

SUMMARY
At an injury-site, efficient clearance of apoptotic cells by wound macrophages or efferocytosis is a pre-requisite for the timely resolution of inflammation. Emerging evidence indicates that miR-21 may regulate the inflammatory response. In this work, we sought to elucidate the significance of miR-21 in the regulation of efferocytosis mediated suppression of innate immune response, a key process implicated in resolving inflammation following injury. An increased expression of inducible miR-21 was noted in post-efferocytotic peripheral blood monocyte-derived macrophages (MDM). Such induction of miR-21 was associated with silencing of its target genes PTEN and PDCD4. Successful efferocytosis of apoptotic cells by MDM resulted in the suppression of LPS-induced NF-κB activation and TNFα expression. Interestingly, bolstering of miR-21 levels alone using miR mimic resulted in significant suppression of LPS-induced TNFα expression and NFκB activation. We report that efferocytosis-induced miR-21, by silencing PTEN and GSK3β, tempers LPS-induced inflammatory response. Macrophage efferocytosis is known to trigger the release of anti-inflammatory cytokine IL-10. This study demonstrates that following successful efferocytosis, miR-21 induction in macrophages silence PDCD4 favoring cJun-AP1 activity which in turn results in elevated production of anti-inflammatory IL-10. In summary, this work provides direct evidence implicating miRNA in the process of turning-on an anti-inflammatory phenotype in the post-efferocytotic macrophage. Elevated macrophage miR-21 promotes efferocytosis and silences target genes PTEN and PDCD4 which in turn accounts for a net anti-inflammatory phenotype. Findings of this study highlight the significance of miRNAs in the resolution of wound inflammation.
INTRODUCTION
Efferocytosis, a term coined by deCathelineau and Henson (1) and Gardai et al (2), refers to phagocytosis of apoptotic cells (3). Efferocytosis is the final fate of apoptotic cells at an injury site. Successful efferocytosis drives timely resolution of inflammation (4–7). Defective clearance of apoptotic cells has been linked to autoimmunity and persistent inflammatory diseases (8). In contrast to uptake of pathogens or FcR-mediated phagocytosis, the engulfment of apoptotic cells does not lead to pro-inflammatory cytokine production by macrophages (9). Thus, efferocytosis is non-inflammatory and non-immunogenic (10).
MicroRNAs (miRNAs), 19- to 22-nucleotide long, are noncoding RNAs found in all eukaryotic cells (11). These non-coding small RNA regulate roughly 30% of the human genome primarily through translational repression (12). miRNAs were linked with immune responses in a study where miRNA expression profiling was performed in a monocytic cell line treated with the lipopolysaccharide (LPS), a ligand for TLR4 (13). The expression of miR-146a, miR-155 and miR-132 were induced in response to LPS stimulation (13, 14). While the role of miRNA in inflammatory response associated with cancer has been extensively studied (15–18), information on their role in regulating efferocytosis mediated immune suppression and resolution of inflammation is scanty. It has been commonly noted that inflammatory stimuli induce miR-21 (19, 20). A single primary transcript containing miR-21 (pri-miR-21) is transcribed from an evolutionarily conserved promoter that resides in an intron of an overlapping coding gene, TMEM49 (21). PTEN and the tumor suppressor PDCD4 have been identified as one of the first validated direct targets that are translationally silenced by miR-21 (22, 23). Recent evidences indicate that miR-21 may serve as an rheostat to control the inflammatory response (24). In one of the first works that noted the anti-inflammatory properties of miR-21 in macrophages, it was reported that miR-21 silences the pro-inflammatory interleukin (IL)-12 (25). In the lungs, miR-21 inhibited toll-like receptor 2 agonist-induced lung inflammation in mice (26). miR-21 is inducible by resolvin D1, an endogenous lipid mediator generated during the resolution phase of acute inflammation. Thus, miR-21 has been proposed to a play a role in resolving acute inflammation (27). Beyond its direct effects on macrophages, miR-21 acts on a number of biological targets validated in a variety of cell types pointing to an overall anti-inflammatory role (24). As an anti-inflammatory agent, miR-21 silences PTEN as well as PDCD4 (24, 28). In this work, we sought to elucidate the significance of miR-21 in the regulation of efferocytosis mediated suppression of innate immune response, a key process implicated in resolving inflammation following injury.
MATERIALS IN METHODS
Peripheral Blood Monocyte Derived Macrophages (MDM)
Human peripheral blood mononuclear cells were isolated from fresh blood leukocyte source packs (American Red Cross, Columbus, OH) by density gradient centrifugation using a Ficoll-Hypaque density gradient (GE Healthcare, formerly Amersham Biosciences, Piscataway, NJ). Positive selection for monocytes was performed using CD14 antibody conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA). Purity of these preparations of monocytes was >90% as determined by fluorescence-activated cell sorting analyses using CD14 antibodies. Differentiation of these cells to macrophages (MDM) was performed as described (29).
Apoptotic cell clearance (efferocytosis) assay
MDM were seeded in 6-well plates. Apoptosis in Jurkat cells was induced by treating the cells with anti-Fas Antibody (human, activating), clone CH11 (250 ng/ml, Millipore, Temecula, CA). Apoptotic Jurkat cells (Clone E6-1, ATCC, Manassas, VA) were added to MDM cultures at a ratio of (1:10) macrophage:Jurkat cell. The co-culture and efferocytosis assay was performed as described previously (4). Following completion of efferocytosis assay, LPS was added to the culture media as specified in figure legends.
ELISA
For measurement of IL-10 and TNF-α produced by macrophages, cells were seeded in 6-well or 12-well plates and cultured in RPMI 1640 medium containing 10% heat-inactivated bovine serum under standard culture conditions. After specified duration, the culture media was collected and IL-10 and TNF-α levels were measured using commercially available ELISA kits (R & D Systems, Minneapolis, MN) as per manufacturer’s instructions (4, 29).
Reverse transcription and quantitative RT-PCR (qPCR)
Total RNA was extracted using the mirVana RNA isolation kit (Ambion, Austin, TX), according to manufacturer’s instructions. mRNA was quantified by real-time or quantitative (Q) PCR assay using the double-stranded DNA binding dye SYBR Green-I as described previously (29–31). For determination of miR expression, specific TaqMan assays for miRs and the TaqMan Micro-RNA Reverse Transcription Kit were used, followed by real time PCR using the Universal PCR Master Mix (Applied Biosystems, Foster City, CA)(22, 32, 33).
miRIDIAN miRNA mimic/inhibitor and siRNA delivery
DharmaFECT™ 1 transfection reagent (Dharmacon RNA technologies, Lafayette, CO) was used to transfect cells with miRIDIAN mimic-miR-21 (Dharmacon RNA technologies, Lafayette, CO) for 72h as per the manufacturer’s instructions. miRIDIAN miRNA mimic/inhibitor negative controls (Dharmacon RNA Technologies, Lafayette, CO) were used for control transfections. siRNA transfections were performed as described (29, 31). In brief, DharmaFECT™ 1 was used to transfect cells with 100nM siRNA pool of PTEN, PDCD4 or cJun (Dharmacon RNA technologies, Lafayette, CO) for 72h. For control, siControl non-targeting siRNA pool (mixture of 4 siRNA, designed to have ≥4 mismatches with the gene) was used. Using this approach, the transfection efficiency was ~70%.
Western blot
Western blot was performed using primary antibody against PDCD4, PTEN, phospho-p65, phospho-IκB-α, IκB-α, phospho-IKK-β, IKK-β, phospho-c-Jun (Cell Signaling) and c-Jun (Santa Cruz Biotechnology) as described previously(31, 34, 35). Membranes were probed with anti-GAPDH or β-actin antibody to control for sample loading.
Adenoviral delivery of PTEN, NFκB luciferase reporter and AP1 luciferase reporter
Primary human macrophages were infected with adenovirus encoding for PTEN (Applied Biological Materials Inc., Canada), NFκB promoter luciferase reporter or AP1 luciferase reporter gene (Vector Biolabs, Philadelphia, PA) as described previously (22, 36). After 72-h infection, cells were harvested for protein, RNA, NFκB reporter or AP1 reporter luciferase assay.
DNA binding of NFκB
Nuclear protein extracts of cells were prepared using the nuclear extraction kit (Active Motif, Carlsbad, CA) according to manufacturer’s instructions. Binding of NFκB family of proteins to their consensus sites was determined using an ELISA-based Trans-AM NFκB kit (Active Motif, Carlsbad, CA).
miR-target 3′-UTR luciferase reporter assay
miRIDIAN mimic-miR-21 were transfected to HEK293 cells followed by transfection with pGL3-PTEN-3′-UTR plasmid or lenti luc-PDCD4–3′UTR (SA Biosciences). Luciferase assay were performed using the reporter assay system (Promega) as described (32, 33).
AP-1 reporter assay
For AP-1 transcriptional activation assay HEK293 TLR4/IL-1R1/MD-2 cells were provided by Dr. Mikhail Gavrilin at The Ohio State University (37). Cells were transfected with 500 ng of AP-1 plasmid (Stratagene, CA) using Lipofectamine LTX/Plus reagent (Invitrogen, NY) according to the manufacturer’s protocol. After 48 h, cells were transfected with control or miR-21 mimic for 72 h. Luciferase activity was determined using the luciferase reporter assay system (Promega, WI).
Statistics
Data are reported as mean ± SD of 3–4 experiments as indicated in the respective figure legends. Comparisons among multiple groups were tested using analysis of variance (ANOVA). p ≤ 0.05 was considered statistically significant.
RESULTS
Increased expression of LPS-inducible miR-21 following efferocytosis
We determined whether successful efferocytosis or engulfment of apoptotic cells by macrophages regulate the expression of miR-21. For efferocytosis assay, MDM were co-cultured with apoptotic (effrhi) or viable (effrlo) Jurkat T cells. Such co-culture resulted in successful engulfment of apoptotic Jurkat cells but not the viable cells (Fig 1A). The current study addressed efferocytosis associated with inflammatory settings. Inflammatory response in engulfing MDM was induced by treating cells with the TLR-4 agonist lipopolysaccharide (LPS). Following LPS treatment (6h or 24h), the expression of miR-21 expression was increased in MDM that engulfed apoptotic cells compared to the MDM that were co-cultured with viable cells (Fig 1B). In the absence of TLR-4 agonist, miR-21 expression in MDMs co-cultured with viable or apoptotic cells remained unaltered (Fig 1C). To test whether the LPS-induced miR-21 expression response is specific to efferocytosis, cytoskeleton was disrupted using cytochalasin D. Cytochasin D is known to block efferocytosis by disrupting actin polymerization (38). Pre-incubation with cytochasin D blocked efferocytosis mediated miR-21 induction (Fig 1D). Furthermore, miR-21 expression in macrophages remained unaltered in response to phagocytosis of bacteria (not shown). These two lines of evidence support that induction of miR-21 is a response that is specifically caused by efferocytosis. Finally, induction of miR-21 expression was associated with silencing of its target genes PTEN and PDCD4 (Fig 1E–F).
Figure 1. Increased expression of miR-21 and silencing of target proteins PTEN & PDCD4 in MDM following engulfment of apoptotic cells (efferocytosis).
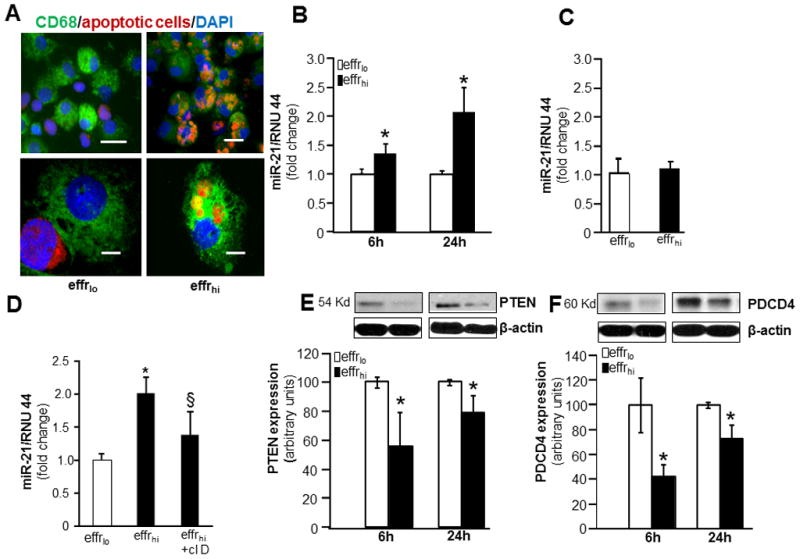
For efferocytosis assay, blood monocyte derived macrophages (MDM) were co-cultured with apoptotic or viable Jurkat T cells. Non-efferocytosed Jurkat cells were removed by washing with saline. A, Representative images of MDM (green, CD68) cultured with either viable (effrlo) or apoptotic (effrhi) labeled (red, CMTMR™ cell tracker). Cells were counterstained with DAPI (nuclear, blue); B, MiR-21 expression in effrlo or effrhi groups following 6 or 24h of LPS (1 μg/ml) treatment. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with effrlo group. C, miR-21 expression in effrlo or effrhi groups post-efferocytosis. Data are mean ±SD (n = 6); D, miR expression in cells pretreated with cytochalasin D (1 mg/ml, 1h) followed by efferocytosis and LPS treatment. miR-21 expression was measured 24h post-LPS treatment. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with effrlo; §, p<0.05 compared to macrophage cultured with effrhi group. E–F, Expression of miR-21 target proteins, E, PTEN and F, PDCD4 in effrlo or effrhi groups following 6 or 24h of LPS (1 μg/ml) treatment measured using Western blot. Quantification of PTEN or PDCD4 levels was performed using densitometry. Data were normalized to β-actin. Data are mean ± SD (n=3). *, p<0.05 compared to macrophage cultured with effrlo group.
Efferocytosis-induced miR-21 suppressed the pro-inflammatory NFκB-TNFα pathway
Under pro-inflammatory conditions such as presence of pathogenic microbial stimuli, the engulfment of apoptotic cells by macrophage suppressed production of the pro-inflammatory cytokine TNFα and induced the production of anti-inflammatory cytokine IL-10 (39–41). Successful efferocytosis of apoptotic Jurkat cells by MDM resulted in suppression of LPS-induced TNFα levels both at protein as well as mRNA levels (Fig 2A–B). Interestingly, isolated bolstering of miR-21 levels in MDM using miR mimic (miRIDIAN hsa-miR-21, Fig 2F) resulted in significant suppression of LPS-induced TNFα expression (Fig 2C). Lenti-miR-000-zip or lenti-miR-21-zip vectors and puromycin selection were utilized to generate THP-1 cells with stable knockdown of miR-21 (Fig G-H). Such THP-1 cells with stable knockdown of miR-21 expression were differentiated to macrophages as described (29). In these cells, LPS-induced TNFα levels were further potentiated as compared to that of LPS treated lenti-miR-000-zip THP-1 cells (Figure 2D). Finally, efferocytosis dependent suppression of LPS-induced TNFα expression was significantly blocked in cells with stable knockdown of miR-21 levels (Fig 2E). In summary, these data establish that elevated miR-21 causes efferocytosis-induced suppression of inducible TNFα expression.
Figure 2. miR-21 is required for efferocytosis induced suppression of pro-inflammatory TNFα expression.
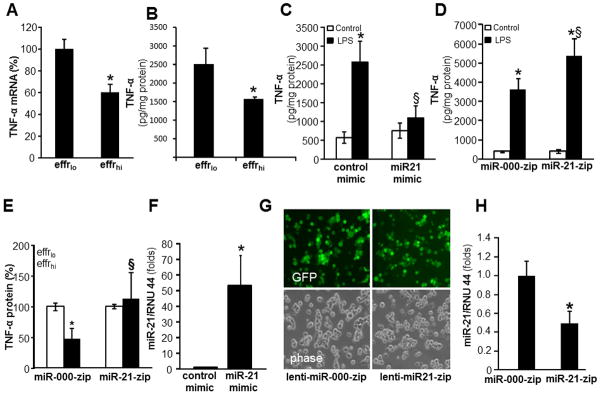
A–B, For efferocytosis assay, blood monocyte derived macrophages (MDM) were co-cultured either viable (effrlo) or apoptotic (effrhi) Jurkat T cells for 1h. Non-efferocytosed cells were removed by washing with saline. Cells were treated with LPS (1ug/ml) for 6h (mRNA) or 24h (protein) post efferocytosis. TNFα A, mRNA and B, protein expression were measured using quantitative PCR and ELISA, respectively. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with effrlo group. C, MDM were transfected with miRIDIAN hsa-miR-21 mimic or control-mimic to increase miR-21 levels. Cells were activated with LPS (1ug/ml) for 24h after forced expression of miR-21 in MDM. The TNFα protein levels in cells were determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with control-mimic group. §, p<0.05 compared to control-mimic LPS treated group. D, Stable knock down of miR-21 in THP-1 monocytic cells was achieved following lentiviral transduction with lenti-miR-000-zip or lenti-miR-21-zip vectors and puromycin selection. The THP-1 cells with stable knockdown of miR-21 were cultured and differentiated to macrophages with phorbol-12-myristate-13-acetate (PMA, 20 ng/ml, 48h). Cells were activated with LPS (1ug/ml) for 24h. TNFα protein released by the cells in culture media was determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with miR-000-zip (control) group. §, p<0.05 compared to miR-000-zip LPS treated group. E, Loss of miR-21 in macrophages result in abrogation of efferocytosis mediated suppression of LPS-induced TNFα expression. MiR-000-zip or miR-21-zip cells were subjected to efferocytosis followed by treatment with LPS for 24h. TNFα protein levels in cells were determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with effrlo group. §, p0.05 compared to miR-000-zip cells. F, miR-21 expression in miRIDIAN hsa-miR-21 mimic or control-mimic transfected cells. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with control-mimic cells. G, GFP (green) and phase contrast (phase) images of THP-1 cells showing transduction efficiency following lentiviral transduction with lenti-mi-000-zip or lenti-miR-21-zip vectors and puromycin selection showing over 80% cells were GFP positive. H, miR-21 expression in lenti-miR-000-zip or lenti-miR-21-zip cells. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with miR-000-zip cells.
NF-κB is one of the major transcription factors that drive inducible TNFα expression in macrophages (42). We tested whether efferocytosis may influence LPS-induced NF-κB activation. Both DNA binding activity of NF-κB in nuclear extracts of MDM as well as NF-κB transcriptional activation as measured using NF-κB-dependent luciferase reporter gene (Ad5NFκB-LUC) was significantly inhibited in MDM co-cultured with apoptotic cells (effrhi)as compared to that in MDM co-cultured with viable cells (effrlo, Fig 3A–B). LPS induced phosphorylation of IκB as well as of the NF-κB subunit p65 in macrophages play a critical role in NF-κB transactivation (43). Efferocytosis significantly inhibited LPS-induced p65 phosphorylation (Fig 3C). Comparable to the effect of efferocytosis, increase or knockdown in miR-21 levels in MDM was inversely related to phosphorylation of p65 and IκB indicating direct regulation of NF-κB activation by miR-21 in MDM (Fig 3E–G). Bolstering miR-21 in MDM by miR mimic delivery did not influence TLR-4 expression suggesting that miR-21 acts downstream of TLR4 (Fig 3D). The delivery of miR-21 mimic to MDM, however, did enhance efferocytosis (Fig 3H).
Figure 3. Efferocytosis-induced miR-21 suppressed the pro-inflammatory NFκB-activation.
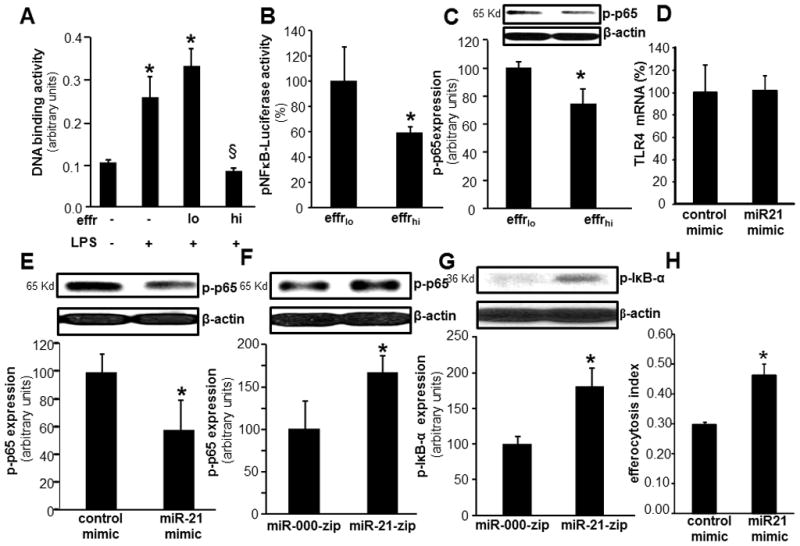
A–C, For efferocytosis assay, blood monocyte derived macrophages (MDM) were co-cultured either viable (effrlo) or apoptotic (effrhi) Jurkat T cells for 1h. Non-efferocytosed cells were removed by washing with saline. Cells were treated with LPS (1ug/ml) for 6h post efferocytosis. A, DNA binding activity of NF-κB in MDM measured using an ELISA-based (Trans-AM®) method. B, NFκB transcription activity in MDMs transiently transfected with NF-κB-dependent luciferase reporter gene (Ad5NFκB-LUC) followed by co-culture with either viable (effrlo) or apoptotic (effrhi) Jurkat T cells for 1h and LPS treatment (6h). Luciferase activity was determined. Data are mean ±SD (n = 4). C, Phosphorylation of p65 protein was measured using Western blot in human macrophages following efferocytosis and LPS treatment (1ug/ml). D, TLR4 mRNA expression in MDM transfected with miRIDIAN hsa-miR-21 mimic or control-mimic. Data are mean ± SD (n=3). E, Phosphorylation of NF-κB p65 in MDM transfected with miRIDIAN hsa-miR-21 mimic or control-mimic to increase miR-21 levels. Cells were activated with LPS after forced expression of miR-21 in MDM. Serine 536 phosphorylation on p65 was determined using Western blot. Quantification of phospho-p65 level was performed using densitometry. Data were normalized to β-actin. Data are mean ± SD (n=3). *, p<0.05 compared to control mimic transfected cells. F, LPS induced phosphorylation of p65 in THP-1 monocytic cells where stable knock down of miR-21 was achieved following lentiviral transduction with lenti-miR-000-zip or lenti-miR-21-zip vectors. Data are mean ± SD (n=3). *, p<0.05 compared to control lenti-mi-000-zip cells. G, LPS induced phosphorylation of IκB (Serine 32) in THP-1 monocytic cells where stable knock down of miR-21 was achieved following lentiviral transduction with lenti-mi-000-zip or lenti-miR-21-zip vectors. Data are mean ± SD (n=3). *, p<0.05 compared to control lenti-miR-000-zip cells. H, Efferocytosis index of MDMs after delivery of miR-21 mimic. Data is presented as mean ± SD (n = 3). *, p<0.05 compared to control.
miR-21 target PTEN exacerbated LPS-induced TNFα expression by potentiating NFκB activation
Using miR mimic, knockdown and PTEN-3′-UTR firefly luciferase expression construct we observed that PTEN is a direct target of miR-21 in MDM (Fig 4A–C). Overexpression of PTEN in MDM using adenoviral-PTEN vector (Fig 5F) resulted in increased LPS-induced TNFα production (Fig 5A). Vanadate derivatives such the bisperoxovanadium (bpV) function as phosphatase inhibitors in micromolar concentration (44). bpV(phen) specifically inhibits PTEN in nanomolar concentrations (44). Potent inhibition of LPS-induced TNFα production was noted with in MDM treated with bpV(phen) (100 nM) (Fig 5B) indicating a supporting role of PTEN in LPS-induced TNFα production. Furthermore PTEN inhibition using siPTEN or bpV(phen) blocked inducible TNFα production under conditions of miR-21 depletion (Fig 5C–D). This data suggests that PTEN plays a critical role in miR-21 mediated regulation of TNFα. Next, we determined the effect of PTEN on LPS-induced NFκB activation. Both LPS-induced NFκB transactivation using NFκB-Luc reporter construct as well as phospho-p65 induction was further potentiated in MDM where forced expression of PTEN was achieved in MDM using adPTEN. These findings support that high PTEN levels in cell increases LPS-induced NF-κB activation and therefore, TNFα expression (Fig 5E–F). Thus, the PTEN silencing effects of miR-21 may account for its anti-inflammatory function.
Figure 4. PTEN is a direct target of miR-21 in human macrophages.
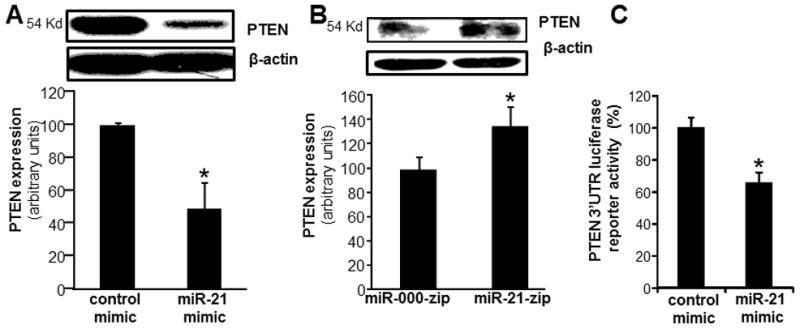
A, PTEN expression in MDM transfected with miRIDIAN hsa-miR-21 mimic or control-mimic to increase miR-21 expression. PTEN levels were determined using Western blot. Quantification of PTEN level was performed using densitometry. Data were normalized to β-actin. Data are mean ± SD (n=3). *, p<0.05 compared to control mimic transfected cells. B, PTEN expression in stably knocked down in miR-21 THP-1 monocytic cells. Data are mean ± SD (n=3). *, p<0.05 compared to control lenti-miR-000-zip cells. PTEN protein expression were measured using Western blot; C, To demonstrate that PTEN is a direct target for miR-21, HEK293 cells were transfected with a pGL3-PTEN-3′-UTR firefly luciferase expression construct and co-transfected with pRL-TK Renilla luciferase expression construct along with either miR-21 mimic or control mimic. Data represent mean ± SD (n=3). *, p<0.05 compared to control transfected cells.
Figure 5. miR-21 silences LPS induced NF-κB signaling and TNFα expression via a PTEN dependent mechanism.
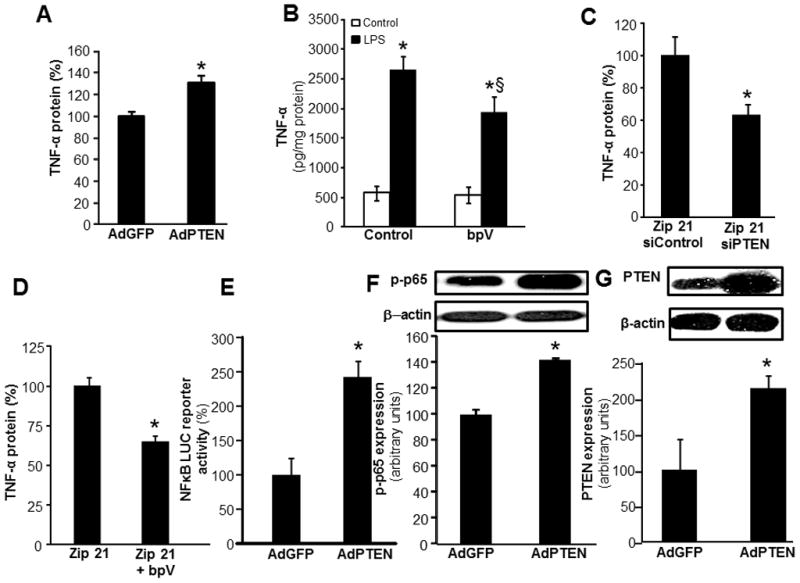
A, TNFα protein expression was measured in MDM infected with adenovirus-PTEN vector or adenovirus GFP vectors followed by LPS treatment (1ug/ml) for 24h. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with infected with adenovirus GFP vectors B, TNFα protein expression was measured in MDM treated with bpV(phen) (100nM), a PTEN inhibitor, followed by LPS treatment. Data are mean ± SD (n=4). *, p<0.05 compared to LPS non-treated cells; §, p<0.05 compared to bpV(phen) treated group. C–D, Effect of PTEN knockdown using C, siPTEN or D, PTEN inhibitor bpV(phen) on LPS-inducible TNFα protein expression in THP-1 monocytic cells where stable knock down of miR-21 was achieved following lentiviral transduction with lenti-miR-000-zip or lenti-miR-21-zip vectors. TNFα protein expression was determined in cells PTEN knockdown cells after LPS (1ug/ml) for 24h. Data are mean ± SD (n=4). *, p<0.05 compared to control Zip-21 cells. E, LPS inducible NFκB transcription activity in MDMs transiently transfected with NF-κB-dependent luciferase reporter gene (Ad5NFκB-LUC) following infection with adenovirus-PTEN or adenovirus GFP (control) vectors Luciferase activity was determined. Data are mean ±SD (n = 4). F, Phosphorylation of p65 protein was measured using Western blot in MDM infected with adenovirus-PTEN or adenovirus GFP vectors followed by LPS treatment (1ug/ml). Data are mean ± SD (n=3). *, p<0.05 compared to control adenovirus GFP infected cells. G, PTEN protein expression was measured using Western blot in MDM infected with adenovirus-PTEN vector or adenovirus GFP vectors. Quantification of PTEN level was performed using densitometry. Data are mean ± SD (n=3). *, p<0.05 compared to control adenovirus GFP infected cells.
miR-21 silencing of PTEN inhibited GSK3β implicated in NFκB activation and inducible TNFα expression
PTEN blocks the action of PI3-K by dephosphorylating the signaling lipid PIP3. Thus, PTEN antagonizes signaling through the PI3-K pathway (45). PI3-K/Akt signaling pathway is a major regulator of glycogen synthase kinase 3 (GSK3). GSK3 isoforms are normally constitutively active in a cell, and they are regulated through inhibition (46). GSK3β activity can be downregulated by phosphorylation at the N-terminal region serine 9 which leads to the inhibition of this isoform (46). In general, phosphorylation at serine- 9 has been used as a marker for inactive GSK3β (46). Knockdown of miR-21 and overexpression of PTEN both resulted in strong inhibition in the phosphorylation GSK3β. Thus, lowering of miR-21 levels in human macrophages resulted in increased GSK3β activity via a PTEN dependent mechanism (Fig 6A–B). Inhibition of GSK3β activity using a specific inhibitor, SB 216763, lowered the abundance of phospho-p65 as well as lowered phospho-IκB and phopsho-IKKβ abundance (Fig 6C–E). Thus, LPS–induced NFκB activation is dependent on GSK3β activity. Pharmacological inhibition of GSK3β activity resulted in significant inhibition of LPS-induced TNFα expression (Fig 6F). These findings support a role of GSK3β in miR-21-PTEN mediated regulation of LPS-induced NFκB activation and TNFα expression (Fig 10). Finally, pharmacological inhibition of GSK3β negated the ability of efferocytosis to blunt inducible TNFα expression supporting a key role of GSK3β in the efferocytosis dependent resolution of inflammation pathway (Fig 6G).
Figure 6. miR-21 silencing of PTEN inhibited GSK3β that in turn inhibited inducible NFκB activation.

A, Phosphorylation of GSK3β (Serine 9) in THP-1 monocytic cells where stable knock down of miR-21 was achieved following lentiviral transduction with lenti-miR-000-zip or lenti-miR-21-zip vectors followed by LPS treatment (1ug/ml). Data are mean ± SD (n=3). *, p<0.05 compared to control lenti-miR-000-zip cells. B, Phosphorylation of GSK3β protein was measured using Western blot in MDM infected with adenovirus-PTEN or adenovirus GFP vectors followed by LPS treatment. Data are mean ± SD (n=3). *, p<0.05 compared to control adenovirus GFP infected cells. C, Phosphorylation of NF-κB p65 protein was measured using Western blot in MDM treated with 10 μM SB 216763 (GSK3β inhibitor) followed by LPS treatment. Quantification was performed using densitometry. Data are mean ± SD (n=3). *, p<0.05 compared to control cells. D, Phosphorylation of IκB-α protein in MDM treated with 10 μM SB 216763, followed by LPS treatment. Data are mean ± SD (n=3). *, p<0.05 compared to control cells. E, Phosphorylation of IKKβ protein (Serine 181) in MDM treated with 10 μM SB 216763, followed by LPS treatment. Data are mean ± SD (n=3). *, p<0.05 compared to control cells. F, TNFα protein expression was measured in MDM treated with 10 μM SB 216763 followed by LPS treatment. Data are mean ± SD (n=4). *, p<0.05 compared to LPS treated cells; §, p<0.05 compared to SB 216763 treated group. G, TNFα protein expression in MDM pretreated with 10μM SB216763 followed by efferocytosis and LPS treatment. Data are mean ± SD (n=3). *, p<0.05 compared to Effrlo group; §, p<0.05 compared to SB 216763 non-treated group.
Figure 10. Central role of miR-21 in effecrocytosis dependent resolution of wound inflammation.
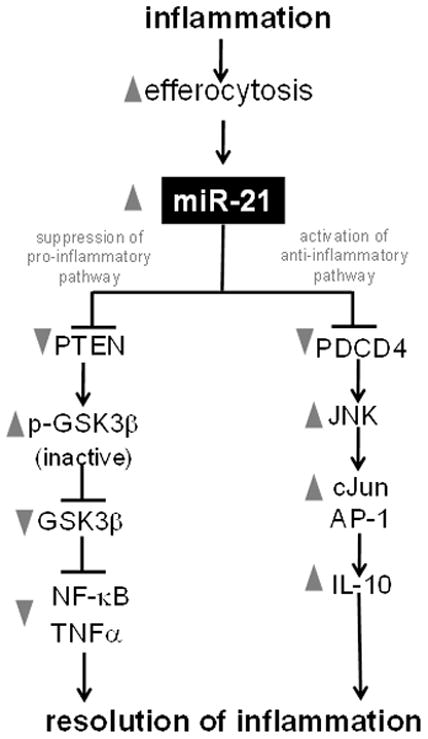
miR-21 downregulates pro-inflammatory response via blocking the TNFα-NFκB pathway, and upregulates anti-inflammatory IL-10 via the AP-1 pathway.
Successful efferocytosis potentiates inducible IL-10 expression via a miR-21 dependent mechanism
IL-10 is an anti-inflammatory cytokine (39–41). After successful efferocytosis, human MDM showed enhanced IL-10 expression (Fig 7A–B). MDM transfected with miRIDIAN hsa-miR-21 mimic to increase miR-21 levels also showed increased IL-10 protein levels compared to MDM transfected with control mimic (Fig 7C). These observations support that elevated cellular miR-21 level is sufficient to potentiate inducible IL-10 in macrophages. Efferocytosis dependent induction of IL-10 was attenuated under conditions of miR-21 inhibition demonstrating a role of miR-21 (Fig 7D). IL-10 is known to inhibit the production of LPS-induced pro-inflammatory cytokines by macrophages (47). We observe that adding IL-10 to LPS-induced MDM dose dependently inhibited inducible TNFα release from cells supporting a potent anti-inflammatory activity of IL-10 (Fig 7E).
Figure 7. Successful efferocytosis potentiates inducible IL-10 expression via a miR-21 dependent mechanism.
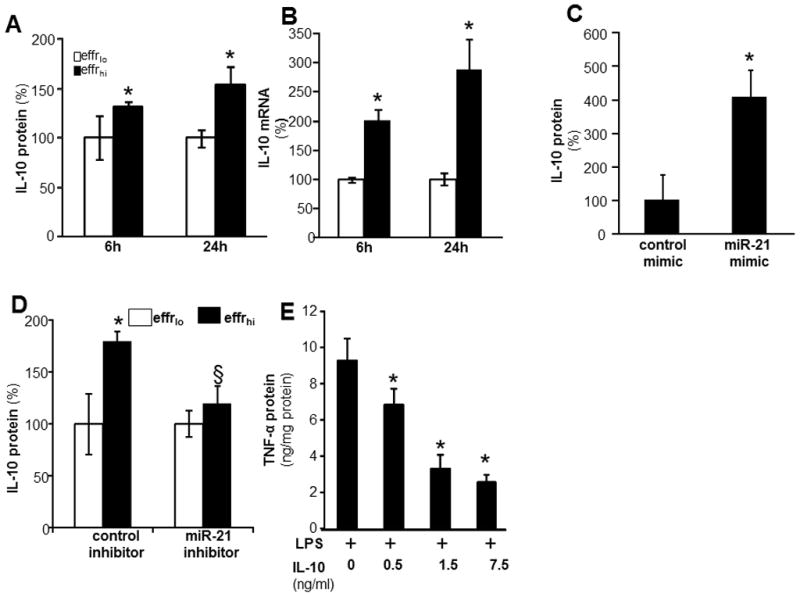
A–B, For efferocytosis assay, blood monocyte derived macrophages (MDM) were co-cultured either viable (effrlo) or apoptotic (effrhi) Jurkat T cells for 1h. Non-efferocytosed cells were removed by washing with saline. Cells were treated with LPS (1ug/ml) for 6 and 24h post efferocytosis. IL-10 A, protein and B, mRNA expression were measured using ELISA and quantitative PCR, respectively. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with effrlo group. C, MDM were transfected with miRIDIAN hsa-miR-21 mimic or control-mimic to increase miR-21 levels. Cells were activated with LPS (1ug/ml) for 24h after forced expression of miR-21 in MDM. The IL-10 protein levels in cells were determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage cultured with control-mimic group. D, MDM were transfected with miRIDIAN hsa-miR-21 hairpin single stranded inhibitor or control-inhibitor to knockdown miR-21 levels. Cells were subjected to efferocytosis followed by activation with LPS (1μg/ml) for 24h after knockdown miR-21 in MDM. The IL-10 protein levels in cells were determined using ELISA. Data are mean ±SD (n = 3); *, p<0.05 compared to macrophage cultured with effrlo group. §, compared to macrophage cultured with control inhibitor group. E, MDM were treated with recombinant human IL-10 for 1h followed by LPS (1ug/ml) activation for 24 h. The TNF-α protein levels in cells were determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to IL-10 untreated macrophage.
MiR-21 potentiated inducible IL-10 expression via a PDCD4- cJun-AP-1 pathway
To determine the mechanisms of miR-21 mediated potentiation of inducible IL-10 expression, MDM were transfected with miRIDIAN hsa-miR-21 mimic to increase cellular miR-21 abundance. PDCD4 is a confirmed target of miR-21 in macrophages (48). Our results support that finding and show that elevation of miR-21 levels inhibited PDCD4 expression in MDM (Fig 8A). Elevated cellular miR-21 also inhibited luciferase reporter activity in cells transfected with PDCD4 3′UTR-luciferase reporter construct (Fig 8B) establishing PDCD4 as a direct target of miR-21. To test for a direct role of PDCD4 in LPS-induced IL-10 expression, knock down of PDCD4 by siRNA was achieved (Fig 8C). Such knockdown resulted in ~80% lowering of PDCD4 protein levels (Fig 8C) and augmented LPS-induced IL-10 expression response (Fig 8D–E). These data establish that PDCD4 is directly implicated in LPS-induced IL-10 expression.
Figure 8. MiR-21 regulates IL-10 expression via PDCD4.
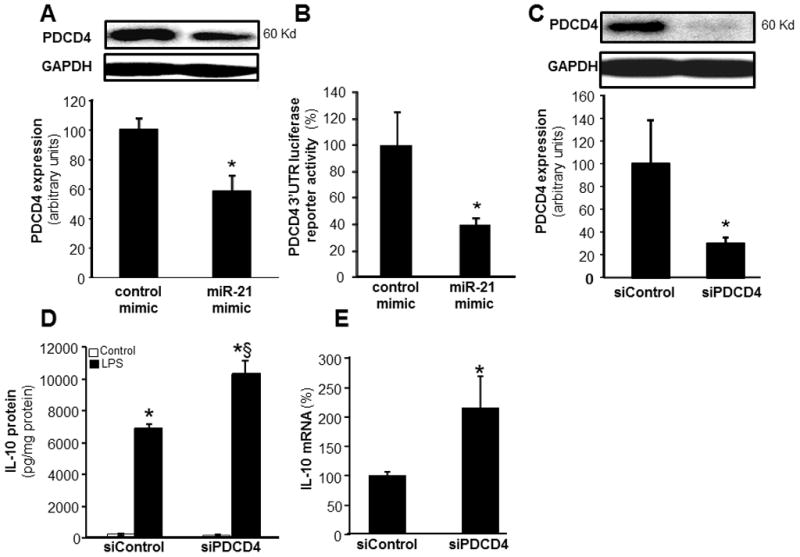
A, PDCD4 expression in MDM transfected with miRIDIAN hsa-miR-21 mimic or control-mimic. PDCD4 levels were determined using Western blot. Quantification of PDCD4 level was performed using densitometry and the data were normalized to β-actin. Data are mean ± SD (n=3). *, p<0.05 compared to control mimic transfected cells. B, To demonstrate that PDCD4 is a direct target for miR-21, HEK 293 cells were infected with lenti Luc-PDCD4–3′UTR plasmid along with either miR-21 mimic or control mimic. Data represent mean ± SD (n=4). *, p<0.05 compared to control transfected cells. C, PDCD4 protein expression was measured using Western Blot in MDM transfected with SiPDCD4 (100nM) or siControl. Quantification of PDCD4 level was performed using densitometry. Data are mean ± SD (n=3). *, p<0.05 compared to siControl transfected cells. D–E, MDM were transfected with siPDCD4 or siControl. Cells were activated with LPS (1ug/ml) for 24h after knockdown of PDCD4 in MDM. D, IL-10 protein and E, IL-10 mRNA were determined using ELISA and quantitative PCR respectively. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage transfected with siControl non-LPS. §, p<0.05 compared to siControl LPS treated group.
Pharmacological inhibition of JNK (420119 JNK Inhibitor II) significantly inhibited LPS induced IL-10 protein expression indicating the involvement of JNK in IL-10 expression (Fig 9A). Knockdown of cellular cJun using siRNA (Fig 9C, ~75% ↓cJun) also resulted in significant downregulation of inducible IL-10 protein expression demonstrating a direct role of cJun and JNK in LPS-induced IL-10 expression in human MDM (Fig 9B). Efferocytosis or delivery of miR-21 mimic to cells induced the transcriptional activity of AP-1 (Fig 9D–E). Likewise, knockdown of PDCD4 increased phospho-cJun levels (Fig 9F) establishing that efferocytosis, miR-21 and PDCD4 can regulate the c-Jun-AP1 pathway which in turn controls inducible IL-10 expression (Fig 10).
Figure 9. MiR-21 potentiated inducible IL-10 expression via a PDCD4- cJun-AP-1 pathway.

A, IL-10 protein expression was measured in MDM treated with 20 μM JNK Inhibitor II, followed by LPS treatment. Data are mean ± SD (n=4). *, p<0.05 compared to control non-LPS treated cells. §, p<0.05 compared to control LPS treated group. B, MDM were transfected with sicJun or siControl. Cells were activated with LPS (1ug/ml) for 24h after knockdown of PDCD4 in MDM. IL-10 protein was determined using ELISA. Data are mean ±SD (n = 4); *, p<0.05 compared to macrophage transfected with non-LPS siControl. §, p<0.05 compared to siControl LPS treated group. C, Representative image of c-Jun protein expression in MDM transfected with sicJun (100nM) or siControl. D, AP-1 transcription activity in MDMs transiently transfected with AP1-dependent luciferase reporter gene (Ad-AP1-Luc) followed by co-culture with either viable (effrlo) or apoptotic (effrhi) Jurkat T cells for 1h and LPS treatment for 24h. Data represent mean ± SD (n=3). *, p<0.05 compared to control transfected cells. E, HEK293 TLR4/IL-1R1/MD-2 cells were co-transfected with a pAP-1 luciferase and pRL-TK Renilla luciferase plasmids along with either miR-21 mimic or control mimic followed by LPS stimulation. Data represent mean ± SD (n=3). *, p<0.05 compared to control transfected cells. F, Phosphorylation of c-Jun (Serine 73) in MDM transfected with siPDCD4 or siControl followed by activation with LPS. Data are mean ± SD (n=3). *, p<0.05 compared to macrophage transfected with siControl.
DISCUSSION
At the injury-site, efficient dead cell clearance or efferocytosis is a pre-requisite for the timely resolution of inflammation (4–7). Successful engulfment of apoptotic cells by activated macrophages triggers potent anti-inflammatory and immunosuppressive mechanisms. Following efferocytosis, wound-associated activated macrophages produce anti-inflammatory cytokines such as IL-10 and suppress the release of pro-inflammatory mediators including TNFα (41, 49, 50). The current study recognizes miR-21 as being directly implicated in switching wound-associated macrophages to an anti-inflammatory mode following successful engulfment of apoptotic cells at the site of injury.
Lipopolysaccharide (LPS) engagement of TLR4 is known to initiate a cascade of signaling events that culminate in the production of inflammatory cytokines by macrophages. Recent studies suggest that negative regulatory control mechanisms exist to limit the toxic effects of LPS (48). Identified as one of the first mammalian microRNAs (miRs), the miR-21 sequence is strongly conserved throughout evolution (24). miR-21 initially described as “oncomir”, is known to be a common inflammation-inducible miR (19, 20, 24). Suggestive evidence supports that LPS-induced miR-21 expression serves as a negative regulatory mechanism to curb the deleterious effects of LPS (48). The current study demonstrates that potentiation of LPS–induced miR-21 expression following efferocytosis may function as an effective anti-inflammatory response that limits LPS-induced inflammation.
PTEN is validated as a target gene for miR-21 (22, 51). The role of PTEN in infection and inflammation has been addressed (52–54). Of note in the context of this study is the observation that PTEN facilitates LPS-induced TNF-α production. In PTEN™/™ macrophages, LPS-induced TNF-α production was blunted (53, 54). PTEN is a dual protein–lipid phosphatase which dephosphorylates the secondary messenger produced by PI3K and interrupts the downstream activation of Akt (55–57). Thus, downregulation of PTEN activity favors sustained activation of PI3K/AKT pathway. Activated Akt phosphorylates and inhibits the activity of glycogen synthase kinase-3β (GSK3β), a substrate for Akt (58). Phosphorylation of GSK3β by AKT at the N-terminal region serine 9 renders GSK3β inactive (46). This work demonstrates that efferocytosis-induced miR-21, by silencing PTEN and GSK3β, tempers LPS-induced inflammatory response.
Following successful efferocytosis, inhibition of NF-κB leads to anti-inflammatory responses such as down-regulation of inducible TNFα production (8). Ubiquitously expressed, the NF-κB family of transcription factors regulate the expression of numerous genes implicated in immunity and inflammation (59). Vertebrate Rel/NF-κB transcription factors include RelA, RelB, c-Rel, p50/p105 and p52/p100 (59). NF-κB resides in the cytoplasm of cells in an inactive form bound to the inhibitor, IκB. Activation of NF-κB is initiated through phosphorylation of IκBα by a macromolecular cytoplasmic IκB kinase (IKK) complex (59). Once activated, NF-κB is released from IκB and translocate to the nucleus where it can drive gene expression such as that of TNFα (42). Inducible activation of NF-κB is further controlled by post-translational modifications such as phosphorylation of the NF-κB subunit p65 as well as interaction with transcriptional co-activators (43). Multiple controls in the regulation of NF-κB activity suggest a complex and microenvironment-dependent function for this transcription factor. It has been proposed that GSK3β is a point of convergence of many signaling pathways, including that of the NF-κB signaling pathway (60). GSK3β inhibits NF-κB activity by lowering DNA binding (60). This work demonstrates that miR-21 controls NF-κB activation via silencing of GSK3β. This observation unveils a novel pathway wherein miR-21 blunts LPS-induced NFκB activation by silencing PTEN and GSK3β.
Efferocytosis triggers release of anti-inflammatory cytokine IL-10 in macrophages (49). IL-10 is amongst the most prominent anti-inflammatory cytokines released following inflammation (61). The notion that IL-10 acts as an anti-inflammatory molecule originated from studies showing blunted production of a large spectrum of pro-inflammatory cytokines by cells of monocytic lineage (47, 61). Although a number of studies described the release of IL-10 following efferocytosis (7, 41, 62), underlying mechanisms remain obscure. In this work, stimulation of TLR4 by LPS after efferocytosis resulted in increased abundance of miR-21 which in turn silenced PDCD4 (programmed cell death 4) and elevated IL-10 protein level. These findings indicated that miR-21-PDCD4 pathway may be involved in efferocytosis-induced anti-inflammatory IL-10 production in macrophages. Initially identified as a protein the abundance of which was increased by apoptotic stimuli and later characterized as a tumor suppressor, PDCD4 regulates both tumorigenesis and inflammation (63). The suppressive effect of PDCD4 on LPS-induced IL-10 expression was suggested to occur at the translational level (48). In the current study, knock-down of PDCD4 up-regulated IL-10. This observation prompted us to look for miR-21 and PDCD4 dependent transcriptional control of IL-10. PDCD4 is known to block c-Jun activation by inhibiting the expression of mitogen-activated protein kinase kinase kinase kinase 1 (MAP4K1; also known as hematopoietic progenitor kinase 1), a kinase upstream of Jun N-terminal kinase (JNK) (64). Jun/AP-1 proteins are known to be involved in transcriptional activation of IL-10 in monocytic cells (65). Results of this work demonstrate that the miR-21-PDCD4 pathway favors cJun expression and AP-1 transactivation. Furthermore, it is established that cJun plays a critical role in supporting inducible IL-10 expression. Taken together these observations demonstrate that following efferocytosis, miR-21 induction in macrophages silence PDCD4 therefore favoring cJun-AP1 activity resulting in higher production of anti-inflammatory IL-10.
The current work recognizes a regulatory loop wherein efferocytosis induces miR-21 which in turn promotes efferocytosis. Delivery of miR-21 to MDM bolstered efferocytosis. This observation is consistent with the report that PTEN, a direct target of miR-21, down-regulates engulfment of apoptotic cells (52). Furthermore, inducible TNF-α known to inhibit efferocytosis (66), is repressed by miR-21. In conclusion, this work provides first evidence directly implicating miRNA in the process of turning on an anti-inflammatory phenotype in the post-efferocytotic macrophage. Specifically, miR-21 is recognized as efferocytosis-inducible in macrophages. Elevated macrophage miR-21 promotes efferocytosis and silences target genes such as PTEN and PDCD4 which in turn accounts for a net anti-inflammatory phenotype. Findings of this study underscore the significance of miRNAs in the resolution of inflammation.
Footnotes
Wound healing research in the authors’ laboratory is funded by NIDDK R01 DK076566 to SR, NIGMS GM069589, GM 077185 & NR013898 to CKS.
References
- 1.deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays in Biochemistry. 2003;39:105–117. doi: 10.1042/bse0390105. [DOI] [PubMed] [Google Scholar]
- 2.Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. Journal of Leukocyte Biology. 2006;79:896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- 3.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 4.Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5:e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manfredi AA, Iannacone M, D’Auria F, Rovere-Querini P. The disposal of dying cells in living tissues. Apoptosis. 2002;7:153–161. doi: 10.1023/a:1014366531885. [DOI] [PubMed] [Google Scholar]
- 6.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nature Reviews Immunology. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 7.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 8.Tibrewal N, Wu Y, D’Mello V, Akakura R, George TC, Varnum B, Birge RB. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283:3618–3627. doi: 10.1074/jbc.M706906200. [DOI] [PubMed] [Google Scholar]
- 9.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 10.Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–1181. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 11.Sen CK. MicroRNAs as new maestro conducting the expanding symphony orchestra of regenerative and reparative medicine. Physiol Genomics. 2011;43:517–520. doi: 10.1152/physiolgenomics.00037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen CK, Gordillo GM, Khanna S, Roy S. Micromanaging vascular biology: tiny microRNAs play big band. J Vasc Res. 2009;46:527–540. doi: 10.1159/000226221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asirvatham AJ, Magner WJ, Tomasi TB. miRNA regulation of cytokine genes. Cytokine. 2009;45:58–69. doi: 10.1016/j.cyto.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 16.Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008;111:478–486. doi: 10.1016/j.ygyno.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Okada H, Kohanbash G, Lotze MT. MicroRNAs in immune regulation--opportunities for cancer immunotherapy. Int J Biochem Cell Biol. 2010;42:1256–1261. doi: 10.1016/j.biocel.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–3127. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 19.Roy S, Sen CK. MiRNA in innate immune responses: novel players in wound inflammation. Physiol Genomics. 2011;43:557–565. doi: 10.1152/physiolgenomics.00160.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy S, Sen CK. miRNA in wound inflammation and angiogenesis. Microcirculation. 2012;19:224–232. doi: 10.1111/j.1549-8719.2011.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 22.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 24.Sen CK, Roy S. MicroRNA 21 in tissue injury and inflammation. Cardiovasc Res. 2012;96:230–233. [Google Scholar]
- 25.Lu TX, Munitz A, Rothenberg ME. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009;182:4994–5002. doi: 10.4049/jimmunol.0803560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Case SR, Martin RJ, Jiang D, Minor MN, Chu HW. MicroRNA-21 inhibits toll-like receptor 2 agonist-induced lung inflammation in mice. Exp Lung Res. 2011;37:500–508. doi: 10.3109/01902148.2011.596895. [DOI] [PubMed] [Google Scholar]
- 27.Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. 2011;25:544–560. doi: 10.1096/fj.10-169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunzl P, Schabbauer G. Recent advances in the genetic analysis of PTEN and PI3K innate immune properties. Immunobiology. 2008;213:759–765. doi: 10.1016/j.imbio.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 29.Ganesh K, Das A, Dickerson R, Khanna S, Parinandi NL, Gordillo GM, Sen CK, Roy S. Prostaglandin E(2) induces oncostatin M expression in human chronic wound macrophages through Axl receptor tyrosine kinase pathway. J Immunol. 2012;189:2563–2573. doi: 10.4049/jimmunol.1102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy S, Khanna S, Rink C, Biswas S, Sen CK. Characterization of the acute temporal changes in excisional murine cutaneous wound inflammation by screening of the wound-edge transcriptome. Physiol Genomics. 2008;34:162–184. doi: 10.1152/physiolgenomics.00045.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy S, Khanna S, Rink T, Radtke J, Williams WT, Biswas S, Schnitt R, Strauch AR, Sen CK. P21waf1/cip1/sdi1 as a central regulator of inducible smooth muscle actin expression and differentiation of cardiac fibroblasts to myofibroblasts. Mol Biol Cell. 2007;18:4837–4846. doi: 10.1091/mbc.E07-03-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan YC, Khanna S, Roy S, Sen CK. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem. 2011;286:2047–2056. doi: 10.1074/jbc.M110.158790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan YC, Roy S, Huang Y, Khanna S, Sen CK. The microRNA miR-199a-5p down-regulation switches on wound angiogenesis by de-repressing the v-ets erythroblastosis virus E26 oncogene homolog 1 -matrix metalloproteinase-1 pathway. J Biol Chem. 2012 doi: 10.1074/jbc.M112.413294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roy S, Khanna S, Bickerstaff AA, Subramanian SV, Atalay M, Bierl M, Pendyala S, Levy D, Sharma N, Venojarvi M, Strauch A, Orosz CG, Sen CK. Oxygen sensing by primary cardiac fibroblasts: a key role of p21(Waf1/Cip1/Sdi1) Circ Res. 2003;92:264–271. doi: 10.1161/01.res.0000056770.30922.e6. [DOI] [PubMed] [Google Scholar]
- 35.Roy S, Khanna S, Nallu K, Hunt TK, Sen CK. Dermal wound healing is subject to redox control. Mol Ther. 2006;13:211–220. doi: 10.1016/j.ymthe.2005.07.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, Sen CK. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci U S A. 2010;107:6976–6981. doi: 10.1073/pnas.1001653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seshadri S, Kannan Y, Mitra S, Parker-Barnes J, Wewers MD. MAIL regulates human monocyte IL-6 production. J Immunol. 2009;183:5358–5368. doi: 10.4049/jimmunol.0802736. [DOI] [PubMed] [Google Scholar]
- 38.Wan E, Yeap XY, Dehn S, Terry RL, Novak ML, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney JW, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB. Enhanced Efferocytosis of Apoptotic Cardiomyocytes Through MER Tyrosine Kinase Links Acute Inflammation Resolution to Cardiac Repair After Infarction. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.113.301198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2007 doi: 10.1038/sj.cdd.4402184. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 40.Fadok VA. Clearance: the last and often forgotten stage of apoptosis. J Mammary Gland Biol Neoplasia. 1999;4:203–211. doi: 10.1023/a:1011384009787. [DOI] [PubMed] [Google Scholar]
- 41.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang F, Tang E, Guan K, Wang CY. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5635. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- 44.Schmid AC, Byrne RD, Vilar R, Woscholski R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004;566:35–38. doi: 10.1016/j.febslet.2004.03.102. [DOI] [PubMed] [Google Scholar]
- 45.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 46.Sutherland C, I, Leighton A, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 49.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 50.Huynh ML, V, Fadok A, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. Journal of Clinical Investigation. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mondal S, Ghosh-Roy S, Loison F, Li Y, Jia Y, Harris C, Williams DA, Luo HR. PTEN negatively regulates engulfment of apoptotic cells by modulating activation of Rac GTPase. J Immunol. 2011;187:5783–5794. doi: 10.4049/jimmunol.1100484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao X, Wei G, Fang H, Guo J, Weinstein M, Marsh CB, Ostrowski MC, Tridandapani S. The inositol 3-phosphatase PTEN negatively regulates Fc gamma receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851–4857. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 54.Gunzl P, Bauer K, Hainzl E, Matt U, Dillinger B, Mahr B, Knapp S, Binder BR, Schabbauer G. Anti-inflammatory properties of the PI3K pathway are mediated by IL-10/DUSP regulation. J Leukoc Biol. 2010;88:1259–1269. doi: 10.1189/jlb.0110001. [DOI] [PubMed] [Google Scholar]
- 55.Rosivatz E. Inhibiting PTEN. Biochem Soc Trans. 2007;35:257–259. doi: 10.1042/BST0350257. [DOI] [PubMed] [Google Scholar]
- 56.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- 57.Georgescu MM. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer. 2010;1:1170–1177. doi: 10.1177/1947601911407325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 60.Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pretolani M. Interleukin-10: an anti-inflammatory cytokine with therapeutic potential. Clin Exp Allergy. 1999;29:1164–1171. doi: 10.1046/j.1365-2222.1999.00456.x. [DOI] [PubMed] [Google Scholar]
- 62.McDonald PP, V, Fadok A, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. Journal of Immunology. 1999;163:6164–6172. [PubMed] [Google Scholar]
- 63.Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, Schaefer L. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011;4:ra75. doi: 10.1126/scisignal.2001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Zhang Y, Yang HS. Pdcd4 knockdown up-regulates MAP4K1 expression and activation of AP-1 dependent transcription through c-Myc. Biochim Biophys Acta. 2012;1823:1807–1814. doi: 10.1016/j.bbamcr.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang ZY, Sato H, Kusam S, Sehra S, Toney LM, Dent AL. Regulation of IL-10 gene expression in Th2 cells by Jun proteins. J Immunol. 2005;174:2098–2105. doi: 10.4049/jimmunol.174.4.2098. [DOI] [PubMed] [Google Scholar]
- 66.McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, Bratton DL, Kang JL, Henson P. TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol. 2007;178:8117–8126. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]