained with Alexa Fluor 488-labeled rabbit anti-mouse LC3 antibody (Novus Biologicals) together with Cy3-conjugated rabbit anti-mouse LAMP1 antibody. The samples were analyzed under the Zeiss LSM 510 confocal microscope.
Wound-healing Assays
Male 8-week-old Tollip−/− and WT mice were injected intraperitoneally with either PBS or LPS (5 ng/kg of body weight) every 3 days for 10 days. At day 10, mice were anesthetized as recommended. The backs of the mice were shaved with an Oster Mark II animal clipper (Sunbeam-Oster Co., Fort Lauderdale, FL), followed by treatment with cooling hair removal gel (Church & Dwight Co., Ewing, NJ). After disinfection with 7.5% iodine, followed by 70% ethanol to remove the iodine, four full-thickness punch biopsies were obtained using an Acu·Punch (6 mm; Acuderm Inc., Fort Lauderdale, FL). The skin incision was immediately covered with a transparent dressing (3.8 × 3.8 cm; Systagenix, North Yorkshire, United Kingdom). The mice were monitored and photographed on a daily basis. The area of the wound was measured daily using NIH ImageJ. Wound sizes at day 9 post-wounding were expressed as ratio of the initial (day 0) wound area. Wound tissues were collected at day 9 post-wounding and processed for Western blotting.
Statistical Analysis
Results are expressed as mean ± S.D. Statistical significances between groups were determined using a two-tailed Student's t test and are indicated by asterisks in the figures; p values <0.05 were considered statistically significant.
RESULTS
Super-low-dose LPS Disrupts Lysosome Fusion and Causes Low-grade Stress in Macrophages
We first tested the endosome acidification process in primary macrophages harvested from WT mice treated with super-low-dose LPS. As shown in Fig. 1A, a higher dose of LPS (100 ng/ml) induced significant acidification of late endosomes as determined by flow cytometry analyses of the phagocytosed pH-sensitive dextran. In contrast, super-low-dose LPS (50 pg/ml) failed to induce any noticeable late endosome acidification. Next, we monitored the fusion of lysosomes with endosomes through sequential incubation of fluorescently labeled dextran and zymosan. As shown in Fig. 1B, macrophages challenged with 50 pg/ml LPS exhibited significantly reduced fusion of endosomes and lysosomes compared with control macrophages. To further corroborate our conclusion, we performed electron microscopy analyses of WT macrophages treated with and without 50 pg/ml LPS. As shown in Fig. 1C, macrophages treated with 50 pg/ml LPS exhibited elevated levels of densely stained lysosome structures compared with fused gray-colored endolysosomes. We further examined the cellular levels of p62, the accumulation of which is correlated with the disruption of lysosome fusion with late endosomes or autophagosomes (32). As shown in Fig. 1D, super-low-dose LPS induced a rapid accumulation of p62. Next, we determined the activation status of key stress kinases such as JNK and p38. As shown in Fig. 1E, 50 pg/ml LPS induced mild yet sustained phosphorylation of JNK and p38. In contrast, 100 ng/ml LPS triggered robust and transient phosphorylation of JNK and p38. Intriguingly, 50 pg/ml LPS not only failed to induce ERK phosphorylation, but also caused a slight reduction.
FIGURE 1.

Super-low-dose LPS suppresses lysosome fusion. A, WT macrophages treated with either PBS or LPS (50 pg/ml or 100 ng/ml) were subjected to endosome acidification assays by incorporating pH-sensitive dextran double-labeled with FITC and tetramethylrhodamine. Flow cytometry analyses were performed to monitor the increase in fluorescence intensity, which indicates a decrease in endosomal pH. *, p < 0.05 compared with untreated controls. B, WT macrophages treated with either PBS or 50 pg/ml LPS were subjected to endosome-lysosome fusion assays by sequential incorporation of Alexa Fluor 546-labeled dextran and FITC-labeled zymosan BioParticles. Fluorescence images were obtained with the Zeiss LSM 510 laser-scanning confocal microscope to observe isolated lysosomes (red), endosomes (green), and fused endolysosomes (orange). The percentage of cells with fused endolysosomes is represented in the graph. *, p < 0.05 compared with untreated controls. C, WT macrophages treated with PBS or 50 pg/ml LPS for 2 h were subjected to analyses by transmission electron microscopy. Dark electron-dense lysosome structures can be seen in the samples challenged with 50 pg/ml LPS. D, WT macrophages were treated with either 50 pg/ml or 100 ng/ml LPS for the indicated times. The levels of p62 and GAPDH controls were determined by Western blot analyses. Data represent three experiments. *, p < 0.05 compared with untreated controls. E, WT macrophages were treated with either 50 pg/ml or 100 ng/ml LPS for the indicated times. The levels of phosphorylated (p) and total JNK, ERK, and p38 were determined by Western blotting. Data represent three experiments.
Tollip Facilitates the Process of Lysosome Fusion
Given previous findings that Tollip interacts with PI(3)P and Tom1, and localizes at late endosomes/lysosomes (29, 33), we tested the hypothesis that Tollip may be involved in the process of lysosome fusion. As shown in Fig. 2A, Tollip-deficient macrophages had a constitutive defect in the acidification of late endosomes. Furthermore, the lysosome fusion with endosomes/autophagosomes was constitutively disrupted in Tollip-deficient macrophages, as visualized by the clear separation of dextran and zymosan in Tollip-deficient cells under a confocal microscope (Fig. 2B). Western blot analyses demonstrated that cellular p62 levels were constitutively and significantly elevated in resting macrophages from Tollip-deficient mice, a clear sign of disrupted fusion of lysosomes with autophagosomes (Fig. 2C). We observed that super-low-dose LPS has no further effects on p62 in Tollip-deficient macrophages. Electron microscopy analyses also revealed elevated lysosomes in resting Tollip-deficient macrophages (Fig. 2D).
FIGURE 2.

Tollip facilitates lysosome fusion. A, WT and Tollip-deficient macrophages were treated with 50 pg/ml LPS for the indicated times. Endosome acidification assays were performed, followed by flow cytometry analysis. B, WT and Tollip-deficient macrophages were treated with 50 pg/ml LPS. Assays that measure lysosome fusion with endosome were performed, followed by confocal fluorescence microscopy analysis. The percentage of cells with fused endolysosomes are represented in the graph. *, p < 0.05 compared with untreated controls. C, WT and Tollip-deficient macrophages were treated with 50 pg/ml for the indicated times. The expression levels of p62 and GAPDH controls were determined by Western blot analyses. Data represent three experiments. *, p < 0.05 compared with untreated controls. D, Tollip-deficient macrophages treated with PBS or 50 pg/ml LPS for 2 h were subjected to transmission electron microscopy analyses. Dark electron-dense lysosome structures can be seen in the samples challenged with or without LPS challenge.
Suppression of AMPK and VPS34 by Super-low-dose LPS Is Dependent on Tollip
To further examine the underlying molecular mechanisms, we determined the phosphorylation status of AMPK and VPS34. AMPK phosphorylation is required for the initial assembly of autophagosomes (19), whereas VPS34 phosphorylation inhibits lysosome fusion with endosomes/autophagosomes (25). As shown in Fig. 3, WT macrophages treated with 50 pg/ml LPS exhibited a rapid and dramatic reduction in AMPK phosphorylation. On the other hand, 50 pg/ml LPS significantly induced the inhibitory phosphorylation of VPS34 (Fig. 3).
FIGURE 3.

Super-low-dose LPS suppresses AMPK and VPS34. WT and Tollip-deficient macrophages were treated with 50 pg/ml LPS for the indicated times. The levels of phosphorylated (p) and total AMPK (A) and VPS34 (B) were determined by Western blot analyses. The relative levels of phosphorylated AMPK and VPS34 were quantified from three experiments. *, p < 0.05 compared with untreated controls.
Because we determined that Tollip is involved in the process of lysosome fusion with endosomes/autophagosomes, we further determined the phosphorylation status of AMPK and VPS34 in Tollip-deficient cells. As shown in Fig. 3, Tollip-deficient cells had constitutively reduced levels of AMPK phosphorylation compared with WT cells. LPS failed to further alter the phosphorylation status in Tollip-deficient cells. On the other hand, the inhibitory phosphorylation of VPS34 was constitutively elevated in Tollip-deficient macrophages compared with WT macrophages. LPS failed to further alter the phosphorylation status of VPS34 in Tollip-deficient cells.
Clearance of Tollip by Super low-dose LPS
Our data suggest that super-low-dose LPS may disrupt lysosome fusion by compromising the function of Tollip. We previously reported that super-low-dose LPS causes translocation of Tollip to mitochondria (12). A separate study reported that Tollip may undergo ubiquitination and degradation (31). On the basis of these studies, we further tested whether super-low-dose LPS reduces and/or clears Tollip away from late endosomes/lysosomes and subsequently disrupts lysosome fusion with endosomes/autophagosomes. Indeed, as shown in Fig. 4A, we observed that macrophages treated with 50 pg/ml LPS reduced Tollip protein levels in WT macrophages. Furthermore, we tested whether LPS treatment affects Tollip subcellular localization. To test this, WT BMDMs treated with PBS or 50 pg/ml LPS were stained with specific antibodies against Tollip, the late endosome/lysosome marker LAMP1, or the mitochondrial dye MitoTracker Red. Consistent with previous findings (33, 34), Tollip co-localized with late endosomes/lysosomes in resting cells as shown by confocal microscopy, enabling lysosome fusion and cellular homeostasis (Fig. 4B). In sharp contrast, a clear separation of Tollip from LAMP1-positive endosomes was observed in cells treated with 50 pg/ml LPS. Instead, we observed increased co-localization of Tollip with mitochondria in LPS-treated cells (Fig. 4C), consistent with our previous report (12).
Next, we aimed to further confirm that the disruption of lysosome fusion by super-low-dose LPS is mediated by Tollip removal from late endosomes/lysosomes. To achieve this objective, we employed a Tollip mutant MEF cell line. Tollip fulfills its function in lysosome fusion through its critical PI(3)P-binding potential of the C2 domain (35, 36). Notably, the Tollip CUE domain compromises the PI(3)P-binding potential of the Tollip C2 domain (35). The Tollip M240A/F241A mutant lacks a functional CUE domain and thus may have constitutive lysosome localization. We hypothesized that super-low-dose LPS may fail to translocate the Tollip mutant away from lysosomes to mitochondria and fail to block the fusion of lysosomes with endosomes/autophagosomes.
To test this hypothesis, we treated WT and Tollip mutant MEFs with either PBS or 50 pg/ml LPS. The distribution of WT and mutant Tollip MEFs was visualized by immunohistochemical staining and confocal analyses. As shown in Fig. 5A, super-low-dose LPS treatment caused translocation of WT Tollip away from late endosomes/lysosomes to mitochondria, as manifested by the separation of Tollip staining from LAMP1 staining. In contrast, mutant Tollip retained its co-localization with LAMP1 in the presence or absence of super-low-dose LPS (Fig. 5, A and B).
FIGURE 5.

Cells with mutant Tollip constitutively localized at late endosomes/lysosomes exhibit lysosome fusion in the presence of super-low-dose LPS. WT and Tollip M240A/F241A mutant MEF cells were treated with PBS or 50 pg/ml LPS for 24 h. A and B, the intracellular distribution of Tollip in late endosomes/lysosomes or mitochondrial compartments was visualized by confocal fluorescence microscopy. Cells were stained with goat anti-mouse Tollip antibody, followed by Alexa Fluor 488-labeled rabbit anti-goat IgG together with either Cy3-conjugated LAMP1-specific antibody (A) or MitoTracker Red (B). C, fusion of lysosomes with autophagosomes was visualized by staining autophagosomes with Alexa Fluor 488-labeled LC3 (LCIII) antibody and Cy3-conjugated LAMP1-specific antibody.
We functionally tested the fusion of lysosomes with autophagosomes in WT and Tollip mutant MEF cells because MEFs are not proficient in endocytic uptake of zymosan particles. To test the fusion of lysosomes with autophagosomes, we co-stained the cells with the autophagosome marker LC3 and the lysosome marker LAMP1. Indeed, we observed that super-low-dose LPS caused a separation of LC3 and LAMP1 in WT MEFs, an indication of the disruption of lysosome fusion with autophagosomes (Fig. 5B). In contrast, LC3 and LAMP1 were co-localized in Tollip mutant MEF cells with or without LPS treatment (Fig. 5C).
Impaired Wound Healing and Elevated Inflammation in Tollip-deficient Mice
We next tested the in vivo relevance of our above observation by using a full skin-deep excision wound-healing model in WT and Tollip-deficient mice. Mice injected with super-low-dose LPS had delayed wound closure compared with mice injected with PBS. Furthermore, Tollip-deficient mice demonstrated significantly reduced wound closure compared with WT mice (Fig. 6A).
FIGURE 6.

Impaired wound repair and increased inflammatory monocytes in wound tissues in Tollip-deficient mice. A, WT and Tollip-deficient mice were injected intraperitoneally with either PBS or 100 pg/mouse LPS every 3 days for 10 days. On day 10, the lower backs of the mice were punctured with 4-mm full skin-depth wounds and monitored daily for wound size. The ratios of the unhealed wound sizes to the original wounds are plotted (n > 7). *, p < 0.05; ***, p < 0.001. B, total protein lysates were harvested from wound tissues from WT and Tollip-deficient mice. The levels of phospho-JNK (p-JNK), p62, and Tollip were determined by Western blotting. Representative blots are shown. The relative intensities of p62 and phospho-JNK (12 WT samples and six Tollip-deficient samples) were adjusted with GAPDH loading controls and are plotted. *, p < 0.05; ***, p < 0.001.
To determine whether the in vitro mechanisms remain valid in vivo, we examined the levels of phospho-JNK, p62, and phospho-VPS34 in wound tissues. As shown in Fig. 6B, wound tissues in mice injected with super-low-dose LPS had elevated levels of phospho-JNK and p62 compared with wound tissues in mice injected with PBS, suggesting a disruption of lysosome fusion with autophagosomes and elevated inflammation in vivo. Super-low-dose LPS also reduced the total protein levels of Tollip in wound tissues (Fig. 6B). Consistent with our in vitro observation, wound tissues from Tollip knock-out mice exhibited elevated levels of phospho-JNK and p62.
This study reveals novel mechanisms that underlie low-grade chronic inflammation induced by super-low-dose endotoxin. Our findings suggest that super-low-dose LPS may selectively induce cell stress and propagate nonresolving low-grade inflammation through the disruption of lysosome fusion with endosomes/autophagosomes. We have demonstrated that super-low-dose LPS inhibited lysosome fusion through the clearance of Tollip from late endosomes/lysosomes and the subsequent suppression of AMPK and VPS34 (Fig. 7).
FIGURE 7.
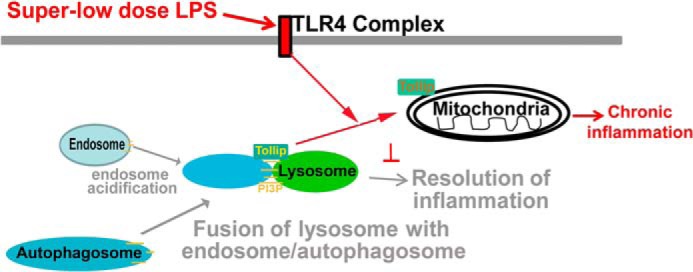
Schematic diagram of the role and regulation of Tollip in innate macrophages challenged with super-low-dose endotoxin.
DISCUSSION
Emerging studies point to the concept that an orderly fusion of lysosomes with endosomes/autophagosomes is essential for resolving inflammation (14, 15). Disruption of this process has been shown to cause elevated activation of inflammatory macrophages through exacerbated activation of stress kinases and chronic inflammatory diseases such as atherosclerosis (37). Our data reveal that super-low-dose LPS is highly potent in disrupting lysosome fusion and causing low-grade inflammation. Given the emerging recognition of low-grade endotoxemia in chronic diseases, our work provides a novel mechanism reconciling the pathological impacts of super-low-dose endotoxemia in humans and experimental animals.
Our study reveals a critical role of Tollip in maintaining proper homeostasis in resting cells through facilitating the fusion of lysosomes with endosomes/autophagosomes. We demonstrated that a reduction in Tollip through either gene knock-out or TLR-mediated clearance renders macrophages deficient in lysosome fusion. Our data provide functional relevance to previous biochemical studies reporting the interaction of Tollip with PI(3)P near late endosome/lysosome compartments (29, 33). Tollip was also shown to interact with Tom1, a key molecule involved in lysosome fusion (34). Consistent with our observation, a recent study suggests that Tollip may be involved in autophagy (36). Our study not only extends these previous works, but also provides a clear role of Tollip in the completion of lysosome fusion with endosomes/autophagosomes in the context of innate macrophages. Our data further reveal that Tollip facilitates lysosome fusion through affecting the activation status of the lipid phosphatase VPS34. Proper lysosome fusion is critically important during the resolution of inflammation through facilitating the clearance of cell debris, microorganisms, and other stress signals (6, 15, 37, 38). Our findings suggests that Tollip may serve as a key target for the modulation of cell stress and inflammation. In the context of super-low grade endotoxemia, a phenomenon widely present in humans with chronic infection or inflammation (39), we speculate that Tollip may be degraded and/or cleared away from late endosomes/lysosomes. Supporting this hypothesis, our data reveal that super-low-dose LPS can indeed reduce cellular Tollip levels, clear Tollip away from late endosomes/lysosomes to mitochondria, and disrupt lysosome fusion. Thus, depending on its cellular location and binding partner, Tollip may serve as a double-edged sword in inflammation through its lipid-binding and ubiquitin-binding domains (40). Our biochemical data further clarify the inhibitory nature of the Tollip ubiquitin-binding CUE domain in lysosome fusion. We found that the Tollip mutant protein with a modified CUE domain preferentially localizes at late endosomes/lysosomes, and cells carrying this Tollip mutant can maintain proper lysosome fusion when challenged with super-low-dose LPS. Future strategies that explore compounds capable of stabilizing Tollip or retaining Tollip at late endosomes/lysosomes may hold therapeutic potential.
Our work provides a mechanistic explanation for recent clinical studies regarding Tollip polymorphisms in humans (41, 42). For example, humans with a rare Tollip haplotype have reduced Tollip levels and elevated risks for infection and tuberculosis (41). Other studies suggest that Tollip variants may be associated with elevated risks of sepsis, dermatitis, and pulmonary fibrosis (43–45). Our wound-healing model clearly demonstrates a key role for Tollip in the resolution of inflammation during tissue repair. Collectively, our results suggest that reduced lysosome fusion due to Tollip deficiency may be responsible for reduced bacterial clearance and/or inflammatory complications.
Acknowledgments
We thank laboratory members for helpful discussions and technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL115835 and AI106287 (to H. W. C. and L. L.).
- AMPK
- AMP-activated protein kinase
- PI(3)P
- phosphatidylinositol 3-phosphate
- BMDM
- bone marrow-derived macrophage
- MEF
- murine embryonic fibroblast.
REFERENCES
- 1. Andreasen A. S., Kelly M., Berg R. M., Møller K., Pedersen B. K. (2011) Type 2 diabetes is associated with altered NF-κB DNA binding activity, JNK phosphorylation, and AMPK phosphorylation in skeletal muscle after LPS. PLoS ONE 6, e23999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim W. G., Mohney R. P., Wilson B., Jeohn G. H., Liu B., Hong J. S. (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J. Neurosci. 20, 6309–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maitra U., Parks J. S., Li L. (2009) An innate immunity signaling process suppresses macrophage ABCA1 expression through IRAK-1-mediated downregulation of retinoic acid receptor α and NFATc2. Mol. Cell. Biol. 29, 5989–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shoelson S. E., Lee J., Goldfine A. B. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S., Zhang X., Song L., Le W. (2012) Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 22, 110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang P., Mizushima N. (2014) Autophagy and human diseases. Cell Res. 24, 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolfe D. M., Lee J. H., Kumar A., Lee S., Orenstein S. J., Nixon R. A. (2013) Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 37, 1949–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Q., Mao Z. (2010) Dysregulation of autophagy and Parkinson's disease: the MEF2D link. Apoptosis 15, 1410–1414 [DOI] [PubMed] [Google Scholar]
- 9. Lassenius M. I., Pietiläinen K. H., Kaartinen K., Pussinen P. J., Syrjänen J., Forsblom C., Pörsti I., Rissanen A., Kaprio J., Mustonen J., Groop P. H., Lehto M., and FinnDiane Study Group (2011) Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 34, 1809–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang S., Li L. (2011) Metabolic endotoxemia: a novel concept in chronic disease pathology. J. Med. Sci. 31, 191–209 [Google Scholar]
- 11. Wiesner P., Choi S. H., Almazan F., Benner C., Huang W., Diehl C. J., Gonen A., Butler S., Witztum J. L., Glass C. K., Miller Y. I. (2010) Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor κB and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 107, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maitra U., Deng H., Glaros T., Baker B., Capelluto D. G., Li Z., Li L. (2012) Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J. Immunol. 189, 1014–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parkinson-Lawrence E. J., Shandala T., Prodoehl M., Plew R., Borlace G. N., Brooks D. A. (2010) Lysosomal storage disease: revealing lysosomal function and physiology. Physiology 25, 102–115 [DOI] [PubMed] [Google Scholar]
- 14. Cannizzo E. S., Clement C. C., Morozova K., Valdor R., Kaushik S., Almeida L. N., Follo C., Sahu R., Cuervo A. M., Macian F., Santambrogio L. (2012) Age-related oxidative stress compromises endosomal proteostasis. Cell Rep. 2, 136–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer M. L., Blohmke C. J., Falsafi R., Fjell C. D., Madera L., Turvey S. E., Hancock R. E. (2013) Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. J. Immunol. 190, 1227–1238 [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Alvarez R., Roderiquez G., Guan E., Caldwell Q., Wang J., Phelan M., Norcross M. A. (2005) CpG-independent synergistic induction of β-chemokines and a dendritic cell phenotype by orthophosphorothioate oligodeoxynucleotides and granulocyte-macrophage colony-stimulating factor in elutriated human primary monocytes. J. Immunol. 174, 6113–6121 [DOI] [PubMed] [Google Scholar]
- 17. Miura K., Matsuo J., Rahman M. A., Kumagai Y., Li X., Rikihisa Y. (2011) Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-κB but not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or Toll-like receptors. Infect. Immun. 79, 4947–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Egan D., Kim J., Shaw R. J., Guan K.-L. (2011) The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao K., Klionsky D. J. (2011) AMPK activates autophagy by phosphorylating ULK1. Circ. Res. 108, 787–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong P. M., Puente C., Ganley I. G., Jiang X. (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Devereaux K., Dall'Armi C., Alcazar-Roman A., Ogasawara Y., Zhou X., Wang F., Yamamoto A., De Camilli P., Di Paolo G. (2013) Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PLoS ONE 8, e76405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaber N., Dou Z., Chen J.-S., Catanzaro J., Jiang Y.-P., Ballou L. M., Selinger E., Ouyang X., Lin R. Z., Zhang J., Zong W.-X. (2012) Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U.S.A. 109, 2003–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Juhász G., Hill J. H., Yan Y., Sass M., Baehrecke E. H., Backer J. M., Neufeld T. P. (2008) The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J. Cell Biol. 181, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noda T., Matsunaga K., Taguchi-Atarashi N., Yoshimori T. (2010) Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 21, 671–676 [DOI] [PubMed] [Google Scholar]
- 25. Furuya T., Kim M., Lipinski M., Li J., Kim D., Lu T., Shen Y., Rameh L., Yankner B., Tsai L. H., Yuan J. (2010) Negative regulation of Vps34 by Cdk-mediated phosphorylation. Mol. Cell 38, 500–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J., Kim Y. C., Fang C., Russell R. C., Kim J. H., Fan W., Liu R., Zhong Q., Guan K.-L. (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Funderburk S. F., Wang Q. J., Yue Z. (2010) The beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tumbarello D. A., Waxse B. J., Arden S. D., Bright N. A., Kendrick-Jones J., Buss F. (2012) Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 14, 1024–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li T., Hu J., Li L. (2004) Characterization of Tollip protein upon lipopolysaccharide challenge. Mol. Immunol. 41, 85–92 [DOI] [PubMed] [Google Scholar]
- 30. Piao W., Song C., Chen H., Diaz M. A., Wahl L. M., Fitzgerald K. A., Li L., Medvedev A. E. (2009) Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-β-dependent pathways and increases expression of negative regulators of TLR signaling. J. Leukoc. Biol. 86, 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang G., Ghosh S. (2002) Negative regulation of Toll-like receptor-mediated signaling by Tollip. J. Biol. Chem. 277, 7059–7065 [DOI] [PubMed] [Google Scholar]
- 32. Puri R., Suzuki T., Yamakawa K., Ganesh S. (2012) Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 21, 175–184 [DOI] [PubMed] [Google Scholar]
- 33. Katoh Y., Shiba Y., Mitsuhashi H., Yanagida Y., Takatsu H., Nakayama K. (2004) Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. J. Biol. Chem. 279, 24435–24443 [DOI] [PubMed] [Google Scholar]
- 34. Katoh Y., Imakagura H., Futatsumori M., Nakayama K. (2006) Recruitment of clathrin onto endosomes by the Tom1-Tollip complex. Biochem. Biophys. Res. Commun. 341, 143–149 [DOI] [PubMed] [Google Scholar]
- 35. Mitra S., Traughber C. A., Brannon M. K., Gomez S., Capelluto D. G. (2013) Ubiquitin interacts with the Tollip C2 and CUE domains and inhibits binding of Tollip to phosphoinositides. J. Biol. Chem. 288, 25780–25791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu K., Psakhye I., Jentsch S. (2014) Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 158, 549–563 [DOI] [PubMed] [Google Scholar]
- 37. Emanuel R., Sergin I., Bhattacharya S., Turner J. N., Epelman S., Settembre C., Diwan A., Ballabio A., Razani B. (2014) Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler. Thromb. Vasc. Biol. 34, 1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madsen D. H., Leonard D., Masedunskas A., Moyer A., Jürgensen H. J., Peters D. E., Amornphimoltham P., Selvaraj A., Yamada S. S., Brenner D. A., Burgdorf S., Engelholm L. H., Behrendt N., Holmbeck K., Weigert R., Bugge T. H. (2013) M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 202, 951–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morris M., Li L. (2012) Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch. Immunol. Ther. Exp. 60, 13–18 [DOI] [PubMed] [Google Scholar]
- 40. Capelluto D. G. (2012) Tollip: a multitasking protein in innate immunity and protein trafficking. Microbes Infect. 14, 140–147 [DOI] [PubMed] [Google Scholar]
- 41. Shah J. A., Vary J. C., Chau T. T., Bang N. D., Yen N. T., Farrar J. J., Dunstan S. J., Hawn T. R. (2012) Human TOLLIP regulates TLR2 and TLR4 signaling and its polymorphisms are associated with susceptibility to tuberculosis. J. Immunol. 189, 1737–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montoya-Buelna M., Fafutis-Morris M., Tovar-Cuevas A. J., Alvarado-Navarro A., Valle Y., Padilla-Gutierrez J. R., Muñoz-Valle J. F., Figuera-Villanueva L. E. (2013) Role of Toll-interacting protein gene polymorphisms in leprosy Mexican patients. BioMed Res. Int. 2013, 459169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noth I., Zhang Y., Ma S. F., Flores C., Barber M., Huang Y., Broderick S. M., Wade M. S., Hysi P., Scuirba J., Richards T. J., Juan-Guardela B. M., Vij R., Han M. K., Martinez F. J., Kossen K., Seiwert S. D., Christie J. D., Nicolae D., Kaminski N., Garcia J. G. (2013) Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir. Med. 1, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song Z., Yin J., Yao C., Sun Z., Shao M., Zhang Y., Tao Z., Huang P., Tong C. (2011) Variants in the Toll-interacting protein gene are associated with susceptibility to sepsis in the Chinese Han population. Crit. Care 15, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schimming T. T., Parwez Q., Petrasch-Parwez E., Nothnagel M., Epplen J. T., Hoffjan S. (2007) Association of Toll-interacting protein gene polymorphisms with atopic dermatitis. BMC Dermatol. 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]