

Abstract
Background: The purpose of this study was to explore the effect of experimental sleep deprivation (SD) on the temporomandibular joint (TMJ) of rats and the possible mechanism related to abnormal bone metabolism. Material and methods: SD was induced by a modified multiple platform method and assessed by serum adrenocorticotropic hormone (ACTH) level. TMJs were detached and stained with hematoxylin and eosin (H&E). Expression of interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), osteoprotegerin (OPG) and receptor activator of nuclear factor kappa B ligand (RANKL) was evaluated by quantitative reverse transcription polymerase chain reaction, H&E staining, immunohistochemical staining and enzyme linked immunosorbent assay. Results: Compared with controls, SD significantly increased serum ACTH, indicating that the SD model was successful. In the SD group, H&E staining revealed greater vessel hyperplasia in the synovial membrane and thicker hypertrophic layers in condylar cartilages. Compared with controls, RNA and protein expression of the inflammatory factors IL-1β and TNF-α and the bone metabolism-related factor RANKL increased in condylar cartilage in the SD group, whereas OPG and the OPG/RANKL ratio decreased. Immunohistochemical staining revealed that OPG/RANKL immunopositive cells were mainly located in hypertrophic layers. Conclusions: These results suggest that sleep deprivation might play an important role in the occurrence and development of temporomandibular disorders, which may occur through abnormal secretion of inflammatory and bone metabolism-related factors.
Keywords: Sleep deprivation, temporomandibular joint, bone metabolism, inflammation
Introduction
Temporomandibular disorders (TMDs) are common conditions in the oral and maxillofacial region, with predominant clinical manifestations of structural damage to and dysfunction of the temporomandibular joint (TMJ) [1,2]. The etiology of TMD is complex, and remains both controversial and unclear. In addition to occlusal problems, it is widely recognized that psychosomatic factors play a role in TMD, and factors such as sex, age and sleep disorders may also be involved [3-5]. Sleep disorders can trigger various problems, including memory deficits and hormonal abnormalities [6]. As a form of chronic stress, poor sleep quality has been reported to be significantly correlated with the occurrence of TMD in clinical retrospective analyses [7]. However, there is little direct evidence from animal experiments to confirm whether sleep deprivation (SD) can lead to TMD and the exact mechanism of how SD causes the pathologic changes of TMD remains unclear.
Inflammatory infiltration and damage to cartilage and subchondral bone are two important clinical manifestations of TMD [8]. Tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β) appear to be the major inflammatory cytokines involved in almost all of the pathologic processes in TMD, inducing abnormal secretion of osteoprotegerin (OPG) and receptor activator of nuclear factor kappa B (NF-κB) ligand (RANKL) [9]. Imbalances in the OPG/RANKL system can cause abnormal bone metabolism and subchondral bone damage [10]. In previous studies, we found that SD can induce overexpression of pain-related factors in the synovial membrane of the TMJ through activation of estrogen and NF-κB signaling [11]. Whether SD can also induce damage to the condyle and the possible mechanism of this are unknown.
In the present study, we used a modified multiple platform method to induce experimental SD in rats and evaluated correlations between morphologic changes and the expression of inflammatory and bone metabolism-related factors. We aimed to investigate the possible mechanism by which TMD is induced by SD and to provide experimental evidence to aid the clinical treatment of TMD.
Material and methods
Laboratory animals and animal groups
One hundred and fifty male 8-week-old Sprague-Dawley rats weighing 180 ± 20 g were provided and raised by the Laboratory Animal Center of Shandong University. Before the experiments, the animals were housed in a room maintained at 26°C with a 12 h/12 h light/dark cycle and all received the same standardized diet. All procedures and the care administered to the animals were approved by the University Ethics Committee and performed according to institutional guidelines.
The rats were divided randomly into a cage control (CC) group, a SD group and a tank control (TC) group. Each group was further divided into five subgroups of 10 rats each according to the duration of SD; namely, 1 day, 3 days, 5 days, 7 days or 9 days. The CC group rats were raised in the standardized environment without any stress.
Experimental establishment of SD
As described previously, a modified multiple platform method (Jinan General Military Hospital, Jinan, China) was applied to induce experimental SD. Fifteen narrow (6.3 cm diameter) or wide (18.0 cm width) platforms were placed inside a water tank constructed from organic glass (110.0 cm long × 40.0 cm wide × 70.0 cm high). The narrow platforms were set 15 cm apart. In the SD procedure, the tanks were filled with water to a level 1.0 cm below the upper surface of the platforms. The narrow and wide platforms were assigned to the SD group and the TC group, respectively. On the narrow platforms, when the rats entered paradoxical sleep, which is characterized by muscle atonia, they would fall into the water and wake up. On the wide platforms, the rats were able to lie down without falling into the water, although their tails might get wet. The rats in the CC group were maintained in cages in the same room as those in the SD and TC groups. All rats had free access to food and water.
Tissue preparation
At each experimental time point, rats were sacrificed after intraperitoneal injection of 0.6% pentobarbital sodium (30 mg/kg) and blood samples were immediately collected from the cardiac ventricle. The plasma was then separated by centrifugation (12000 g, 15 min, 4°C) and stored immediately at -20°C for subsequent hormone assays. Serum levels of adrenocorticotropic hormone (ACTH) were measured using enzyme linked immunosorbent assay (ELISA) kits (Huijia Biotechnology, Xiamen, China) following the manufacturer’s instructions. The left condyles of six of the 10 rats in each subgroup were dissected out, frozen in liquid nitrogen and stored for isolation of RNA; the six right condyles of these rats were prepared for hematoxylin and eosin (H&E) staining and immunohistochemistry. The detached condyles were fixed with 10% paraformaldehyde overnight at 4°C and decalcified for 7 days with formalin/nitric acid solution (40% formaldehyde 5 ml, concentrated nitric acid 10 ml and distilled water 85 ml). After dehydration and embedding in paraffin, 5 μm serial sections were cut on the sagittal plane for H&E staining and immunohistochemistry. The entire cartilage of the other four rats in each group was dissected out and used for ELISA.
H&E staining and immunohistochemistry
H&E and immunohistochemical staining were used for histologic assessment. Deparaffinized sections were incubated in Harris hematoxylin (0.75% w/v) for 12 min, then immersed in acid alcohol for 30 s and in Scott’s Tap Water for 2 min, and finally stained with 1% (w/v) aqueous eosin for 5 min. The sections were washed with running tap water before and after each solution, dehydrated in serial alcohol and mounted with gum. A standard three-step avidin-biotin complex immunohistochemical staining protocol was performed. The primary antibodies were anti-OPG (sc-8468; 1:75 dilution) and anti-RANKL (sc-7628; 1:75 dilution), obtained commercially from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For negative-control slides, non-immune goat serum was substituted for the primary antibody.
RNA isolation, reverse transcription and real-time quantitative polymerase chain reaction (RT-qPCR)
Samples were immersed in liquid nitrogen for 10 min and broken into pieces with a non-enzymatic masher. Total RNA was isolated with TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) and then homogenized in a gentleMACSTM Dissociator (Miltenyl Biotech, BergischGladbach, Germany). For RT-qPCR, we used an Ultra SYBR Two Step RT-qPCR Kit (CW Biotech, Beijing, China) following the manufacturer’s instructions. Reactions were performed in 20 μl volumes of a solution containing 10 μl mix, 0.4 μl ROX, 2 μl cDNA, 6 μl dd H2O and 1.6 μl primers (Takara, Dalian, China). Primer sequences were as follows: IL-1β: forward 5-CTCGT GCTGT CTGAC CCAT-3 and reverse 5-CAAAC CGCTT TTCCA TCTTC-3; TNF-α: forward 5-AGGCG CTCCC CAAAA AGATG-3 and reverse 5-TGGCG GAGAG GAGGC TGACT-3; OPG: forward 5-GCAAC CGCAC CCACA AC-3 and reverse 5-GAACC CATCC GGACA TCTTT-3; RANKL: forward 5-GAGCG CAGAT GGATC CTAAC AGAA-3 and reverse 5-CTTTG CACGG CCCCT TGAA-3; and GAPDH: forward 5-CACGG CAAGT TCAAC GGCAC A-3 and reverse 5-AGCGG AAGGG GCGGA GATG-3. Assays were performed on a RT-qPCR system (ABI 7500; Applied Biosystems, Foster City, CA, USA) and analyzed using ABI 7500 software. All RT-qPCR reactions were performed in triplicate. Relative gene levels were calculated using the 2−ΔΔCt method.
ELISA
Samples were immersed in liquid nitrogen for 10 min, broken into pieces with a masher and then mixed with 10-fold phosphate buffered saline. Cells were disrupted using a Soniprep 150 Plus ultrasound generator (MSE Ltd, London, UK) at 14 μm for 30 s. After centrifugation at 12,000 rpm for 10 min at 4°C, IL-1β and TNF-α levels were measured by ELISA (FangCheng Biotech Co. Ltd, Beijing, China) according to the manufacturer’s instructions. Absorbance was read at 450 nm. Semiquantification was performed using standard curves.
Statistical analysis
Quantitative analysis was not performed on the immunohistochemistry findings, which were recorded as the location of immunopositive cells. RT-qPCR and ELISA results were compared by two-factor analysis of variance with full interaction using SPSS 17.0 software (IBM, Armonk, NY, USA). When significant differences were found, the Student-Newman-Keuls post hoc test was used. p values were considered statistically significant when less than 0.05.
Results
Serum ACTH level
As shown in Table 1, the concentration of serum ACTH in the SD1 subgroup was significantly higher than that in the CC group (P < 0.05) but not significantly different from that in the TC group (P > 0.05). Serum ACTH in the SD3, SD5, SD7 and SD9 subgroups was significantly higher than that in both the CC group and the TC group (P < 0.01). There was no significant difference between the TC subgroups and the CC subgroups at any time point (P > 0.05).
Table 1.
Serum levels of ACTH in each group (n = 10, ng/ml, x̅ ± SEM)
| CC group | SD group | TC group | |
|---|---|---|---|
| 1 d | 10.37 ± 0.96 | 12.36 ± 0.94* | 11.35 ± 0.42 |
| 3 d | 12.81 ± 0.91 | 19.58 ± 0.93*,# | 12.14 ± 1.28 |
| 5 d | 11.71 ± 1.54 | 24.2 ± 2.67*,# | 12.02 ± 1.46 |
| 7 d | 12.01 ± 0.91 | 21.52 ± 1.63*,# | 12.18 ± 1.97 |
| 9 d | 12.36 ± 0.17 | 19.26 ± 0.96*,# | 12.31 ± 1.17 |
Data were expressed as “mean ± SEM” and analyzed from each group (n = 10). CC: cage control, SD: sleep deprivation, TC: tank control.
refers to P < 0.05, significantly different compared with time matched CC group;
refers to P < 0.05, significantly different compared with time matched TC group.
H&E staining of synovial tissues
The surface of the condylar cartilage in each subgroup was smooth and intact, with no obvious histopathologic changes (Figure 1). As shown in Figure 2, compared with the CC group (Figure 2A), edema of the synovial membranes and neovascularization were observed in the synovial tissues of rats in the SD3 subgroup (Figure 2B). Greater proliferation of small blood vessels was found in the synovial membranes of the SD5 subgroup (Figure 2C). Greater synovitis and intercellular edema with significant dilatation of small blood vessels were seen in the SD9 subgroup (Figure 2D).
Figure 1.

The modified multiple platform method.Sleep deprivation was achieved by depriving the rats of paradoxical sleep. A. Photos of circular narrow platforms with rats inside. B. Photos of a grid floor with rats inside. The tank is filled with water up to approximately 1 cm from the surface of the platform or grid.
Figure 2.
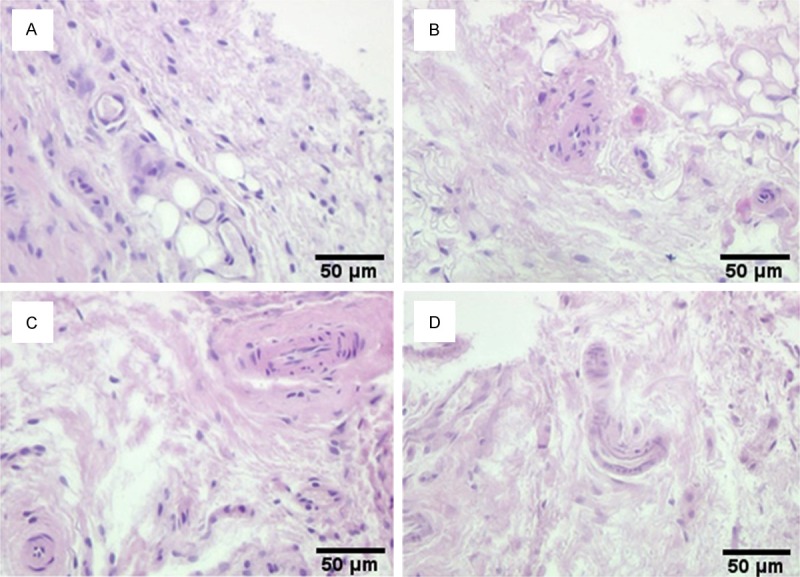
Synovial tissues of rat temporomandibular joint stained with hematoxylin and eosin. A. Synovial tissues of control group rats: The surface of the condylar cartilage in each subgroup was smooth and intact, with no obvious histopathologic changes. B. Synovial tissues of SD group rats at Day 3: edema of the synovial membranes and neovascularization were observed in the synovial tissues of rats in the SD3 subgroup. C. Synovial tissues of SD group rats at Day 5: Greater proliferation of small blood vessels was found in the synovial membranes of the SD5 subgroup. D. Synovial tissues of SD group rats at Day 9: Greater synovitis and intercellular edema with significant dilatation of small blood vessels were seen in the SD9 subgroup. (Original magnification × 100; scale bar = 50 μm).
Expression of cytokines in condylar cartilage
As shown in Figure 3, compared with those in the time-matched TC subgroups, mRNA levels of IL-1β and TNF-α were significantly increased in the SD subgroups at day 5 (D5), D7 and D9 after the start of SD (all P < 0.05) (Figure 3A, 3B), with peaks at D7. There were no differences between time points in the CC and TC groups (all P > 0.05).
Figure 3.
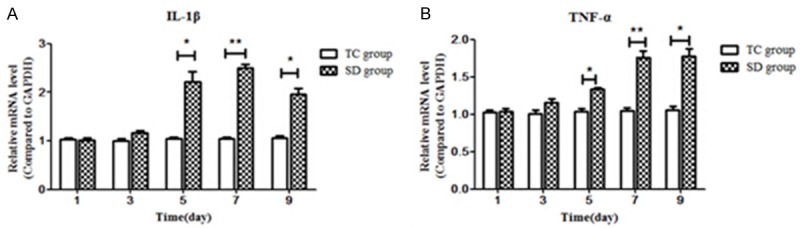
Comparison of the mRNA expression of IL-1β (A) and TNF-α (B) in the condylar cartilage between the SD and TC groups. Compared with those in the time-matched TC subgroups, mRNA levels of IL-1β and TNF-α were significantly increased in the SD subgroups at day 5 (D5), D7 and D9 after the start of SD (all P < 0.05), with peaks at D7. Data were expressed as “mean ± SEM” and analyzed from each group. SD: sleep deprivation, TC: Tank control. *refers to P < 0.05, and **refers to P < 0.01, versus time matched CC groups.
Bone metabolism in cartilage
mRNA levels of OPG increased significantly with experimentally induced SD compared with the time-matched TC subgroups at D5 and D9 (both P < 0.05) (Figure 4A). RANKL was significantly increased compared with the time-matched TC subgroups at D5, D7 and D9 (all P < 0.05) (Figure 4B). The RANKL/OPG mRNA ratio was also significantly increased at D5, D7 and D9 compared with the TC subgroups (all P < 0.05) (Figure 4C).
Figure 4.
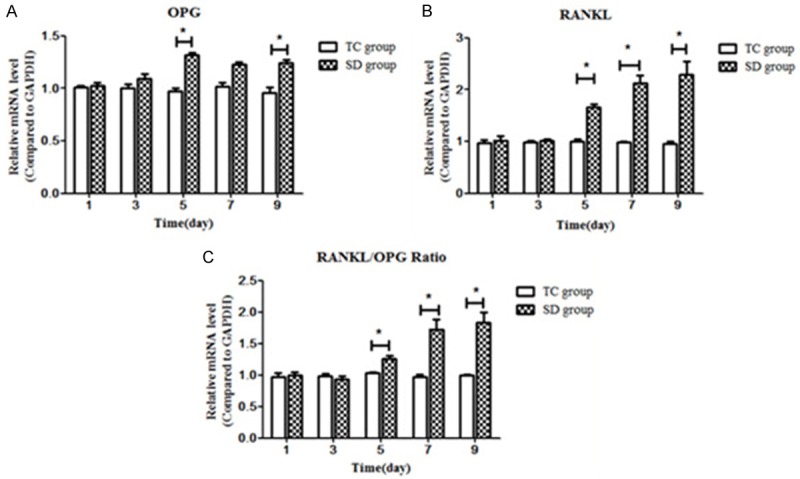
Comparison of mRNA level of Bone metabolism related cytokines in cartilage. A. mRNA levels of osteoprotegerin (OPG) differed significantly among the SD5 and SD9 subgroups (P < 0.05) but no significant difference was found among the TC subgroups (P > 0.05). B. There were significant differences in the mRNA levels of receptor activator of nuclear factor kappa B ligand (RANKL) among the subgroups of the SD group (P < 0.01) but not among the subgroups of the TC group (P > 0.05). C. Expression of RANKL tended to increase later in SD, and the RANKL/OPG ratio showed an increasing trend with prolonged SD. Data were expressed as “mean ± SEM” and analyzed from each group. SD: sleep deprivation, TC: tank control. *refers to P < 0.05, and **refers to P < 0.01, versus time matched CC groups.
Immunohistochemistry showed that, compared with the TC group (Figure 5A, 5C), significantly more cells were immunopositive for OPG and RANKL in the SD group (Figure 5C, 5D); these cells were mainly located in the hypertrophic layer of chondrocytes. ELISA showed that the concentrations of OPG and RANKL increased with SD and were significantly higher than those in the time-matched TC subgroups at D5, D7 and D9 (all P < 0.05) (Figure 5E, 5F).
Figure 5.
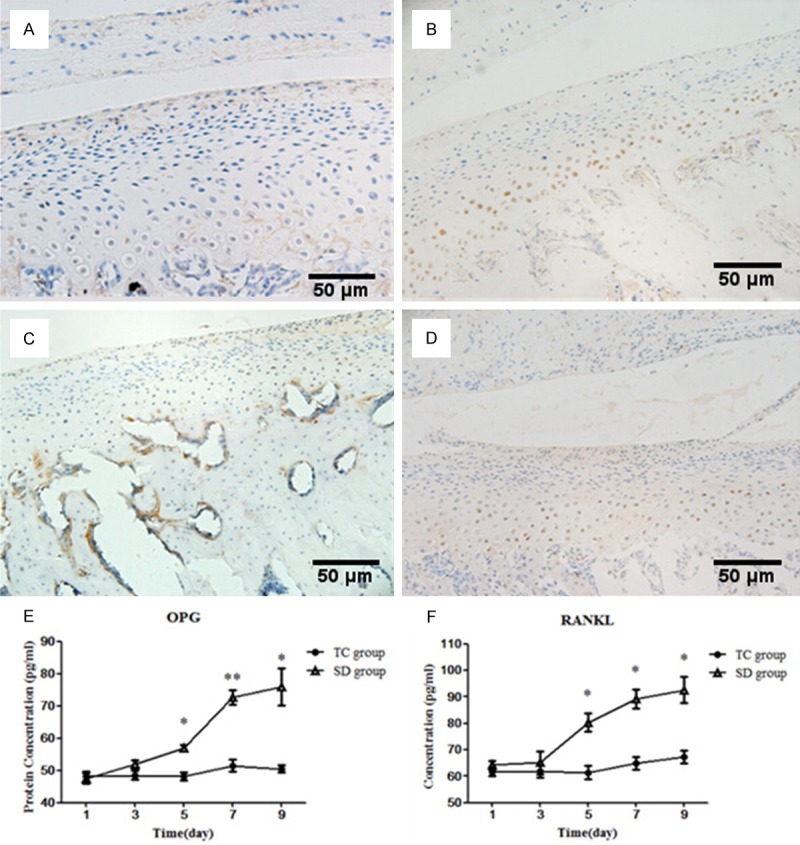
Protein expression of OPG and RANKL in cartilage. (A-D) Serial sections of condylar cartilage stained by immunostaining for OPG (A and C) and RANKL (B and D). Immunohistochemistry showed that, compared with the TC group (A and C), significantly more cells were immunopositive for OPG and RANKL in the SD group (B and D); these cells were mainly located in the hypertrophic layer of chondrocytes. (E and F) ELISA showed that the concentrations of OPG (E) and RANKL (F) increased with SD and were significantly higher than those in the time-matched TC subgroups at D5, D7 and D9 (all P < 0.05). Data were expressed as “mean ± SEM” and analyzed from each group. SD: sleep deprivation, TC: tank control. *refers to P < 0.05, and **refers to P < 0.01, versus time matched CC groups. (Original magnification × 100; scale bar = 50 μm).
Discussion
Using an effective animal model of SD stress, this study observed a direct effect of long term SD on TMJ metabolic activity. Our results indicate that SD induces the expression of cytokines (IL-1β and TNF-α) and bone metabolism factors (OPG and RANKL) in the TMJ, suggesting that experimentally induced SD may lead to pathologic changes in the TMJ.
The cytokines IL-1 and TNF-α serve as initiating factors and effectors in immune function and the regulation of inflammatory processes, and their coordination can inhibit proteoglycan production by chondrocytes [12]. In addition, IL-1 plays an important role in the destruction of joint cartilage. Regulation of bone metabolism occurs through the classical OPG/RANK/RANKL pathway, the direct and indirect functions of which are influenced by many factors, including cytokines (e.g. IL-1β, TNF-α) and hormones. OPG plays an important role in inhibiting osteoclastogenesis by blocking the effects of RANKL, which activates osteoclastogenesis [13,14]. In the present study, we found that the OPG/RANKL ratio and IL-1 and TNF-α expression initially exhibited an increasing trend and then decreased during prolonged SD.
Other studies have found that an increase in the permeability of blood vessels may cause the release of many inflammatory factors (including IL-1β and TNF-α) into the articular cavity, which impairs the lubrication and nutrition of the cartilage and cartilage disc and leads to a positive feedback effect that can result in the development of arthropathy [15]. In addition, there is an important relationship between the expression level of vascular endothelial growth factor (VEGF) and synovial fluid changes [16]. In the present study, H&E staining of the TMJ revealed vessel hyperplasia in the synovial membrane, suggesting that SD induced these changes.
Studies have shown that IL-1β stimulates the production of factors including matrix metalloproteinase (MMP)-1, MMP-13 and nitric oxide [17], and prove that the ERK1/2, p38 [18], MAPK and JNK signaling pathways and the NF-κB and activator protein 1 transcription factors [19] participate in IL-1-mediated increased expression of downstream proteolytic enzymes, including MMPs. Binding of RANK to RANKL stimulates the differentiation and signaling pathways of osteoclastogenesis and odontoclastogenesis [20]. Furthermore, the RANK-RANKL axis is a key pathway in the activation of seven signaling pathways (JNK, NF-κB, src, c-myc, calcineurin, ERK and p38) [21-23]. These pathways influence one another and have an impact on cartilage metabolism though a complicated signaling pathway network.
Conclusions
In this exploratory study of the relationship between SD and TMD, we investigated factors in the early and middle stages of the OPG/RANK/RANKL pathway (IL-1, VEGF, OPG and RANKL) and bone metabolism factors (MMP-13). Our results indicate that SD induces alterations in the target genes of the OPG/RANK/RANKL pathway and plays an important role in the occurrence and development of TMD.
Acknowledgements
The authors are deeply thankful to Dr. Haiying Chen of Liaocheng People’s Hospital of China (Liaocheng, China) for her earnest guidance. This work was supported by grant from National Science Foundation of China (No. 81400573; 61471384) and Lu Cai Jiao Zhi (2013) 171.
Disclosure of conflict of interest
None.
References
- 1.Moulton RE. Emotional factors in non-organic temporomandibular joint pain. Dent Clin North Am. 1966:609–620. [PubMed] [Google Scholar]
- 2.Kafas P, Leeson R. Assessment of pain in temporomandibular disorders: the bio-psychosocial complexity. Int J Oral MaxillofacSurg. 2006;35:145–149. doi: 10.1016/j.ijom.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Glaros AG, Williams K, Lausten L. The role of parafunctions, emotions and stress in predicting facial pain. J Am Dent Assoc. 2005;136:451–458. doi: 10.14219/jada.archive.2005.0200. [DOI] [PubMed] [Google Scholar]
- 4.List T, Wahlund K, Larsson B. Psychosocial functioning and dental factors in adolescents with temporomandibular disorders: a case-control study. J Orofac Pain. 2001;15:218–227. [PubMed] [Google Scholar]
- 5.Smith MT, Wickwire EM, Grace EG, Edwards RR, Buenaver LF, Peterson S, Klick B, Haythornthwaite JA. Sleep disorders and their association with laboratory pain sensitivity in temporomandibular joint disorder. Sleep. 2009;32:779–790. doi: 10.1093/sleep/32.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yatani H, Studts J, Cordova M, Carlson CR, Okeson JP. Comparison of sleep quality and clinical and psychologic characteristics in patients with temporomandibular disorders. J Orofac Pain. 2002;16:221–228. [PubMed] [Google Scholar]
- 7.Everson CA. Sustained sleep deprivation impairs host defense. Am J Physiol. 1993;265:R1148–1154. doi: 10.1152/ajpregu.1993.265.5.R1148. [DOI] [PubMed] [Google Scholar]
- 8.How JM, Foo SC, Low E, Wong TM, Vijayan A, Siew MG, Kanapathy R. Effects of sleep deprivation on performance of Naval seamen: I. Total sleep deprivation on performance. Ann Acad Med Singapore. 1994;23:669–675. [PubMed] [Google Scholar]
- 9.Suchecki D, Duarte Palma B, Tufik S. Sleep rebound in animals deprived of paradoxical sleep by the modified multiple platform method. Brain Res. 2000;875:14–22. doi: 10.1016/s0006-8993(00)02531-2. [DOI] [PubMed] [Google Scholar]
- 10.Suchecki D, Tufik S. Social stability attenuates the stress in the modified multiple platform method for paradoxical sleep deprivation in the rat. Physiol Behav. 2000;68:309–316. doi: 10.1016/s0031-9384(99)00181-x. [DOI] [PubMed] [Google Scholar]
- 11.Wu G, Chen L, Wei G, Li Y, Zhu G, Zhao Z, Huang F. Effects of sleep deprivation on pain-related factors in the temporomandibular joint. J Surg Res. 2014;192:103–11. doi: 10.1016/j.jss.2014.05.035. [DOI] [PubMed] [Google Scholar]
- 12.Arend WP, Dayer JM. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum. 1990;33:305–315. doi: 10.1002/art.1780330302. [DOI] [PubMed] [Google Scholar]
- 13.Aida Y, Maeno M, Suzuki N, Shiratsuchi H, Motohashi M, Matsumura H. The effect of IL-1beta on the expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human chondrocytes. Life Sci. 2005;77:3210–3221. doi: 10.1016/j.lfs.2005.05.052. [DOI] [PubMed] [Google Scholar]
- 14.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 15.Kubota E, Kubota T, Matsumoto J, Shibata T, Murakami KI. Synovial fluid cytokines and proteinases as markers of temporomandibular joint disease. J Oral MaxillofacSurg. 1998;56:192–198. doi: 10.1016/s0278-2391(98)90868-0. [DOI] [PubMed] [Google Scholar]
- 16.Sato J, Segami N, Nishimura M, Kaneyama K, Demura N, Yoshimura H. Relation between the expression of vascular endothelial growth factor in synovial tissues and the extent of joint effusion seen on magnetic resonance imaging in patients with internal derangement of the temporomandibular joint. Br J Oral Maxillofac Surg. 2003;41:88–94. doi: 10.1016/s0266-4356(02)00295-4. [DOI] [PubMed] [Google Scholar]
- 17.Schwab W, Schulze-Tanzil G, Mobasheri A, Dressler J, Kotzsch M, Shakibaei M. Interleukin-1beta-induced expression of the urokinase-type plasminogen activator receptor and its co-localization with MMPs in human articular chondrocytes. Histol Histopathol. 2004;19:105–112. doi: 10.14670/HH-19.105. [DOI] [PubMed] [Google Scholar]
- 18.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 19.Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291:124–130. [PubMed] [Google Scholar]
- 20.Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene. 1998;17:173–178. doi: 10.1038/sj.onc.1201941. [DOI] [PubMed] [Google Scholar]
- 21.Huang TS, Lee SC, Lin JK. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1991;88:5292–5296. doi: 10.1073/pnas.88.12.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh S, Aggarwal BB. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected] . J Biol Chem. 1995;270:24995–25000. doi: 10.1074/jbc.270.42.24995. [DOI] [PubMed] [Google Scholar]
- 23.Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, Sartor RB. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol. 1999;163:3474–3483. [PubMed] [Google Scholar]
- 24.Gepstein A, Shapiro S, Arbel G, Lahat N, Livne E. Expression of matrix metalloproteinases in articular cartilage of temporomandibular and knee joints of mice during growth, maturation, and aging. Arthritis Rheum. 2002;46:3240–3250. doi: 10.1002/art.10690. [DOI] [PubMed] [Google Scholar]