

Abstract
Importance
Cocaine addiction is associated with altered resting-state functional connectivity among regions of the mesocorticolimbic dopamine pathways. Methylphenidate hydrochloride, an indirect dopamine agonist, normalizes task-related regional brain activity and associated behavior in cocaine users; however, the neural systems–level effects of methylphenidate in this population have not yet been described.
Objective
To use resting-state functional magnetic resonance imaging to examine changes in mesocorticolimbic connectivity with methylphenidate and how connectivity of affected pathways relates to severity of cocaine addiction.
Design
Randomized, placebo-controlled, before-after, crossover study.
Setting
Clinical research center.
Participants
Eighteen nonabstaining individuals with cocaine use disorders.
Interventions
Single doses of oral methylphenidate (20 mg) or placebo were administered at each of 2 study sessions. At each session, resting scans were acquired twice: immediately after drug administration (before the onset of effects [baseline]) and 120 minutes later (within the window of peak effects).
Main outcomes and Measures
Functional connectivity strength was evaluated using a seed voxel correlation approach. Changes in this measure were examined to characterize the neural systems–level effects of methylphenidate; severity of cocaine addiction was assessed by interview and questionnaire.
Results
Short-term methylphenidate administration reduced an abnormally strong connectivity of the ventral striatum with the dorsal striatum (putamen/globus pallidus), and lower connectivity between these regions during placebo administration uniquely correlated with less severe addiction. In contrast, methylphenidate strengthened several corticolimbic and corticocortical connections.
Conclusions and Relevance
These findings help elucidate the neural systems–level effects of methylphenidate and suggest that short-term methylphenidate can, at least transiently, remodel abnormal circuitry relevant to the pathophysiologic characteristics of cocaine addiction. In particular, the effects of methylphenidate within striatal and cortical pathways constitute a potentially viable mechanism by which methylphenidate could facilitate control of behavior in cocaine addiction.
Resting-state functional connectivity is a noninvasive and replicable method for assessing neural circuitry function in neuropsychiatric disorders.1 This method captures the synchronicity of low-frequency, spontaneous fluctuations in blood oxygen level–dependent signals that reflect fluctuations in neuronal activity2 between brain regions in the absence of external stimulation.3 These synchronous fluctuations are confined to gray matter and can be observed for mono-synaptic or polysynaptic anatomic connections.2,4 More important, resting-state connectivity is linked to task-related functioning of discrete brain regions comprising the same circuits5 and to corresponding behavior.5,6 Thus, resting-state connectivity can be useful for advancing neural systems–level understanding of the functional and behavioral abnormalities that characterize neuropsychiatric disorders such as addiction, with the potential to also serve as a target for therapeutic interventions.
Perturbations in resting-state connectivity within and between functional brain networks that subserve attentional, emotional, and inhibitory control processes have been observed in individuals addicted to nicotine,7,8 opioids,9-11 and cocaine.12,13 Specifically, individuals with cocaine use disorders (CUDs) exhibit reduced connectivity of the dorsal frontoparietal attention network13; in addition, and especially pertinent to the current study, cocaine users exhibit reduced connectivity of the mesocorticolimbic dopamine pathways.12 However, thus far it has not been shown whether this abnormal connectivity could be modified in individuals with chronic, severe CUD.
Methylphenidate hydrochloride is a psychostimulant widely used to treat attention-deficit/hyperactivity disorder. Like cocaine, methylphenidate competitively blocks dopamine and norepinephrine transporters, thereby increasing extracellular concentration of these neuromodulatory neurotransmitters.14 However, unlike cocaine, the rate of clearance of orally administered methylphenidate from the brain is substantially slower (90-minute half-life compared with 20-minute half-life for cocaine), contributing to its lower abuse potential15 and possible viability as a therapeutic agent in treating cocaine addiction.16 Methylphenidate improves task-related regional brain activation in the dopamine-innervated ventral medial prefrontal and dorsal anterior cingulate cortices17 and corresponding stop signal reaction times in CUD.18 Oral methylphenidate also has been shown19 to attenuate changes in glucose metabolism in the lateral orbitofrontal and inferior frontal cortices, hippocampus, and nucleus accumbens when cocaine users are exposed to drug cues.
In the present study, we used a placebo-controlled, before-after, crossover experimental design to examine the effects of short-term methylphenidate administration on resting-state connectivity in active cocaine users. To our knowledge, the only study20 that assessed drug-induced changes in resting-state connectivity in individuals with CUD included a small sample size, used intravenous cocaine, and focused on the sensorimotor cortices. We examined mesocorticolimbic connectivity using a seed correlation approach based on a more recent study12 reporting that cocaine addiction was associated with reduced connectivity in several pathways involved in emotion processing. Given the normalizing effects of methylphenidate on task-related activation and behavior in CUD,17,18 we hypothesized that methylphenidate would strengthen connectivity of the medial frontal cortex in pathways underlying emotion regulation (eg, with amygdala and hippocampus21-23) and inhibitory control (eg, with dorsal and lateral prefrontal cortex18,19). In addition, because methylphenidate attenuated brain metabolism following exposure to cocaine cues,19 we expected reduced connectivity between regions underlying cue-induced craving (eg, orbitofrontal cortex, ventral and dorsal striatum, and amygdala24-26). Finally, to examine whether methylphenidate modified connectivity in pathways directly associated with addictive behavior, we inspected correlations between connectivity strength and severity of cocaine addiction.
Methods
Participants
Participants were 18 nontreatment-seeking individuals with CUD, recruited from advertisements in local newspapers and by word of mouth. All participants were right-handed and native English speakers and provided their written consent to participate in the study in accordance with the Stony Brook University institutional review board. Participants were otherwise healthy and not taking any medications, as ascertained during a full physical and neurologic examination by a neurologist and a diagnostic interview by a clinical psychologist. This latter interview included the Structured Clinical Interview for DSM-IV Axis I disorders (research version27,28) and the Addiction Severity Index.29 All participants were currently using cocaine and identified cocaine as their primary drug of choice, meeting criteria for current cocaine dependence (n = 17) or abuse (n = 1) (eAppendix in the Supplement). Current comorbidities included heroin dependence (n = 1), marijuana abuse (n = 1), alcohol abuse (n = 1), and nicotine dependence (n = 14). Exclusion criteria were (1) history of head trauma or loss of consciousness (>30 minutes) or other neurologic disease, (2) abnormal vital signs at the time of screening, (3) history of major medical conditions, (4) history of major psychiatric disorder (other than substance abuse or dependence), (5) pregnancy as confirmed with a urine test in all women, (6) contraindications to the magnetic resonance imaging (MRI) environment, (7) history of glaucoma, and (8) except for cocaine, positive urine screen results for psychoactive drugs or their metabolites (amphetamine or methamphetamine, phencyclidine, benzodiazepines, cannabis, opiates, barbiturates, and inhalants). Nine participants tested positive for cocaine on methylphenidate day and 8 tested positive for cocaine on placebo day (see the eAppendix in the Supplement for additional information). Demographic information is reported in Table 1.
Table 1. Demographic and Clinical Characteristics of 18 Individuals With Cocaine Use Disorders.
| Characteristic | Data |
|---|---|
| Sex, No. | |
| Male | 16 |
| Female | 2 |
| Race, No. | |
| African American | 15 |
| Othera | 3 |
| Age, mean (SD), y | 45.6 (7.3) |
| Educational level, mean (SD), y | 12.9 (1.8) |
| Verbal IQ, mean (SD)b | 91.8 (9.2) |
| Nonverbal IQ, mean (SD)c | 9.3 (3.1) |
| Socioeconomic status: Hollingshead Index, mean (SD) | 35.8 (8.4) |
| Drug use history | |
| Cigarette smokers, No. | |
| Current or past | 14 |
| Nonsmokers | 4 |
| Cigarettes per day, mean (SD), No. (12 current smokers only) | 8.0 (4.2) |
| Age of onset of cocaine use, mean (SD), y | 26.9 (6.3) |
| Duration of cocaine use, mean (SD), y | 15.3 (7.5) |
| Preferred route of cocaine administration,d No. | |
| Smoking | 14 |
| Intranasal | 1 |
| Intravenous | 1 |
| Days of cocaine use per week, mean (SD), No. in past 30 de | 2.7 (2.1) |
Abbreviation: WASI, Wechsler Abbreviated Scale of Intelligence.
White, Hispanic, or Asian.
Determined with the Wide Range Achievement Test III–Reading Scale.30
Determined with the WASI–Matrix Reasoning Scale.31
Missing data for 2 participants.
Variables used to compute the addiction severity composite score.
Cocaine withdrawal symptoms were assessed prior to medication administration at each study day with the Cocaine Selective Severity Assessment scale32; participants also completed the Cocaine Craving Questionnaire33 and the Severity of Dependence Scale,34 which captures perceived control over drug-taking and difficulty with stopping drug use during the past year. For each participant, the severity of addiction was quantified as a composite score (average Z value) of the severity of withdrawal, craving, and dependence as assessed on placebo day and the frequency of use of cocaine in the past 30 days as assessed during the clinical interview. The severity of withdrawal and dependence did not differ significantly between the study days (P > .11); however, participants reported more recent use of cocaine and more severe craving on methylphenidate day than on placebo day (Table 2).
Table 2. Daily Baseline Assessment.
| Characteristic | Methylphenidate | Placebo |
|---|---|---|
| Cocaine urine status, No. | ||
| Positive | 9 | 8 |
| Negative | 9 | 10 |
| Current cocaine abstinence, mean (SD), d since last usea | 5.4 (6.5) | 7.8 (9.5) |
| Depression score, mean (SD)b | 6.9 (5.5) | 6.1 (4.8) |
| Withdrawal symptomsc,d | 15.9 (10.6) | 12.2 (7.8) |
| Severity of Dependence Scaled,e | 7.4 (2.5) | 7.3 (2.6) |
| Cocaine cravingd,f,g | 22.0 (13.4) | 17.4 (12.7) |
P < .05.
Determined with the Beck Depression Inventory II35 (score range, 0-63).
Determined with the 18-item Cocaine Selective Severity Assessment32 (score range, 0-126).
Variables used to compute the addiction severity composite score.
Determined with the Severity of Dependence Scale34 (score range, 0-15).
Determined with the 5-item Craving Questionnaire33 (score range, 0-45).
P < .01.
Study Sessions
At each of the 2 study sessions (conducted at mean [SD], 8.9 [4.0] days apart), participants were randomized to receive a single oral dose of methylphenidate (20 mg) or placebo (lactose). This methylphenidate dose has been shown to affect task-related brain activation and behavior in CUD.17,19 Because the effects of methylphenidate in individuals with CUD are not well characterized (particularly vis-à-vis potential cardiovascular complications), the study was initially performed such that only participants were blinded to the administered challenge (n = 13). Once it became clear that risks were minimal, we transitioned to double-blind administration (n = 5), with study personnel also blinded to the medication.
The methylphenidate and placebo sessions consisted of identical study procedures. Resting scans were acquired twice, shortly after medication administration (before the onset of drug effects) and approximately 120 minutes later (after completion of 2 functional MRI tasks, a rewarded drug cue sustained attention task36 and a classic color word Stroop task37), within the window of peak methylphenidate effects.38 To inspect whether methylphenidate affected cardiovascular reactivity and mood state, we assessed participants' heart rate, blood pressure, desire for methylphenidate, and feelings of being alert, anxious, high, or restless throughout the study sessions (eAppendix in the Supplement).
Image Acquisition
Functional MRI was performed (4-T Varian/Siemens MRI scanner) using a coronal T2*-weighted single-shot gradient-echo echo planar imaging sequence (echo time/repetition time, 20/1600 milliseconds; 3.125 × 3.125 mm2 in-plane resolution; 4-mm slice thickness; 1-mm gap; 33 coronal slices; 20-cm field of view; 64 × 64 matrix size; 90°-flip angle; and 200-kHz bandwidth with ramp sampling). Padding was used to minimize motion, and earplugs and headphones were used to minimize the influence of scanner noise on brain activation.39,40 Participants were instructed to keep their eyes open, lie as still as possible, and remain awake during the resting scans. No video corroboration that participants adhered to the instructions could be obtained. Each resting scan was approximately 8 minutes in duration (representing 311 consecutive data points after the first 10 data points were removed to account for signal stabilization). This length is comparable to that in previous studies of this type7,8 and within the recommended range for optimal assessment of seed-based connectivity.41 Of 21 participants initially scanned, 3 were excluded because of missing data (n = 2) or signal dropout (n = 1) on at least 1 of the 4 scans.
Image Processing and Construction of the Functional Connectivity Maps
Image processing and analyses were performed in SPM8 (Wellcome Trust Centre for Neuroimaging; http://www.fil.ion.ucl.ac.uk/spm/software/spm8/). The data were first realigned, slice time–corrected, and spatially normalized to a standard Montreal Neurological Institute (MNI) frame, resulting in a final voxel size of 3 × 3 × 3mm. Other preprocessing steps were carried out in Interactive Data Language (Exelis Visual Information Solutions; http://www.exelisvis.com/ProductsServices/IDL.aspx) and included motion correction using the 6 time-varying realignment parameters (3 translations and 3 rotations), global signal normalization, and band pass filtering (0.01-0.10 Hz) to remove magnetic field drifts of the scanner and minimize physiologic noise of high-frequency components.42 Because a recent study43 showed that movement at a finer time scale can increase the variability of functional connectivity measures, we additionally computed the mean absolute displacement of the brain from every time frame to the next. Displacement was in the minimal range for all 4 resting scans (0.14-0.21 mm); however, displacement was higher overall on placebo day than on methylphenidate day (F1,17 = 5.56, P = .03; for all other effects, P > .68) (eAppendix in the Supplement).
Six functional seed regions were defined by centering bilateral 9-mm cubes (27 voxels) at the coordinates shown in Figure 1. The size of the seed regions was chosen on the basis of our previous studies44-46 and was kept constant across anatomic regions to minimize systematic bias in averaging signals across larger vs smaller volumes. After conversion to MNI space, these seeds were identical to those used by Gu et al12 (ventral tegmental area, nucleus accumbens, amygdala, hippocampus, thalamus, and rostral anterior cingulate). Control seeds were also placed in the primary motor (coordinates in MNI space: x = ±44, y = −10, and z = 40), auditory (x = ±44, y = −36, and z = 13), and visual (x = ±10, y = −91, and z = 1) cortices to determine the specificity of effects to mesocorticolimbic regions. Whole-brain cross-correlation maps were calculated separately for each seed and for each participant for each of the 4 scans (methylphenidate peak effects, placebo peak effects [ie, after the parallel time has elapsed, reflecting an active baseline], methylphenidate baseline [ie, before the expected onset of medication effects], and placebo baseline), reflecting correlations over time between average blood oxygen level–dependent signal fluctuations in the respective seed region (averaged time series across all voxels in the seed) and those in all other voxels of the brain. These cross-correlation coefficient maps were converted to Z-score maps using the Fisher Z transform and smoothed with an 8-mm full-width at half-maximum gaussian kernel prior to group-level analyses in SPM8.
Figure 1. Center Coordinates and Locations of the Mesocorticolimbic Seed Regions (Color Boxes).
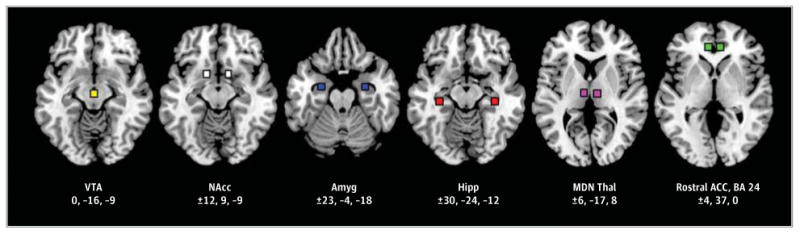
The listed x, y, z coordinates follow Montreal Neurological Institute convention. ACC indicates anterior cingulate cortex; Amyg, amygdala; BA, Brodmann area; Hipp, hippocampus; MDN Thal, mediodorsal nucleus thalamus; NAcc, nucleus accumbens; and VTA, ventral tegmental area.
Functional Connectivity Analysis
A 2 (medication: methylphenidate, placebo) × 2 (time: baseline, peak drug effects at 120 minutes) repeated-measures analysis of covariance in SPM8 was used to analyze differences in the strength of each seed's connectivity as a function of methylphenidate administration. In these analyses, to control for differences between the methylphenidate and placebo days in abstinence, craving, the potential influence of the medication administration paradigm, and micromotion, we included as covariates days since last cocaine use, the Craving Questionnaire scores, a dummy regressor indicating whether participants received single- or double-blind medication administration, and mean absolute head displacement.
The primary analysis involved a comparison of methylphenidate peak effects vs placebo peak effects. Two strategies were used. First, using region of interest analyses, we tested for restored connectivity with methylphenidate of regions previously reported to exhibit reduced connectivity with our seed regions in CUD.12 The following anatomic regions were created in PickAtlas (ANSIR Laboratory; http://fmri.wfubmc.edu/software/PickAtlas): (1) bilateral thalamus (for analyses with the ventral tegmental area seed); (2) Brodmann areas (BAs) 10, 9, and 24 in the medial prefrontal cortex (for analyses with the amygdala seeds); (3) bilateral putamen (for analyses with the thalamus seeds); (4) BAs 6, 8, and 9 in the superior and lateral frontal cortex (for analyses with the hippocampus seeds); (4) BAs 41 and 13 in the temporal gyrus/insula (for analyses with the rostral anterior cingulate seeds); and (5) right parahippocampal gyrus, hippocampus, and amygdala (for analyses with the rostral anterior cingulate seeds) (eFigure 1 in the Supplement). These region of interest analyses were thresholded at P < .05 family wise error (FWE)–corrected at the voxel level. Second, we explored whole-brain changes in connectivity using a cluster-level P < .05 FWE-corrected threshold (see the eAppendix in the Supplement). Here, we selected a priori a minimum height (P < .005 uncorrected) and cluster extent (20 adjacent voxels) threshold. We then applied the FWE correction (at the cluster level) as implemented in SPM8 to determine the probability of obtaining a given cluster size assuming a random gaussian field distribution. Significant regions from the methylphenidate vs placebo peak contrast were extracted as 3-mm radius spheres, chosen according to the image smoothness (ie, the volume of the resolution elements47), using the EasyROI toolbox (http://www.sbirc.ed.ac.uk/LCL/LCL_M1.html). The extracted average signal in these regions was used for visual representation of the data, comparison with healthy controls, and multiple regression analysis with addiction severity, described below. Anatomic specificity was determined with the Anatomy toolbox.48
Secondary (control) analyses compared methylphenidate peak effects vs same-day baseline and methylphenidate same-day baseline vs placebo same-day baseline, both used to rule out ancillary factors particular to each study session (eg, pre-medication administration cocaine craving and days since last cocaine use) (Table 2). Results from the control analyses were masked by the methylphenidate peak effects vs placebo peak effects contrast (P < .05 uncorrected) and are reported at P < .005 uncorrected with a minimum cluster extent of 20 adjacent voxels.
Finally, we sought to determine whether methylphenidate modified connectivity in participants with CUD to a level that no longer significantly differs from that of healthy individuals. For this purpose, we compared connectivity strength during placebo and, separately, during methylphenidate with that of an independent sample of 16 healthy control participants for whom we acquired resting-state data under placebo conditions (see the eAppendix in the Supplement for additional information).
Association of Connectivity Strength With Severity of Cocaine Addiction
To determine whether methylphenidate modified connectivity between regions that, under active baseline conditions (ie, placebo peak effects), were directly associated with severity of cocaine addiction, we used the composite addiction severity score (Table 2) as the dependent variable in a multiple regression analysis in SPSS 18.0 (SPSS, Inc) restricting the predictors to include connectivity measures in regions that both changed after methylphenidate and differed significantly from healthy controls (either in the present study [eTable 2 in the Supplement] or as reported by Gu et al12) (6 total predictors).
Results
Effects of Methylphenidate on Cardiovascular Reactivity and Subjective Mood
Except for diastolic blood pressure (which was higher after methylphenidate than after placebo), changes in cardiovascular reactivity and mood did not differ significantly between the methylphenidate and placebo study sessions (eTable 1 in the Supplement). Consistent with prior studies using 20 mg of oral methylphenidate,17,19 self-reports of cocaine wanting also did not differ significantly (eAppendix in the Supplement).
Effects of Methylphenidate on Connectivity Strength
Primary Analyses: Methylphenidate vs Placebo Peak Effects
The region of interest analyses indicated that methylpheni-date modified 2 corticolimbic connections that were previously reported to be disrupted in CUD12: compared with placebo, methylphenidate increased the connectivity of (1) the right and left hippocampus with the left postcentral gyrus (BAs 4, 6) (peak coordinate in MNI space: x = −60, y = −3, and z = 33, Z = 4.39, FWE-corrected P = .008; and x = −63, y = 3, and z = 30, Z = 4.05, FWE-corrected P = .03, respectively) (Figure 2) and (2) the left rostral anterior cingulate with the right parahippocampal gyrus (x = 15, y = −36, and z = −9, Z = 3.57, FWE-corrected P = .02) (Figure 3A and B).
Figure 2. Changes in Hippocampal (Hipp) Connectivity With Methylphenidate.
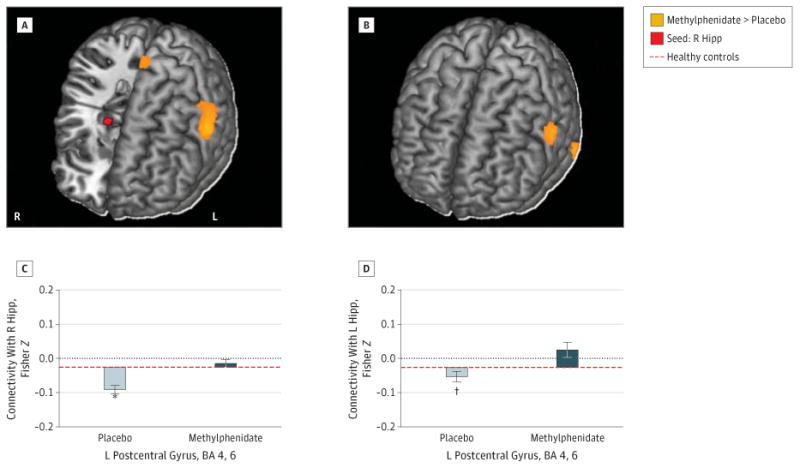
Increased left (L) postcentral gyrus (Brodmann areas [BAs] 4, 6) functional connectivity with the right (R) (A and C) and L (B and D) Hipp seeds (shown in red) following a single dose of oral methylphenidate hydrochloride, 20 mg. Color maps (A and B) show increased connectivity strength with methylphenidate vs placebo (orange) in a T-score window from ±3.0 to ±7.0. Bar plots (C and D) show the Fisher Z values for placebo peak effects (light gray) and methylphenidate peak effects (dark gray) plotted from values of healthy control participants scanned during placebo conditions. Error bars represent SEM. *P < .005. †P < .05.
Figure 3. Changes in Rostral Anterior Cingulate Connectivity With Methylphenidate.
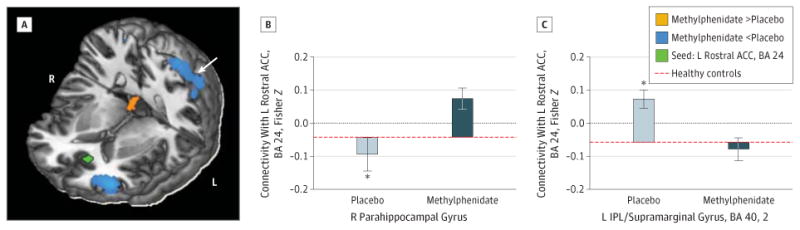
Increased left (L) rostral anterior cingulate cortex (ACC) (Brodmann area [BA] 24) (seed shown in green) with right (R) parahippocampal gyrus connectivity and decreased L rostral anterior cingulate with inferior parietal cortex connectivity following a single dose of oral methylphenidate hydrochloride, 20 mg. Color map (A) shows increased (orange) or decreased (cyan) connectivity strength with methylphenidate vs placebo in a T-score window from ±3.0 to ±7.0. Bar plots (B and C) show the Fisher Z values for placebo peak effects (light gray) and methylphenidate peak effects (dark gray) plotted from values of healthy control participants scanned during placebo conditions. Error bars represent SEM. IPL indicates inferior parietal lobule. *P < .05.
Whole-brain analyses revealed additional effects of methylphenidate in connections relevant to self-control and craving (Table 3 and eFigure 2 in the Supplement). Specifically, compared with placebo, methylphenidate increased the connectivity of (1) the right rostral anterior cingulate with the left dorsal cingulate (eFigure 3 in the Supplement), (2) the right thalamus with the medial orbitofrontal cortex, and (3) the right nucleus accumbens with the medial orbitofrontal cortex and right superior temporal gyrus extending to the postcentral gyrus and rolandic operculum (Figure 4A and C). Methylpheni-date also reduced the connectivity of (1) the ventral tegmental area with the right caudate and putamen; (2) the left hippocampus with the left insula, thalamus, and putamen; (3) the right thalamus with the bilateral putamen; and (4) the right nucleus accumbens with the left putamen/globus pallidus (Figure 4A and D). Other methylphenidate-induced changes included reduced connectivity of the ventral tegmental area, right hippocampus, and left rostral anterior cingulate with the cerebellum and reduced connectivity of the left rostral anterior cingulate (Figure 3C), right hippocampus, and right nucleus accumbens with the inferior parietal cortex extending to the angular gyrus and precuneus. No significant changes in connectivity strength were observed for the bilateral amygdala, left thalamus, or left nucleus accumbens seeds in either the region of interest or the whole-brain analyses.
Table 3. Whole-Brain Changes in Resting-State Functional Connectivity With Methylphenidatea.
| Characteristic | BA | Side | Cluster Size, mm3 | Peak Z Valueb | P Value, Correctedc | MNI Coordinates | ||
|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||
| Seed: VTA | ||||||||
| Caudate/putamend | R | 385 | −4.3 | .03 | 15 | 15 | 6 | |
| 18 | 6 | 15 | ||||||
| Cerebellumd | L | 540 | −3.7 | .008 | −6 | −45 | −45 | |
| R | −6 | −48 | −33 | |||||
| Seed: R hippocampus | ||||||||
| Cerebellumd | R | 943 | −4.3 | <.001 | 3 | −60 | −36 | |
| 15 | −66 | −27 | ||||||
| Superior frontal gyrus | 10 | R | 397 | −3.9 | .04 | 15 | 57 | 18 |
| L | 9 | 60 | 27 | |||||
| Middle occipital/angular gyrus/precuneusd | 40, 7, 23 | L | 436 | −3.5 | .03 | −39 | −66 | 39 |
| −9 | −60 | 30 | ||||||
| Seed: L hippocampus | ||||||||
| Superior temporal gyrus/postcentral gyrusd | 43, 22 | R | 417 | +3.6 | .03 | 60 | −6 | −3 |
| 63 | −21 | 6 | ||||||
| Insula/thalamus/putamend | L | 702 | −4.0 | .003 | −33 | 27 | −3 | |
| −15 | −9 | 6 | ||||||
| Seed: R MDN thalamus | ||||||||
| Medial orbitofrontal cortex/gyrus rectusd | 11 | L | 408 | +4.6 | .04 | −18 | 48 | −15 |
| R | 0 | 48 | −21 | |||||
| Putamen/thalamusd | L | 1148 | −4.1 | <.001 | −30 | −6 | −9 | |
| R | 30 | −9 | −6 | |||||
| Seed: R rostral ACC (BA 24) | ||||||||
| Dorsal ACCd | 32, 24 | L | 335 | +4.0 | .058 | −3 | 18 | 24 |
| 0 | 39 | 27 | ||||||
| Seed: L rostral ACC (BA 24) | ||||||||
| Cerebellumd | R | 593 | −4.3 | .007 | 12 | −24 | −30 | |
| L | −12 | −45 | −33 | |||||
| Inferior parietal/supramarginal gyrusd,e | 40, 2 | L | 501 | −3.7 | .016 | −33 | −45 | 69 |
| −51 | −51 | 54 | ||||||
| Seed: R NAcc | ||||||||
| Superior temporal gyrus/postcentral gyrus/ rolandic operculumd,e | 20, 22, 38 | R | 1292 | +4.7 | <.001 | 57 | 3 | 0 |
| 63 | −18 | −27 | ||||||
| Medial orbitofrontal cortex/gyrus rectusd | 11 | R | 823 | +4.5 | .001 | 6 | 30 | −15 |
| 9 | 45 | −24 | ||||||
| Inferior parietal/angular gyrusd | 40, 39 | L | 841 | −5.2 | <.001 | −54 | −51 | 48 |
| −54 | −60 | 33 | ||||||
| Putamen/globus pallidus/thalamusd,e,f | L | 2671 | −5.0 | <.001 | −27 | −3 | 0 | |
| −6 | −6 | −3 | ||||||
Abbreviations: ACC, anterior cingulate cortex; BA, Brodmann area; L, left; MDN, mediodorsal nucleus; MNI, Montreal Neurological Institute; NAcc, nucleus accumbens; R, right; VTA, ventral tegmental area.
Analysis of covariance (covariates: medication administration paradigm, baseline craving, days since last cocaine use, and micromotion).
Positive Z score indicates increased connectivity strength with methylphenidate hydrochloride (methylphenidate > placebo peak effects). Negative Z score indicates decreased connectivity strength with methylphenidate (methylphenidate < placebo peak effects).
Statistical threshold: cluster-level P < .05 familywise error–corrected with a voxel-level P < .005 uncorrected height threshold and k = 20 voxels (T = 2.65). No significant effects of methylphenidate were observed for the bilateral amygdala, left thalamus, or left NAcc seeds at the set significance threshold.
Region also showing effects for the methylphenidate peak effects vs same-day baseline contrast (voxel-level P < .005 uncorrected and k = 20 voxels within a methylphenidate vs placebo peak effects P < .05 uncorrected inclusive mask). In all regions, no differences were observed for the methylphenidate same-day baseline vs placebo same-day baseline contrast using the same statistical threshold and masking procedure.
Region significantly different from healthy controls during placebo peak effects (eTable 1 in eAppendix in the Supplement).
Region associated with the addiction severity composite score.
Figure 4. Changes in Nucleus Accumbens (NAcc) Connectivity With Methylphenidate and Relationship to Addiction Severity.
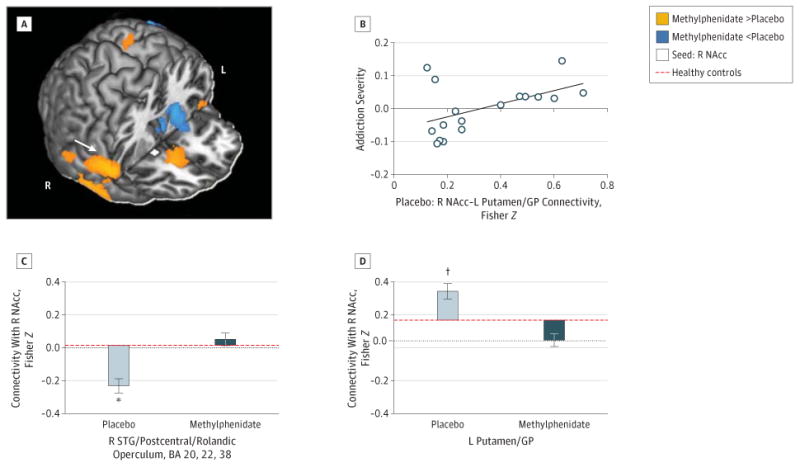
Increased right (R) NAcc (A) (seed shown in white) with R superior temporal gyrus (STG) extending to the postcentral gyrus and rolandic operculum connectivity (C) and decreased R NAcc with left (L) putamen/globus pallidus (GP) connectivity (D) following a single dose of oral methylphenidate hydrochloride, 20 mg. Color map (A) shows increased (orange) or decreased (cyan) connectivity strength with methylphenidate vs placebo in a T-score window from ±3.0 to ±7.0. Bar plots (C and D) show the Fisher Z values for placebo peak effects (light gray) and methylphenidate peak effects (dark gray) plotted from values of healthy control participants scanned during placebo conditions. Error bars represent SEM. The connectivity strength between the R NAcc and L putamen/GP during placebo (C) was uniquely positively correlated with the severity of cocaine addiction composite scores (B). *P < .005. †P < .05.
Control Analyses
Supporting the idea that these changes in connectivity were due to the pharmacologic effects of methylphenidate (and not differences between the study days or the participants' expectation to receive methylphenidate), baseline connectivity did not differ significantly between the 2 study days for any of the seeds (as inspected with the methylphenidate baseline vs placebo baseline contrast, masked by the methylphenidate peak > placebo peak or methylphenidate peak < placebo peak contrasts reported above). The methylphenidate peak effect vs same-day baseline contrast was similarly examined and, in contrast, revealed significant results in most brain regions identified by the methylphenidate peak vs placebo peak contrast (regions identified in both analyses are indicated in Table 3).
Notably, methylphenidate generally did not modify connectivity with our control seeds, with the exception of an increase in connectivity between the primary motor cortex and the cerebellum. This exception is a well-established motor pathway that also depends on dopamine (eAppendix in the Supplement).
Comparison With Health
Within these connections, relative to healthy controls who were studied during active baseline (ie, during placebo peak effects), individuals with CUD showed reduced connectivity of (1) the bilateral hippocampus with the left postcentral gyrus (corroborating findings by Gu et al12) and (2) the right nucleus accumbens with the right superior temporal gyrus, extending to the postcentral gyrus and rolandic operculum, and increased connectivity of (1) the left rostral anterior cingulate with the left inferior parietal cortex and (2) the right nucleus accumbens with the left putamen/globus pallidus (eTable 2 in the Supplement). More important, after methylphenidate administration, group differences in all of these connections were no longer significant, suggesting that methylphenidate normalized connectivity strength between these regions.
Relationship to Severity of Cocaine Addiction
To determine whether connectivity modified by methylphenidate was directly associated with cocaine addiction severity, we conducted a multiple regression analysis in SPSS with addiction severity as the dependent variable. The predictors (ie, the extracted average connectivity measures during placebo peak effects) in this analysis included the 6 connectivity pathways that significantly differed from those of healthy controls during placebo administration in the present study or as reported by Gu et al12 and that were normalized in strength with methylphenidate (regions in bold in eTable 2 in the Supplement plus left rostral anterior cingulate-right parahippocampal gyrus connectivity). These 6 predictors accounted for 24% of the variance in addiction severity (adjusted R2 = 0.24, P = .18). However, only connectivity between the right nucleus accumbens and left putamen/globus pallidus accounted for significant unique variance (β = 0.61, P = .04; for all others, P > .16) (Figure 4B). That is, methylphenidate reduced connectivity strength between these regions (described above), and lower connectivity strength during placebo was associated with less severe addiction. The severity of addiction did not interact with the effects of methylphenidate, suggesting that changes in connectivity with methylphenidate did not differ in cocaine users of different severity (eAppendix in the Supplement).
Discussion
Using short-term oral administration of methylphenidate, the present study showed that mesocorticolimbic connectivity is susceptible to dopaminergic manipulation in CUD. Taking into account differences in baseline connectivity from healthy individuals and correlations between these measures and severity of addiction, the direction of change in connectivity strength with methylphenidate is consistent with a beneficial response to the drug, extending its previously reported efficacy in normalizing task-related brain activation and behavior in this population.17,18 Moreover, our study design enabled us to show that connectivity with our seed regions was stable such that the observed changes in connectivity were due to the effects of methylphenidate and not ancillary factors particular to each study session.
Using predefined regions of interest, we found that methylphenidate strengthened connectivity of the bilateral hippocampus with the postcentral gyrus and of the rostral anterior cingulate with the parahippocampal gyrus–corticolimbic connections suggested to underlie successful emotion regulation21-23 that were reported to be disrupted in cocaine addiction.12 In particular, the postcentral gyrus is involved in craving suppression,49 and both the postcentral gyrus and the hippocampus fail to normally activate when cocaine users are exposed to stress, abnormalities that could underlie stress-related vulnerability to cocaine relapse.50 Strengthened connectivity in emotion processing and memory formation path-ways with methylphenidate may contribute to enhanced retention of emotional associative learning (as has been shown in rodents51,52) and better control over emotional disturbances and emotional memories (eg, when combined with exposure therapy), particularly those associated with withdrawal symptoms and conditioned responses that frequently lead to relapse in addiction.53
In addition to strengthened corticolimbic connectivity, methylphenidate strengthened connectivity of the rostral anterior cingulate with the dorsal cingulate. These cingulate regions have differential anatomic connections with emotion and cognitive control networks,54 and reduced correlations have been observed between these regions during processing of salient cues in cocaine users.55 Therefore, strengthened frontal cortical connectivity with methylphenidate may point to the mechanism that contributes to the methylphenidate-induced improvements in behavioral and neural measures of self-control on both neutral18 and salient17 tasks of executive function in CUD. This finding is also important in view of tractography studies in CUD, in which higher fractional anisotropy in regions of the frontal cortex and the rostral corpus callosum linking these regions predicts longer abstinence.56 Although methylphenidate increased connectivity of the rostral with dorsal cingulate, an executive control and attention network region, methylphenidate reduced connectivity of the rostral cingulate with the inferior parietal cortex/supramarginal gyrus, a “default mode” region, which may play a role in methylphenidate's attention-enhancing properties.57
Most notably, methylphenidate reduced the connectivity of several subcortical regions, including the ventral tegmental area, hippocampus, thalamus, and nucleus accumbens, with the dorsal striatum. In particular, connectivity within striatal circuits, possibly instantiated via the spiraling dopamine connections through the midbrain that link the nucleus accumbens with the dorsolateral striatum or via other nodes of the cortico-thalamic-striatal loops,58 is strongly implicated in drug-seeking.59 Because the progression of cocaine addiction involves a shift in striatal circuits from ventral to dorsal,26,60-62 the strength of connectivity between these regions may be marking individual differences in disease severity. Indeed, higher baseline connectivity of the nucleus accumbens with the putamen/globus pallidus uniquely correlated with more severe addiction. The finding that short-term methylphenidate can modify this connection may be clinically relevant given that blocking striato-midbrain-striatal serial connectivity selectively decreased drug-seeking in rats trained to habitually self-administer cocaine.59 Future longer-term intervention studies should test whether systematic, prolonged weakening of this connection helps restore control over drug-seeking behavior in humans. Other effects of methylphenidate may also contribute to increased control over craving and drug-seeking, including methylphenidate-strengthened connectivity of the nucleus accumbens with a region encompassing the superior temporal and postcentral gyri and the rolandic operculum. Indeed, cognitive control of craving is associated with inverse coupling between these cortical regions and the nucleus accumbens in addicted individuals using different strategies to resist craving.63,64
Although not hypothesized a priori or identified as abnormal relative to healthy controls, connectivity in other pathways modified by methylphenidate also may be relevant to addiction; further research is needed to clarify the precise mechanisms and consequences of these changes. For example, because the locus ceruleus, which is in close proximity to our ventral tegmental area seed, is the main source of nor-adrenergic innervation to the cerebellum,65 connectivity reductions in this pathway could be the result of differential effects of methylphenidate on norepinephrine. Other effects may also have noradrenergic underpinnings, including methylphenidate-strengthened connectivity of the thalamus with the medial orbitofrontal cortex, potentially signaling normalization of noradrenergic deficits in this region in cocaine abusers.66
Several caveats should be considered when interpreting the current results. First, like most stimulants, methylpheni-date produced cardiovascular changes that differed from those with placebo. However, because blood oxygen level– dependent fluctuations correlating with heart rate and respiration are global,67,68 these changes are not likely to significantly influence our results. At the neurochemical level, a second concern is that the effects of methylphenidate are not specific to dopamine because methylphenidate also blocks the norepinephrine transporter69 (and may account for some of its effects, as discussed above). Nevertheless, even if effects are the result of norepinephrine transporter blockade, the underlying mechanism could still be dopaminergic, since dopamine also has high affinity for norepinephrine transporters— particularly in regions where norepinephrine transporters are more abundant than dopamine transporters, such as the frontal cortex.70 Third, similar to prior studies, we detected a number of negative relationships with our seed regions. One concern that arises is the possible contribution of analytical procedures (eg, global signal normalization).41,71,72 However, prior work suggests that negative relationships cannot simply be attributed to correction for the global signal73 and that removing global signal from the data, which is primarily localized to gray matter, can actually improve neuroanatomic specificity of positive relationships.74 Never the less, because our contrast of interest was a within-subject change in connectivity, this issue is unlikely to modify our conclusions. Fourth, although seed-based methods for connectivity are one of the most commonly used approaches, in part because these methods are reliable75 and especially appropriate when researchers have specific a priori hypotheses,76 they are limited by (1) the univariate nature of the analysis, which limits the scope of network-level conclusions that can be drawn,76 and (2) susceptibility to biases related to variations in seed positioning.76,77 We addressed this latter limitation by using seeds identical to those used in previous research.12 Future studies could also apply complementary methods (eg, those based on graph theory42 or independent component analysis) to capture the effects of methylphenidate on brain networks. Last, we cannot at present speak to whether methyl-phenidate-induced modulation of connectivity is a viable target for treatment approaches in CUD. Future studies would need to examine the effects of methylphenidate using dose-dependent and/or prolonged administration (ie, occurring over days or weeks). In addition, because methylphenidate as a stand-alone treatment may be insufficient to achieve a positive clinical outcome in cocaine addiction,78,79 except for in instances of comorbidity with attention-deficit/hyperactivity disorder,80 the therapeutic effects of methylphenidate should be explored in conjunction with behavioral interventions targeted toward increasing emotion regulation and self-control and decreasing conditioned associations.
In summary, our findings provide novel evidence that mesocorticolimbic connectivity is susceptible to modification by pharmacologic agents targeting dopamine in individuals with chronic, severe CUD. Methylphenidate primarily strengthened connections between regions underlying emotion regulation and cognitive control and reduced connections between regions underlying habits, including compulsive drug-seeking and craving. Although the precise mechanism of these effects remains to be determined, our data suggest that methylphenidate may transiently, and independently of task demands, modify striatal and cortical synchronous activity with connected brain regions. These changes could serve to facilitate behavior or make cortical processing underlying behavior more efficient57,81-83 or less difficult to override.84 By highlighting effects in mesocorticolimbic pathways in CUD, our results extend prior work that has shown pharmacologic modulation of default mode and executive control network circuitry in nicotine dependence (via nicotine patch7,8) and limbic circuitry in depression (via the antidepressant sertraline). A better understanding of the potentially therapeutic effects of methylphenidate on neural circuitry function in CUD (eg, with future studies evaluating the clinical efficacy of methylphenidate) may promote the development of improved treatment options for stimulant addictions.
Supplementary Material
Acknowledgments
Funding/Support: This research was conducted with grant support from the National Institute on Drug Abuse (1R01DA023579 to Dr Goldstein and 1F32DA030017-01 to Dr Moeller).
Footnotes
Author Contributions: Ms Konova and Drs Moeller, Tomasi, and Goldstein had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Tomasi, Volkow, Goldstein.
Acquisition of data: Tomasi.
Analysis and interpretation of data: Konova, Moeller, Tomasi, Goldstein.
Drafting of the manuscript: Konova, Moeller.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Konova, Moeller, Tomasi.
Obtained funding: Goldstein.
Administrative, technical, and material support: Tomasi, Volkow, Goldstein.
Study supervision: Tomasi, Volkow, Goldstein.
Conflict of Interest Disclosures: None reported.
Additional Contributions: Nelly Alia-Klein, PhD; Daniel Carrero, MS; Tom Maloney, PhD; Muhammad A. Parvaz, PhD; Patricia A. Woicik, PhD; Gene-Jack Wang, MD; Frank Telang, MD; Barbara Hubbard, RN; and the entire Neuropsychoimaging and PET group provided assistance with participant recruitment and study procedures.
Contributor Information
Anna B. Konova, Departments of Psychiatry and Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York; Department of Psychology, Stony Brook University, Stony Brook, New York.
Scott J. Moeller, Departments of Psychiatry and Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York.
Dardo Tomasi, National Institute on Alcohol Abuse and Alcoholism, Rockville, Maryland.
Nora D. Volkow, National Institute on Alcohol Abuse and Alcoholism, Rockville, Maryland; National Institute on Drug Abuse, Bethesda, Maryland.
Rita Z. Goldstein, Departments of Psychiatry and Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York.
References
- 1.Rosazza C, Minati L. Resting-state brain networks: literature review and clinical applications. Neurol Sci. 2011;32(5):773–785. doi: 10.1007/s10072-011-0636-y. [DOI] [PubMed] [Google Scholar]
- 2.Shmuel A, Leopold DA. Neuronal correlates of spontaneous fluctuations in fMRI signals in monkey visual cortex: implications for functional connectivity at rest. Hum Brain Mapp. 2008;29(7):751–761. doi: 10.1002/hbm.20580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8(9):700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- 4.Damoiseaux JS, Greicius MD. Greater than the sum of its parts: a review of studies combining structural connectivity and resting-state functional connectivity. Brain Struct Funct. 2009;213(6):525–533. doi: 10.1007/s00429-009-0208-6. [DOI] [PubMed] [Google Scholar]
- 5.Hampson M, Driesen NR, Skudlarski P, Gore JC, Constable RT. Brain connectivity related to working memory performance. J Neurosci. 2006;26(51):13338–13343. doi: 10.1523/JNEUROSCI.3408-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly AM, Uddin LQ, Biswal BB, Castellanos FX, Milham MP. Competition between functional brain networks mediates behavioral variability. Neuroimage. 2008;39(1):527–537. doi: 10.1016/j.neuroimage.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Cole DM, Beckmann CF, Long CJ, Matthews PM, Durcan MJ, Beaver JD. Nicotine replacement in abstinent smokers improves cognitive withdrawal symptoms with modulation of resting brain network dynamics. Neuroimage. 2010;52(2):590–599. doi: 10.1016/j.neuroimage.2010.04.251. [DOI] [PubMed] [Google Scholar]
- 8.Hong LE, Gu H, Yang Y, et al. Association of nicotine addiction and nicotine's actions with separate cingulate cortex functional circuits. Arch Gen Psychiatry. 2009;66(4):431–441. doi: 10.1001/archgenpsychiatry.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma N, Liu Y, Li N, et al. Addiction related alteration in resting-state brain connectivity. Neuroimage. 2010;49(1):738–744. doi: 10.1016/j.neuroimage.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Upadhyay J, Maleki N, Potter J, et al. Alterations in brain structure and functional connectivity in prescription opioid-dependent patients. Brain. 2010;133(pt 7):2098–2114. doi: 10.1093/brain/awq138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma N, Liu Y, Fu XM, et al. Abnormal brain default-mode network functional connectivity in drug addicts. PLoS One. 2011;6(1):e16560. doi: 10.1371/journal.pone.0016560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu H, Salmeron BJ, Ross TJ, et al. Mesocorticolimbic circuits are impaired in chronic cocaine users as demonstrated by resting-state functional connectivity. Neuroimage. 2010;53(2):593–601. doi: 10.1016/j.neuroimage.2010.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly C, Zuo XN, Gotimer K, et al. Reduced interhemispheric resting state functional connectivity in cocaine addiction. Biol Psychiatry. 2011;69(7):684–692. doi: 10.1016/j.biopsych.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem. 1997;68(5):2032–2037. doi: 10.1046/j.1471-4159.1997.68052032.x. [DOI] [PubMed] [Google Scholar]
- 15.Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9(6):557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- 16.Levin FR, Evans SM, Brooks DJ, Garawi F. Treatment of cocaine dependent treatment seekers with adult ADHD: double-blind comparison of methylphenidate and placebo. Drug Alcohol Depend. 2007;87(1):20–29. doi: 10.1016/j.drugalcdep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein RZ, Woicik PA, Maloney T, et al. Oral methylphenidate normalizes cingulate activity in cocaine addiction during a salient cognitive task. Proc Natl Acad Sci USA. 2010;107(38):16667–16672. doi: 10.1073/pnas.1011455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li CS, Morgan PT, Matuskey D, et al. Biological markers of the effects of intravenous methylphenidate on improving inhibitory control in cocaine-dependent patients. Proc Natl Acad Sci U S A. 2010;107(32):14455–14459. doi: 10.1073/pnas.1002467107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volkow ND, Wang GJ, Tomasi D, et al. Methylphenidate attenuates limbic brain inhibition after cocaine-cues exposure in cocaine abusers. PLoS One. 2010;5(7):e11509. doi: 10.1371/journal.pone.0011509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li SJ, Biswal B, Li Z, et al. Cocaine administration decreases functional connectivity in human primary visual and motor cortex as detected by functional MRI. Magn Reson Med. 2000;43(1):45–51. doi: 10.1002/(sici)1522-2594(200001)43:1<45::aid-mrm6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 21.Diekhof EK, Geier K, Falkai P, Gruber O. Fear is only as deep as the mind allows: a coordinate-based meta-analysis of neuroimaging studies on the regulation of negative affect. Neuroimage. 2011;58(1):275–285. doi: 10.1016/j.neuroimage.2011.05.073. [DOI] [PubMed] [Google Scholar]
- 22.Mak AK, Hu ZG, Zhang JX, Xiao ZW, Lee TM. Neural correlates of regulation of positive and negative emotions: an fMRI study. Neurosci Lett. 2009;457(2):101–106. doi: 10.1016/j.neulet.2009.03.094. [DOI] [PubMed] [Google Scholar]
- 23.Kanske P, Heissler J, Schönfelder S, Bongers A, Wessa M. How to regulate emotion? neural networks for reappraisal and distraction. Cereb Cortex. 2011;21(6):1379–1388. doi: 10.1093/cercor/bhq216. [DOI] [PubMed] [Google Scholar]
- 24.Chase HW, Eickhoff SB, Laird AR, Hogarth L. The neural basis of drug stimulus processing and craving: an activation likelihood estimation meta-analysis. Biol Psychiatry. 2011;70(8):785–793. doi: 10.1016/j.biopsych.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kühn S, Gallinat J. Common biology of craving across legal and illegal drugs–a quantitative meta-analysis of cue-reactivity brain response. Eur J Neurosci. 2011;33(7):1318–1326. doi: 10.1111/j.1460-9568.2010.07590.x. [DOI] [PubMed] [Google Scholar]
- 26.Volkow ND, Wang GJ, Telang F, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci. 2006;26(24):6583–6588. doi: 10.1523/JNEUROSCI.1544-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.First MB, Spitzer RL, Gibbon M, Williams J. Structured Clinical Interview for DSM-IV Axis I Disorders–Patient Edition (SCID-I/P, Version 2.0) New York: Biometrics Research Dept, New York State Psychiatric Institute; 1996. [Google Scholar]
- 28.Ventura J, Liberman RP, Green MF, Shaner A, Mintz J. Training and quality assurance with the Structured Clinical Interview for DSM-IV (SCID-I/P) Psychiatry Res. 1998;79(2):163–173. doi: 10.1016/s0165-1781(98)00038-9. [DOI] [PubMed] [Google Scholar]
- 29.McLellan AT, Kushner H, Metzger D, et al. The fifth edition of the Addiction Severity Index. J Subst Abuse Treat. 1992;9(3):199–213. doi: 10.1016/0740-5472(92)90062-s. [DOI] [PubMed] [Google Scholar]
- 30.Wilkinson G. The Wide-Range Achievement Test 3–Administration Manual. Wilmington, DE: Wide Range Inc; 1993. [Google Scholar]
- 31.Wechsler D. Wechsler Abbreviated Scale of Intelligence. San Antonio, TX: Psychological Corp; 1999. [Google Scholar]
- 32.Kampman KM, Volpicelli JR, McGinnis DE, et al. Reliability and validity of the Cocaine Selective Severity Assessment. Addict Behav. 1998;23(4):449–461. doi: 10.1016/s0306-4603(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 33.Tiffany ST, Singleton E, Haertzen CA, Henningfield JE. The development of a cocaine craving questionnaire. Drug Alcohol Depend. 1993;34(1):19–28. doi: 10.1016/0376-8716(93)90042-o. [DOI] [PubMed] [Google Scholar]
- 34.Gossop M, Griffiths P, Powis B, Strang J. Severity of dependence and route of administration of heroin, cocaine and amphetamines. Br J Addiction. 1992;87(11):1527–1536. doi: 10.1111/j.1360-0443.1992.tb02660.x. [DOI] [PubMed] [Google Scholar]
- 35.Beck AT, Steer RA, Brown GK. Beck Depression Inventory Manual. 2nd. San Antonio, TX: Psychological Corp; 1996. [Google Scholar]
- 36.Goldstein RZ, Tomasi D, Rajaram S, et al. Role of the anterior cingulate and medial orbitofrontal cortex in processing drug cues in cocaine addiction. Neuroscience. 2007;144(4):1153–1159. doi: 10.1016/j.neuroscience.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moeller S, Tomasi D, Honorio J, Volkow ND, Goldstein RZ. Dopaminergic involvement during mental fatigue in health and cocaine addiction. Transl Psychiatr. 2012;2:e176. doi: 10.1038/tp.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkow ND, Wang GJ, Fowler JS, et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155(10):1325–1331. doi: 10.1176/ajp.155.10.1325. [DOI] [PubMed] [Google Scholar]
- 39.Tomasi D, Caparelli EC, Chang L, Ernst T. fMRI-acoustic noise alters brain activation during working memory tasks. Neuroimage. 2005;27(2):377–386. doi: 10.1016/j.neuroimage.2005.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomasi DG, Ernst T. Echo planar imaging at 4 Tesla with minimum acoustic noise. J Magn Reson Imaging. 2003;18(1):128–130. doi: 10.1002/jmri.10326. [DOI] [PubMed] [Google Scholar]
- 41.Van Dijk KR, Hedden T, Venkataraman A, Evans KC, Lazar SW, Buckner RL. Intrinsic functional connectivity as a tool for human connectomics: theory, properties, and optimization. J Neurophysiol. 2010;103(1):297–321. doi: 10.1152/jn.00783.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomasi D, Volkow ND. Functional connectivity density mapping. Proc Natl Acad Sci USA. 2010;107(21):9885–9890. doi: 10.1073/pnas.1001414107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Dijk KR, Sabuncu MR, Buckner RL. The influence of head motion on intrinsic functional connectivity MRI. Neuroimage. 2012;59(1):431–438. doi: 10.1016/j.neuroimage.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomasi D, Volkow ND. Language network: segregation, laterality and connectivity. Mol Psychiatry. 2012;17(8):759. doi: 10.1038/mp.2012.99. [DOI] [PubMed] [Google Scholar]
- 45.Tomasi D, Volkow ND. Abnormal functional connectivity in children with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;71(5):443–450. doi: 10.1016/j.biopsych.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomasi D, Volkow ND, Wang R, et al. Disrupted functional connectivity with dopaminergic midbrain in cocaine abusers. PLoS One. 2010;5(5):e10815. doi: 10.1371/journal.pone.0010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Worsley KJ, Evans AC, Marrett S, Neelin P. A three-dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab. 1992;12(6):900–918. doi: 10.1038/jcbfm.1992.127. [DOI] [PubMed] [Google Scholar]
- 48.Eickhoff SB, Stephan KE, Mohlberg H, et al. A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage. 2005;25(4):1325–1335. doi: 10.1016/j.neuroimage.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 49.Brody AL, Mandelkern MA, Olmstead RE, et al. Neural substrates of resisting craving during cigarette cue exposure. Biol Psychiatry. 2007;62(6):642–651. doi: 10.1016/j.biopsych.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinha R, Lacadie C, Skudlarski P, et al. Neural activity associated with stress-induced cocaine craving: a functional magnetic resonance imaging study. Psychopharmacology (Berl) 2005;183(2):171–180. doi: 10.1007/s00213-005-0147-8. [DOI] [PubMed] [Google Scholar]
- 51.Bethancourt JA, Camarena ZZ, Britton GB. Exposure to oral methylphenidate from adolescence through young adulthood produces transient effects on hippocampal-sensitive memory in rats. Behav Brain Res. 2009;202(1):50–57. doi: 10.1016/j.bbr.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Abraham AD, Cunningham CL, Lattal KM. Methylphenidate enhances extinction of contextual fear. Learn Mem. 2012;19(2):67–72. doi: 10.1101/lm.024752.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- 54.Margulies DS, Kelly AM, Uddin LQ, Biswal BB, Castellanos FX, Milham MP. Mapping the functional connectivity of anterior cingulate cortex. Neuroimage. 2007;37(2):579–588. doi: 10.1016/j.neuroimage.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 55.Goldstein RZ, Alia-Klein N, Tomasi D, et al. Anterior cingulate cortex hypoactivations to an emotionally salient task in cocaine addiction. Proc Natl Acad Sci USA. 2009;106(23):9453–9458. doi: 10.1073/pnas.0900491106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, DeVito EE, Worhunsky PD, Carroll KM, Rounsaville BJ, Potenza MN. White matter integrity is associated with treatment outcome measures in cocaine dependence. Neuropsychopharmacology. 2010;35(7):1541–1549. doi: 10.1038/npp.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomasi D, Volkow ND, Wang GJ, et al. Methylphenidate enhances brain activation and deactivation responses to visual attention and working memory tasks in healthy controls. Neuroimage. 2011;54(4):3101–3110. doi: 10.1016/j.neuroimage.2010.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20(6):2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57(3):432–441. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 60.Porrino LJ, Smith HR, Nader MA, Beveridge TJ. The effects of cocaine: a shifting target over the course of addiction. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(8):1593–1600. doi: 10.1016/j.pnpbp.2007.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3125–3135. doi: 10.1098/rstb.2008.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8(11):1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 63.Volkow ND, Fowler JS, Wang GJ, et al. Cognitive control of drug craving inhibits brain reward regions in cocaine abusers. Neuroimage. 2010;49(3):2536–2543. doi: 10.1016/j.neuroimage.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kober H, Mende-Siedlecki P, Kross EF, et al. Prefrontal-striatal pathway underlies cognitive regulation of craving. Proc Natl Acad Sci USA. 2010;107(33):14811–14816. doi: 10.1073/pnas.1007779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogawski MA, Aghajanian GK. Modulation of lateral geniculate neurone excitability by noradrenaline microiontophoresis or locus coeruleus stimulation. Nature. 1980;287(5784):731–734. doi: 10.1038/287731a0. [DOI] [PubMed] [Google Scholar]
- 66.Ding YS, Singhal T, Planeta-Wilson B, et al. PET imaging of the effects of age and cocaine on the norepinephrine transporter in the human brain using (S,S)-[11C]O-methylreboxetine and HRRT. Synapse. 2010;64(1):30–38. doi: 10.1002/syn.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Birn RM, Diamond JB, Smith MA, Bandettini PA. Separating respiratory-variation-related fluctuations from neuronal-activity-related fluctuations in fMRI. Neuroimage. 2006;31(4):1536–1548. doi: 10.1016/j.neuroimage.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 68.Chang C, Glover GH. Effects of model-based physiological noise correction on default mode network anti-correlations and correlations. Neuroimage. 2009;47(4):1448–1459. doi: 10.1016/j.neuroimage.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hannestad J, Gallezot JD, Planeta-Wilson B, et al. Clinically relevant doses of methylphenidate significantly occupy norepinephrine transporters in humans in vivo. Biol Psychiatry. 2010;68(9):854–860. doi: 10.1016/j.biopsych.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morón JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22(2):389–395. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murphy K, Birn RM, Handwerker DA, Jones TB, Bandettini PA. The impact of global signal regression on resting state correlations: are anti-correlated networks introduced? Neuroimage. 2009;44(3):893–905. doi: 10.1016/j.neuroimage.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saad ZS, Gotts SJ, Murphy K, et al. Trouble at rest: how correlation patterns and group differences become distorted after global signal regression. Brain Connect. 2012;2(1):25–32. doi: 10.1089/brain.2012.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fransson P. Spontaneous low-frequency BOLD signal fluctuations: an fMRI investigation of the resting-state default mode of brain function hypothesis. Hum Brain Mapp. 2005;26(1):15–29. doi: 10.1002/hbm.20113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fox MD, Zhang D, Snyder AZ, Raichle ME. The global signal and observed anticorrelated resting state brain networks. J Neurophysiol. 2009;101(6):3270–3283. doi: 10.1152/jn.90777.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shehzad Z, Kelly AM, Reiss PT, et al. The resting brain: unconstrained yet reliable. Cereb Cortex. 2009;19(10):2209–2229. doi: 10.1093/cercor/bhn256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cole DM, Smith SM, Beckmann CF. Advances and pitfalls in the analysis and interpretation of resting-state FMRI data. Front Syst Neurosci. 2010;4:8. doi: 10.3389/fnsys.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 78.Gawin F, Riordan C, Kleber H. Methylphenidate treatment of cocaine abusers without attention deficit disorder: a negative report. Am J Drug Alcohol Abuse. 1985;11(3-4):193–197. doi: 10.3109/00952998509016861. [DOI] [PubMed] [Google Scholar]
- 79.Grabowski J, Roache JD, Schmitz JM, Rhoades H, Creson D, Korszun A. Replacement medication for cocaine dependence: methylphenidate. J Clin Psychopharmacol. 1997;17(6):485–488. doi: 10.1097/00004714-199712000-00008. [DOI] [PubMed] [Google Scholar]
- 80.Collins SL, Levin FR, Foltin RW, Kleber HD, Evans SM. Response to cocaine, alone and in combination with methylphenidate, in cocaine abusers with ADHD. Drug Alcohol Depend. 2006;82(2):158–167. doi: 10.1016/j.drugalcdep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 81.Volkow ND, Fowler JS, Wang GJ, et al. Methylphenidate decreased the amount of glucose needed by the brain to perform a cognitive task. PLoS One. 2008;3(4):e2017. doi: 10.1371/journal.pone.0002017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marquand AF, De Simoni S, O'Daly OG, Williams SC, Mourão-Miranda J, Mehta MA. Pattern classification of working memory networks reveals differential effects of methylphenidate, atomoxetine, and placebo in healthy volunteers. Neuropsychopharmacology. 2011;36(6):1237–1247. doi: 10.1038/npp.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mehta MA, Owen AM, Sahakian BJ, Mavaddat N, Pickard JD, Robbins TW. Methylphenidate enhances working memory by modulating discrete frontal and parietal lobe regions in the human brain. J Neurosci. 2000;20(6):RC65. doi: 10.1523/JNEUROSCI.20-06-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Epstein JN, Casey BJ, Tonev ST, et al. ADHD- and medication-related brain activation effects in concordantly affected parent-child dyads with ADHD. J Child Psychol Psychiatry. 2007;48(9):899–913. doi: 10.1111/j.1469-7610.2007.01761.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.