

Abstract
Background
Arginine is a high-value product, especially for the pharmaceutical industry. Growing demand for environmental-friendly and traceable products have stressed the need for microbial production of this amino acid. Therefore, the aim of this study was to improve arginine production in Escherichia coli by metabolic engineering and to establish a fermentation process in 1-L bioreactor scale to evaluate the different mutants.
Results
Firstly, argR (encoding an arginine responsive repressor protein), speC, speF (encoding ornithine decarboxylases) and adiA (encoding an arginine decarboxylase) were knocked out and the feedback-resistant argA214 or argA215 were introduced into the strain. Three glutamate independent mutants were assessed in bioreactors. Unlike the parent strain, which did not excrete any arginine during glucose fermentation, the constructs produced between 1.94 and 3.03 g/L arginine. Next, wild type argA was deleted and the gene copy number of argA214 was raised, resulting in a slight increase in arginine production (4.11 g/L) but causing most of the carbon flow to be redirected toward acetate. The V216A mutation in argP (transcriptional regulator of argO, which encodes for an arginine exporter) was identified as a potential candidate for improved arginine production. The combination of multicopy of argP216 or argO and argA214 led to nearly 2-fold and 3-fold increase in arginine production, respectively, and a reduction of acetate formation.
Conclusions
In this study, E. coli was successfully engineered for enhanced arginine production. The ∆adiA, ∆speC, ∆speF, ∆argR, ∆argA mutant with high gene copy number of argA214 and argO produced 11.64 g/L of arginine in batch fermentation, thereby demonstrating the potential of E. coli as an industrial producer of arginine.
Keywords: Escherichia coli, L-arginine, Metabolic engineering, Fermentation
Background
L-arginine has gained considerable interest from the pharmaceutical industry, notably because it is a precursor to nitric oxide, a blood vessel dilator [1]. Arginine is also commonly used in cosmetics, dental care products, dietary supplements and flavoring agents. It has also recently been shown that arginine can be used as an efficient nitrogen source and a potential alternative to inorganic nitrogen in plant fertilizers [2].
Given the wide utilization of arginine, there is a significant industrial demand for this amino acid, especially from sources that can guarantee an environmentally and economically sustainable production. Biotechnology processes, encompassing microbial biosynthesis, for the production of arginine from renewable resources need to be further explored to enable an industrial vital production. L-arginine can be synthesized de novo from L-glutamate by a large group of microorganisms. Members of the genus Corynebacterium are well-known L-glutamic acid producers [3] and are widely used for commercial production of amino acids [4], therefore they have been the organism of choice for microbial L-arginine production [5]. Recently, Park et al. [6] engineered a C. glutamicum strain able to produce 92.5 g/L L-arginine during fed-batch fermentation in 5 L bioreactors. However, other organisms can also be considered as arginine producers; for a review on the production of L-arginine by different microorganisms, including members of the Corynebacterium family, see [7]. Favorable traits of Escherichia coli, such as fast growth in inexpensive media, robust organism for industrial processes, and its well characterized metabolism and available molecular tools for genetic engineering, render it an organism of interest for the production of arginine. A number of patents regarding arginine production by E. coli strains exist (e.g. [8-10]). The inventors claim to have obtained up to 19.3 g/L in batch-fermentations with an acetate utilizing mutant derived from an arginine producing E. coli strain [8] and 11.6 g/L in 2 mL test tubes by attenuating the expression of genes encoding the lysine/arginine/ornithine ABC transporter [9]. However, the literature concerning E. coli and arginine biosynthesis has mainly been focused on the genetics and regulation systems rather than the production. To the best of our knowledge, no study has been published on the fermentative production of arginine by E. coli.
In E. coli, arginine biosynthesis follows a linear pathway starting from the precursors glutamate and acetyl-CoA (Figure 1). The first enzyme in the biosynthetic pathway, N-acetylglutamate synthase (NAGS) encoded by the argA gene, is inhibited by arginine through feedback inhibition [11]. In addition, the arginine responsive repressor protein ArgR, encoded by the argR gene, negatively regulates transcription of arginine biosynthesis genes [12]. E. coli also possesses machineries for the export of some amino acids, including arginine. The arginine export pump ArgO, encoded by the argO gene, is transcriptionally regulated by ArgP [13-15]. The latter is responsive to intracellular arginine levels and activates the transcription of argO accordingly [15-17]. In addition, E. coli has degradative pathways for both L-arginine and its precursor L-ornithine. Two ornithine decarboxylases (encoded by the speC and speF genes) are responsible for the conversion of ornithine to putrescine [18,19], whereas arginine is first degraded into agmatine by an arginine decarboxylase (encoded by adiA), which is subsequently converted to putrescine and urea [18]. In summary, the biosynthesis pathway of arginine is constrained by several layers of metabolic and transcriptional regulations resulting in a complex network to engineer for arginine overproduction. Arginine overproducing E. coli strains have been classically obtained by selection of canavanine-resistant mutants [20]. Canavanine, an arginine analogue, inhibits growth by competing for arginine in protein synthesis [21]. The rationale for using this selection system is that mutants resistant to canavanine are likely to be derepressed for arginine synthesis, as over-production of arginine will release the inhibition caused by canavanine. When characterized, these mutants have subsequently been found to carry mutations in argA, argR and in some instances argP [12,22,23]. Not surprisingly, mutations in argA commonly resulted in an ArgA feedback resistant to arginine, which led some workers to derive further mutants by directed selection [24]. Similarly, mutations in argP resulted in ArgP acting in a constitutive manner, independent of the presence of arginine [14,17].
Figure 1.
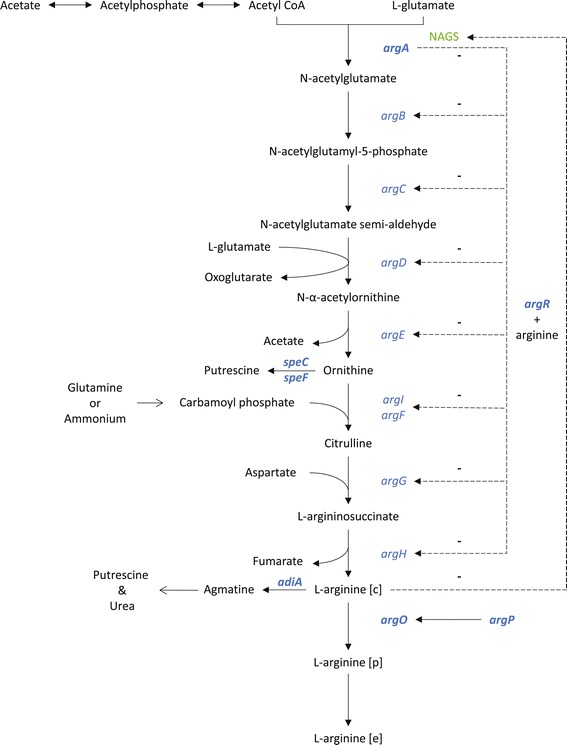
Arginine biosynthesis pathway in Escherichia coli . NAGS: N-acetylglutamate synthase, −: inhibition/negative regulation, [c]: cytoplasm, [p]: periplasm, [e]: extracellular. Targeted genes are indicated in bold.
In this study we used both rational design based on known regulatory and metabolic information and selection procedures aiming for E. coli strains with enhanced production of arginine. Experimental validation of the engineered strains was carried out in 1-L fermenters under controlled conditions. We examined first the impact of deleting the speC, speF, adiA and argR genes and introducing a feedback-resistant argA. Then, the wild type argA was knocked-out and the feedback-resistant variant overexpressed. Finally, arginine production was significantly increased by overexpression of either argP or argO.
Results & discussion
Effect of introduction of feedback resistant variants of argA and selection of glutamate producing strains
In the first rate-limiting step of the arginine synthesis, NAGS, encoded by argA, catalyzes the acetylation of glutamate. To block the feedback inhibition of NAGS the plasmids pKH15 and pKH19, derived from the ASKA- plasmid pCA24N, were transferred into C600+Δ4 (see Table 1) to over-express the feedback resistant variants of argA (H15Y for argA214 and Y19C for argA215) under the control of an IPTG-inducible promoter [25] (Table 1). The strain C600+Δ4 carrying either plasmid pKH15 or pKH19 could not be grown on M9 minimal media containing IPTG without exogenous glutamate supplementation and only weak growth was observed on the same medium when both glutamate and IPTG were absent. This suggested that over-expression of the feedback resistant argA in this strain resulted in glutamate starvation. To overcome this limitation, spontaneous mutants able to grow in the absence of glutamate were selected by plating washed and diluted cell cultures on M9 medium supplemented with IPTG without glutamate. Twelve colonies were picked at random and screened for arginine production based on the bioassay method. The three clones with the highest arginine production were chosen for subsequent work (SJB001, 003 and 004).
Table 1.
Plasmids and strains used in this study
| Plasmid/Strain | Relevant characteristics/genotype | Source/Reference |
|---|---|---|
| Plasmids | ||
| pKH15 | pCA24N (clone JW2786), argA214 | ASKA- collection [26], this work |
| pKH19 | pCA24N (clone JW2786), argA215 | ASKA- collection [26], this work |
| pTrc99a | Amp-R, lacIq | Lab stock |
| pTrcArgP216 | pTrc99a with a mutant argP216 allele | This work |
| pJB044 | pBR322 derived, infA, rop- | Lab stock [27] |
| pJB044argA15 | pJB044 with argA214 downstream of infA | This work |
| pJB044p1argA15 | Same as pJB044argA15 but with rrsBp1 | This work |
| pArgObla | Arginine bio-sensor plasmid with bla (Amp-R) under transcriptional control of argOp | This work |
| pArgObla10C | Arginine bio-sensor plasmid with bla (Amp-R) under transcriptional control of argOp with mutation in RBS | This work |
| pTrcArgP216 | pTrc99a with argP216 cloned under transcriptional control of trc promoter | This work |
| pJB044argAO | pJB044argA15 with argO cloned downstream of argA214 | This work |
| pJB044argAP | pJB044argA15 with argp216 cloned downstream of argA214 | This work |
| Strains | ||
| E. coli K-12 C600 | thr-,1 leuB6, thi-1, lacY1, glnV44, supE44, rfbD1, mcrA1 | Lab stock [28] |
| MG1655 | ilvG-, rfb-50, rph-1 | Lab stock |
| C600+ | Same as C600 but thr + , leu + | This work |
| C600+∆4 | Same as C600+ but ∆adiA, ∆speC, ∆speF, ∆argR | This work |
| pTrcArgP216/C600+∆4 | C600+ ∆4 with a mutant argP216 allele | This work |
| JW3932 | Auxotrophic for arginine ∆argH | [29] |
| SJB001 | Glutamate independent mutant of C600+∆4 with pKH15 (clone 2) | This work |
| SJB003 | Glutamate independent mutant of C600+∆4 with pKH19 (clone 2) | This work |
| SJB004 | Glutamate independent mutant of C600+∆4 with pKH19 (clone 4) | This work |
| SJB003A | SJB003 but no plasmid | This work |
| SJB005 | SJB003A but ∆argA | This work |
| SJB015 | SJB005 with pJB044argA15 | This work |
| SJB025 | SJB005 with pJB044p1argA15 | This work |
| SJB006 | Arginine producing mutant of C600+∆4 from biosensor selection, argP216 | This work |
| SJB007 | Derivative of SJB006 from second round biosensor selection | This work |
| SJB009 | SJB005 with pJB044argAO | This work |
| SJB010 | SJB005 with pJB044argAP | This work |
Although these three strains were constructed in the same way, fermentations revealed very different growth behavior and arginine production abilities (Table 2). Indeed, SJB003 produced more arginine, with a productivity of 0.14 g/L/h and a final arginine concentration (3.03 g/L) significantly higher than that of the other similar mutants. The higher arginine producing capability of SJB003 compared to that of SJB001and 004 indicates that this strain had acquired beneficial mutations during growth under glutamate limitation. SJB003 was therefore chosen as a chassis for further genetic manipulation, although its beneficial mutations were not characterized.
Table 2.
Comparison of the performances of the different E. coli strains for arginine production by fermentation
| E. coli strain | Yields | |||||||
|---|---|---|---|---|---|---|---|---|
| μ (1/h) | Y X/S (g dcw/g glc) | Y P/S (g arg/g glc) | Y P/X (g arg/g dcw) | Q P (g/L/h) | Arginine (g/L) | Acetic acid (g/L) | Ac/Arg (mol ac/mol arg) | |
| SJB001 | 0.14 ± 0.02 | 0.26 ± 0.02 | 0.03 ± 0.00 | 0.10 ± 0.00 | 0.08 ± 0.00 | 1.94 ± 0.12 | 5.57 ± 0.11 | 8.5 ± 0.36 |
| SJB003 | 0.14 ± 0.00 | 0.27 ± 0.01 | 0.04 ± 0.01 | 0.15 ± 0.03 | 0.14 ± 0.02 | 3.03 ± 0.59 | 6.12 ± 1.14 | 6.43 ± 2.36 |
| SJB004 | 0.13 ± 0.02 | 0.27 ± 0.01 | 0.03 ± 0.00 | 0.11 ± 0.00 | 0.09 ± 0.00 | 2.04 ± 0.00 | 6.15 ± 0.24 | 8.90 ± 0.36 |
| SJB015 | 0.04 ± 0.00 | 0.11 ± 0.00 | 0.07 ± 0.00 | 0.50 ± 0.02 | 0.08 ± 0.00 | 4.11 ± 0.49 | 15.85 ± 1.60 | 11.37 ± 0.22 |
| SJB006 | 0.17 ± 0.01 | 0.35 ± 0.01 | 0.03 ± 0.00 | 0.09 ± 0.00 | 0.11 ± 0.00 | 2.03 ± 0.05 | 6.24 ± 0.45 | 9.07 ± 0.42 |
| SJB007 | 0.16 ± 0.02 | 0.36 ± 0.01 | 0.04 ± 0.00 | 0.11 ± 0.00 | 0.14 ± 0.00 | 2.74 ± 0.21 | 5.31 ± 1.73 | 5.90 ± 2.31 |
| SJB009 | 0.04 ± 0.00 | 0.12 ± 0.01 | 0.17 ± 0.01 | 1.18 ± 0.01 | 0.24 ± 0.01 | 11.64 ± 0.75 | 14.56 ± 0.93 | 3.72 ± 0.48 |
| SJB010 | 0.09 ± 0.00 | 0.25 ± 0.01 | 0.11 ± 0.00 | 0.44 ± 0.02 | 0.29 ± 0.01 | 7.95 ± 0.04 | 3.14 ± 0.87 | 1.17 ± 0.33 |
Aerobic batch fermentations were performed in 1 L bioreactors at 32°C and pH 7; the initial glucose concentration was 70 g/L. μ, growth rate; YX/S, biomass yield vs. glucose; YP/S, product yield vs. glucose; YP/X, product yield vs. cell mass; QP, volumetric productivity. Results are given as means ± standard deviations.
Control fermentations with the parent strain C600+ were also performed. This strain did not yield any arginine (data not shown), which confirmed that the arginine productions displayed by the other strains are the result of their genetic modifications.
Effect of overexpression of a feedback resistant argA on arginine production
To avoid the use of IPTG in an industrial process, it is of interest to place the feedback resistant argA gene under a constitutive promoter. First the SJB003 was cured of the pKH19 plasmid, harboring argA215, by repeated streaking on Luria Agar (LA) medium without antibiotic, giving rise to SJB003A. The argA214 allele was chosen as the argA variant to be introduced in the strain since we found this allele to be slightly better for arginine productivity in preliminary shake flask experiments (data not shown). To avoid potential recombination with the new argA214 plasmid, the chromosomal copy of the wild type argA gene was deleted in the SJB003A strain, resulting in SJB005.
The feedback resistant argA214 was cloned into a high copy number plasmid pJB044 downstream of the infA gene encoding the translation initiation factor IF1. pJB044 carries a tetracycline resistance gene that can be removed by homologous recombination due to the presence of direct repeats flanking the gene, as previously described [27]. The argA214 gene was placed downstream of the ribosome binding site (RBS) (AGGAGG) either with or without a strong constitutive rRNA promoter (rrsBp1) upstream (Table 1). The argA start codon GUG was changed by site-directed mutagenesis to the more efficient AUG codon in both constructs, termed pJB044argA15 and pJB044p1argA15. Strain SJB005 was the host of the pJB044 derived plasmids, resulting in the two IPTG-independent strains SJB0015 and SJB025 that differ only by the absence or presence of a strong rRNA promoter upstream of the argA214 gene respectively.
When cultivated in bioreactors, SJB015 displayed a slightly improved arginine production compared to the previous constructs (Table 2). In particular, YP/x was relatively high (0.50 g arg/g dcw). However, cell growth was seriously hampered for this strain and SJB015 had the lowest μ and YX/S and the final cell density was lower than that of the other strains (data not shown). Consequently the volumetric productivity of SJB015 was relatively low (0.08 g/L/h). In addition, SJB015 produced high levels of acetate. When a DCW of about 7 g/L was reached (Figure 2d), cell growth stopped, arginine production drastically decreased and the remaining sugar (approximately 30% of the initial glucose) was mainly used for acetate formation (up to 28 g/L).
Figure 2.
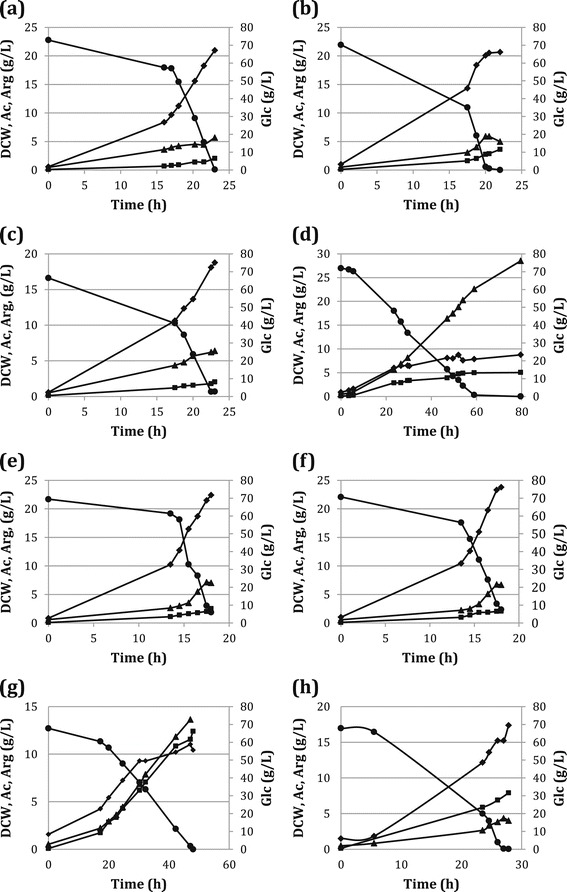
Fermentation profile of (a) SJB001, (b) SJB003, (c) SJB004, (d) SJB015, (e) SJB006, (f) SJB007, (g) SJB009 and (h) SJB010. ● glucose, ▲ acetic acid, ■ arginine and ◆ DCW.
SJB025 exhibited slow growth on both rich (LA) and minimal medium (M9) with the appearance of some large colonies (data not shown). This suggested that the strong promoter driven argA214 was toxic for the strain, with large colonies representing revertants. 50 of these colonies were screened for fast growing mutants with enhanced ability to produce arginine. However, none had retained this capacity (ascertained by the bioassay method) and consequently, neither these clones nor the parental SJB025 were used for further work.
For this strain it is likely that the rate of arginine formation exceeds the capacity of the arginine export system due to the overexpression of argA214 in combination with the presence of a strong promoter upstream of argA214. The resulting accumulation of arginine inside the cell might have a variety of negative effects on cellular processes, which could explain why cell growth was seriously hampered in SJB025 and the ability for enhanced arginine production easily lost. This is consistent with previous reports, where mutants of C. glutamicum with deletion of lysE, encoding an exporter similar to ArgO [30,31], were growth inhibited in the presence of intracellular arginine [30-32]. Growth arrest due to intracellular arginine in ∆argO and ∆argP mutants of E. coli has also been reported [14]. The increased nitrogen flow towards arginine production might also hinder the biosynthesis of other metabolites required for cell growth.
Identification of novel mutations for enhanced arginine production using a biosensor
To complement the above described rational strain improvement strategies, a selection procedure was employed to select novel or previously unidentified mutants with increased arginine production. C600+Δ4 carrying the biosensor plasmid pArgObla10C was used for direct selection and screening of arginine accumulating mutants on M9 plates supplemented with 2, 3 and 4 mg/mL Amp. Only mutants with increased expression of the bla gene, most likely through increased transcription of the argOp promoter, can grow on media with Amp concentration higher than 0.6 mg/mL.
High Amp resistant mutants were randomly chosen from each Amp concentration and assayed for arginine production using the bioassay method. The isolated mutant showing the highest production was cured of the plasmid by repeated streaking on M9 plates without antibiotic (resulting in SJB006). After the biosensor plasmid removal an improved arginine production was retained, indicating that the acquired increased arginine accumulation was due to chromosomal mutations. Chromosomal sequencing of argA and argP genes in this clone revealed wild type argA and a T647C mutation in argP resulting in a valine to alanine mutation in position 216 (V216A).
To assess the effect of the V216A mutation on arginine production the mutant allele argP216 was cloned into a high copy number plasmid downstream of an IPTG inducible promoter (pTrc99a) to give pTrcArgP216. Even without IPTG induction, the arginine accumulation of the strain pTrcArgP216 / C600+Δ4 was equivalent to SJB006, as based on the bioassay method (data not shown). We thus concluded that the increase in arginine accumulation observed in SJB006 was at least partly due to the presence of the argP216 allele.
Selection of mutants with increased arginine accumulation was extended by transforming SJB006 with pArgObla10C anew, and screening on LA plates supplemented with 6, 8 and 10 mg/mL of Amp. Several colonies were assayed for arginine production; the best clone was cured of the biosensor plasmid and used for further work (SJB007). Sequencing of argA and argP showed that SJB007 also carried wild-type argA and no other mutation on the argP gene, other than the V216A mutation present in the parent strain SJB006.
Even with only the wild type argA, SJB006 produced similar amounts of arginine as SJB001 and SJB004 during fermentation (Table 2). Further, the productivity of SJB006 (0.11 g/L/h) was even slightly higher due to a faster growth. SJB007, which results from a second level biosensor selection, displayed increased arginine production compare to its parent SJB006. This demonstrates the potential effects of the mutation V216A carried by these two strains on the argP gene, but also that there might be some additional unknown mutation in SJB007 promoting arginine production.
Effect of co-overexpression of a feedback resistant argA and argP or argO on arginine production
The mutant allele argP216 resulted in increased accumulation of arginine. Amongst other physiological functions in the cells, ArgP also controls the transcription of argO. It was therefore of interest to combine overexpression of each of these two genes with the known feedback resistant argA214 allele.
The plasmid pJB044argAP was constructed such that the argP216 allele was placed downstream of argA214, under the control of the RBS sequence AGGAGG. The plasmid pJB044argAO was constructed by placing an argO ORF with the RBS sequence AGGAGG, downstream of argA214. In addition the inefficient start codon GUG was changed for the canonical AUG. The plasmids pJB044argAO and pJB044argAP were transferred to SJB005, to yield SBJ009 and SJB010 respectively.
Slow growth was also observed in the strains having a gene involved in arginine transport overexpressed in combination with the argA214 allele (Figure 2d, g and h). In particular, SJB009 had almost the same μ and YX/S as SJB015 and also produced significant amounts of acetate. Nevertheless, cells grew to a somewhat higher density and arginine was steadily formed throughout the whole fermentation. Furthermore, SJB009 had the highest arginine production per amount of cells (1.18 g arg/g DCW), 2 to 13-fold that of the other strains. Consequently this strain yielded the highest final arginine titer (11.64 g/L) at a fair production rate (0.24 g/L/h). Also, a low growth associated with a high YP/X means that a large part of the glucose is used for arginine formation. SJB009 therefore showed the highest YP/S (0.17 g arg/g glc) of all strains evaluated. ArgO is directly responsible for the transport of arginine outside the cytoplasm and the high YP/S might be the result of an immediate excretion, enhanced by ArgO, of the large amount of arginine produced, due to argA214.
Interestingly, despite overexpression of the argP gene, responsible for argO transcription, SJB010 had significantly lower product yields than SJB009, yet higher than the other mutants. However, the cells of this strain grew twice as fast as cells of SJB009 and therefore SJB010 had the highest productivity of all strains (0.29 g/L/h). Unlike SJB015 and SJB009, SJB010 did not form high levels of acetate but produced both acetate and succinate (4–5 g/L).
SJB009 and SJB010 are similar to SJB015 except that one of their genes responsible of arginine export has been altered (argO and argP, respectively). This resulted in an important increase in the final arginine concentration (+183% and +93%), productivity (+200% and +262%) and product yield YP/S (+143% and +57%) for SJB009 and SJB010, respectively, compared to SJB015. This positive effect of argO and argP overexpression has previously been observed, showing that E. coli strains carrying multicopy yggA+ (argO) and argPd (S94L mutation) had a greatly increased arginine production as determined from cross-feeding ability on agar plate [14]. Export has been identified as the rate-limiting step for the production of different amino acids when using C. glutamicum [33-35]. Similarly, it seems that the arginine export system plays a major role for the arginine production by E. coli.
Formation of acetate during arginine fermentation
All mutants produced acetate as the main by-product. Acetate is formed during the 5th step of L-arginine biosynthesis from L-glutamate (Figure 1). However, for most strains the ratio of ac:arg produced was higher than 1:1 (Table 2), which means that acetate was also formed via another pathway.
The accumulation of acetate by E. coli, even in aerobic environment, when growing under conditions of high glucose consumption is known as overflow metabolism. It occurs when the rate of glucose consumption is greater than the capacity of the cell to reoxidize the reduced equivalents, i.e. NAD(P)H, generated by glycolysis. Instead of entering the tricarboxylic acid (TCA) cycle, the carbon flux from acetyl-CoA is diverted to acetate, likely to prevent any further NAD(P)H accumulation as only ATP is formed during acetate formation while the TCA cycle generates several reducing equivalents [36,37].
As fermentations were run in batch mode with high initial glucose concentration (70 g/L) overflow metabolism is to be expected. The acetate production depends on the specific glucose uptake rate, with acetate formation occurring only after a certain threshold [36]. SJB001;3;4;6 and 7 indeed produced large amounts of acetate compared to arginine, which allowed them to have a high glucose uptake (0.44 to 0.54 mol glc/mol dcw/h) and a fast growth (μ > 0.13 h−1). SJB009 and SJB010 however had a considerably lower ac/arg ratio, i.e. 3.72 and 1.17 mol/mol, respectively, compared to at least 5.9 mol/mol for the other strains. The glucose uptake was also reduced (0.33 and 0.36 mol glc/mol dcw/h) as well as the growth (μ < 0.09 h−1). It is possible that the redirection of carbon and nitrogen toward arginine results in a shortage of other essential amino acids, thereby limiting the growth and the need for fast glucose assimilation. This could also be because the carbon flow from acetyl-CoA is forced toward arginine biosynthesis by the overexpressed argA214, thereby limiting the formation of acetate from acetyl-CoA.
However SJB015 had the highest ac/arg ratio (11.37 mol/mol) despite having a low specific glucose uptake (0.36 mol glc/mol dcw/h). This strain produced 15.85 g/L of acetate, which is comparable to the 14.56 g/L produced by SJB009. It is therefore likely that a large part of the acetate produced by SJB015 comes from the increased carbon flux through the arginine pathway, but that mainly acetate was excreted while arginine accumulated inside the cell due to the export limitation.
Conclusion
We reported the development E. coli strains overproducing arginine, by targeting genes regulating repression of arginine biosynthesis and competing degradation pathways in addition to amplification of genes for N-acetylglutamate formation and arginine export. The two final strains obtained (SJB009 and SJB010) had the highest arginine yield (1.18 and 0.44 g arg/g glc, respectively) and productivity (0.24 and 0.29 g arg/L/h, respectively) and will be used for further genetic improvement and/or process optimization. The fermentation process developed for the comparison of the different constructs needs to be further optimized regarding fermentation medium, process conditions and process control.
Materials and methods
Strain construction
Bacterial strains and plasmids
Table 1 lists the plasmids and bacterial strains used in this study. In addition, the E. coli strain DH5α was used as a primary host for all cloning work.
Cultivation media
Strains were grown for 24–48 h in 15 mL tubes at 37°C and 220 rpm on Luria Bertani (LB) or in M9 minimal media supplemented with thiamine 5 μg/mL and with the addition of 1.5% agar when solid media was used. When necessary, glutamate was supplemented at 0.8 mM. Unless otherwise indicated, antibiotics were used at the following concentrations: tetracycline 20 μg/mL; chloramphenicol 50 μg/mL; kanamycin 50 μg/mL; ampicillin (Amp) 200 μg/mL. For induction experiments, IPTG was used at a final concentration of 0.1 mM.
DNA manipulation
Cloning of genes and regulatory elements was achieved either through standard restriction enzyme based cloning methods, or with the use of the sequence- and ligation- independent cloning method [24]. Site directed mutagenesis was used in order to introduce single nucleotide alterations into plasmids [38]. All gene deletions were done according to the in-frame gene excision method described by Link et al. [39]. Deletions were verified by PCR and DNA sequencing.
Construction of a base strain for arginine production (C600+Δ4)
A base strain with deletions of key genes involved in the arginine and ornithine degradation pathways and the arginine repression system was first constructed. The threonine and leucine auxotrophic E. coli strain K-12 C600 [18] was used as a starting strain. C600 was made autotrophic by moving wild type alleles from MG1655 donor strain via P1 transduction and subsequent selection on M9 plates without any amino acids. The resultant strain was termed C600+. One target for increased arginine production was inactivation of the arginine repressor argR and genes in the arginine and ornithine degradation pathways. The genes argR, adiA, speC and speF were subsequently deleted in order to obtain the strain termed C600+Δ4.
Biosensor plasmid construction
To select for arginine producing mutants with previously unidentified mutations, a biosensor plasmid termed pArgObla, was engineered based on one previously described [40]. The arginine sensitive promoter argOp was used to control the transcription of the ampicillin resistance gene bla. In addition, the T1T2 terminator was placed upstream to prevent any transcription from upstream promoters. Plasmid sensitivity to arginine was decreased by mutating the core RBS AAGGA upstream of the bla gene to ACGGA to reduce the efficiency of the translation initiation. The Amp minimum inhibitory concentration was considerably reduced for C600+Δ4 carrying this new biosensor variant pArgObla10C, compared to pArgObla (from > 5 mg/mL to 0.6 mg/mL).
Bioassay for screening of arginine producing mutants
Spent M9 growth media from cultures of candidate arginine producers were separated from the cell biomass by centrifugation at 10500 × g for 5 minutes. The supernatant was then sterilized with a 0.2 μm filter and diluted into the assay medium, i.e. M9 supplemented with Kan. The strain JW3932, which was obtained from the Keio collection [29], is auxotrophic for arginine (ΔargH). This strain was inoculated into the assay medium and its growth (measured as OD590) was used to determine the arginine concentration, based on a reference curve constructed from known concentrations of arginine.
Fermentations
Seed cultures preparation
Cultures of the strains constructed as described above were stored in LB at −80°C as 15% glycerol stocks. The medium used for cultivation of the strains consisted of (per liter): 70 g glucose, 15 g corn steep liquor, 15 g (NH4)2SO4, 1 g KH2PO4, 0.5 g MgSO4 · 7 H2O, 20 mg FeSO4 · 7 H2O, 12 mg MnSO4 · H2O, 0.5 mg thiamine · HCl and appropriate concentrations of antibiotics. 1 mL of the stock culture was inoculated to a 500-mL shake flask containing 100 mL of sterile medium and 4 g CaCO3 to maintain a pH of about 7. The seed culture was incubated at 32°C in an orbital shaker at 200 rpm until an optical density at 562 nm (OD562) between 1.6 and 2.5 was reached.
Fermentations
Batch fermentations were conducted in 1 L bioreactors (Biobundle 1 L, Applikon Biotechnology, the Netherlands). The medium used was the same as described above, supplemented with 0.4 g/L of antifoam. 100 mL of seed culture were added in a sterile bioreactor containing 600 mL of medium. Prior to inoculation, pH was adjusted to 7 and thereafter maintained at that pH by automatic addition of NH3 solution (14-15% v/v). Fermentations were performed at 32°C and initial stirring was set at 500 rpm. The dissolved oxygen concentration (DO) was controlled with pulses of air (at 10 vvm) and to maintain a DO level above 30% the stirring was gradually increased from 500 rpm to 1000 rpm. All fermentations were carried out in duplicates.
Samples were regularly taken throughout the fermentations for analysis of optical density, glucose, organic acids and arginine concentrations.
Cell growth measurement
Cell growth was estimated by measuring the OD562 of appropriately diluted samples. Dry cell weight (DCW, in g/L) was determined from the optical density using a linear relationship between OD562 and DCW established in our laboratory under similar conditions: DCW = 0.4507 × OD562 + 0.6095.
Glucose and organic acids analysis
The samples were centrifuged for 10 min at 10621 × g and 4°C. The supernatant was then diluted five times with water and filtered through a 0.2 μm syringe filter. Quantification of glucose and organic acids was performed by HPLC using a guard column (Micro-Guard IG Cation H Cartridge, BioRad), a cation exchange column (Aminex HPX87-H, BioRad) and a Series 200 refractive index detector (PerkinElmer). The column was kept at 65°C and 0.005 M H2SO4 at a flow rate of 0.6 mL/min was used as mobile phase.
Arginine analysis
Samples were centrifuged at 10621 × g for 10 min at 4°C and appropriately diluted with water. Arginine concentration was measured using the UPLC-AccQTag method (UPLC Amino Acid Analysis System Solution, Waters). The amino acids were first derivatized using AccQ · Fluor™ UltraReagent, then eluted with AccQ · Tag Ultra Eluents A and B and separated on a bridged ethyl hybrid C18 column (AccQ · Tag Ultra Column, 2.1 × 100 mm, 1.7 μm, Waters) with UV detection at 260 nm (ACQUITY UPLC System, Waters).
Acknowledgements
The authors would like to thank Carl Trygger Foundation, Kempestiftelserna and Bio4Energy, a strategic research environment appointed by the Swedish government, for supporting this work.
Footnotes
Mireille Ginesy and Jaroslav Belotserkovsky contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MG designed and carried out the fermentation studies, performed the analysis and drafted the manuscript. JB designed and performed the molecular genetic work and drafted the manuscript. LI, JE and UR conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Mireille Ginesy, Email: mireille.ginesy@ltu.se.
Jaroslav Belotserkovsky, Email: jaroslav.belotserkovsky@su.se.
Josefine Enman, Email: josefine.enman@ltu.se.
Leif Isaksson, Email: leif.isaksson@su.se.
Ulrika Rova, Email: ulrika.rova@ltu.se.
References
- 1.Loscalzo J. L-arginine and atherothrombosis. J Nutr. 2004;134(10 Suppl):2798S–800. doi: 10.1093/jn/134.10.2798S. [DOI] [PubMed] [Google Scholar]
- 2.Ohlund J, Nasholm T. Low nitrogen losses with a new source of nitrogen for cultivation of conifer seedlings. Environ Sci Technol. 2002;36(22):4854–9. doi: 10.1021/es025629b. [DOI] [PubMed] [Google Scholar]
- 3.Kinoshita S, Udaka S, Shimono M. Studies on the amino acid fermentation part I. Production of L-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 1957;3(3):193–205. doi: 10.2323/jgam.3.193. [DOI] [PubMed] [Google Scholar]
- 4.Hermann T. Industrial production of amino acids by coryneform bacteria. J Biotechnol. 2003;104(1–3):155–72. doi: 10.1016/S0168-1656(03)00149-4. [DOI] [PubMed] [Google Scholar]
- 5.Utagawa T. Production of arginine by fermentation. J Nutr. 2004;134(10 Suppl):2854S–7. doi: 10.1093/jn/134.10.2854S. [DOI] [PubMed] [Google Scholar]
- 6.Park SH, Kim HU, Kim TY, Park JS, Kim S, Lee SY. Metabolic engineering of Corynebacterium glutamicum for L-arginine production. Nat Commun. 2014;5:4618. doi: 10.1038/ncomms5618. [DOI] [PubMed] [Google Scholar]
- 7.Shin J, Lee S. Metabolic engineering of microorganisms for the production of L-arginine and its derivatives. Microb Cell Fact. 2014;13(1):166. doi: 10.1186/s12934-014-0166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gusyatiner MM, Leonova TV, Ptitsyn LR, Yampolskaya TA. L-Arginine Producing Escherichia Coli and Method of Producing L-Arginine. 2005. [Google Scholar]
- 9.Rostova YG, Voroshilova EB, Gusyatiner MM. Method for Producing an L-Amino Acid Using a Bacterium of the Enterobacteriaceae Family Having Attenuated Expression of Genes Encoding a Lysine/Arginine/ Ornithine Transporter. 2013. [Google Scholar]
- 10.Kiryukhin MY, Gusyatiner MM. Method for Producing an L-Amino Acid Using a Bacterium of the Enterobacteriaceae Family. 2013. [Google Scholar]
- 11.Vyas S, Maas WK. Feedback inhibition of acetylglutamate synthetase by arginine in Escherichia coli. Arch Biochem Biophys. 1963;100(3):542–6. doi: 10.1016/0003-9861(63)90124-3. [DOI] [PubMed] [Google Scholar]
- 12.Maas WK. The arginine repressor of Escherichia coli. Microbiol Rev. 1994;58(4):631–40. doi: 10.1128/mr.58.4.631-640.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Celis RT. Repression and activation of arginine transport genes in Escherichia coli K 12 by the ArgP protein. J Mol Biol. 1999;294(5):1087–95. doi: 10.1006/jmbi.1999.3308. [DOI] [PubMed] [Google Scholar]
- 14.Nandineni MR, Gowrishankar J. Evidence for an arginine exporter encoded by yggA (argO) that is regulated by the LysR-type transcriptional regulator ArgP in Escherichia coli. J Bacteriol. 2004;186(11):3539–46. doi: 10.1128/JB.186.11.3539-3546.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laishram RS, Gowrishankar J. Environmental regulation operating at the promoter clearance step of bacterial transcription. Genes Dev. 2007;21(10):1258–72. doi: 10.1101/gad.1520507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peeters E, Nguyen Le Minh P, Foulquie-Moreno M, Charlier D. Competitive activation of the Escherichia coli argO gene coding for an arginine exporter by the transcriptional regulators Lrp and ArgP. Mol Microbiol. 2009;74(6):1513–26. doi: 10.1111/j.1365-2958.2009.06950.x. [DOI] [PubMed] [Google Scholar]
- 17.Marbaniang CN, Gowrishankar J. Role of ArgP (IciA) in lysine-mediated repression in Escherichia coli. J Bacteriol. 2011;193(21):5985–96. doi: 10.1128/JB.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Applebaum D, Sabo DL, Fischer EH, Morris DR. Biodegradative ornithine decarboxylase of Escherichia coli. Purification, properties, and pyridoxal 5′-phosphate binding site. Biochemistry. 1975;14(16):3675–81. doi: 10.1021/bi00687a025. [DOI] [PubMed] [Google Scholar]
- 19.Applebaum DM, Dunlap JC, Morris DR. Comparison of the biosynthetic and biodegradative ornithine decarboxylases of Escherichia coli. Biochemistry. 1977;16(8):1580–4. doi: 10.1021/bi00627a008. [DOI] [PubMed] [Google Scholar]
- 20.Kubota K, Onoda T, Kamijo H, Yoshinaga F, Okumura S. Micorbial production of L-arginine; I. Production of L-ariginine by mutants of glutamic acid-producing bacteria. J Gen Appl Microbiol. 1973;19(5):339–52. doi: 10.2323/jgam.19.339. [DOI] [Google Scholar]
- 21.Schwartz JH, Maas WK. Analysis of the inhibition of growth produced by canavanine in Escherichia coli. J Bacteriol. 1960;79:794–9. doi: 10.1128/jb.79.6.794-799.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celis TF, Rosenfeld HJ, Maas WK. Mutant of Escherichia coli K-12 defective in the transport of basic amino acids. J Bacteriol. 1973;116(2):619–26. doi: 10.1128/jb.116.2.619-626.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Celis TF. Independent regulation of transport and biosynthesis of arginine in Escherichia coli K-12. J Bacteriol. 1977;130(3):1244–52. doi: 10.1128/jb.130.3.1244-1252.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong JY, Yim HS, Ryu JY, Lee HS, Lee JH, Seen DS, et al. One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl Environ Microbiol. 2012;78(15):5440–3. doi: 10.1128/AEM.00844-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajagopal BS, DePonte J,3rd, Tuchman M, Malamy MH. Use of inducible feedback-resistant N-acetylglutamate synthetase (argA) genes for enhanced arginine biosynthesis by genetically engineered Escherichia coli K-12 strains. Appl Environ Microbiol. 1998;64(5):1805–11. doi: 10.1128/aem.64.5.1805-1811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12(5):291–9. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 27.Hagg P, de Pohl JW, Abdulkarim F, Isaksson LA. A host/plasmid system that is not dependent on antibiotics and antibiotic resistance genes for stable plasmid maintenance in Escherichia coli. J Biotechnol. 2004;111(1):17–30. doi: 10.1016/j.jbiotec.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Appleyard RK. Segregation of Lambda Lysogenicity during Bacterial Recombination in Escherichia Coli K12. Genetics. 1954;39(4):429–39. doi: 10.1093/genetics/39.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellmann A, Vrljic M, Patek M, Sahm H, Kramer R, Eggeling L. Expression control and specificity of the basic amino acid exporter LysE of Corynebacterium glutamicum. Microbiology. 2001;147(Pt 7):1765–74. doi: 10.1099/00221287-147-7-1765. [DOI] [PubMed] [Google Scholar]
- 31.Vrljic M, Sahm H, Eggeling L. A new type of transporter with a new type of cellular function: L-lysine export from Corynebacterium glutamicum. Mol Microbiol. 1996;22(5):815–26. doi: 10.1046/j.1365-2958.1996.01527.x. [DOI] [PubMed] [Google Scholar]
- 32.Kennerknecht N, Sahm H, Yen MR, Patek M, Saier MH, Jr, Jr EL. Export of L-isoleucine from Corynebacterium glutamicum: a two-gene-encoded member of a new translocator family. J Bacteriol. 2002;184(14):3947–56. doi: 10.1128/JB.184.14.3947-3956.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simic P, Willuhn J, Sahm H, Eggeling L. Identification of glyA (encoding serine hydroxymethyltransferase) and its use together with the exporter ThrE to increase L-threonine accumulation by Corynebacterium glutamicum. Appl Environ Microbiol. 2002;68(7):3321–7. doi: 10.1128/AEM.68.7.3321-3327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morbach S, Sahm H, Eggeling L. l-Isoleucine production with Corynebacterium glutamicum: further Flux increase and limitation of export. Appl Environ Microbiol. 1996;62(12):4345–51. doi: 10.1128/aem.62.12.4345-4351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua Q, Yang C, Shimizu K. Metabolic control analysis for lysine synthesis using Corynebacterium glutamicum and experimental verification. J Biosci Bioeng. 2000;90(2):184–92. doi: 10.1016/S1389-1723(00)80108-5. [DOI] [PubMed] [Google Scholar]
- 36.El-Mansi EM, Holms WH. Control of carbon flux to acetate excretion during growth of Escherichia coli in batch and continuous cultures. J Gen Microbiol. 1989;135(11):2875–83. doi: 10.1099/00221287-135-11-2875. [DOI] [PubMed] [Google Scholar]
- 37.Hollywood N, Doelle HW. Effect of specific growth rate and glucose concentration on growth and glucose metabolism of Escherichia coli K-12. Microbios. 1976;17(67):23–33. [PubMed] [Google Scholar]
- 38.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32(14):e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 1997;179(20):6228–37. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Binder S, Schendzielorz G, Stabler N, Krumbach K, Hoffmann K, Bott M, et al. A high-throughput approach to identify genomic variants of bacterial metabolite producers at the single-cell level. Genome Biol. 2012;13(5):R40. doi: 10.1186/gb-2012-13-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]