

Abstract
K-Ras is a well-validated cancer target but is considered to be “undruggable” due to the lack of suitable binding pockets. We previously discovered small molecules that bind weakly to K-Ras but wanted to improve their binding affinities by identifying ligands that bind near our initial hits that we could link together. Here we describe an approach for identifying second site ligands that uses a cysteine residue to covalently attach a compound for tight binding to the first site pocket followed by a fragment screen for binding to a second site. This approach could be very useful for targeting Ras and other challenging drug targets.
Keywords: K-Ras, Fragment-based drug discovery, Second-site screen, Cysteine tethering
K-Ras is a highly validated cancer target (Pylayeva-Gupta et al. 2011). It is mutated in 90% of pancreatic cancers, 50% of colon cancers, and 30% of lung cancers (Laghi et al. 2002; Lau et al. 2009; Riely et al. 2009; Bos et al. 1989; Yen et al. 2010), and tumors that contain Ras activating mutations are usually resistant to chemotherapy and radiotherapy (Bernhard et al. 2000). In addition, inhibition of K-Ras activity in established tumors or cancer cell lines has been shown to result in the reversal of the malignant phenotype and suppression of tumorigenicity in human cancer cells (Chin et al. 1999; Podsypanina et al. 2008). However, it is extremely difficult to target Ras due to the lack of suitable small molecule binding sites on its surface. Although K-Ras is thought to be “undruggable”, a great deal of effort has been expended by major pharmaceutical companies as well as academic labs to identify lead molecules to inhibit this protein over the past decade. However, to date, no potent lead molecules have yet been discovered.
One possible approach for targeting challenging proteins such as K-Ras is to use fragment-based methods. Fragment-based drug design (FBDD) has emerged as a popular and productive route for discovering lead compounds for therapeutically important targets, including those that have been considered to be “undruggable”(Oltersdorf et al. 2005; Bollag et al. 2010). Compared to traditional high-throughput screening, FBDD covers more chemical space. Also, FBDD approaches often result in small molecules that have better ligand efficiencies than molecules identified through conventional methods (Erlanson et al. 2006).
Although the initial hits identified from a fragment-based screen generally have low affinities due to their small size, the potency of the initial hits could be dramatically improved by linking them to other fragments that bind to nearby pockets using the “SAR by NMR” paradigm (Shuker et al. 1996). The general method used to identify second-site ligands, involves a screen carried out in the presence of saturating amounts of the first-site ligand. The second-site ligands can be discovered using multiple methods, such as monitoring chemical shift perturbations of the protein (Hajduk et al. 2000), observing intermolecular NOEs between bound ligands (Li et al. 1999) or by attaching a spin-label to the first-site ligand and following the paramagnetic relaxation enhancement of the bound second-site ligands (Wolfgang et al. 2000). However, in some cases, the results of a second-site screen can be difficult or impossible to interpret if the first-site ligand does not bind tight enough to saturate the primary binding pocket.
Previously, we conducted a fragment-based screen against GDP-bound K-Ras using NMR (Sun et al. 2012). The hits identified in the screen occupied a hydrophobic pocket between the switch II helix and a central beta sheet. We were able to improve the affinity of these compounds using structure-based design and demonstrated that they inhibit Sos-catalyzed nucleotide exchange of K-Ras. However, when we attempted to conduct a second-site screen to further improve their binding affinities, we encountered a problem. We were unable to find a suitable first-site ligand that could fully saturate the first-site pocket due to its weak binding and poor aqueous solubility. As a result, many of the compounds in the second-site screen competed for binding to the first-site pocket.
Inspired by the “cysteine tethering” strategy of Erlanson et al. (2004), we developed an approach that involved the preparation of cysteine mutants of K-Ras that could be used to covalently attach a first-site ligand. This approach would enable us to saturate the first-site pocket so that any newly identified hits from the screen would bind to a distinct site and the modification would lock K-Ras in a state that mimics the conformation when the primary pocket is occupied by our previously identified inhibitors. To accomplish this, we designed a panel of 6 cysteine mutants (Fig.1), targeting the region surrounding the primary binding site. Each mutant K-Ras protein was expressed and purified. The 1H/15N HMQC spectra of the proteins showed that all of the mutants were folded properly. Purified proteins were then mixed with thiol-reactive compounds to covalently modify the protein.
Fig. 1.
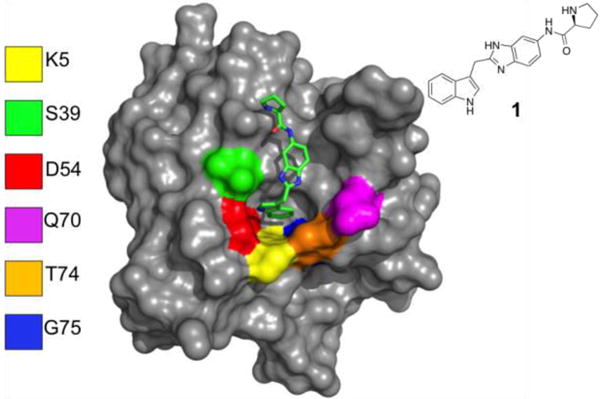
Six residues around the primary binding pocket were mutated to cysteine. A previously identified K-Ras inhibitor (1) is depicted as a stick figure to indicate the location of the primary binding pocket
Since a large number of our screening hits contain an indole, we started with an indole-containing compound (compound 2, Fig.2). This compound was allowed to react with the cysteine mutants, and mass spectrometry was used to confirm that the reaction was complete. We obtained crystal structures of tethered mutant proteins to determine where the attached indole was bound. For the T74C mutant, the indole moiety was found to point out of the pocket (Fig.2a), while in the S39C mutant, the indole moiety occupies the pocket, but it does not bind in the same conformation as the original ligand and is unable to form a hydrogen bond with D54 (Fig.2b). The Q70C mutant attached to 2 was also not suitable for screening since the indole moiety binds in a pocket of the neighboring protein molecule in the unit cell, and induces the formation of a crystallographic dimer (Fig.2c). Full occupancy of the first site pocket should block binding of compound 1. However, when we added compound 1 and recorded HMQC spectra, all of the 15N-labeled K-Ras mutants that were covalently modified experienced chemical shift changes indicating that the primary pocket was not fully blocked.
Fig. 2.
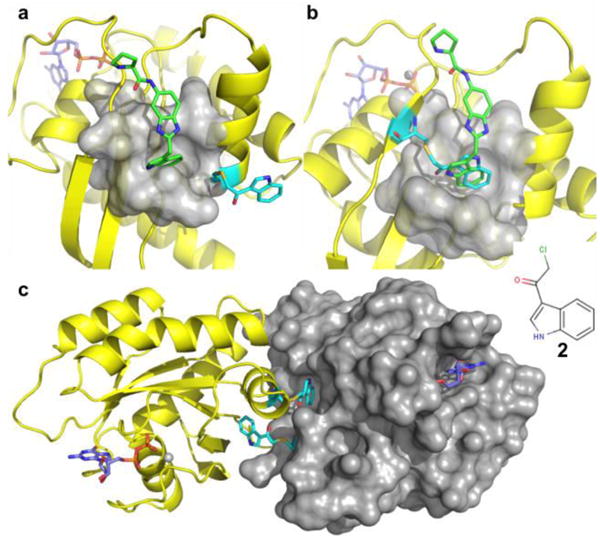
Crystal structures of various K-Ras mutants covalently attached to compound 2 (cyan). a) In mutant T74C, the covalently attached compound 2 is pointing towards the solvent. b) In the mutant S39C, the linked compound is sitting inside of the primary pocket, but the NH group of the indole is pointing in the opposite direction compared to compound 1 (green). c) The modified Q70C mutant crystallized as a dimer with the indole portion of the compound occupying the pocket of a neighboring molecule. (PDB code 4PZY) None of these modifications blocks the probe compound 1 from binding to the first-site
After these initial failures, we prepared a small library of 32 thiol-reactive compounds (Supplementary Material Fig. S1) to identify the desired combination of cysteine mutants and thiol-reactive compounds. Each compound was reacted with each of the cysteine mutants, and the reaction was monitored using mass spectrometry and NMR. To test whether the covalently attached molecule could be displaced from the primary site, the probe compound 1 was added, and 1H/15N HMQC spectra were recorded.
Using this approach, we were able to identify four suitable mutant/compound combinations in which the primary pocket was fully occupied and saturated (Fig.3). As confirmed by NMR, no further chemical shift changes were observed upon addition of compound 1. Superposition of the structure of K-Ras S39C/benzimidazolethiomethane (3) covalent complex with that of an indole-containing compound 1, showed a perfect overlap between the indole and benzimidazole (Fig.3a). The latter blocks the pocket without interfering with potential nearby binding sites. Thus, we chose the modified protein K-Ras S39C/3 to be used in our second-site screen. Unlike the initial first-site screen, which yielded over 140 hits, only 20 hits were identified in a second-site screen from a library containing ∼13,000 compounds. Figure 4 depicts an HMQC spectrum of the K-Ras S39C mutant covalently attached to compound 3 in the presence (red) and absence (blue) of a fragment hit. The second-site hits that we identified bind to both modified and native K-Ras protein with affinities ranging from 0.3-3 mM.
Fig. 3.
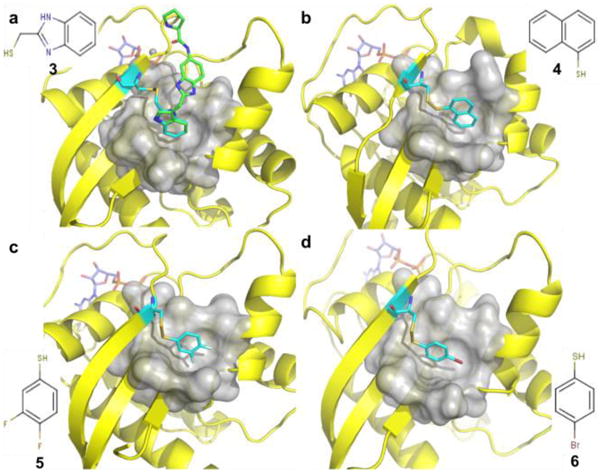
Ribbon and molecular surface representations of the crystal structures of GDP-bound K-Ras S39C that was reacted with thiol-reactive compounds (cyan) a) 3, (PDB 4PZZ) b) 4, (PDB 4Q01) c) 5, (PDB 4Q02) and d) 6. (PDB 4Q03) All these compounds completely block the pocket and prevent the probe compound from interacting with the protein. Among them, compound 3 perfectly overlays with the probe compound 1 (green)
Fig. 4.
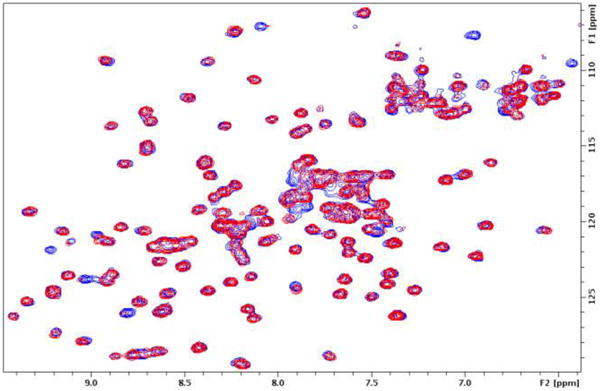
1H/15N HMQC spectra of the uniformly 15N-labeled K-Ras S39C mutant covalently modified by compound 3 recorded in the absence (blue) and presence (red) of a fragment hit
In summary, we have described a useful approach for identifying small molecules that bind to a protein at a second-site that differs from the primary binding pocket. The method solves the problem of the need to saturate the first-site pocket, which is a major issue when conducting a second-site screen. The challenging step of this approach is to identify an ideal combination between a cysteine mutated protein and a thiol-reactive compound. The covalently attached compounds must have the correct orientation and linker length to fully saturate the first-site pocket. For K-Ras, thiol-based compounds were generally more successful than alkyl-halides because the disulfide bond provides more flexibility to correctly position the hydrophobic portion of the compounds in the pocket. Based on our experience, the best way to find an optimum covalently attached first-site ligand is to screen a small library of well-designed thiol-reactive compounds in combination with several cysteine mutant proteins. Once a suitable combination of mutant/compound is found, the modified protein can be used to screen for second-site ligands using conventional methods. This differs from the tethering approach using mass spectrometry that requires a large library of reactive compounds (Erlanson et al. 2004). Our approach may serve as a useful method for identifying second-site hits of protein targets in which the first-site screening hits bind only weakly and cannot saturate the protein, which complicates the interpretation of the screening results.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health (NIH) grants DP1OD006933/DP1CA174419 (NIH Director's Pioneer Award; S.W.F), P50A095103-12 (NCI SPORE in GI Cancer; R.J. Coffey), and RC2CA148375 to L.J. Marnett as well as The Lustgarten Foundation Award (S.W.F.). The use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The use of mass spectrometry core was supported by scholarship S000003067 from the Vanderbilt Ingram Cancer Center. The use of Vanderbilt NMR facility was supported in part by grants for NMR instrumentation from the US National Science Foundation (NSF) (0922862), NIH (S10 RR025677) and Vanderbilt University matching funds. A portion of the experiments described here used the Vanderbilt robotic crystallization facility, which was supported by NIH grant S10 RR026915. The authors would like to thank Dr. Wade Calcutt in the Vanderbilt Mass Spectrometry Core Laboratory for his help in characterizing the covalently modified intact proteins by LC-MS.
Footnotes
Electronic supplementary material The online version of this article (doi:xxxxxxxx) contains supplementary material, which is available to authorized users.
References
- Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, Muschel RJ, McKenna WG. Direct Evidence for the Contribution of Activated N-ras and K-ras Oncogenes to Increased Intrinsic Radiation Resistance in Human Tumor Cell Lines. Cancer Res. 2000;60:6597–6600. [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D'Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, Horner JW, Cordon-Cardo C, Yancopoulos GD, DePinho RA. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annu Rev Biophys Biomol Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Erlanson DA. Fragment-based lead discovery: a chemical update. Curr Opin Biotechnol. 2006;17:643–52. doi: 10.1016/j.copbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Gomtsyan A, Didomenico S, Cowart M, Bayburt EK, Solomon L, Severin J, Smith R, Walter K, Holzman TF, Stewart A, McGaraughty S, Jarvis MF, Kowaluk EA, Fesik SW. Design of adenosine kinase inhibitors from the NMR-based screening of fragments. J Med Chem. 2000;43:4781–4786. doi: 10.1021/jm000373a. [DOI] [PubMed] [Google Scholar]
- Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo A, Boland CR, Malesci A. Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene. 2002;21:4301–4306. doi: 10.1038/sj.onc.1205533. [DOI] [PubMed] [Google Scholar]
- Lau KS, Haigis KM. Non-redundancy within the RAS oncogene family: insights into mutational disparities in cancer. Mol Cells. 2009;28:315–320. doi: 10.1007/s10059-009-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, DeRose EF, London RE. The inter-ligand Overhauser effect: a powerful new NMR approach for mapping structural relationships of macromolecular ligands. J Biomol NMR. 1999;15:71–76. doi: 10.1023/a:1008360208627. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6:201–5. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang J, Lawrence BP, Paris CG, Strauss A, Fendrich G, Nalin CM. Second-Site NMR Screening with a Spin-Labeled First Ligand. J Am Chem Soc. 2000;122:7394–7395. [Google Scholar]
- Yen LC, Uen YH, Wu DC, Lu CY, Yu FJ, Wu IC, Lin SR, Wang JY. Activating KRAS mutations and overexpression of epidermal growth factor receptor as independent predictors in metastatic colorectal cancer patients treated with cetuximab. Ann Surg. 2010;251:254–60. doi: 10.1097/SLA.0b013e3181bc9d96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.