

Wilms’ tumor, or nephroblastoma, is the most common pediatric renal cancer. The tumors morphologically resemble embryonic kidneys with a disrupted architecture and are associated with undifferentiated metanephric precursors. In this review, Hohenstein et al. discuss genetic and epigenetic findings in Wilms’ tumor in the context of renal development as well as the recent identification of Wilms’ tumor cancer stem cells.
Keywords: Wilms’ tumor, Wt1, β-catenin, kidney development, miRNA, nephron progenitor cells
Abstract
Wilms’ tumor, or nephroblastoma, is the most common pediatric renal cancer. The tumors morphologically resemble embryonic kidneys with a disrupted architecture and are associated with undifferentiated metanephric precursors. Here, we discuss genetic and epigenetic findings in Wilms’ tumor in the context of renal development. Many of the genes implicated in Wilms’ tumorigenesis are involved in the control of nephron progenitors or the microRNA (miRNA) processing pathway. Whereas the first group of genes has been extensively studied in normal development, the second finding suggests important roles for miRNAs in general—and specific miRNAs in particular—in normal kidney development that still await further analysis. The recent identification of Wilms’ tumor cancer stem cells could provide a framework to integrate these pathways and translate them into new or improved therapeutic interventions.
Wilms’ tumors and kidney development
Wilms’ tumors, or nephroblastomas, are pediatric cancers that develop in young children, usually before the age of 5 years. Despite being relatively rare in absolute numbers (they affect one in 10,000 children), they are extensively studied for a variety of reasons. First, although treatment outcome has improved over recent decades, there remains a strong clinical need to improve therapies to both minimize secondary effects later in life due to current treatments and find more effective therapies for high-risk histological subtypes and relapsed patients (Perotti et al. 2013). Second, it was one of the three types of cancer on which Knudson (Knudson and Strong 1972) based his two-hit model for tumor suppressor genes, and the loss of Wilms’ tumor 1 (WT1) in a subset of Wilms’ tumor cases remains an archetypal example of a classic tumor suppressor gene, as originally proposed. Since then, many variations in classifications and the genetics and mechanics of tumor suppressor genes have been found (Berger et al. 2011), and the biological basis of the multiple tumors that arise in genetically predisposed individuals may clearly involve genes other than WT1. Third, Wilms’ tumors are the direct result of maldevelopment of the embryonic kidney, an important experimental model that has been studied for over six decades and has led to many fundamental insights that were afterward applied to other systems. As such, Wilms’ tumors are a model to study the link between normal development and tumorigenesis. Understanding normal kidney development will help our understanding and treatment of Wilms’ tumors, and understanding Wilms’ tumors genetically and mechanistically will help the understanding of normal kidney development. In this review, we focus on this last aspect and provide an update on the many recent developments in the Wilms’ tumor field.
Wilms’ tumors recapitulate normal nephrogenic differentiation, but whereas normal developing nephrons are beautifully structured, nephrogenic structures in Wilms’ tumors are disorganized (Fig. 1). The metanephric kidney develops from the intermediate mesoderm (IM). This structure gives rise to three cell types and structures that will form the kidney: the epithelial nephric or Wolffian duct, Six2-positive mesenchymal cells that will form the nephrons, and Foxd1-positive cells that will give rise to the stromal cells (Costantini and Kopan 2010; Li et al. 2014b). Kidney development starts when an epithelial outgrowth of the Wolffian duct forms and invades the IM (Fig. 2). This invasion process will set off bidirectional communication between the epithelial cells, by now called the ureteric bud, and the mesenchymal cells, now called the metanephric mesenchyme. This leads to the first bifurcation of the ureteric bud and the condensation of the mesenchymal cells to form the cap mesenchyme around the tips of the ureteric bud. The cells that form the cap mesenchyme are marked by Six2 and are the nephron progenitor cells. Lineage tracing has shown that the complete nephron is derived from these cells (Kobayashi et al. 2008), and loss of Six2 leads to a rapid exhaustion of these progenitors and ectopic epithelization (Self et al. 2006). Responding to signals from the bud and stroma, these progenitor cells will form a pretubular aggregate and undergo a mesenchymal-to-epithelial transition (MET) to form the renal vesicle. This structure will fuse with its distal end to the ureteric bud to form a continuous open connection to the ureteric bud. Via the comma-shaped and S-shaped body stages, it will form the tubule that, as differentiation progresses, gets patterned into the proximal tubule, loop of Henle, and distal tubule that make up the nephron. Each of these parts will express specific transporters to allow the proper function of the nephron. Moreover, from early in the patterning process, podocyte precursors can be found at the most proximal end of the nephron. Together with invading endothelial and mesangial cells and the cells that make up Bowman’s capsule, they will eventually form the glomerulus, in which the mature podocytes perform the actual filtering functions of the nephron (Schell et al. 2014). As this nephrogenic program is executed, the ureteric bud will keep on growing and branching. Each time new bud tips are formed, the induction process is repeated, and new nephrons start to form, connect to the bud, and pattern to form a functional nephron. In the final, mature nephron, the blood will enter through the capillaries and is filtered by the podocytes, and valuable electrolytes, water, sugars, and other molecules are transported back into the blood. The remainder is drained into the ureteric bud, which by then has formed the collecting duct system, and goes to the bladder for disposal. In humans, metanephric kidney development starts around week 5 of gestation and continues until week 36, whereas in mice, the ureteric bud invades the metanephric mesenchyme at embryonic day 10.5 (E10.5), and initiation of new nephrons continues until day 3 after birth. The repetitive nature of branching and nephron induction will yield ∼13,000 nephrons in a mouse kidney and 1 million in a human kidney, and the centrifugal branching pattern will also lead to a tissue architecture with older nephrons on the inside and younger nephrons on the outside of the renal cortex.
Figure 1.

Wilms’ tumors resemble fetal kidneys. (A) Human fetal kidney (18-wk gestation) showing blastema (metanephric mesenchyme), epithelial differentiation (S-shaped body), stroma, and a mature glomerulus. (B) A triphasic Wilms’ tumor showing all three cell types: blastema, epithelia, and stroma. (Bl) Blastema; (SSB) S-shaped body; (St) stroma; (Gl) glomerulus; (IES) immature epithelial structure.
Figure 2.

Schematic representation of kidney development. The ureteric bud (gray) extends from the Wolffian duct, and, upon contact with the metanephric mesenchyme (red), reciprocal signaling induces both bud bifurcation and condensation of the mesenchyme to generate the cap mesenchyme. The blastemal mesenchyme then undergo a MET to generate the renal vesicle, which continues to form the comma-shaped and S-shaped body before the distal end fuses with the ureteric bud (which forms the collecting duct), and the proximal end joins to form the glomerulus, generating the mature nephron (dark orange).
From early on, it has been clear that Wilms’ tumor development is tightly linked to this developmental process. Wilms’ tumors are found associated with so-called nephrogenic rests, of which there are two types. Intralobar nephrogenic rests (ILNRs) are located toward the renal medulla and are thought to arise early in renal development, whereas perilobar nephrogenic rests (PLNRs) are located toward the periphery of the kidney, the region that retains nephrogenic activity until late in gestation. These nephrogenic rests are histologically recognizable structures that resemble embryonic developmental stages normally not found in the postnatal kidney (Beckwith et al. 1990), suggesting that a block in the nephrogenic process is an early, maybe even initiating, step in the formation of Wilms’ tumors. This was further emphasized by genome-wide expression analysis of Wilms’ tumor xenografts (Dekel et al. 2006) and recently in more detail by the study of Gadd et al. (2012), which described an expression analysis of a larger set of Wilms’ tumors that not only identified five subsets of tumors based on expression patterns but could also couple this to different stages at which nephron development was blocked. It is therefore not surprising that many genes found mutated in Wilms’ tumors have essential roles in nephron development.
Wilms’ tumor genetics
Overall, there are very few genes commonly mutated in Wilms’ tumor, and all show relatively low mutation frequencies. The discovery of many Wilms’ tumor-associated genes began with studying constitutional syndromes that confer predisposition. For many years, Wilms’ tumors have been recognized as occurring at a highly elevated rate in patients with WAGR (Wilms’ tumor, aniridia, genitourinary anomalies, and mental retardation) syndrome, Beckwith-Wiedemann syndrome (BWS), hemihypertrophy, Denys-Drash, and Perlman syndrome. However, these were not associated with genomic aberration until much later, when methods for detecting chromosomal aberrations improved. Constitutional chr11p13 deletions were initially found in patients with Aniridia-associated Wilms’ tumor (Francke et al. 1979) and then later identified in sporadic nonsyndrome-associated cases (Kaneko et al. 1981; Slater and de Kraker 1982). In 1990, the candidate gene at 11p13, WT1, was finally characterized (Call et al. 1990; Gessler et al. 1990) and shown to be a zinc finger DNA-binding transcription factor involved in genitourinary development (Pritchard-Jones et al. 1990). Germline mutations in WT1 are associated with WAGR (Grundy et al. 1995) and Denys-Drash (Pelletier et al. 1991) syndromes and are more often seen in tumors with a stromal subtype (Schumacher et al. 1997). A somatic inactivating mutation or deletion of WT1 is found in ∼12% of Wilms’ tumors and is considered an “early” event, as mutations are also seen in associated ILNRs (Park et al. 1993; Fukuzawa et al. 2007).
The next Wilms’ tumor gene identified, also with a role in renal development, was CTNNB1, which is mutated somatically in 15% of tumors, often alongside a WT1 mutation (Koesters et al. 1999; Maiti et al. 2000). CTNNB1 encodes β-catenin, a key protein involved in the Wnt signaling pathway, which is critical for epithelialization after MET (Koesters et al. 1999). In Wilms’ tumor, the CTNNB1 mutation commonly occurs at a specific activating residue (Ser45) (Kusafuka et al. 2002), causing nuclear accumulation of β-catenin (Koesters et al. 2003). CTNNB1 mutations have been defined as a “late” event in Wilms’ tumorigenesis, as they have been observed in Wilms’ tumors but not nephrogenic rests (Fukuzawa et al. 2007; Grill et al. 2011). Other potential mechanisms for increased Wnt signaling in Wilms’ tumor have been identified, including overexpression of human Dickkopf-1, a Wnt signaling antagonist (Wirths et al. 2003), as well as loss of WT1, which negatively regulates Wnt signaling via transcriptional activation of WID (WT1-induced inhibitor of Dishevelled) (Kim et al. 2010).
WTX (Wilms’ tumor on chromosome X, also known as AMER1) further links both the WT1 and CTNNB1 genes together, as the protein negatively regulates Wnt signaling (Major et al. 2007) and has been associated with WT1 transcriptional control (Rivera et al. 2009). WTX is inactivated by mutation or deletion in ∼18% of tumors (Rivera et al. 2007; Ruteshouser et al. 2008) and has been described as a tumor suppressor gene. WTX mutations have been observed in both tumor and nephrogenic rest tissue; however, its role in tumor initiation has been disputed as an initiating event due to the heterogeneity of the mutation within a tumor (Wegert et al. 2009; Fukuzawa et al. 2010), the fact that germline WTX mutations do not predispose to Wilms’ tumor (Jenkins et al. 2009), and a lack of linkage between polymorphic SNPs at WTX and Wilms’ tumor compared with controls (Guertl et al. 2010). Some reports couple WTX function to the genes directly involved in the control of nephron progenitors (which is discussed in more detail below). It was shown to modulate Wt1 activity (Rivera et al. 2009) and negatively regulates β-catenin being part of the degradation complex (Major et al. 2007). Wtx knockout mice are perinatal lethal with phenotypes in multiple tissues of mesodermal origin (Moisan et al. 2011), consistent with the germline mutations found in sclerosing skeletal dysplasia (Jenkins et al. 2009). The kidney phenotype in these mice is confusing, being either agenesis or overgrowth, often in the same animal. These data therefore cannot confirm a causative role in Wilms’ tumor development, although multiple roles and phenotypes during kidney development—reminiscent of the multiple Wt1 phenotypes—remains a possibility. Additional conditional analyses are required to shed light on this possibility.
Besides genes already known to be central players in renal development, mutations in other cancer-associated genes have been identified, including TP53 (Lemoine et al. 1992). TP53 mutation or loss of copy number occurs in a low proportion of Wilms’ tumor (∼5%) but is particularly associated with tumors that show anaplasia. These tumors show characteristic copy number profiles with multiple focal areas of gain or loss that are generally absent in tumors with wild-type TP53, showing that TP53 loss is associated with genomic instability; these cases often have a poor prognosis (Bardeesy et al. 1994; Maschietto et al. 2014). WTX mutation may play a role in TP53 inactivation, as in vitro evidence suggests that WTX modulates CBP/p300-mediated TP53 acetylation and hence expression (Kim et al. 2012). It has become clear that p53 is functional in the development of multiple tissues; although, in general, Tp53-deficient mice are viable, on certain genetic backgrounds, increased embryonic mortality is found (El-Dahr et al. 2014). On a C57BL/6 background, multiple congenital abnormalities in the kidneys can be found (Saifudeen et al. 2009), but none of these have an obvious link to Wilms’ tumor development. However, it should be noted that the mutations found in Wilms’ tumors are missense rather than loss-of-function alleles. Mouse models with missense alleles are available (Frese and Tuveson 2007), and appropriate in vivo modeling with these might determine whether the role of p53 in Wilms’ tumors reflects a developmental role for TP53 in the developing kidney after all or a more general role in overseeing genomic stability in proliferating cells in general and control of the response to any problems that occur as cells divide.
High expression and copy number gain of MYCN have been associated with Wilms’ tumor for a long time (Nisen et al. 1986; Norris et al. 1988; Williams et al. 2010), and, recently, mutations in MYCN were identified by exome sequencing (Rakheja et al. 2014), including a recurrent mutation (P44L) previously identified in neuroblastoma and believed to be a gain-of-function mutation (Williams et al. 2015). MYCN is a target of ubiquitination and degradation by FBXW7, which is deleted or mutated in ∼4% of tumors (Williams et al. 2010). Together, this evidence suggests the combined role of different genetic events leading to MYCN overexpression in Wilms’ tumor and supports studies that describe elevated expression of MYCN in tumors with (Norris et al. 1988) and without (Nisen et al. 1986) gene amplification.
The most recent evidence highlighting novel Wilms’ tumor genes was generated by exome sequencing in which mutations in microRNA (miRNA) processor genes DICER1, DROSHA, XPO5, DGCR8, and TARBP2 were identified (Torrezan et al. 2014). The effect of mutation in miRNA processor genes in Wilms’ tumor is discussed later. These genome-wide mutation analyses also identified low-frequency somatic mutations in epigenetic remodelers SMARCA4 and ARID1A (Rakheja et al. 2014). Very recently, mutations in DROSHA and DGCR8 were also identified in exome sequencing studies of high-risk blastemal Wilms’ tumors (Wegert et al. 2015) and favorable histology Wilms’ tumors (Walz et al. 2015). These studies also identified a recurring Q177R missense mutation in both SIX1 and SIX2.
CTR9 was only recently identified as a Wilms’ tumor predisposition gene in families (Hanks et al. 2014). It is part of the PAF1c complex, which associates with the RNA polymerase II (Pol II) complex (Jaehning 2010). It is important in embryonic stem cells (ESCs) to maintain their pluripotent state (Ding et al. 2009), but specific kidney functions or even its expression pattern have not been described. Again, it remains to be seen whether the mutations found in Wilms’ tumor families reflect a specific kidney development function or a general requirement for the PAF1c complex in many processes and cell types, including the developing kidney.
Wilms’ tumor epigenetics
From early on, it was realized that, on a chromosomal scale, pediatric cancers, including Wilms’ tumor, had relatively stable genomes (Cox 1966), and this has been confirmed by the low mutation frequencies reported. Around the same time that WT1 was characterized, interest in a second region on 11p, the 11p15.5 locus, showed that BWS was associated with IGF2 overexpression and that tumors commonly lose the maternal allele, resulting in homozygosity (Scott et al. 1985; Schroeder et al. 1987; Koufos et al. 1989). These phenomena were not associated until later, when genomic imprinting was identified (Wilkins 1988; Ogawa et al. 1993): where one allele is expressed in a parent of origin-dependent manner that is controlled by DNA methylation.
At 11p15.5, an H19 imprinting control region (ICR) is located that controls expression of IGF2 and H19 and is differentially methylated depending on the chromosome parent of origin. Maternal alleles are methylated, preventing CTCF binding and IGF2 expression; however, H19 is actively expressed. Conversely, the paternal chromosome is not methylated, allowing CTCF binding, which blocks H19 expression and allows IGF2 expression. In BWS and Wilms’ tumor, loss of the maternal allele and duplication of the paternal allele by either loss of heterozygosity (LOH) or aberrant gain of methylation at the H19-ICR results in biallelic IGF2 expression (Ogawa et al. 1993). LOH has been detected outside of tumor tissue, in the normal kidney and blood cells of BWS patients (Schneid et al. 1991), which provided the first evidence that IGF2 overexpression does not necessarily result in Wilms’ tumorigenesis. Furthermore, mosaic loss of imprinting (LOI) was seen in the surrounding kidney and nephrogenic rests (Ohlsson et al. 1999). Transgenic overexpression of the genomic mouse Igf2 locus in mouse ESCs followed by the production of chimeric mice resulted in BWS-like phenotypes, such as overgrowth fetuses with overgrown kidneys, but no renal abnormalities that support a causative role in Wilms’ tumors (Sun et al. 1997). The same was found in a knockout study of H19, where only maternal inherited alleles were affected, and overgrowth was observed in the offspring (Leighton et al. 1995; Ripoche et al. 1997). Other data based on “imprint-free” ESCs have also argued against the oncogenic potential of IGF2 LOI on its own (Holm et al. 2005). However, the same lack of Wilms’ tumors is seen in the Wt1 knockout mouse unless additional mutations are introduced, even though it is clearly a bone fide Wilms’ tumor gene. A recent mouse model has provided strong evidence for a driver role in Wilms’ tumor of the combination of Wt1 mutation and Igf2 overexpression (Hu et al. 2011). While WT1 mutation is often associated with maternal 11p allele loss that spans the IGF2 locus, this combination accounts for only ∼5%–20% of somatic mutations in human Wilms’ tumor, and additional events must accompany IGF2 expression changes to drive Wilms’ tumor in the majority of cases with LOH/LOI at this locus that lack WT1 mutation.
Many other site-specific and global epigenetic events have been described in Wilms’ tumor that may be associated with development of the tumor, including promoter methylation-associated gene silencing, methylation aberration at imprinted loci, long-range epigenetic silencing, and satellite DNA hypomethylation. These include genes known to be involved in tumorigenesis as well as development, growth, and Wnt signaling (see Table 1); however, very few epigenetic abnormalities (other than IGF2/H19) have the full range of experimental evidence to substantiate mechanistic involvement. Still, the available evidence seems to suggest a close relationship between the epigenetic controls of normal kidney development and epigenetic aberrations in Wilms’ tumorigenesis (Charlton et al. 2015).
Table 1.
Epigenetic aberrations associated with Wilms’ tumor
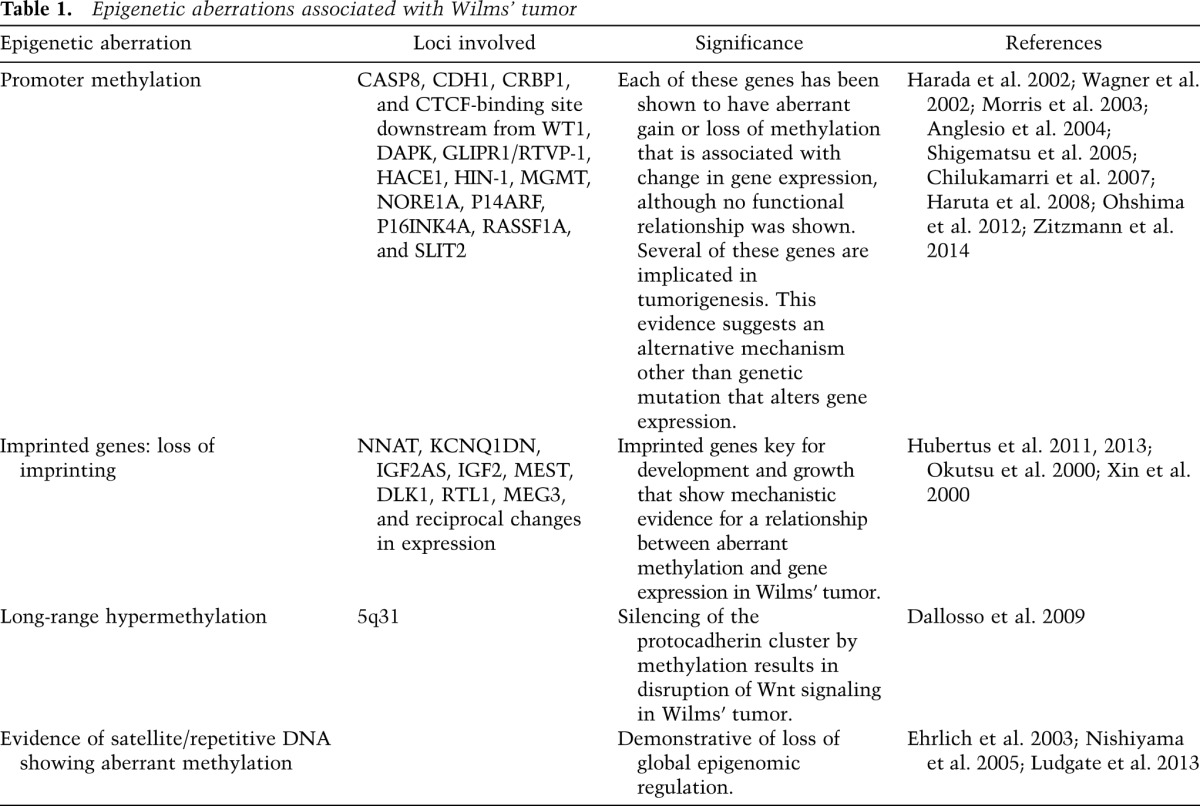
There are several lines of evidence suggesting that the WT1 gene can directly influence the epigenome; for example, WT1 transcriptionally regulates an enzyme that sets de novo DNA methylation marks, DNMT3A, by binding the DNMT3A promoter (Szemes et al. 2013). This interaction thereby influences the methylation status and reciprocal gene expression levels of many target genes. This was demonstrated by overexpression of WT1 in human embryonic kidney cells that resulted in acquired DNA methylation at specific gene promoters where DNMT3A recruitment was increased and resulted in gene silencing. Depletion of WT1 in Wilms’ tumor cells led to reactivation of gene expression from methylated promoters. WT1 has also been shown to affect chromatin structures around the Wnt4 gene during renal development (see below) and specifically binds and recruits Polycomb complex proteins SUZ12 and EZH2 as well as another DNA methylation regulator, DNMT1, to repress Pax2 expression (Xu et al. 2011). Given this evidence, it is possible that in Wilms’ tumors with WT1 mutation, the loss of normal WT1 activity leads to loss of tight epigenetic regulation, giving cells increased epigenetic plasticity and therefore growth advantage. Wilms’ tumor animal models might help to elucidate the mechanism by which WT1 mutation or deletion leads to global or site-specific epigenetic alterations.
Besides DNA methylation, chromatin modification also alters gene expression. ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing) has been performed on three Wilms’ tumors as well as embryonic kidney and normal kidney tissues using antibodies against H3K4me3 (marking transcriptional initiation when present at promoters), H3K36me3 (marking active genes), and H3K27me3 (marking silent promoters) and compared with data from ESCs (Aiden et al. 2010). The results of this study supported previous evidence that Wilms’ tumors are governed by a set of transcription factors essential for normal renal development and confirmed SIX2 as a key master transcription factor. Furthermore, by studying H3K4me3 at gene promoters, a set of genes enriched for epigenetic regulators that were positively expressed in ESCs and Wilms’ tumor but not expressed in normal kidneys was identified. This evidence suggests that Wilms’ tumors maintain the activity of key epigenetic remodelers, allowing them to adapt to their microenvironment to overcome growth inhibition (as discussed in Timp and Feinberg 2013). Indeed, reacquisition of epigenetic plasticity gives cancer cells self-renewal and proliferation capacity, similar to normal progenitor populations.
The same study further described the presence of “bivalent domains” (combined H3K4me3 and H3K27me3 chromatin) in Wilms’ tumors, which identified a subset of genes expressed in normal kidney but with low expression in the tumors. In ESCs, bivalent domains mark developmental loci that are considered essential for normal development and are resolved during cellular differentiation by losing either chromatin mark, thereby causing the underlying gene to become either expressed or repressed. Unsurprisingly, a subset of the genes that aberrantly retained their bivalent chromatin structure in Wilms’ tumor is strongly expressed in mouse kidney epithelial structures, indicating that Wilms’ tumor cells are prevented from expressing key epithelial genes and therefore differentiating properly. As other domains that are bivalent in ESCs showed H3K27me3 and no expression in Wilms’ tumors, Aiden et al. (2010) suggested that Wilms’ tumors show partial lineage commitment. They concluded that developmental arrest occurs in undifferentiated metanephric mesenchyme, as genes required for differentiation (FOXD1 and LHX1, which are involved in stromal and epithelial differentiation, respectively) are among the subset of genes with bivalent chromatin that is not expressed. As polycomb proteins are involved in establishing H3K27me3, another (earlier) study examined levels of polycomb proteins (EZH2, BMI-1, EED, and SUZ12) in embryonic kidneys, adult kidneys, and Wilms’ tumors (Metsuyanim et al. 2008). Of these polycomb genes, only EZH2 was developmentally regulated during renal development, whereas in progressive Wilms’ tumor xenografts, EZH2, EED, SUZ12, and BMI-1 were all up-regulated compared with pooled mid-gestation embryonic kidneys, suggesting an increase in activity in the tumor tissue.
These data give important insights into the biology of Wilms’ tumor cells, which, due to their developmentally disrupted state, maintain an undifferentiated epigenome. If the maintained bivalent domains could be manipulated to resolve, expression of these underlying genes may force differentiation and result in a less malignant phenotype. This hypothesis requires testing, but resetting epigenetic states in other cancers has already been proven successful (Stricker et al. 2013).
Wilms’ tumors and the control of nephron progenitor cells
WT1 is expressed throughout kidney development and has essential functions at multiple stages during nephrogenesis (for review, see Ozdemir and Hohenstein 2014). In the IM, it has a prosurvival role, as, in the conventional knockout mouse, massive apoptosis of these cells leads to renal agenesis (Kreidberg et al. 1993). In the cap mesenchyme, Wt1 is essential for the MET (Davies et al. 2004), the developmental stage believed to be disrupted in the development of Wilms’ tumors (Huff 2011). This step has long been known to be controlled by Wnt4, which is essential (Stark et al. 1994) and sufficient (Kispert et al. 1998) for nephron induction. Wt1 has now been shown to directly control Wnt4 expression in a dichotomous manner at the chromatin level using a “chromatin flip-flop.” Via this mechanism, Wt1 controls histone modifications of the complete Wnt4 locus, as delineated by CTCF insulator elements, in a cell type-dependent and two-directional manner (Essafi et al. 2011), thereby providing a rationale for the inactivating mutations in Wilms’ tumors through control of the nephron progenitor cell population. In the cap mesenchyme, Wt1 keeps the Wnt4 locus in an open/active state characterized by H3K4me3 histone modifications, while in the epicardium, Wt1 keeps the locus in a repressed/inactive state with H3K27me3 modifications. Loss of Wt1 results in the Wnt4 locus getting repressed and its expression being lost in the kidney mesenchyme, while the opposite was found in the epicardium. The molecular details via which Wt1 controls these histone modifications remain unclear, but the flip-flop mechanism could explain how Wt1 can exert its different functions in different stages of the developing nephron (Ozdemir and Hohenstein 2014). Wt1 is essential in later stages of nephrogenesis, as shown using heterozygous mutants, additional conditional models, and specific missense and splicing mutants (Hammes et al. 2001; Guo et al. 2002; Menke et al. 2003; Gao et al. 2004; Chau et al. 2011; Gebeshuber et al. 2013). These data illustrate the role of Wt1 mutations in glomerular sclerosis rather than Wilms’ tumors (Miller-Hodges and Hohenstein 2012) and emphasize the importance of developmental stage-specific disruption of nephron development (Gadd et al. 2012), as too early or too late loss of Wt1 will not lead to tumor formation. WT1 mutant Wilms’ tumors are usually stromal predominant (Miyagawa et al. 1998; Schumacher et al. 2003), and this is likely linked to the separation of the mesenchymal renal lineage in Foxd1+ stromal cells and Six2+ nephron progenitor cells early in renal development and the apparent earlier stage of origin of WT1 mutant tumors as described by Gadd et al. (2012).
The strong correlation between WT1 loss and activating CTNNB1 mutations in tumors strongly suggests that any role for β-catenin in control of the MET will be tightly linked to the role of CTNNB1 and WT1 in Wilms’ tumors. Indeed, β-catenin is a key regulator of the choice of nephron progenitor cells to remain in their stem cell state or be induced, go through the MET, and form an epithelialized nephron. The ureteric bud-derived signal that induces the nephron formation is mediated by Wnt9b that acts on the Six2-positive nephron progenitor cells in the cap mesenchyme. In these cells, it signals via the canonical, β-catenin-mediated Wnt pathway to control expression of Six2 and Wnt4 (Karner et al. 2011; Park et al. 2012). This genetic network is further controlled by a Yap/Taz-mediated signal downstream from Fat4 in the stromal cells (Das et al. 2013). Fat4 expressed on the surface of stromal cells will activate the Hippo pathway in the outer layer of the cap mesenchyme. This will result in phosphorylation of Yap and Taz in these cells, keeping these proteins cytoplasmic and inactive. In the absence of nuclear Yap/Taz, β-catenin signaling downstream from Wnt9b will activate target genes involved in nephron induction and differentiation. The inner layer of the cap mesenchyme receives the same Wnt9b signal as the outer layer, but since there is no direct contact with the stroma, the Hippo pathway will not get activated, and Yap/Taz will not be phosphorylated, translocate to the nucleus, and shift the β-catenin response from differentiation target genes to progenitor self-renewal targets (Das et al. 2013). Interestingly, Fat4-deficient kidneys show a developmental block that resembles nephrogenic rests, and the cytoplasmic retention of phosphorylated YAP is disturbed specifically in anaplastic Wilms’ tumors (Murphy et al. 2014).
MYCN is coexpressed with WT1 in the developing kidney, and WT1 has been shown to mediate MYCN expression, at least in vitro, causing overexpression of MYCN when the WT1 DNA-binding domain is mutated (Zhang et al. 1999; Udtha et al. 2003). The gene is expressed at high levels in the nephron progenitor cells (Mugrauer et al. 1988). Mycn-deficient embryos die at E10.5–E12.5, with a reduction in the number of mesonephric tubules (Charron et al. 1992). When analyzed in organ culture, Mycn mutant metanephroi and kidneys with reduced Mycn levels using a hypomorphic allele were smaller due to reduced proliferation but could undergo the normal MET (Bates et al. 2000). Xu et al. (2014) have now shown that Eya1 functions genetically upstream of Six2 and that the two proteins directly bind the Mycn protein. This leads to dephosphorylation of T58 of Mycn (using the phosphatase activity of Eya1). Phosphorylation of this residue leads to degradation of the protein and exit from the cell cycle. It is conceivable that uncontrolled activation of MYCN through increased expression, increased copy number, or gain-of-function alleles like the recurrent P44L mutation can lead to increased proliferation of the nephron progenitors and thereby favor the self-renewal and stemness of these cells (Laurenti et al. 2009).
The recent identification of Q177R missense mutations in SIX1 and SIX2 (Walz et al. 2015; Wegert et al. 2015) fit neatly with a central role for disturbed control of nephron progenitors in Wilms’ tumors. The mutations are somatic, and the majority was found to be in a heterozygous state, in which case wild-type and mutant alleles showed equal expression. All of this points to a dominant effect of these mutations that, at least for SIX2, is fully consistent with the observed phenotypes. Whereas loss of Six2 in mice leads to ectopic epithelialization and rapid exhaustion of the nephron progenitors (Self et al. 2006), these dominant mutations are giving the opposite effect, as they are found in blastemal-type Wilms’ tumors (Walz et al. 2015; Wegert et al. 2015). Six1 acts upstream of Six2, and loss of Six1 in mice leads to an earlier phenotype than loss of Six2; the metanephric mesenchyme is formed but reduced in size, and condensates are not formed (Xu et al. 2003). Since Six1 and Eya1 work together in a transcriptional complex (Xu 2013), we hypothesize that the mutations found in SIX1 and SIX2 feed back to the control of MYCN, further supported by the fact that the unsupervised clustering of the blastemal tumors placed the SIX1/2 mutant samples in a group that is characterized by up-regulation of cell cycle genes. The exact molecular effect of the Q177R mutation in both genes remains unclear, but genome-wide ChIP data for SIX1-Q177R suggested a shift in DNA-binding specificity and corresponding gene expression changes rather than loss of DNA binding (Wegert et al. 2015).
Combined, the data above provide a mechanistic rationale for disturbed control of nephron progenitor cells in the origin of Wilms’ tumors (Fig. 3). Careful modeling of Wilms’ tumor-associated mutations in animal models could explain the occurrence and combinations of these mutations in these tumors and provide a unique insight into the earliest events in a tumorigenic process. The data also suggest that other genes involved in the nephron progenitor control process will be strong candidate genes for Wilms’ tumorigenesis.
Figure 3.

Control of nephron progenitor cells is disturbed in many Wilms’ tumors. Model for the control of the fate of nephron progenitor cells. Eya1 lies genetically upstream of Six2. Six2 labels the nephron progenitor cells, which can either maintain a progenitor state and self-renew or differentiate via the Wnt4-mediated MET. Wnt4 expression is under the direct control of Wt1. β-Catenin is involved in both progenitor cell fates through activation of different transcriptional programs. Active nuclear phosphorylated Yap/Taz shifts the progenitor balance toward the self-renewal fate. Eya1 and Six2 interact directly with Mycn, leading to dephosphorylation of Mycn pT58, stabilization of the protein, increased proliferation, and potentially a shift of the nephron progenitor toward self-renewal. Genes activated in Wilms’ tumors are depicted in green, and inactivated genes are in blue. Deregulation of Yap/Taz in Wilms’ tumors results in phosphorylated Yap not being retained in the cytoplasm as it should, but it translocates to the nucleus and thus shifts the progenitor cell balance toward self-renewal. This model is likely a simplification, as it presumes that all Wilms’ tumors, regardless of causative mutation, are caused by the same mechanism.
Wilms’ tumor cancer stem cells
Disturbance of the control of nephron progenitor cells as an early event in Wilms’ tumor development has direct implications for the cancer stem cell model. This model describes how a small subset of cells with stem cell properties in a tumor gives rise to the bulk of the tumor. As these stem cell properties make these stem cell-like cells insensitive to DNA damage-inducing agents, current therapies often fail to hit this essential target population and can even enrich for these cancer stem cells, making relapse more likely. The cancer stem cell is closely linked but not necessarily identical to the cell of origin of the tumor (Visvader 2011). A variety of genetic and epigenetic studies had identified a strong stemness phenotype in Wilms’ tumors (for review, see Pode-Shakked and Dekel 2011), but, for a long time, the putative Wilms’ tumor cancer stem cell remained unknown. It took optimization of xenograft procedures for Wilms’ tumors to identify an NCAM1+/ALDH1+ population that showed all characteristics of a Wilms’ tumor cancer stem cell (Pode-Shakked et al. 2013). Transplantation of just 200 double-positive cells in immune-compromised mice is sufficient to induce tumors that recapitulate the triphasic nature of the original tumor, and specific targeting of this cell population with anti-NCAM1 antibodies conjugated with a cytotoxic drug effectively stopped the growth of tumors in the Wilms’ tumor xenograft model.
At present, it is not yet clear how these NCAM1+/ALDH1+ cells relate to the nephron progenitor population. In human embryonic kidneys, where cell isolation methods are currently limited to cell surface markers, NCAM1-positive cells have been identified as nephron progenitor cells with potential stem cell therapeutic potential (Harari-Steinberg et al. 2013). NCAM1 is expressed in the Six2-positive nephron progenitors but also in the first few epithelialized stages, and it is not clear which NCAM1 subpopulation is responsible for the progenitor phenotype. Even more confusing is the ALDH1+ phenotype of the cells. ALDH1 status is usually determined using an enzymatic activity assay (ALDEFLUOR) instead of an expression analysis. There are three human ALDH1 paralogs (four in mice), each with its own cell type-specific expression pattern in the developing kidney, and it is not clear which of these is responsible for the ALDH1 activity in Wilms’ tumor (and other) cancer stem cells.
Surprisingly, recent data suggest that the NCAM1+/ALDH1+ cancer stem cells dedifferentiate to a more mesenchymal state from which the bulk of the tumor forms (Shukrun et al. 2014). This clearly has consequences for the interpretation of the identity of the Wilms’ tumor cancer stem cells in the context of the normal nephron progenitor cells. It also emphasizes potential limitations of deducing the developmental stage of origin of different subgroups of tumors from expression patterns and histology. As different subgroups of Wilms’ tumors could have a different developmental stage of origin (Gadd et al. 2012), it is possible that the different groups have different cancer stem cells. However, despite these limitations, at least for the moment, the identification of NCAM1+/ALDH1+ cancer stem cells in at least some Wilms’ tumors will provide new insights in the developmental origins of the tumors.
The miRNA pathway in Wilms’ tumorigenesis
Multiple lines of evidence support the significance of the miRNA processing pathway in Wilms’ tumorigenesis. This pathway (shown in Fig. 4) generates mature miRNAs that negatively regulate protein expression by targeting mRNAs. Recent excitement surrounding the disruption of the miRNA processing pathway in Wilms’ tumor arose after identification of inactivating DIS3L2 mutations in the Wilms’ tumor susceptibility disease Perlman syndrome (Astuti et al. 2012). DIS3L2 is involved in recognition and degradation of polyuridylated mRNA and miRNAs (Chang et al. 2013). Specific targets of DIS3L2 include uridylated pre-let-7 miRNAs, and this process of uridylation is performed by LIN28-activated TUT4 and TUT7 enzymes (see Fig. 4; Heo et al. 2009; Thornton et al. 2012; Chang et al. 2013). By binding to the Let-7 pre-miRNA loop, LIN28 blocks DICER processing and therefore expression of mature let-7 miRNAs in undifferentiated cells during development, maintaining cell renewal and pluripotency. Let-7 miRNAs are considered to have tumor suppressor activity, and therefore, not surprisingly, overexpression of LIN28 is oncogenic. Indeed, a mouse model with kidney-specific Lin28 overexpression generated Wilms’ tumor-like lesions, further discussed below. Of note, LIN28 can also bind to pri-Let-7 miRNA in the nucleus, preventing DROSHA/DGCR8 processing; however, the mechanism of degradation of LIN28-bound pri-Let-7 in this instance is unknown. Therefore, common hits to the same Let-7 biogenesis pathway (LIN28 overexpression or inactivation of DIS3L2) result in aberrant mature Let-7 miRNA expression and are associated with Wilms’ tumor.
Figure 4.

The miRNA processing pathway is commonly mutated in Wilms’ tumor. Expression of mature miRNA is initiated by RNA polymerase-mediated transcription of DNA-encoded sequences into pri-miRNA, which form a long double-stranded hairpin. This structure is then cleaved by a complex of Drosha and DGCR8 (termed Pasha) into a smaller pre-miRNA hairpin, which is exported from the nucleus and then cleaved by Dicer (an RNase) and TRBP (with specificity for dsRNA) to remove the hairpin loop and leave two single-stranded miRNAs. The functional strand binds to Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides the complex to its target mRNA, while the nonfunctional strand is degraded. Targeting of mRNAs by this method results in mRNA silencing by mRNA cleavage, translational repression, or deadenylation. Let-7 miRNAs are a family of miRNAs highly expressed in ESCs with tumor suppressor properties. In cases in which LIN28 is overexpressed, LIN28 binds to pre-Let-7 miRNA, preventing DICER from binding and resulting in LIN28-activated polyuridylation by TUT4 or TUT7, causing reciprocal DIS3L2-mediated degradation of Let-7 pre-miRNAs. Genes involved in miRNA processing that have been associated with Wilms’ tumor are highlighted in blue (inactivating) and green (activating) and include DROSHA, DGCR8, XPO5 (encoding exportin-5), DICER1, TARBP2, DIS3L2, and LIN28.
Other miRNAs have been specifically associated with Wilms’ tumor, including miRNAs that can predict chemoresponsiveness in blastemal Wilms’ tumors (Watson et al. 2013). For example, miR-192, miR-215, and miR-194 all showed significantly lower expression in Wilms’ tumors compared with normal kidneys, whereas other miRNAs show cell type-specific up-regualtion or down-regulation (Senanayake et al. 2012). Several of these miRNAs were shown to target the activin receptor type 2B (ACVR2B, expressed in the cap mesenchyme) (Metsuyanim et al. 2009), a member of the TGF-β pathway, and loss of their expression in Wilms’ tumors resulted in up-regulation of the receptor, suggesting that the TGF-β pathway may be important in tumorigenesis. Expression of both transcription factor E2F3 and Oncomir-1 (an oncogenic cluster of miRNAs on chr13 regulated by E2F3) was increased in Wilms’ tumor compared with normal tissue, with the increase correlating with high stage and metastatic disease (Kort et al. 2008). This indicates the role of specific miRNAs in tumor progression.
The final piece of evidence, as briefly mentioned above, was the frequent number of mutations found in several miRNA processor genes in Wilms’ tumor, including DROSHA, DICER1, DGCR8, XPO5, and TARBP2 (see Fig, 4; Torrezan et al. 2014). Overall, 33% of Wilms’ tumors (22 out of 66) examined had mutations or deletions in miRNA processor genes, and these often occurred in combination, resulting in multiple hits to the same pathway in one tumor, with XPO5 and TARBP2 mutations frequently occurring together (Torrezan et al. 2014). The most commonly mutated miRNA processor gene is DROSHA, in 12% of Wilms’ tumors (26 out of 222), most frequently occurring as a heterozygous E1147K mutation altering a Mg2+-binding residue of the RNase IIIb domain. Although one study found mutually exclusive mutations in WT1/CTNNB1 and miRNA processors (Rakheja et al. 2014), another found that in four out of 10 tumors with DROSHA mutation, WT1 mutation was also seen (Torrezan et al. 2014).
Germline DICER1 mutations were first found in patients with renal tumors in 2010, but DICER1 mutations were not identified in the germline of any of 50 patients with sporadic Wilms’ tumors (Bahubeshi et al. 2010). This shows that although DICER1 mutations were later associated with Wilms’ (Foulkes et al. 2011; Slade et al. 2011), they are not fully penetrant. This may be associated with the fact that more than one hit is required for tumorigenesis—either biallelic DICER1 mutation (Wu et al. 2013) or a hit to another miRNA processor.
Dicer has been shown to be important for maintaining the viability of self-renewing progenitors in renal development, demonstrated by the premature termination of nephron formation with Six2-Cre-mediated removal of Dicer activity (Nagalakshmi et al. 2011). This evidence appears contrary to the findings in Wilms’ tumor indicating that mutation of DICER1 is associated with tumorigenesis. Furthermore, whereas common mutations in the DICER1 RIIIb domain showed that 5p miRNAs are mainly affected, DROSHA and DGCR8 mutations result in failure to generate mature miRNAs in general (Torrezan et al. 2014). So how does dysregulation of a protein involved in such a general process lead to tumorigenesis?
The answer is unclear but may again be associated with the developmental nature of Wilms’ tumor. It can be hypothesized that if the miRNA processor mutation was to occur somatically within a renal progenitor cell, the cell stage-specific array of pre-miRNAs expressed would fail to be correctly processed, resulting in loss of mature miRNAs required for gene silencing. Several studies have identified miRNAs that are down-regulated in Wilms’ tumor and impact on expression of key genes involved in renal development. For example, miR-23a shows low expression in Wilms’, and, as miR-23a regulates HOXB4, the decrease in the tumor was associated with HOXB4 overexpression (Koller et al. 2013). HOXB4 was shown in developing chicks to mediate expression of kidney-specific genes in the nonkidney intermediate mesenchyme (Preger-Ben Noon et al. 2009); therefore, a decrease in miR-23a may result in reactivation of HOXB4 and kidney-specific genes, resulting in persistent embryonic renal tissue. Similar findings in several other studies showed that down-regulation of miR-562, miR-204, and miR-185 correlated with increased expression of EYA1 (Drake et al. 2009), MEIS1, and SIX1 (Imam et al. 2010), respectively. In particular, miR-185 was shown to inhibit anchorage-independent growth and cell migration in addition to suppressing tumor growth in vivo, implicating it as a potent tumor suppressor silenced in Wilms’ tumor (Imam et al. 2010). Therefore, by failing to generate mature miRNAs, the normal nephrogenic differentiation process cannot continue, as primordial renal genes cannot be silenced, and cells remain stuck in a precursor-like state. Germline mutations in miRNA processor genes very rarely lead to Wilms’ tumor; therefore, it is more likely that timing is key to the resulting phenotype.
Wilms’ tumor animal models
Animal models for cancer are an essential tool to understand the genetic, molecular, and cellular events that lead to cancer initiation and progression and for the development and testing of therapeutic intervention. In particular, for studying the developmental aspects of Wilms’ tumor, they are indispensable. Animal models can be either xenograft models of human tumors in immune-compromised mice or genetically modified animals. As this review focuses on the developmental aspects of Wilms’ tumors, we limit the description of animal models to the latter group.
The first Wilms’ tumor gene, WT1, was cloned in 1990, and this was rapidly (for those days) followed by the description of a Wt1-deficient mouse model (Kreidberg et al. 1993). Its embryonic-lethal phenotype at mid-gestation showed the importance of the gene in many different developing tissues. However, mutant embryos showed complete renal agenesis due to massive apoptosis of the IM. Not only did this severely limit the usefulness of this model for Wilms’ tumor development, an apoptotic phenotype was completely counterintuitive to the loss of a tumor suppressor gene. Other Wt1 mouse models were generated that showed a pivotal role of Wt1 in other stages of nephron development and analyzed the role of specific isoforms (for review, see Ozdemir and Hohenstein 2014), but it was not until a breakthrough in 2011 that the first Wt1 mutant mouse tumor model was described (Hu et al. 2011). This model combined two genetic events found in Wilms’ tumor patients—loss of Wt1 (in mice through a conditional allele) and activation of Igf2 (in mice through loss of H19)—and was the first clear evidence that the same genes that cause Wilms’ tumors in patients can do so in mice as well.
Whereas the Wt1 mouse models were directly based on mutations found in patients, the Lin28 tumor model took the opposite route. Almost serendipitously, it was found that a Vasa-Cre mouse used to drive Lin28a overexpression in primordial germ cells led, through leaky expression in the kidney, to the development of Wilms’ tumors (Urbach et al. 2014). Use of kidney-specific Cre lines confirmed the finding, making Lin28 a genuine Wilms’ tumor gene candidate through overexpression. A subsequent analysis of human Wilms’ tumor samples identified LIN28B overexpression in 18 out of 105 tumors as well as in the NCAM1+/ALDH1+ cancer stem cells (Urbach et al. 2014), making this another patient-relevant Wilms’ tumor mouse model.
The patient relevance of two other mouse models is less clear at present. One study used a conditionally activated β-catenin model with a conditionally activated Kras allele in combination with kidney-specific Cre lines and found metastatic tumors that resembled the epithelial component of Wilms’ tumors (Clark et al. 2011). A potential problem with this model is that there are no reports of KRAS mutations in Wilms’ tumors, making the genetic background of this model difficult to interpret. This is not to say that the model does not have any value; it is very well possible that the pathways that are affected in these mice are the same as deregulated pathways in human tumors, but further study of this is required.
Finally, it was shown recently that interrupted in vivo induced pluripotent stem cell (iPSC) reprogramming of renal tubular cells resulted in tumors that resembled Wilms’ tumors (Ohnishi et al. 2014). Surprisingly, continued reprogramming of tumor cells resulted in normal iPSCs that could contribute to normal kidney structures in chimeric mice, something that would not be expected from cells with mutations that cause Wilms’ tumors. The background of this model makes it difficult to consider this as a model for tumors in patients, but it does highlight the importance of “stemness” in general for Wilms’ tumors and could provide valuable information on the mechanics of Wilms’ tumor development in this way. It also highlights the importance of experienced histopathological analysis of models; the step from blocked development to Wilms’ tumor is subtle, and care should be taken with proper interpretation.
The apparent difficulties in generating mouse models for Wilms’ tumors could lead to the simple question of why mice are used for this in the first place. The simple answer to this is that, until recently, the mouse was the only species in which the genetic modification required to model the genetic events leading to Wilms’ tumors in patients was routinely used. However, other model organisms could be better for developing Wilms’ tumor models. For instance, Wilms’ tumors can be induced in rats with high efficiency using mutagens like ENU, especially in the inbred Nb strain (Hard 1985), but not in mice (Vesselinovitch et al. 1977). Although these tumors show a high incidence of nuclear β-catenin, no mutations in Ctnnb1 or Wt1 were found (Ehrlich et al. 2010), and the patient relevance of these tumors therefore needs to be demonstrated. In contrast, ENU-mediated mutagenesis of outbred Sprague-Dawley rats does result in Wilms’ tumors with mutations in Wt1 (Sharma et al. 1994). Other species in which Wilms’ tumor is found spontaneously or can be induced are rabbits and chickens. Mice might very well be one of the most difficult model organisms to model Wilms’ tumors. However, the recent advances in gene editor technologies, such as zinc finger nucleases, TAL effector nucleases, and CRISPR/Cas9, have opened up genomic engineering possibilities in these species to comparable levels that so far have only been possible in mice (Li et al. 2013a,b; Honda et al. 2014; Park et al. 2014; Yang et al. 2014). Maybe the time is ripe to reconsider the best possible experimental model for Wilms’ tumor research.
Another species that has gained much interest as a cancer model is pigs (Flisikowska et al. 2013). Especially for embryonal tumors like Wilms’ tumors, pigs have potential advantages. If Wilms’ tumor development is dependent on the right mutations being acquired at the right developmental stage, it could be simple statistics that the target cell population size and time during development when these cells are present are rate-limiting steps for Wilms’ tumor mouse models. Kidneys with the same size as human kidneys and a gestation time of several months could make pigs another promising new model. Moreover, Wilms’ tumors are one of the most frequently found spontaneous tumors found in pigs in slaughterhouses (Cotchin 1975), and pigs can efficiently be modified using gene editors (Lillico et al. 2013; Li et al. 2014a).
It is unlikely that these species will ever replace mice in the fundamental analysis of Wilms’ tumors, its genetics, and its developmental background, but the availability of new experimental species that can more efficiently couple genetic aberrations to tumor development would be a welcome addition to Wilms’ tumor research.
Outlook
The last few years have seen a wealth of new developments in the Wilms’ tumor field, such as the description of the first Wilms’ tumor mouse model, the identification of Wilms’ tumor cancer stem cells, and the recent identification of mutations in miRNA processing genes in Wilms’ tumors. At the same time, the increased understanding of early kidney development, especially of the control of the nephron progenitor cells, has provided a detailed developmental framework in which the origins of Wilms’ tumors can be studied and explained. Conversely, the role of multiple Wilms’ tumor genes in this process illustrates the value of studying fundamental Wilms’ tumor biology for the better understanding of normal kidney development.
As the role of the miRNA processor pathway in Wilms’ tumor is now known to be important in tumorigenesis, future research should focus on whether there are any potential therapeutic avenues arising and also on the mechanistic roles of miRNAs and the genes that process them during normal renal development. The few studies available on this are likely only the tip of the iceberg. As most genetic aberrations discovered to date appear to overlap with either the CTNNB1/WT1/WTX pathway or the miRNA processor pathways, there may be two distinct groups of tumors present, as indicated by targeted sequencing of a cohort of blastemal-predominant Wilms’ tumors (Rakheja et al. 2014). However, a much larger study is required, including other Wilms’ tumor subtypes, to fully assess this. Furthermore, the relationship between epigenetic abnormalities (such as IGF2) and mutations in miRNA processors is unknown.
The identification of miRNA processor mutations in Wilms’ tumors emphasizes the importance of miRNAs in normal kidney development, in particular in the nephron progenitor cells. The Six2-Cre-mediated knockout of Dicer1 (Nagalakshmi et al. 2011) has already demonstrated the importance of miRNAs in these cells, and specific miRNAs found deregulated in Wilms’ tumors would be strong candidates to analyze for their roles in nephron progenitors. A mouse ESC resource for the generation of miRNA reporter and conditional knockout alleles has been described (Prosser et al. 2011), and, combined with Cre drivers active in many different cell types in the developing kidney, this would provide the experimental platform to analyze this in more detail.
Although epigenetic mechanisms play a clear role in a subset of Wilms’ tumors in which IGF2 and H19 are dysregulated, whether the epigenome plays an initiating role in tumor formation is yet to be elucidated. Thus far, mutations in two key epigenome remodelers (SMARCA4 and ARID1A) have been identified, and with further sequencing studies, this number may increase. As WT1 and MYCN both interact with common epigenome remodelers, the plasticity and epigenomic events detected may indeed be a secondary event. Studies of the epigenome within Wilms’ tumor with well-characterized genomes will be the next step in understanding the relationship between the two. Such data might subsequently lead to new or better insights into the epigenetic mechanisms underlying normal kidney development.
Acknowledgments
J.C. was funded by the University College London (UCL) Grand Challenges Scheme, the Olivia Hodson fund, and the European Commission FP7 project reference 270089: p-Medicine. K.P.J. is in part supported by the National Institute for Health Research Great Ormond Street Hospital UCL Biomedical Research Centre, Cancer Research UK (C1188/A4614), Great Ormond Street Hospital Children’s Charity, Children with Cancer (grant reference 113), and the European Commission FP7 project reference 261474: European Network for Cancer Research in Children and Adolescents (ENCCA). The Roslin Institute receives Institute Strategic Programme grant funding from the Biotechnology and Biological Sciences Research Council (BBSRC grant BB/J004316/1).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.256396.114.
References
- Aiden AP, Rivera MN, Rheinbay E, Ku M, Coffman EJ, Truong TT, Vargas SO, Lander ES, Haber DA, Bernstein BE. 2010. Wilms tumor chromatin profiles highlight stem cell properties and a renal developmental network. Cell Stem Cell 6: 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE, Leach S, Marra MA, Brooks-Wilson AR, Penninger J, et al. 2004. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet 13: 2061–2074. [DOI] [PubMed] [Google Scholar]
- Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, et al. 2012. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44: 277–284. [DOI] [PubMed] [Google Scholar]
- Bahubeshi A, Bal N, Rio Frio T, Hamel N, Pouchet C, Yilmaz A, Bouron-Dal Soglio D, Williams GM, Tischkowitz M, Priest JR, et al. 2010. Germline DICER1 mutations and familial cystic nephroma. J Med Genet 47: 863–866. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Falkoff D, Petruzzi M-J, Nowak N, Zabel B, Adam M, Aguiar MC, Grundy P, Shows T, Pelletier J. 1994. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7: 91–97. [DOI] [PubMed] [Google Scholar]
- Bates CM, Kharzai S, Erwin T, Rossant J, Parada LF. 2000. Role of N-myc in the developing mouse kidney. Dev Biol 222: 317–325. [DOI] [PubMed] [Google Scholar]
- Beckwith JB, Kiviat NB, Bonadio JF. 1990. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr Pathol 10: 1–36. [DOI] [PubMed] [Google Scholar]
- Berger AH, Knudson AG, Pandolfi PP. 2011. A continuum model for tumour suppression. Nature 476: 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al. 1990. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60: 509–520. [DOI] [PubMed] [Google Scholar]
- Chang HM, Triboulet R, Thornton JE, Gregory RI. 2013. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28–let-7 pathway. Nature 497: 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton J, Williams RD, Sebire NJ, Popov S, Vujanic G, Chagtai T, Alcaide-German M, Morris T, Butcher LM, Guilhamon P, et al. 2015. Comparative methylome analysis identifies new tumour subtypes and biomarkers for transformation of nephrogenic rests into Wilms tumour. Genome Medicine 7: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, Robertson EJ, Alt FW. 1992. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev 6: 2248–2257. [DOI] [PubMed] [Google Scholar]
- Chau YY, Brownstein D, Mjoseng H, Lee WC, Buza-Vidas N, Nerlov C, Jacobsen SE, Perry P, Berry R, Thornburn A, et al. 2011. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet 7: e1002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilukamarri L, Hancock AL, Malik S, Zabkiewicz J, Baker JA, Greenhough A, Dallosso AR, Huang TH, Royer-Pokora B, Brown KW, et al. 2007. Hypomethylation and aberrant expression of the glioma pathogenesis-related 1 gene in Wilms tumors. Neoplasia 9: 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PE, Polosukhina D, Love H, Correa H, Coffin C, Perlman EJ, de Caestecker M, Moses HL, Zent R. 2011. β-Catenin and K-RAS synergize to form primitive renal epithelial tumors with features of epithelial Wilms’ tumors. Am J Pathol 179: 3045–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini F, Kopan R. 2010. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell 18: 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotchin E. 1975. Spontaneous tumours in young animals. Proc R Soc Med 68: 653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. 1966. Chromosome constitution of nephroblastomas. Cancer 19: 1217–1224. [DOI] [PubMed] [Google Scholar]
- Dallosso AR, Hancock AL, Szemes M, Moorwood K, Chilukamarri L, Tsai HH, Sarkar A, Barasch J, Vuononvirta R, Jones C, et al. 2009. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet 5: e1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Tanigawa S, Karner CM, Xin M, Lum L, Chen C, Olson EN, Perantoni AO, Carroll TJ. 2013. Stromal–epithelial crosstalk regulates kidney progenitor cell differentiation. Nat Cell Biol 15: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JA, Ladomery M, Hohenstein P, Michael L, Shafe A, Spraggon L, Hastie N. 2004. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet 13: 235–246. [DOI] [PubMed] [Google Scholar]
- Dekel B, Metsuyanim S, Schmidt-Ott KM, Fridman E, Jacob-Hirsch J, Simon A, Pinthus J, Mor Y, Barasch J, Amariglio N, et al. 2006. Multiple imprinted and stemness genes provide a link between normal and tumor progenitor cells of the developing human kidney. Cancer Res 66: 6040–6049. [DOI] [PubMed] [Google Scholar]
- Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, et al. 2009. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 4: 403–415. [DOI] [PubMed] [Google Scholar]
- Drake KM, Ruteshouser EC, Natrajan R, Harbor P, Wegert J, Gessler M, Pritchard-Jones K, Grundy P, Dome J, Huff V, et al. 2009. Loss of heterozygosity at 2q37 in sporadic Wilms’ tumor: putative role for miR-562. Clin Cancer Res 15: 5985–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Hopkins NE, Jiang G, Dome JS, Yu MC, Woods CB, Tomlinson GE, Chintagumpala M, Champagne M, Dillerg L, et al. 2003. Satellite DNA hypomethylation in karyotyped Wilms tumors. Cancer Genet Cytogenet 141: 97–105. [DOI] [PubMed] [Google Scholar]
- Ehrlich D, Bruder E, Thome MA, Gutt CN, von Knebel Doeberitz M, Niggli F, Perantoni AO, Koesters R. 2010. Nuclear accumulation of β-catenin protein indicates activation of wnt signaling in chemically induced rat nephroblastomas. Pediatr Dev Pathol 13: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Dahr S, Hilliard S, Aboudehen K, Saifudeen Z. 2014. The MDM2–p53 pathway: multiple roles in kidney development. Pediatr Nephrol 29: 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essafi A, Webb A, Berry RL, Slight J, Burn SF, Spraggon L, Velecela V, Martinez-Estrada OM, Wiltshire JH, Roberts SG, et al. 2011. A Wt1-controlled chromatin switching mechanism underpins tissue-specific Wnt4 activation and repression. Dev Cell 21: 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flisikowska T, Kind A, Schnieke A. 2013. The new pig on the block: modelling cancer in pigs. Transgenic Res 22: 673–680. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, Choong CS, Charles A, Frieder RP, Dishop MK, et al. 2011. Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32: 1381–1384. [DOI] [PubMed] [Google Scholar]
- Francke U, Holmes LB, Atkins L, Riccardi VM. 1979. Aniridia-Wilms’ tumor association: evidence for specific deletion of 11p13. Cytogenet Cell Genet 24: 185–192. [DOI] [PubMed] [Google Scholar]
- Frese KK, Tuveson DA. 2007. Maximizing mouse cancer models. Nat Rev Cancer 7: 645–658. [DOI] [PubMed] [Google Scholar]
- Fukuzawa R, Heathcott RW, More HE, Reeve AE. 2007. Sequential WT1 and CTNNB1 mutations and alterations of β-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60: 1013–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzawa R, Holman SK, Chow CW, Savarirayan R, Reeve AE, Robertson SP. 2010. WTX mutations can occur both early and late in the pathogenesis of Wilms tumour. J Med Genet 47: 791–794. [DOI] [PubMed] [Google Scholar]
- Gadd S, Huff V, Huang CC, Ruteshouser EC, Dome JS, Grundy PE, Breslow N, Jennings L, Green DM, Beckwith JB, et al. 2012. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a children’s oncology group study. Neoplasia 14: 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Maiti S, Sun G, Ordonez NG, Udtha M, Deng JM, Behringer RR, Huff V. 2004. The Wt1+/R394W mouse displays glomerulosclerosis and early-onset renal failure characteristic of human Denys-Drash syndrome. Mol Cell Biol 24: 9899–9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebeshuber CA, Kornauth C, Dong L, Sierig R, Seibler J, Reiss M, Tauber S, Bilban M, Wang S, Kain R, et al. 2013. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat Med 19: 481–487. [DOI] [PubMed] [Google Scholar]
- Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. 1990. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 343: 774–778. [DOI] [PubMed] [Google Scholar]
- Grill C, Sunitsch S, Hatz M, Hauser-Kronberger C, Leuschner I, Hoefler G, Guertl B. 2011. Activation of β-catenin is a late event in the pathogenesis of nephroblastomas and rarely correlated with genetic changes of the APC gene. Pathology 43: 702–706. [DOI] [PubMed] [Google Scholar]
- Grundy P, Coppes MJ, Haber D. 1995. Molecular genetics of Wilms tumor. Hematol Oncol Clin North Am 9: 1201–1215. [PubMed] [Google Scholar]
- Guertl B, Leuschner I, Guelly C, Ebner B, Kronberger C, Hoefler G. 2010. Is predisposition for nephroblastoma linked to polymorphisms of the WTX gene?. Pathol Oncol Res 16: 189–191. [DOI] [PubMed] [Google Scholar]
- Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, Hammes A, Hastie ND, Schedl A. 2002. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet 11: 651–659. [DOI] [PubMed] [Google Scholar]
- Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, Gubler MC, Schedl A. 2001. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106: 319–329. [DOI] [PubMed] [Google Scholar]
- Hanks S, Perdeaux ER, Seal S, Ruark E, Mahamdallie SS, Murray A, Ramsay E, Del Vecchio Duarte S, Zachariou A, de Souza B, et al. 2014. Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, et al. 2002. Deregulation of Caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res 62: 5897–5901. [PubMed] [Google Scholar]
- Harari-Steinberg O, Metsuyanim S, Omer D, Gnatek Y, Gershon R, Pri-Chen S, Ozdemir DD, Lerenthal Y, Noiman T, Ben-Hur H, et al. 2013. Identification of human nephron progenitors capable of generation of kidney structures and functional repair of chronic renal disease. EMBO Mol Med 5: 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hard GC. 1985. Differential renal tumor response to N-ethylnitrosourea and dimethylnitrosamine in the Nb rat: basis for a new rodent model of nephroblastoma. Carcinogenesis 6: 1551–1558. [DOI] [PubMed] [Google Scholar]
- Haruta M, Matsumoto Y, Izumi H, Watanabe N, Fukuzawa M, Matsuura S, Kaneko Y. 2008. Combined BubR1 protein down-regulation and RASSF1A hypermethylation in Wilms tumors with diverse cytogenetic changes. Mol Carcinog 47: 660–666. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. 2009. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138: 696–708. [DOI] [PubMed] [Google Scholar]
- Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM 3rd, Jaenisch R. 2005. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 8: 275–285. [DOI] [PubMed] [Google Scholar]
- Honda A, Hirose M, Sankai T, Yasmin L, Yuzawa K, Honsho K, Izu H, Iguchi A, Ikawa M, Ogura A. 2014. Single-step generation of rabbits carrying a targeted allele of the tyrosinase gene using CRISPR/Cas9. Exp Anim doi: 1538/expanim.14-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Gao F, Tian W, Ruteshouser EC, Wang Y, Lazar A, Stewart J, Strong LC, Behringer RR, Huff V. 2011. Wt1 ablation and Igf2 upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J Clin Invest 121: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubertus J, Lacher M, Rottenkolber M, Muller-Hocker J, Berger M, Stehr M, von Schweinitz D, Kappler R. 2011. Altered expression of imprinted genes in Wilms tumors. Oncol Rep 25: 817–823. [DOI] [PubMed] [Google Scholar]
- Hubertus J, Zitzmann F, Trippel F, Muller-Hocker J, Stehr M, von Schweinitz D, Kappler R. 2013. Selective methylation of CpGs at regulatory binding sites controls NNAT expression in Wilms tumors. PLoS ONE 8: e67605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff V. 2011. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer 11: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam JS, Buddavarapu K, Lee-Chang JS, Ganapathy S, Camosy C, Chen Y, Rao MK. 2010. MicroRNA-185 suppresses tumor growth and progression by targeting the Six1 oncogene in human cancers. Oncogene 29: 4971–4979. [DOI] [PubMed] [Google Scholar]
- Jaehning JA. 2010. The Paf1 complex: platform or player in RNA polymerase II transcription?. Biochim Biophys Acta 1799: 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins ZA, van Kogelenberg M, Morgan T, Jeffs A, Fukuzawa R, Pearl E, Thaller C, Hing AV, Porteous ME, Garcia-Minaur S, et al. 2009. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41: 95–100. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Egues MC, Rowley JD. 1981. Interstitial deletion of short arm of chromosome 11 limited to Wilms’ tumor cells in a patient without aniridia. Cancer Res 41: 4577–4578. [PubMed] [Google Scholar]
- Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ. 2011. Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138: 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Yoon SK, Bollig F, Kitagaki J, Hur W, Whye NJ, Wu YP, Rivera MN, Park JY, Kim HS, et al. 2010. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J Biol Chem 285: 14585–14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WJ, Rivera MN, Coffman EJ, Haber DA. 2012. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol Cell 45: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert A, Vainio S, McMahon AP. 1998. Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125: 4225–4234. [DOI] [PubMed] [Google Scholar]
- Knudson AG Jr, Strong LC. 1972. Mutation and cancer: a model for Wilms’ tumor of the kidney. J Natl Cancer Inst 48: 313–324. [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. 2008. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koesters R, Ridder R, Kopp-Schneider A, Betts D, Adams V, Niggli F, Briner J, von Knebel Doeberitz M. 1999. Mutational activation of the β-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res 59: 3880–3882. [PubMed] [Google Scholar]
- Koesters R, Niggli F, von Knebel Doeberitz M, Stallmach T. 2003. Nuclear accumulation of β-catenin protein in Wilms’ tumours. J Pathol 199: 68–76. [DOI] [PubMed] [Google Scholar]
- Koller K, Das S, Leuschner I, Korbelius M, Hoefler G, Guertl B. 2013. Identification of the transcription factor HOXB4 as a novel target of miR-23a. Genes Chromosomes Cancer 52: 709–715. [DOI] [PubMed] [Google Scholar]
- Kort EJ, Farber L, Tretiakova M, Petillo D, Furge KA, Yang XJ, Cornelius A, Teh BT. 2008. The E2F3–Oncomir-1 axis is activated in Wilms’ tumor. Cancer Res 68: 4034–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufos A, Grundy P, Morgan K, Aleck KA, Hadro T, Lampkin BC, Kalbakji A, Cavenee WK. 1989. Familial Wiedemann-Beckwith syndrome and a second Wilms tumor locus both map to 11p15.5. Am J Hum Genet 44: 711–719. [PMC free article] [PubMed] [Google Scholar]
- Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. 1993. WT-1 is required for early kidney development. Cell 74: 679–691. [DOI] [PubMed] [Google Scholar]
- Kusafuka T, Miao J, Kuroda S, Udatsu Y, Yoneda A. 2002. Codon 45 of the β-catenin gene, a specific mutational target site of Wilms’ tumor. Int J Mol Med 10: 395–399. [PubMed] [Google Scholar]
- Laurenti E, Wilson A, Trumpp A. 2009. Myc’s other life: stem cells and beyond. Curr Opin Cell Biol 21: 844–854. [DOI] [PubMed] [Google Scholar]
- Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. 1995. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375: 34–39. [DOI] [PubMed] [Google Scholar]
- Lemoine NR, Hughes CM, Cowell JK. 1992. Aberrant expression of the tumour suppressor gene p53 is very frequent in Wilms’ tumours. J Pathol 168: 237–242. [DOI] [PubMed] [Google Scholar]
- Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, et al. 2013a. Heritable gene targeting in the mouse and rat using a CRISPR–Cas system. Nat Biotechnol 31: 681–683. [DOI] [PubMed] [Google Scholar]
- Li W, Teng F, Li T, Zhou Q. 2013b. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR–Cas systems. Nat Biotechnol 31: 684–686. [DOI] [PubMed] [Google Scholar]
- Li F, Cowley DO, Banner D, Holle E, Zhang L, Su L. 2014a. Efficient genetic manipulation of the NOD–Rag1−/−IL2RγC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep 4: 5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hartwig S, Rosenblum ND. 2014b. Developmental origins and functions of stromal cells in the normal and diseased mammalian kidney. Dev Dyn 243: 853–863. [DOI] [PubMed] [Google Scholar]
- Lillico SG, Proudfoot C, Carlson DF, Stverakova D, Neil C, Blain C, King TJ, Ritchie WA, Tan W, Mileham AJ, et al. 2013. Live pigs produced from genome edited zygotes. Sci Rep 3: 2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludgate JL, Le Mee G, Fukuzawa R, Rodger EJ, Weeks RJ, Reeve AE, Morison IM. 2013. Global demethylation in loss of imprinting subtype of Wilms tumor. Genes Chromosomes Cancer 52: 174–184. [DOI] [PubMed] [Google Scholar]
- Maiti S, Alam R, Amos CI, Huff V. 2000. Frequent association of β-catenin and WT1 mutations in Wilms tumors. Cancer Res 60: 6288–6292. [PubMed] [Google Scholar]
- Major MB, Camp ND, Berndt JD, Yi X, Goldenberg SJ, Hubbert C, Biechele TL, Gingras AC, Zheng N, Maccoss MJ, et al. 2007. Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science 316: 1043–1046. [DOI] [PubMed] [Google Scholar]
- Maschietto M, Williams RD, Chagtai T, Popov SD, Sebire NJ, Vujanic G, Perlman E, Anderson JR, Grundy P, Dome JS, et al. 2014. TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PLoS ONE 9: e109924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke AL, IJpenberg A, Fleming S, Ross A, Medine CN, Patek CE, Spraggon L, Hughes J, Clarke AR, Hastie ND. 2003. The wt1-heterozygous mouse; a model to study the development of glomerular sclerosis. J Pathol 200: 667–674. [DOI] [PubMed] [Google Scholar]
- Metsuyanim S, Pode-Shakked N, Schmidt-Ott KM, Keshet G, Rechavi G, Blumental D, Dekel B. 2008. Accumulation of malignant renal stem cells is associated with epigenetic changes in normal renal progenitor genes. Stem Cells 26: 1808–1817. [DOI] [PubMed] [Google Scholar]
- Metsuyanim S, Harari-Steinberg O, Buzhor E, Omer D, Pode-Shakked N, Ben-Hur H, Halperin R, Schneider D, Dekel B. 2009. Expression of stem cell markers in the human fetal kidney. PLoS ONE 4: e6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Hodges E, Hohenstein P. 2012. WT1 in disease: shifting the epithelial–mesenchymal balance. J Pathol 226: 229–240. [DOI] [PubMed] [Google Scholar]
- Miyagawa K, Kent J, Moore A, Charlieu JP, Little MH, Williamson KA, Kelsey A, Brown KW, Hassam S, Briner J, et al. 1998. Loss of WT1 function leads to ectopic myogenesis in Wilms’ tumour. Nat Genet 18: 15–17. [DOI] [PubMed] [Google Scholar]
- Moisan A, Rivera MN, Lotinun S, Akhavanfard S, Coffman EJ, Cook EB, Stoykova S, Mukherjee S, Schoonmaker JA, Burger A, et al. 2011. The WTX tumor suppressor regulates mesenchymal progenitor cell fate specification. Dev Cell 20: 583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD, Cooper WN, Lee J, Gentle D, Macdonald F, et al. 2003. Multigene methylation analysis of Wilms’ tumour and adult renal cell carcinoma. Oncogene 22: 6794–6801. [DOI] [PubMed] [Google Scholar]
- Mugrauer G, Alt FW, Ekblom P. 1988. N-myc proto-oncogene expression during organogenesis in the developing mouse as revealed by in situ hybridization. J Cell Biol 107: 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Pierce J, de Caestecker C, Libes J, Neblett D, de Caestecker M, Perantoni AO, Tanigawa S, Anderson JR, Dome JS, et al. 2014. Aberrant activation, nuclear localization, and phosphorylation of Yes-associated protein-1 in the embryonic kidney and Wilms tumor. Pediatr Blood Cancer 61: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi VK, Ren Q, Pugh MM, Valerius MT, McMahon AP, Yu J. 2011. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int 79: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisen PD, Zimmerman KA, Cotter SV, Gilbert F, Alt FW. 1986. Enhanced expression of the N-myc gene in Wilms’ tumors. Cancer Res 46: 6217–6222. [PubMed] [Google Scholar]
- Nishiyama R, Qi L, Lacey M, Ehrlich M. 2005. Both hypomethylation and hypermethylation in a 0.2-kb region of a DNA repeat in cancer. Mol Cancer Res 3: 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris MD, Brian MJ, Vowels MR, Stewart BW. 1988. N-myc amplification in Wilms’ tumor. Cancer Genet Cytogenet 30: 187–189. [DOI] [PubMed] [Google Scholar]
- Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, Smith PJ, Reeve AE. 1993. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature 362: 749–751. [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Cui H, He L, Pfeifer S, Malmikumpu H, Jiang S, Feinberg AP, Hedborg F. 1999. Mosaic allelic insulin-like growth factor 2 expression patterns reveal a link between Wilms’ tumorigenesis and epigenetic heterogeneity. Cancer Res 59: 3889–3892. [PubMed] [Google Scholar]
- Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, et al. 2014. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156: 663–677. [DOI] [PubMed] [Google Scholar]
- Ohshima J, Haruta M, Fujiwara Y, Watanabe N, Arai Y, Ariga T, Okita H, Koshinaga T, Oue T, Hinotsu S, et al. 2012. Methylation of the RASSF1A promoter is predictive of poor outcome among patients with Wilms tumor. Pediatr Blood Cancer 59: 499–505. [DOI] [PubMed] [Google Scholar]
- Okutsu T, Kuroiwa Y, Kagitani F, Kai M, Aisaka K, Tsutsumi O, Kaneko Y, Yokomori K, Surani MA, Kohda T, et al. 2000. Expression and imprinting status of human PEG8/IGF2AS, a paternally expressed antisense transcript from the IGF2 locus, in Wilms’ tumors. J Biochem 127: 475–483. [DOI] [PubMed] [Google Scholar]
- Ozdemir DD, Hohenstein P. 2014. Wt1 in the kidney—a tale in mouse models. Pediatr Nephrol 29: 687–693. [DOI] [PubMed] [Google Scholar]
- Park S, Bernard A, Bove KE, Sens DA, Hazen-Martin DJ, Garvin AJ, Haber DA. 1993. Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms’ tumour. Nat Genet 5: 363–367. [DOI] [PubMed] [Google Scholar]
- Park JS, Ma W, O’Brien LL, Chung E, Guo JJ, Cheng JG, Valerius MT, McMahon JA, Wong WH, McMahon AP. 2012. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 23: 637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TS, Lee HJ, Kim KH, Kim JS, Han JY. 2014. Targeted gene knockout in chickens mediated by TALENs. Proc Natl Acad Sci 111: 12716–12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L, et al. 1991. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67: 437–447. [DOI] [PubMed] [Google Scholar]
- Perotti D, Hohenstein P, Bongarzone I, Maschietto M, Weeks M, Radice P, Pritchard-Jones K. 2013. Is Wilms tumor a candidate neoplasia for treatment with WNT/β-catenin pathway modulators?—A report from the renal tumors biology-driven drug development workshop. Mol Cancer Ther 12: 2619–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pode-Shakked N, Dekel B. 2011. Wilms tumor–a renal stem cell malignancy? Pediatr Nephrol 26: 1535–1543. [DOI] [PubMed] [Google Scholar]
- Pode-Shakked N, Shukrun R, Mark-Danieli M, Tsvetkov P, Bahar S, Pri-Chen S, Goldstein RS, Rom-Gross E, Mor Y, Fridman E, et al. 2013. The isolation and characterization of renal cancer initiating cells from human Wilms’ tumour xenografts unveils new therapeutic targets. EMBO Mol Med 5: 18–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preger-Ben Noon E, Barak H, Guttmann-Raviv N, Reshef R. 2009. Interplay between activin and Hox genes determines the formation of the kidney morphogenetic field. Development 136: 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D, et al. 1990. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 346: 194–197. [DOI] [PubMed] [Google Scholar]
- Prosser HM, Koike-Yusa H, Cooper JD, Law FC, Bradley A. 2011. A resource of vectors and ES cells for targeted deletion of microRNAs in mice. Nat Biotechnol 29: 840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, Khokhar S, Wickiser JE, Karandikar NJ, Malter JS, et al. 2014. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoche MA, Kress C, Poirier F, Dandolo L. 1997. Deletion of the H19 transcription unit reveals the existence of a putative imprinting control element. Genes Dev 11: 1596–1604. [DOI] [PubMed] [Google Scholar]
- Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, Kim JC, Feinberg AP, Gerald WL, Vargas SO, et al. 2007. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315: 642–645. [DOI] [PubMed] [Google Scholar]
- Rivera MN, Kim WJ, Wells J, Stone A, Burger A, Coffman EJ, Zhang J, Haber DA. 2009. The tumor suppressor WTX shuttles to the nucleus and modulates WT1 activity. Proc Natl Acad Sci 106: 8338–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruteshouser EC, Robinson SM, Huff V. 2008. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifudeen Z, Dipp S, Stefkova J, Yao X, Lookabaugh S, El-Dahr SS. 2009. p53 regulates metanephric development. J Am Soc Nephrol 20: 2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell C, Wanner N, Huber TB. 2014. Glomerular development—shaping the multi-cellular filtration unit. Semin Cell Dev Biol 36: 39–49. [DOI] [PubMed] [Google Scholar]
- Schneid H, Vazquez MP, Seurin D, le Bouc Y. 1991. Loss of heterozygosity in non-tumoral tissue in two children with Beckwith-Wiedemann syndrome. Growth Regul 1: 168–170. [PubMed] [Google Scholar]
- Schroeder WT, Chao LY, Dao DD, Strong LC, Pathak S, Riccardi V, Lewis WH, Saunders GF. 1987. Nonrandom loss of maternal chromosome 11 alleles in Wilms tumors. Am J Hum Genet 40: 413–420. [PMC free article] [PubMed] [Google Scholar]
- Schumacher V, Schneider S, Figge A, Wildhardt G, Harms D, Schmidt D, Weirich A, Ludwig R, Royer-Pokora B. 1997. Correlation of germ-line mutations and two-hit inactivation of the WT1 gene with Wilms tumors of stromal-predominant histology. Proc Natl Acad Sci 94: 3972–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher V, Schuhen S, Sonner S, Weirich A, Leuschner I, Harms D, Licht J, Roberts S, Royer-Pokora B. 2003. Two molecular subgroups of Wilms’ tumors with or without WT1 mutations. Clin Cancer Res 9: 2005–2014. [PubMed] [Google Scholar]
- Scott J, Cowell J, Robertson ME, Priestley LM, Wadey R, Hopkins B, Pritchard J, Bell GI, Rall LB, Graham CF, et al. 1985. Insulin-like growth factor-II gene expression in Wilms’ tumour and embryonic tissues. Nature 317: 260–262. [DOI] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. 2006. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 25: 5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senanayake U, Das S, Vesely P, Alzoughbi W, Frohlich LF, Chowdhury P, Leuschner I, Hoefler G, Guertl B. 2012. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis 33: 1014–1021. [DOI] [PubMed] [Google Scholar]
- Sharma PM, Bowman M, Yu BF, Sukumar S. 1994. A rodent model for Wilms tumors: embryonal kidney neoplasms induced by N-nitroso-N′-methylurea. Proc Natl Acad Sci 91: 9931–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H, Suzuki M, Takahashi T, Miyajima K, Toyooka S, Shivapurkar N, Tomlinson GE, Mastrangelo D, Pass HI, Brambilla E, et al. 2005. Aberrant methylation of HIN-1 (high in normal-1) is a frequent event in many human malignancies. Int J Cancer 113: 600–604. [DOI] [PubMed] [Google Scholar]
- Shukrun R, Pode-Shakked N, Pleniceanu O, Omer D, Vax E, Peer E, Pri-Chen S, Jacob J, Hu Q, Harari-Steinberg O, et al. 2014. Wilms’ tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Reports 3: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, Barfoot R, Burke A, Chisholm J, Hewitt M, et al. 2011. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 48: 273–278. [DOI] [PubMed] [Google Scholar]
- Slater RM, de Kraker J. 1982. Chromosome number 11 and Wilms’ tumor. Cancer Genet Cytogenet 5: 237–245. [DOI] [PubMed] [Google Scholar]
- Stark K, Vainio S, Vassileva G, McMahon AP. 1994. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372: 679–683. [DOI] [PubMed] [Google Scholar]
- Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y, Watts C, Way M, Dirks P, Bertone P, et al. 2013. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev 27: 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun FL, Dean WL, Kelsey G, Allen ND, Reik W. 1997. Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature 389: 809–815. [DOI] [PubMed] [Google Scholar]
- Szemes M, Dallosso AR, Melegh Z, Curry T, Li Y, Rivers C, Uney J, Magdefrau AS, Schwiderski K, Park JH, et al. 2013. Control of epigenetic states by WT1 via regulation of de novo DNA methyltransferase 3A. Hum Mol Genet 22: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Chang HM, Piskounova E, Gregory RI. 2012. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 18: 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timp W, Feinberg AP. 2013. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13: 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrezan GT, Ferreira EN, Nakahata AM, Barros BD, Castro MT, Correa BR, Krepischi AC, Olivieri EH, Cunha IW, Tabori U, et al. 2014. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udtha M, Lee SJ, Alam R, Coombes K, Huff V. 2003. Upregulation of c-MYC in WT1-mutant tumors: assessment of WT1 putative transcriptional targets using cDNA microarray expression profiling of genetically defined Wilms’ tumors. Oncogene 22: 3821–3826. [DOI] [PubMed] [Google Scholar]
- Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, Shukrun R, Charlton J, Sebire N, Mifsud W, et al. 2014. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev 28: 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesselinovitch SD, Koka M, Rao KV, Mihailovich N, Rice JM. 1977. Prenatal multicarcinogenesis by ethylnitrosourea in mice. Cancer Res 37: 1822–1828. [PubMed] [Google Scholar]
- Visvader JE. 2011. Cells of origin in cancer. Nature 469: 314–322. [DOI] [PubMed] [Google Scholar]
- Wagner KJ, Cooper WN, Grundy RG, Caldwell G, Jones C, Wadey RB, Morton D, Schofield PN, Reik W, Latif F, et al. 2002. Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene 21: 7277–7282. [DOI] [PubMed] [Google Scholar]
- Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen Q-R, Hsu CH, Yan C, et al. 2015. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JA, Bryan K, Williams R, Popov S, Vujanic G, Coulomb A, Boccon-Gibod L, Graf N, Pritchard-Jones K, O’Sullivan M. 2013. miRNA profiles as a predictor of chemoresponsiveness in Wilms’ tumor blastema. PLoS ONE 8: e53417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegert J, Wittmann S, Leuschner I, Geissinger E, Graf N, Gessler M. 2009. WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48: 1102–1111. [DOI] [PubMed] [Google Scholar]
- Wegert J, Ishaque N, Vardapour R, Georg C, Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, et al. 2015. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27: 298–311. [DOI] [PubMed] [Google Scholar]
- Wilkins RJ. 1988. Genomic imprinting and carcinogenesis. Lancet 1: 329–331. [DOI] [PubMed] [Google Scholar]
- Williams RD, Al-Saadi R, Chagtai T, Popov S, Messahel B, Sebire N, Gessler M, Wegert J, Graf N, Leuschner I, et al. 2010. Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms’ tumor. Clin Cancer Res 16: 2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, Vujanic G, Van Tinteren H, van den Heuvel-Eibrink MM, Kool M, et al. 2015. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Waha A, Weggen S, Schirmacher P, Kuhne T, Goodyer CG, Albrecht S, Von Schweinitz D, Pietsch T. 2003. Overexpression of human Dickkopf-1, an antagonist of wingless/WNT signaling, in human hepatoblastomas and Wilms’ tumors. Lab Invest 83: 429–434. [DOI] [PubMed] [Google Scholar]
- Wu MK, Sabbaghian N, Xu B, Addidou-Kalucki S, Bernard C, Zou D, Reeve AE, Eccles MR, Cole C, Choong CS, et al. 2013. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230: 154–164. [DOI] [PubMed] [Google Scholar]
- Xin Z, Soejima H, Higashimoto K, Yatsuki H, Zhu X, Satoh Y, Masaki Z, Kaneko Y, Jinno Y, Fukuzawa R, et al. 2000. A novel imprinted gene, KCNQ1DN, within the WT2 critical region of human chromosome 11p15.5 and its reduced expression in Wilms’ tumors. J Biochem 128: 847–853. [DOI] [PubMed] [Google Scholar]
- Xu PX. 2013. The EYA–SO/SIX complex in development and disease. Pediatr Nephrol 28: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Zheng WM, Huang L, Maire P, Laclef C, Silvius D. 2003. Six1 is required for the early organogenesis of mammalian kidney. Development 130: 3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zeng D-q, Wu Y, Zheng R, Gu L, Lin X, Hua X, Jin G-H. 2011. Tumor suppressor menin represses paired box gene 2 expression via Wilms tumor suppressor protein-polycomb group complex. J Biol Chem 286: 13937–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wong EM, Cheng C, Li J, Sharkar MTK, Xu CY, Chen B, Sun J, Jing D, Xu PX. 2014. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell 31: 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Xu J, Zhu T, Fan J, Lai L, Zhang J, Chen YE. 2014. Effective gene targeting in rabbits using RNA-guided Cas9 nucleases. J Mol Cell Biol 6: 97–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xing G, Saunders GF. 1999. Proto-oncogene N-myc promoter is down regulated by the Wilms’ tumor suppressor gene WT1. Anticancer Res 19: 1641–1648. [PubMed] [Google Scholar]
- Zitzmann F, Mayr D, Berger M, Stehr M, von Schweinitz D, Kappler R, Hubertus J. 2014. Frequent hypermethylation of a CTCF binding site influences Wilms tumor 1 expression in Wilms tumors. Oncol Rep 31: 1871–1876. [DOI] [PubMed] [Google Scholar]