

Abstract
Phylogenetic signal quantifies the degree to which resemblance in continuously-valued traits reflects phylogenetic relatedness. Measures of phylogenetic signal are widely used in ecological and evolutionary research, and are recently gaining traction in viral evolutionary studies. Standard estimators of phylogenetic signal frequently condition on data summary statistics of the repeated trait observations and fixed phylogenetics trees, resulting in information loss and potential bias.
To incorporate the observation process and phylogenetic uncertainty in a model-based approach, we develop a novel Bayesian inference method to simultaneously estimate the evolutionary history and phylogenetic signal from molecular sequence data and repeated multivariate traits. Our approach builds upon a phylogenetic diffusion framework that model continuous trait evolution as a Brownian motion process and incorporates Pagel’s λ transformation parameter to estimate dependence among traits. We provide a computationally efficient inference implementation in the BEAST software package.
We evaluate the synthetic performance of the Bayesian estimator of phylogenetic signal against standard estimators, and demonstrate the use of our coherent framework to address several virus-host evolutionary questions, including virulence heritability for HIV, antigenic evolution in influenza and HIV, and Drosophila sensitivity to sigma virus infection. Finally, we discuss model extensions that will make useful contributions to our flexible framework for simultaneously studying sequence and trait evolution.
Keywords: comparative approach, Bayesian phylogenetics, virus evolution, adaptation, virulence
1 Introduction
A central premise of comparative biology is that evolutionary history shapes the distribution of phenotypic traits in extant species. The field has long realized that shared ancestry induces statistical dependence among observed trait values and that this correlation structure needs to be taken into account in comparative analyses (Felsenstein, 1985; Harvey & Purvis, 1991). Recent advances in comparative analyses of continuously-valued trait data focus on the tempo and mode of trait evolution, e.g. testing for different evolutionary rates (O’Meara et al., 2006), identifying rate shifts (Revell et al., 2012) and on correlations among traits (Revell & Collar, 2009). However, the question also frequently arises to what extent shared ancestry needs to be controlled for (Freckleton et al., 2002). Different measures have been proposed to quantify and test the tendency for related species to share similar traits, which is generally referred as ‘phylogenetic signal’. This quantification is particularly relevant for traits that are heavily impacted by ecological as well as evolutionary processes (Losos, 2008).
Measures of phylogenetic signal are often classified into indices based on phylogenetic autocorrelation or on Brownian diffusion models of trait evolution (Münkemüller et al., 2012). Building on spatial autocorrelation functions, phylogenetic autocorrelation quantifies the degree of correlation across observations that the evolutionary history explains, but does not provide a generative model for how the traits arise along this history; popular examples include Moran’s I (Moran, 1950) and Abouheif’s Cmean (Abouheif, 1999). On the other hand, Blomberg’s K (Blomberg et al., 2003) and Pagel’s λ (Pagel, 1999) are commonly-used measures that advance a Brownian diffusion process along the history as a data generative model and serve to both quantify and test for phylogenetic correlation. Under both measures, a value of 0 reflects independence across observations, whereas a value of 1 suggests that traits arise according to the generative process. Recent performance evaluations of different indices based on simulations under Brownian diffusion indicate that Pagel’s λ and Abouheif’s Cmean perform well in testing procedures and that Pagel’s λ provides the most reliable quantification of phylogenetic signal (Münkemüller et al., 2012; Shirreff et al., 2013).
Phylogenetic comparative methods are permeating evolutionary biology, but they have only sporadically been adopted in virus evolutionary studies due to a strong genotypic focus and a comparatively lower availability of phenotypic trait data. There is however a growing interest in studying a variety of viral phenotypic measurements ranging from traits that remain close to the genotype, like antigenic properties, to those that may be heavily impacted by the host environment, such as virulence or infections traits (Hartfield et al., 2014). Because of its importance for vaccine selection, antigenic evolution has been extensively studied in human influenza viruses. Although performing assays to measure influenza antigenic properties may be relatively straightforward, they produce challenging data explaining perhaps why antigenic evolutionary studies have largely been divorced from, or at best contrasted against (Smith et al., 2004), sequence evolution (but see Bedford et al. (2014) for recent efforts to integrate both).
The human immunodeficiency virus (HIV) also evades humoral immune responses within a host, but this has far less impact at the population or epidemiological scale as compared to the continuous turnover characteristic for antigenic drift in seasonal influenza. The question still remains to what extent the virus may adapt to humoral immunity at the population level. Current studies addressing this generally do not account for phylogenetic dependence among the viruses for which antigenic neutralization is measured (e.g. Bunnik et al. (2010); Euler et al. (2011)), and it is unclear how this affects the results. The interest in HIV adaptation at the population level also extends to cell-mediated immunity (Kawashima et al., 2009), drug therapy (Little et al., 2002), and viral fitness, all of which can impact virulence and disease severity. This leads to a more general question as to what extent the HIV genotype can control for the rate of progression to AIDS. Disease progression rates vary extensively among HIV patients, but they can be predicted by the level of viraemia in early infection, referred to as set-point viral load (spVL) (Mellors et al., 1996). To assess the heritability of spVL many studies have focused on HIV transmission pairs, which resulted in a broad range of heritability estimates (e.g. Tang et al. (2004); Hollingsworth et al. (2010); van der Kuyl et al. (2010)), although this may be narrowed down by interpreting the results using a consistent measure of heritability (Fraser et al., 2014). Phylogenetic signal estimators have recently been proposed as an alternative approach to study spVL heritability (Alizon et al., 2010; Shirreff et al., 2013). This represents a rare application of the comparative approach to continuously-valued viral characters, but one that in essence also attempts to disentangle the impact of ecological – in this case the host environment – and evolutionary processes.
Virulence may therefore also be addressed from the host perspective, as closely related hosts may exhibit more similarity in their susceptibility to viral infection. This has been elegantly addressed for sigma viruses in fruit flies (Longdon et al., 2011). By experimentally testing the susceptibility of 51 host species to viral infection with three different host-specific viruses, this study showed that the host species phylogeny is a strong determinant of viral persistence and replication in novel Drosophila hosts. Such investigations represent interesting applications for phylogenetic signal estimators that can both test and quantify the degree to which the host phylogeny controls pathogen susceptibility.
The examples of viral phenotypic evolution discussed above are all based on traits that can be subject to considerable quantification error (inherent to the assays) or natural individual variation. To characterize the measurement error many studies produce repeated measures, but then generally condition on the trait means and variances for each strain or taxon in the subsequent analysis. This is akin to how intraspecific variation has been traditionally treated by maximum likelihood estimation procedures in phylogenetic comparative approaches across different species, which raises the problem of error propagation (but see Lynch (1991) and Housworth et al. (2004) for a notable exception). To address this, Revell & Graham Reynolds (2012) have proposed a Bayesian method to accommodate intraspecific variation through simultaneously inferring species means and trait evolutionary model parameters. While taking into account uncertainty, a joint Bayesian inference also allows for cross talk between the different model components (Revell & Graham Reynolds, 2012). Importantly however, the problem of error propagation extends to all aspects of comparative phylogenetic estimation, adding further to its imprecision. For example, there is generally considerable uncertainty in the reconstructed tree, including both branch lengths and tree topology estimates, to which trait evolutionary processes are typically fitted. Although phylogenetic error can be empirically captured by considering a (posterior) distribution of trees (Barker et al., 2007; Longdon et al., 2011; de Villemereuil et al., 2012), all these separate efforts indicate that trait evolutionary analyses would benefit from a general and coherent statistical framework, one that solely conditions on the observed sequence and trait data, rather than on data summary statistics such as trait means and variances of repeated measures and collections of independently inferred phylogenies. This has recently become realistic by the development of an integrated Bayesian inference approach that connects sequence and trait evolutionary processes in a phylogeographic context (Lemey et al., 2009, 2010). Here, we build upon these Bayesian phylogenetic diffusion models and extend them to simultaneously estimate evolutionary history and trait phylogenetic signal. This framework has several additional advantages, including the general applicability to traits of any dimension, the quantification of correlations among them, and the possibility for ancestral state reconstructions. Furthermore, it allows incorporating measurement uncertainty in a natural way by numerically integrating the unobserved average tip trait values based on repeated measures. We use simulations to compare estimator performance of our Bayesian implementation of Pagel’s λ (λB) to standard phylogenetic signal estimators, and demonstrate its use through several applications to viral traits. We revisit heritability estimation for HIV virulence, contrast phylogenetic signal for antigenic evolution in influenza H3N2 and HIV-1 subtype B, and finally return to virulence, but from the perspective of Drosophila hosts challenged by host-specific sigma virus infection. Taken together, we demonstrate how a coherent framework can advance trait evolutionary studies and discuss extensions that may further promote its role in comparative analyses.
2 Methods
We begin with a brief description of Brownian diffusion along a phylogenetic tree and the construction of Pagel’s λ as a measure of phylogenetic signal, parameterized within this diffusion process. Extensive derivations find themselves in Felsenstein (1985) and Pagel (1999).
2.1 Phylogenetic Brownian Process
Let Y = {yij} be the N × K matrix of K-dimensional trait values realized at the tips of a tree
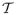 for N taxa. The diffusion process posits that data Y arise from conditionally independent, multivariate normally-distributed displacements along each branch in
. These displacements are centered around the hypothesized trait value at the parent node of the branch and have variance proportional to a K × K positive-definite, symmetric matrix Σ, where the proportionality constant is the branch length. The diagonal elements of Σ describe the (relative) rates at which the different trait dimensions evolve over the tree and the off-diagonal elements reflect the covariation in trait dimensions after controlling for their shared history. Conditioning on the hypothesized ancestral trait values α at the root of
, the joint distribution of vec[Y] falls out as
for N taxa. The diffusion process posits that data Y arise from conditionally independent, multivariate normally-distributed displacements along each branch in
. These displacements are centered around the hypothesized trait value at the parent node of the branch and have variance proportional to a K × K positive-definite, symmetric matrix Σ, where the proportionality constant is the branch length. The diagonal elements of Σ describe the (relative) rates at which the different trait dimensions evolve over the tree and the off-diagonal elements reflect the covariation in trait dimensions after controlling for their shared history. Conditioning on the hypothesized ancestral trait values α at the root of
, the joint distribution of vec[Y] falls out as
| (1) |
where vec[·] is the vectorization operator that stacks the column vectors of its argument below one another, ⊗ is the Kronecker product and X is a NK × K design matrix in which entries in column k contain a 1 for trait k or a 0 otherwise, following Freckleton et al. (2002). More importantly,
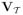 = {vii′} is an N × N variance matrix that is a deterministic function of
; we return to its definition shortly as this relates to Pagel’s λ. Equation (1) relates that vec[Y] is multivariate normally-distributed with NK × NK variance matrix Σ ⊗
, fallaciously suggesting computational order
= {vii′} is an N × N variance matrix that is a deterministic function of
; we return to its definition shortly as this relates to Pagel’s λ. Equation (1) relates that vec[Y] is multivariate normally-distributed with NK × NK variance matrix Σ ⊗
, fallaciously suggesting computational order
 (N3K3) to decompose the variance matrix and evaluate the density. We prefer to work directly with the joint density of Y,
(N3K3) to decompose the variance matrix and evaluate the density. We prefer to work directly with the joint density of Y,
| (2) |
that is a matrix-normal probability density function (Dawid, 1981), where tr[·] is the trace operator and 1 is a N-dimensional column vector of ones. Importantly, Equation (2) clarifies the smaller computational order
(N3 + K3) required to evaluate the density without specialized knowledge of Kronecker product identities. Leading software to estimate Pagel’s λ for multivariate traits, such as the caper package (Orne et al., 2013) in R, rely implicitly on this representation to afford computational efficiency.
However, Pybus et al. (2012) show that is possible to evaluate Equation (2) in computational order
(NK2) by modeling explicitly in terms of precision Σ−1 and, more importantly, exploiting an original dynamic programming algorithm. We adopt this approach here since repeated evaluation of Equation (2) is the rate-limiting step in both profiling the likelihood function in a maximum likelihood framework and numerical integration in a Bayesian framework. This critical insight enables us to scale comparative methods to trees with hundreds or thousands of tips, a situation regularly encountered in viral evolution.
In brief, the dynamic programming algorithm starts with the joint density of Y and the hypothesized trait values at each of the internal and root nodes in
. From this joint density, we recover the marginalized density of Y by integrating out the internal and root node values. We achieve this high-dimensional integration through a post-order tree traversal over the
(N) internal and root nodes. Each nodal visit entails a simple integration of the hypothesized value at the node to arrive at the partial density of the tip trait values descendent to the node given the unobserved value of the parent of the node or the prior distribution assumed on the root trait value. This recursive task has an analytic solution involving
(K2) computational operations.
To complete specification of the Brownian diffusion process along
and introduce Pagel’s λ, we return to our definition of
(λ) that we now explicitly parameterize in terms of λ. Let
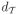 (u, v) equal the sum of branch lengths along the shortest path between node u and node v in
. Then diagonal elements vii =
(ν2N−1, νi), the time-distance between the root node ν2N−1 and tip node i. To interpret these elements, the marginal distribution of Yi given α has variance proportional to the time since the root. For ultrametric trees, these diagonal entries are all equal; for rapidly evolving pathogens, non-contemporaneous sequences are common, leading to different times. Further, the off-diagonal elements
(u, v) equal the sum of branch lengths along the shortest path between node u and node v in
. Then diagonal elements vii =
(ν2N−1, νi), the time-distance between the root node ν2N−1 and tip node i. To interpret these elements, the marginal distribution of Yi given α has variance proportional to the time since the root. For ultrametric trees, these diagonal entries are all equal; for rapidly evolving pathogens, non-contemporaneous sequences are common, leading to different times. Further, the off-diagonal elements
| (3) |
specifying the rescaled time-distance between the root node and the most recent common ancestor of tip nodes i and i′. Intuitively, the covariance between two tip traits is a function of their shared evolutionary history, and the traits become conditionally independent at their most recent common ancestor node in
. Scalar λ exists on the continuum between 0 and 1 (Pagel, 1999), with λ = 1 returning a variance matrix
that perfectly adheres to a Brownian process along
(Felsenstein, 1985). On the other hand, λ = 0 reflects the absence of any phylogenetic correlation. Intermediate values of λ indicate that the phylogeny exerts a weaker effect on the trait evolutionary process than expected from a Brownian motion model.
Although λ operates directly on
given the phylogeny
, it is often convenient to view λ as a transformation of the phylogeny to fit the Brownian motion model to the trait data. Figure 1 shows these transformations for three different values of λ on an example phylogeny relating six taxa as well as their corresponding
. The transformation involves rescaling the internal node heights, with all internal node heights equal to the root node as the most extreme case (λ = 0, starlike tree in Figure 1). Freckleton et al. (2002) demonstrate how to construct, in a maximum likelihood (ML) setting, an estimator λML of λ for hypothesis testing about the strength of phylogenetic signal in fully observed multivariate traits conditional on a known phylogeny.
Figure 1. Phylogenies and corresponding variance-covariance matrices for three different λ values.

The tree on the left (λ = 1, no transformation) provides an example of a phylogeny estimated or hypothesized for six taxa. The numbers above the branches represents the time elapsed on the branches. The corresponding variance-covariance matrix is shown beneath the tree. Under a Brownian motion model of trait evolution, the expected covariances (off diagonals) between each pair of taxa are proportional to shared ancestry for the taxa. The expected variances (diagonal elements) for the tip traits are proportional to the summed branch lengths between the root and each tip. We multiply the off diagonal elements by two different λ values (λ = 0.5 and 0 respectively) in the matrices to the right and show the corresponding tree transformation. The tree for λ = 0 collapses into a starlike tree and therefore represents phylogenetic independence.
2.2 Bayesian Estimator of Repeated Traits Phylogenetic Signal
We develop a coherent estimator λB set in a Bayesian framework for simultaneously estimating the phylogeny and trait signal with potentially missing or repeated measures. Assume that at each tip i in
, we observe Ri trait realizations Xi = (Xi1, …, XiRi). To model these repeated measures, we build upon Guo et al. (2007) by asserting that tip traits Yi are unobserved and characterize a sampling distribution on Xi. Specifically, we posit that each observed value Xir is multivariate normally distributed about Yi with variance Γi = (Ri − 1) × Γ, where Γ is an estimable K × K variance matrix that quantifies the measurement error in the observation process across all taxa. Over all taxa and all trait observations X = (X1, …, XN), we write
| (4) |
Conveniently, when no repeated measures exist for tip i, Ri = 1 and the density function in Equation (4) enforces Xi = Yi, returning the original model.
To simultaneously estimate the phylogeny
and account for its uncertainty, we further consider aligned molecular sequences S from the N taxa and model S using standard Bayesian phylogenetics models parameterized in terms of other phylogenetic and demographic process parameters ϕ. Conditional on
, we assume independence between S and X, enabling us to write down the joint density p(
, ϕ, S) and view it, for the purposes of this paper, as a prior on
after integrating out ϕ. We refer interested readers to, for example, Suchard et al. (2001) and Drummond et al. (2012) for detailed development of p(
, ϕ, S).
Combining the Brownian diffusion process, repeated measures and phylogenetic uncertainty returns the joint posterior distribution
| (5) |
where p(λ), p(Σ), p(Γ) and p(α) are prior distributions, and the integration in the second line of Equation (5) reflects a data augmentation procedure with the unobserved tip traits Y and root node trait α. Lemey et al. (2010) and Pybus et al. (2012) develop priors p(Σ) and p(α) that enable convenient analytic and numerical integration of the augmented data, in the latter case via Markov chain Monte Carlo (MCMC). To construct p(Γ), we structure Γ−1 = diag(γ1, …, γK) as a diagonal matrix, where a priori we assume γj is gamma distributed with expectation 1 and a large variance 1000 for j = 1, …, K. We explore several choices for p(λ) in the Results section; these involve the family of beta distributions that constrain 0 ≤ λ ≤ 1 for consistency with Freckleton et al. (2002).
We define our Bayesian estimator λB as the marginal posterior mean
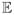 (λ | X, S) and report uncertainty in this estimate via the 95% highest posterior density (HPD) interval of p(λ | X, S). We estimate these quantities by extending MCMC methods implemented in the Bayesian Evolutionary Analysis by Sampling Trees (BEAST) software package. Specifically, we use a random-scan Metropolis-with-Gibbs approach that employs standard transition kernels to integrate over the parameter spaces of
and ϕ. To sample realizations of Σ and Γ, we consider Gibbs samplers, and we develop random-walk Metropolis-Hastings transition kernels on λ and missing entries in X; this latter procedure assumes a missing completely at random structure (Heitjan & Basu, 1996), such that the probability that any particular datum is missing is independent of both the observed trait values and other model parameters. Integrating out the missing entries enables us to continue to draw inference in the presence of partially observed tip trait values, without needing to trim out trait dimensions that are not completely observed across all taxa or taxa that are missing traits. Finally, to compare different restricted models, e.g. λ = 0 (no phylogenetic signal) versus λ is random and estimates phylogenetic signal, we use Bayes factors derived from stepping-stone estimation of the marginal likelihood for each competing model (Baele et al., 2012). We accompany our BEAST implementation with graphical-user interface support in BEAUti for setting up phylogenetic signal analyses with multivariate traits.
(λ | X, S) and report uncertainty in this estimate via the 95% highest posterior density (HPD) interval of p(λ | X, S). We estimate these quantities by extending MCMC methods implemented in the Bayesian Evolutionary Analysis by Sampling Trees (BEAST) software package. Specifically, we use a random-scan Metropolis-with-Gibbs approach that employs standard transition kernels to integrate over the parameter spaces of
and ϕ. To sample realizations of Σ and Γ, we consider Gibbs samplers, and we develop random-walk Metropolis-Hastings transition kernels on λ and missing entries in X; this latter procedure assumes a missing completely at random structure (Heitjan & Basu, 1996), such that the probability that any particular datum is missing is independent of both the observed trait values and other model parameters. Integrating out the missing entries enables us to continue to draw inference in the presence of partially observed tip trait values, without needing to trim out trait dimensions that are not completely observed across all taxa or taxa that are missing traits. Finally, to compare different restricted models, e.g. λ = 0 (no phylogenetic signal) versus λ is random and estimates phylogenetic signal, we use Bayes factors derived from stepping-stone estimation of the marginal likelihood for each competing model (Baele et al., 2012). We accompany our BEAST implementation with graphical-user interface support in BEAUti for setting up phylogenetic signal analyses with multivariate traits.
2.3 Benchmark
We extend a previous simulation study that was modeled after the heritability of set point viral load (spVL) in HIV infection history (Alizon et al., 2010). Briefly, this procedure involves simulating a birth-death infection process in which each branching event represents a new infection event. Starting from an initial value drawn from an empirical distribution of traits (spVL), traits diverge at transmission events. At each branching event, one daughter branch inherits the trait on the parental branch, yP, whereas the trait on the other daughter branch evolves into yD, following
| (6) |
where h2 represents the heritability of the trait and M is a random variable drawn from an empirical log(spVL) trait distribution (taken from one of data sets we analyze in this study). Following the original procedure (Alizon et al., 2010), this process is simulated over 13 generations with a death probability of 1/3 of the transmission probability; each time, a subtree of 128 taxa with associated tip trait values is drawn randomly from the process reflecting incomplete sampling. Whereas the original simulation evolved 20 replicates for four different heritability values (h2 = 0.3, 0.5, 0.7 & 0.9), we also include the same amount of replicates for h2 = 0.1 to ensure symmetric simulation scenarios and because existing methods have difficulties detecting low degrees of heritability (Shirreff et al., 2013).
We also perform simulations according to a more recent procedure proposed by Shirreff et al. (2013). Here, traits are simulated along an empirical phylogeny starting from the mean log(spVL) at the root. Each node (and not just a single daughter branch) inherits a log(spVL) according to the following process with stationary variance:
| (7) |
where M is now drawn from a normal distribution with the mean and variance of the population log(spVL). We iterate through a wider range of heritability values (from 0.05 to 0.95 with a step size 0.05), and simulate 100 replicates for each heritability level. We use both sets of simulations to compare our Bayesian estimate of λ to a maximum likelihood estimate of lambda (λML, for which various implementation exist, e.g. Revell (2012); Harmon et al. (2008); Orme et al. (2013); Paradis et al. (2004); Lavin et al. (2008); Pinheiro et al. (2013)) and Blomberg’s K, both obtained by the phytools R package (Revell, 2012). In order to estimate confidence intervals for λML we extend the phytools estimation procedure to obtain the λ values for 1.92 log likelihood units on either side of the maximum likelihood estimate and construct confidence intervals.
2.4 Viral Examples
We analyze four different data sets that examine the phenotypic evolution in infectious diseases; these phenotypic traits include both virus-specific characteristics and host susceptibility to viral infection. We first focus on the Swiss HIV cohort study (SHCS) data set (Swiss HIV Cohort Study et al., 2010), previously used to investigate the heritability of spVL (Alizon et al., 2010). This study selected HIV-1 subtype B infected participants who had a genotypic drug resistance test and at least three HIV RNA measurements, but remained treatment-naive at entry. In addition to spVL from cohort participants, the study also measured the declining slope of CD4+ T-cell (dsCD4) counts; this quantity also predicts virulence to some extent (Mellors et al., 2007). As a ‘control trait’ in our analyses we followed Alizon et al. (2010) and considered the estimated probability that a treatment-naive virus is resistant to zidovudine (prAZT) from the pol sequence using the geno2pheno system (Beerenwinkel et al., 2003). We examine spVL, dsCD4 and prAZT both independently and jointly as a multivariate trait.
The sequence data for the SHCS consist of population sequences of the HIV-1 polymerase (pol) gene for each patient and we used alignments in which amino acid positions that are strongly associated with antiretroviral drug resistance were removed. Having access to multiple viral load measures per patient after acute infection, but prior to start of antiretroviral therapy, the first CDC C event, or the time when the CD4 count first drops below 200 cells, Alizon et al. (2010) considered two different spVL criteria to distinguish different patients subsets. The ‘strict’ criterion only considers cases where all the viral load measurement fluctuate within a 1-log band around the patient-specific mean, whereas the ‘liberal’ definition applies to all cases where at least three consecutive viral loads measurements are available that fluctuate within a 1-log band of their mean. We also follow Alizon et al. (2010) in studying the transmission group of men who have sex with men (MSM) separately from other transmission groups that are heterosexuals and injection drug users. Because the densely-sampled MSM sequences tend to cluster in phylogenetic trees (Kouyos et al., 2010), it is suggested that they may yield more accurate phylogenies. Furthermore, focusing on the MSM transmission group may remove some confounding factors, such as patient gender, transmission group or age, on infection trait values (Alizon et al., 2010). The distinction between a strict and liberal spVL definition and between MSM and the general population results in four data sets listed in Table 2. For the SHCS analysis, our full probabilistic model included a general-time reversible substitution model with discretized-gamma-distributed rate variation among-sites, an uncorrelated lognormal relaxed molecular clock model and a flexible Gaussian Markov random field model of population size change through time as a tree prior (Minin et al., 2008). For the isolates with unknown sampling times, we integrate out their dates by assuming a uniform prior distribution over a plausible time interval (Shapiro et al., 2011). Because the SHCS provides a large number of taxa (n = 661 for all risk groups and a liberal viral load criterion), we perform the different trait analyses on the full data set using an empirical tree distribution inferred separately from the nucleotide data (cfr. Lemey et al. (2014)). Finally, we estimate marginal likelihoods on a fixed tree topology for the different SHCS data sets because marginal likelihood estimation, for example via stepping stone sampling, requires a series of MCMC simulations for different power posteriors; this is generally much more computationally demanding than a standard MCMC exploration of the posterior (Baele et al., 2012).
Table 2.
Phylogenetic signal estimates for the SHCS data sets
| Data set | n | prAZT
|
log(spVL)
|
dsCD4
|
dsCD4,log(spVL)
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| λB | KL | λB | KL | λB | KL | λB | KL | r | ||
| MSM strict | 134 | 0.964 (0.911,0.999) | 3.421 | 0.501 (0.165,0.857) | 0.036 | 0.315 (0.024,0.628) | 0.398 | 0.368 (0.056,0.727) | 0.215 | −0.195 (−0.377, −0.050) |
| all strict | 230 | 0.986 (0.965,1.000) | 3.725 | 0.303 (0.038,0.596) | 0.599 | 0.303 (0.022,0.632) | 0.513 | 0.206 (0.026,0.431) | 1.380 | −0.221 (−0.344, −0.097) |
| MSM liberal | 404 | 0.997 (0.992,1.000) | 3.858 | 0.228 (0.037,0.352) | 1.397 | 0.181 (0.012,0.366) | 1.631 | 0.113 (0.015,0.256) | 2.398 | −0.235 (−0.323, −0.144) |
| all liberal | 661 | 0.992 (0.974,1.000) | 3.758 | 0.158 (0.050,0.285) | 2.328 | 0.093 (0.038,0.194) | 2.747 | 0.119 (0.023,0.212) | 2.665 | −0.221 (−0.288, −0.148) |
Our second data set explores antigenic evolution in human influenza H3N2 and consists of 1441 hemagglutinin (HA) sequences with known date of sampling and with associated antigenic measurements previously obtained by Russell et al. (2008). These sequences were sampled globally from 2002 to 2007 and represent a subset of strains from a larger antigenic sampling (13,000 isolates) for which HA sequence was available (Russell et al., 2008). Antigenicity was measured using hemagglunination inhibition (HI) assays and mapped into a two-dimensional space using multidimensional scaling (MDS) (Smith et al., 2004). Here, we consider the first two principal antigenic coordinates resulting from a previous MDS fit (Russell et al., 2008) as traits in our phylogenetic diffusion model. Given the size of the data set, we approximate phylogenetic uncertainty by integrating over a set of trees previously reconstructed as part of a phylogeographic study (Lemey et al., 2014).
For the third data set, we return to HIV and study antigenic evolution in the context of enhanced resistance to the broadly neutralizing antibodies (nAb) PG9, PG16 and VRC01 over the course of the HIV-1 epidemic (Euler et al., 2011). The sequence data set encompasses clonally sequenced viral variants from contemporary and historic seroconverters (seroconversion between 2003 and 2006 and between 1985 and 1989, respectively). To focus on the population-level evolution of neutralization resistance and avoid the impact of within-host evolutionary dynamics, we randomly choose one sequence per patient for our analyses and examine the 50% inhibitory concentration (IC50) assay values for the three different antibodies. This assay measures percent neutralization by determining the reduction in p24 production in the presence of neutralizing agent compared to the levels of p24 in the cultures with virus only; a detailed description can be found in (Euler et al., 2011). To constrain the trait values (concentrations) to be strictly positive values under the diffusion process, we model the log-transform of the IC50 values observed at the tips of the tree. When the observed log IC50 value falls outside the tested antibody concentration range, we integrate out the concentration over a plausible IC50 interval. For those IC50s lower then the lowest antibody concentration we set up a uniform prior ranging from the lowest tested ln(nAb) concentration to the 32x diluted lowest tested concentration. The values at the opposite end of the spectrum, where 50% neutralization is not reached at the highest nAb concentration, are integrated out over an appropriately scaled exponential distribution. Other evolutionary models and tree priors follow the same specifications as for the SHCS analyses.
Finally, we return to infection traits and study the heritability of host susceptibility to viral infection. To this purpose, we focus on a data set that has been used to investigate the ability of three host-specific Drosophila sigma viruses to persist and replicate in 51 different species of Drosophilidae (Longdon et al., 2011). In this experimental study, fly species are injected with host-specific sigma virus from D. affinis (DAffSV), D. melanogaster (DMelSV) and D. obscura (DObsSV), and a change in viral titre is measured between day 0 and day 15 post-infection using quantitative reverse-transcription PCR. The copy-number of viral genomic RNA is expressed relative to the endogenous control housekeeping gene RpL32 (Rp49) based on species-specific primers for this gene. The authors aim at performing three replicate measures for each virus per fly species (3 replicates each of the day 0 and day 15 treatments). We incorporate these repeated measures in our analyses. We refer to Longdon et al. (2011) for further details on the experimental procedure. We use the sequence data for the COI, COII, 28S rDNA, Adh, SOD, Amyrel and RpL32 genes to jointly reconstruct the Drosophila host phylogeny with the diffusion process and follow the evolutionary model and analysis settings from Longdon et al. (2011).
3 Results
3.1 Performance
We conduct a simulation study to compare the relative performance of the λB estimator to two standard indices of phylogenetic signal. We extend the simulation study by Alizon et al. (2010) aimed at evaluating the performance of Blomberg’s K (Blomberg et al., 2003) and Pagel’s λ (Pagel, 1999) in capturing the heritability of viral trait evolution. Briefly, the original simulation procedure considers a birth-death infection process with incomplete sampling and evolves a trait along the resulting transmission tree using different degrees of heritability (0.3, 0.5, 0.7 and 0.9). Here, we extend the study to a heritability value of 0.1 to explore a symmetric range of heritability values around 0.5. A comparison of the phylogenetic signal estimates for replicate data generated under different heritability values (Figure 2) suggests that λB captures the underlying heritability with less bias and lower variance compared to Blomberg’s K and λML. This is confirmed by the quantitative bias and mean squared error (MSE), which quantifies the amount by which the estimator differs from the true value, estimates as tabulated across all heritability scenarios (Table 5). Because of the somewhat distinct behavior for small and large heritability values (Figure 2), we also summarize the bias for heritability values smaller and larger than 0.5 (Table 5). This confirms the finding that existing methods have difficulties in estimating relatively low heritability values (h2 < 0.4, Shirreff et al. (2013)); Blomberg’s K and λML result in over- and under-estimation respectively (Table 5). λB follows the sigmoidal pattern for λML to a lesser extent and is characterized by smaller biases for both small and large heritability values.
Figure 2. Estimator performance of Blomberg’sK, Pagel’s λML and λB on simulated data.

Twenty phylogenies are simulated to model the evolution of an infection trait with known heritability (h2 = 0.1, 0.3, 0.5, 0.7 & 0.9). Phylogenetic signal is then estimated on each tree using only 128 leaves to account for incomplete sampling. The box plots shows the median values, the three quartiles and the outliers for Blomberg’s K (blue), Pagel’s λB (red) and λB (grey).
Table 5.
Phylogenetic signal estimates for human influenza H3N2 antigenic evolution.
| Trait | λB | KL | BFf0 | BFf1 |
|---|---|---|---|---|
| PC 1 | 0.821 (0.761,0.879) | 2.611 | 1327.565 | 290.183 |
| PC 2 | 0.420 (0.258,0.563) | 0.921 | 31.773 | 406.558 |
| PC1,PC2 | 0.731 (0.668,0.787) | 2.246 | 1350.579 | 675.260 |
We also report estimator coverage for λB and λML; coverage reflects the probability that the true value from which the data derive falls within the model estimated nominal confidence interval. The uncertainty for the λB estimator is quantified by a 95% Bayesian high posterior density (HPD) interval, the size of which - unlike a frequentist confidence interval - does not necessarily have to correspond to the nominal coverage. To obtain a 95% confidence interval (CI) for λML, we construct a likelihood-ratio test and find its points of rejection using a numerical optimizer in R. The need to independently implement a confidence interval constructor reflects the fact that λML is frequently reported as a point-estimate without quantifying its uncertainty. We note that Blomberg’s K phylogenetic signal statistic is computed as a ratio of two MSE ratios, comparing observed against expected ratios under Brownian motion, and is therefore less amenable to constructing confidence intervals (Blomberg et al., 2003).
We continue to explore the effect of prior specification on λB estimates and compare the β(1, 1) prior to a U-shaped prior (β(0.5, 0.5)) and several bell-shaped priors. Figure 3 shows that bias and MSE can be further minimized while raising coverage close to nominal values for a β(2, 2) prior. This prior may help to linearize the general sigmoidal relationship between λ and the known heritability spectrum as well as reduce the generally large variance of λ estimates for intermediate heritability values. We therefore adhere to this prior specification in further data analyses, but accompany the λ estimates with a posterior divergence measure to quantify the potential prior influence. Specifically, we computed the Kullback-Leibler (KL) divergence between the prior and posterior using the FNN package in R (Boltz et al., 2007; Li, 2012).
Figure 3. Performance of the Bayesian phylogenetic signal estimation under various priors on λB.

Different β(α,β) priors are explored for α = β. We plot bias (filled black circles) and MSE (crosses) according to the primary axis and coverage (open squares) according to the secondary axis. The dotted horizontal line represents zero bias and MSE.
We find largely similar performance differences among the three estimators, and for the different beta priors on λB, in a simulation analysis following a procedure similar to that of Shirreff et al. (2013) (Supporting Information).
3.2 HIV-1 infection traits
We estimate phylogenetic signal for spVL and dsCD4, either as separate traits or in combination as a bivariate trait, in the different SHCS data sets and include prAZT as a control (Table 2). Because the latter is measured directly from the genotype, it is expected to be strongly heritable. We confirm this by estimates of λB that are consistently close to maximum phylogenetic signal for the different SHCS data sets (Table 2). We also compare model fit of the standard diffusion model with estimable λB to a model that represents no phylogenetic signal (λB = 0) and a model with perfect Brownian phylogenetic signal (λB = 1) using Bayes factors in Table 3. This demonstrates that the support for a non-zero phylogenetic signal for prAZT increases as the data set size increases while, in line with the very high λB estimates for prAZT, the support for a perfect Brownian phylogenetic signal increases. For all prAZT λB estimates, there is also a relatively high KL divergence between the prior and posterior distribution, suggesting that the phylogenetic signal estimates are well-informed by the trait data.
Table 3. Comparing different phylogenetic signal models for the SHCS data sets using Bayes factors.
We report Bayes factors for comparing a model in which parameter λB is estimated (or free) versus a model in which it is fixed to 0 (no phylogenetic signal), BFf0, and or comparing a model in which the Pagel’s λB is estimated versus a model in which it is fixed to 1 (phylogenetic signal expectation under Brownian motion), BFf1
| Data set | prAZT
|
log(spVL)
|
dsCD4
|
dsCD4 & log(spVL)
|
||||
|---|---|---|---|---|---|---|---|---|
| BFf0 | BFf1 | BFf0 | BFf1 | BFf0 | BFf1 | BFf0 | BFf1 | |
| MSM strict | 1.637 | 1.998 | 1.363 | 4.536 | −1.696 | 7.359 | −0.870 | 10.249 |
| all strict | 14.068 | −3.790 | 0.601 | 32.837 | −1.452 | 8.037 | −0.661 | 68.446 |
| MSM liberal | 53.682 | −9.713 | −0.921 | 77.527 | −3.111 | 65.486 | −2.931 | 130.696 |
| all liberal | 120.742 | −10.861 | 0.234 | 168.002 | −3.816 | 458.896 | −0.702 | 404.368 |
In accordance with Alizon et al. (2010), we find a relatively high phylogenetic signal for spVL (posterior mean = 0.501, 95% HPD [0.165–0.857]), but only in the MSM subset with the strict spVL criterion (Table 2). In contrast to the prAZT, the spVL phylogenetic signal decreases as the data set increases in size and also the support for a non-zero signal is low, and there is even support in favor of the absence of spVL phylogenetic signal in the MSM liberal data set. The KL divergence indicates that the posterior divergence from the prior is limited, implying that the data does not contribute strongly to the phylogenetic signal estimate in the MSM strict data set. The fact that the posterior stays close to prior distribution for λB is also illustrated in Supplementary Figure 1 under both a β(2, 2) and β(1, 1) prior. Phylogenetic signal estimates for dsCD4 are generally lower, with a similar decrease for larger data sets (and increase in KL), and no data set supports a non-zero signal. The same is true for spVL and dsCD4 as a bivariate trait; in this case the two dimensions show a negative correlation (correlation coefficient posterior mean = −0.22, Table 2).
Phylogenetic signal in the MSM strict data set does not find strong support using BFs or KL information gain. This may be due to the typical star-like nature of HIV-1 subtype phylogenies that is very similar to the null model for phylogenetic signal estimation. The λB estimate may therefore be mostly informed by clusters of closely related viruses from epidemiologically-linked patients. The maximum clade credibility (MCC) tree with ancestral spVL trait estimates for the MSM strict data set in Fig. 4 illustrates this point. In the tree, we indicate a number of clusters with closely related viruses and similar spVL values. Because these clusters have relative recent MRCAs, constituting the density at small node heights in the bimodal node height density plot (Fig. 4), they may offer the strongest resistance against transforming the phylogeny into a fully star-like tree. In fact, it has been suggested that restricting the dataset to MSM patients, which are more densely sampled in the SHCS, yields more accurate phylogenies because of better resolution of transmission chains between patients, and that this may explain a higher spVL heritability in the MSM strict data set (Alizon et al., 2010). We therefore examine whether a differential proportion of such transmission clusters in the different SHCS data sets may be responsible for the differences in λB estimates, by splitting up the data set into one that contains only taxa that share relatively recent nodes with other taxa (< 15 years) and one that exclusively has taxa related by deeper branching patterns (internal node > 15 years), and performing separate analyses on each.
Figure 4. Maximum clade credibility tree for the MSM strict analysis.

We color the nodes in the tree according to the observed (external nodes) or estimated (internal nodes) log spVL trait and the branches with a gradient between the relevant nodes. The arrows point at example clusters of closely related taxa with roughly similar trait values. The density plot in the lower right corner summarizes the marginal node height density from the the full posterior distribution.
Estimates for the prAZT control trait indicate that taxa sharing more recent common ancestry are indeed most informative about phylogenetic signal. Both the λB estimates and the associated KL divergences are high for taxa that descend from relatively recent nodes (< 15 years in Table 4), whereas the λB estimates are much closer to their prior expectation of 0.5 for the remaining taxa yielding considerably lower KL divergences. For spVL, however, the KL divergences remain low for the small data sets composed of taxa descending from more recent nodes, and focusing also on transmission clusters in the other data sets does not lead to noticeably higher phylogenetic signal estimates; this suggests that a higher phylogenetic accuracy for transmission clusters does not explain the higher phylogenetic signal.
Table 4.
Phylogenetic signal estimates for the SHCS data sets
| Data set | n | prAZT
|
log(spVL)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| < 15 | > 15 | < 15
|
> 15
|
< 15
|
> 15
|
|||||
| λB | KL | λB | KL | λB | KL | λB | KL | |||
| MSM strict | 39 | 95 | 0.926 (0.798,1.000) | 2.604 | 0.553 (0.136,0.957) | 0.031 | 0.537 (0.168,0.881) | 0.049 | 0.569 (0.186,0.941) | 0.058 |
| all strict | 99 | 131 | 0.996 (0.903,1.000) | 3.351 | 0.631 (0.199,0.992) | 0.185 | 0.37 (0.098,0.683) | 0.330 | 0.509 (0.111,0.882) | 0.011 |
| MSM liberal | 276 | 128 | 0.996 (0.989,1.000) | 3.858 | 0.462 (0.071,0.856) | 0.017 | 0.243 (0.065,0.421) | 1.376 | 0.464 (0.102,0.841) | 0.029 |
| all liberal | 396 | 265 | 0.997 (0.994,1.000) | 3.856 | 0.363 (0.043,0.783) | 0.193 | 0.184 (0.063,0.319) | 2.036 | 0.341 (0.043,0.658) | 0.338 |
3.3 Human influenza H3N2 antigenic evolution
We study antigenic drift in human influenza H3N2 based on a large sequence data set sampled between 2002 and 2007 with matching HI assay data (Russell et al., 2008). HI assays measure the cross-reactivity of viruses against reference antisera and the resulting table with measurements across a multitude of viruses is frequently mapped in two-dimensional space based on MDS approaches (Smith et al., 2004). Here, we treat the two principal coordinates of a previous MDS analysis on the HI data as traits evolving along the influenza genealogy and estimate their phylogenetic signal (Table 5). We find relatively high signal for the first coordinate, but approximately half of that signal for the second coordinate. These differences are also reflected in the reconstructed trait patterns along the evolutionary history (Figure 5). Despite the much lower phylogenetic signal for the second coordinate, it still receives support for a non-zero estimate (Table 5). The large amount of influenza data included in the analysis may contribute to the more precise λB estimates compared to the smaller SHCS HIV data sets, and probably to a large extent also to the different order of magnitude in BF support. Both the magnitude of the phylogenetic signal for the two coordinates as a bivariate trait and its support appears to be dominated by the signal for the first coordinate. As expected from coordinates resulting from an MDS analysis, we find also little correlation between them when accounting for the ancestral history (r = −0.098 [−0.158,−0.051]).
Figure 5. Influenza H3N2 antigenic evolution based on two MDS coordinates derived from HI assay data.

Ancestral reconstructions for two MDS coordinates (PC1 and PC2) are represented by a color gradient on the same maximum clade credibility (MCC) tree summary.
3.4 HIV-1 resistance to broadly neutralizing antibodies
By comparing viruses isolated from individuals who seroconverted in recent years to viruses from individuals who seroconverted early in the epidemic Bunnik et al. (2010) showed a decreased sensitivity of HIV-1 to polyclonal and monoclonal antibodies. Euler et al. (2011) extend this by examining whether the circulating HIV-1 population has also evolved towards resistance against neutralizing activity of the recently identified broadly neutralizing monoclonal antibodies (MAbs) PG9, PG16, and VRC01 (Walker et al., 2009; Zhou et al., 2010). Here, we adopt a phylogenetic perspective on such studies which traditionally ignore shared ancestry and ask whether these viral evolutionary patterns translate into a noticeable pylogenetic signal for resistance to neutralization. Table 6 lists the λB estimates for the IC50 measurements, both as univariate and combined multivariate traits. The mean posterior phylogenetic signals suggest intermediate heritability of resistance to neutralization for the three MAbs, but the broad credible intervals and low KL divergences indicate that these estimates are poorly informed by the data. For the PG9 IC50 in particular, a non-zero estimate cannot be supported. The lower mean trivariate trait signal is accompanied by a somewhat higher KL and an even stronger support against a non-zero estimate. As expected from the fact that PG9 and PG16 both mainly bind to a quaternary epitope on the second variable loop in the viral envelope trimer (Walker et al., 2009), their log(IC50) values are highly correlated (0.95 [0.831,0.963]). VRC01 on the other hand is directed against the CD4 binding site (Zhou et al., 2010), and its IC50 values are only moderately correlated with those for PG9 (0.362 [0.015,0.649]) and PG16 (0.462 [0.146,0.730]). We illustrate the evolutionary pattern for resistance against PG16 and VRC01 in Fig. 6, and also summarize the mean log(IC50) across all lineages at different points through time below the trees. The latter indicates an overall rise in resistance through time, in particular within the first 5 years since the early samples in the mid to late 1980s. The early time points generally have lower resistance, and although they may indeed have led to less descendants, it is also cautious to bear in mind potential sampling artifacts.
Table 6.
Phylogenetic signal estimates for HIV evolution towards resistance to antibodies.
| Trait | λB | KL | BFf0 | BFf1 |
|---|---|---|---|---|
| IC50PG9 | 0.468 (0.079,0.841) | 0.005 | −0.459 | 5.207 |
| IC50PG16 | 0.570 (0.175,0.958) | 0.044 | 2.359 | 0.243 |
| IC50VRC01 | 0.558 (0.184,0.870) | 0.048 | 2.232 | 9.241 |
| IC50PG9,PG16,VRC01 | 0.364 (0.075,0.677) | 0.230 | −5.052 | 15.743 |
Figure 6. HIV-1 sensitivity to the broadly neutralizing monoclonal antibodies.

Log IC50 measurements for two MAbs, PG16 and VRC01, are shown at the tips of the same same tree (left an right for PG16 and VRC01 respectively), with tip circles areas proportional to these values. Higher log IC50 values represent lower sensitivity of the virus to neutralization. The ancestral reconstruction of the sensitivity to MAb neutralization is depicted using a color gradient along the branches. Below the trees, we plot the average evolution in log IC50 with credible interval intervals (95% highest posterior density intervals) by slicing the tree branches at particular time points and imputing the trait values at that time point (cfr. Bielejec et al. (2011)).
3.5 Drosophila susceptibility to sigma virus infection
In our final example, we investigate the ability of three host-specific sigma viruses (family Rhabdoviridae) to persist and replicate in different species of Drosophilidae. This has been previously studied by infecting 51 Drosophila species with three host-specific viruses and measuring virus titers at fixed time points (Longdon et al., 2011). The authors use a phylogenetic mixed model to demonstrate that host relatedness strongly determines the viral persistence and replication in new hosts. Here we revisit this problem by diffusing the virulence measure, log2 viral load (log2(VL)), over the host phylogeny and measuring its phylogenetic signal while accommodating the multiple measurements through numerical integration of the unobserved average trait values at the tips. Wing size (a proxy for body size) is included as a control as it is expected to be a heritable trait for Drosophila species.
Our analysis indicates that the capacity to infect and replicate in different hosts for three different host-specific sigma viruses (DAffSV, DMelSV and DObsSV)) shows relatively high phylogenetic signal, matching that of wing size in the Drosophila species (Table 7). These estimates are associated with relatively high KL divergences and strong support in favor of a non-zero estimate, but also strong support against a perfect Brownian trait evolutionary process. When combined into a trivariate trait, the virulence evolutionary patterns showed similar, moderate to high positive correlations (0.760 [0.545–0.936], 0.675[0.408,0.954] and 0.661[0.436,0.856] between DAffSV and DMelSV, between DAffSV and DObsSV, and between DMelSV and DObsSV respectively). In Figure 7, we illustrate the trait evolutionary patterns for susceptibility to infection with two viruses from relatively closely related hosts.
Table 7.
Sigma virus infectivity in Drosophila
| Trait | λB | KL | BFf0 | BFf1 |
|---|---|---|---|---|
| wing size | 0.790 (0.588,0.938) | 1.307 | 34.951 | 24.264 |
| DAffSV log2(VL) | 0.770 (0.529,0.969) | 0.971 | 37.811 | 45.629 |
| DMelSV log2(VL) | 0.786 (0.567,0.971) | 1.152 | 24.523 | 7.654 |
| DObsSV log2(VL) | 0.711 (0.494,0.896) | 0.842 | 29.608 | 21.715 |
| DAffSV/DMelSV/DObsSV log2(VL) | 0.781 (0.616,0.921) | 1.313 | 32.813 | 38.680 |
Figure 7. Drosophila evolutionary history with reconstructed susceptibility to infection with different host-specific sigma viruses.

The ancestral reconstruction of log2(VL) measurements for two host-specific viruses (DA3SV in D. affinis, left; DObsSV in D. obscura, right) are shown using a color gradient along the branches of the same MCC tree. Higher log2(VL) values reflect higher levels of viral replication; circles are proportional to these values.
4 Discussion
We present a Bayesian implementation of Pagel’s λ (λB) to quantify phylogenetic signal of multivariate traits. The estimator accommodates the different sources of uncertainty associated with both the sequence and (repeated) trait evolutionary processes and outperforms other estimators, including its maximum likelihood equivalent λML, in terms of accuracy and precision on simulation data under different degrees of heritability. Further exploration of prior specification on λB suggests that a bell-shaped prior (β(2, 2)), preferring intermediate phylogenetic signal a priori, further improves estimator performance. This prior may help to linearize the sigmoidal relationship between λ and the known heritability spectrum as well as reduce the generally large variance of λ estimates for intermediate heritability values. These characteristics are not specific to the relationship between simulated heritability and phylogenetic signal as they have been observed in other simulation studies as well (Münkemüller et al., 2012). Instead of prior specification, it may therefore be useful to examine transformations like to logit function for this type of phylogenetic signal estimators. We did not perform an exhaustive evaluation of different phylogenetic simulators, but more comprehensive simulation studies have shown that λML and Abouheif’s Cmean fulfilled most of the criteria for a good performance (Münkemüller et al., 2012). This, and the fact that λML was also the most robust and sensitive among different methods to estimate spVL heritability (Shirreff et al., 2013), reassures that the λB estimator will generally perform well.
We here focus on different traits related to viral virulence, infectivity and phenotypic evolution. Only recently, phylogenetic signal estimators have been proposed to examine the heritability of viral traits throughout transmission history (e.g. Alizon et al. (2010)). Although we can reproduce a heritability of about 50% for HIV-1 spVL, a predictor of disease progression, in the MSM strict data set from Alizon et al. (2010), we acknowledge that this estimate is not strongly informed by the data. As for the example of HIV-1 resistance to antibody neutralization, the fact that HIV trees represent exponentially growing populations resulting in star-like tree topologies (Lemey et al., 2006), may offer little opportunity to quantify phylogenetic trait association with great precision. However, when focusing on the subset of taxa in clusters with relatively recent common ancestors, we retrieve similar, uncertain estimates for spVL phylogenetic signal in the MSM strict data set, but still lower signal in the other data sets. While in general, phylogenetic signal estimates are likely to benefit from higher phylogenetic accuracy, the potentially higher phylogenetic accuracy gained by focusing on transmission clusters in the SHCS does not appear to explain the higher spVL phylogenetic signal for the MSM strict data set. Other dataset-specific characteristics may therefore be more important. Adhering to a more strict definition for spVL may reduce measurement error and focusing on the MSM risk group may remove the effect of patient sex and to a large extent also age as confounding factors (Alizon et al., 2010). It is for example well-established that spVL is affected by patient sex, with males tending to have a higher spVL (Alizon et al., 2010) (reviewed in Langford et al. (2007)). Ignoring this variability as well as spVL measurement error may of course result in lower estimates of phylogenetic signal. In agreement with the low phylogenetic signal for the larger data sets, Hodcroft et al. (2014) recently found small but significant spVL heritability in a large UK dataset by making use of a phylogenetic mixed modeling, which can also be extended to accommodate intra-specific variation (Lynch, 1991; Housworth et al., 2004). The heritability of spVL therefore requires further investigation, in particular because it has important implications for understanding HIV dynamics (Hool et al., 2013).
Despite the different assays involved, it is clear that the antigenic evolutionary patterns are associated with different phylogenetic signal in human influenza and HIV-1. Although also governed by a limited number of genetic changes (Koel et al., 2013), escaping the antibody response has a strong effect on the influenza population dynamics as reflected by the ladder-like trees for sequences sampled throughout different epidemic seasons. This pattern reflects a continual turnover and a relatively low standing genetic variation at any point in time (akin to the within-host HIV-1 phylodynamics, Lemey et al. (2006))(Grenfell et al., 2004). It is therefore not surprising that this tree structure translates into a strong phylogenetic signal and that major MDS coordinate show a clear drift pattern across the tree (Figure 5). On the other hand, the phylogenetic structure of HIV at the population level, with multiple co-circulating lineages within a particle subtype, does not reflect the action of (humoral) immune selection (Grenfell et al., 2004). It is therefore remarkable to find population evolution towards increased resistance (Euler et al., 2011; Bunnik et al., 2010; Bouvin-Pley et al., 2013). Despite the uncertain phylogenetic signal estimates, the apparently non-random clustering of early and late time point viruses in the example we examined points at the importance of using a phylogenetic approach to address such questions and advocates for caution against potential sampling artifacts.
According to the support against λB = 1 (Table 5), the influenza antigenic evolutionary patterns do not adhere to a perfect Brownian motion process. On the one hand, measurement error may be partly responsible for this because the MDS coordinates are based on sparse HI tables with interval and truncated measurements susceptible to experimental noise. On the other hand, a Brownian trait evolutionary model is unlikely to be appropriate for antigenic drift processes because it assumes zero mean displacement. Therefore, it would be useful to relax this assumption and allow for an unknown estimable drift vector for the mean displacement in the multivariate diffusion model. Such a model extension may also prove useful for the example of HIV-1 resistance against neutralization if there would indeed be a need to model a population evolution process towards increased resistance.
To quantify phylogenetic signal in influenza antigenic evolution, we here focus on the MDS coordinates that were readily available for the data set under investigation (Russell et al., 2008). We note however that the process of mapping antigenic phenotypes, referred to as ‘antigenic cartography’ (Smith et al., 2004), can be integrated with the genetic information by modeling the diffusion of antigenic phenotype over a shared virus phylogeny using the diffusion framework we also adopt here (Bedford et al., 2014). It would therefore be straightforward to use the tree transformation approach, resulting in the phylogenetic signal estimates, in this integrated genetic-antigenic framework. Perhaps the phylogenetic signal estimators may assist in selecting the number of dimensions in the Bayesian MDS approach. We have recently shown that a 2D model yielded optimal predictive power for a different H3N2 data set (Bedford et al., 2014), which seems to be in agreement with the phylogenetic signal estimates we obtain here. The second dimension still results in reasonable phylogenetic signal, but with drastically reduced support compared to the first dimension. It is therefore questionable that a third dimension would still be characterized by tree-based evolutionary patterns.
Our approach is not restricted to viral traits as shown by the sigma virus virulence study, where we in fact measure phylogenetic signal for a host trait (host susceptibility to viral infection). The phylogenetic signal estimates confirm that host phylogeny explains most of the variation in sigma virus replication and persistence in different Drosophila species, in a quantitatively similar way as the host phylogeny controls for the variation in wing sizes. Both measurement error and limitations to the model may again be responsible for deviations from a pure Brownian process, although we attempted to take into account the former by integrating out a mean tip trait value based on the repeated measurements. Longdon et al. (2011) showed that in addition to a ‘phylogenetic effect’, which explains similar levels of susceptibility for related species, there is also a ‘distance effect’ ensuring that viral titer is higher in species that are more closely related to the natural host. This systematic change in viral titer as a function of the distance from the natural host implies that drift, perhaps due to viral adaptation to its natural host, can also play a role in this trait evolutionary history. The correlation among the virulence patterns in the host phylogeny for the three different sigma viruses suggests that, despite a relatively high divergence, they may share similar modes of infection and replication in the same host tissues. As a consequence, genetic changes in different host lineages that impact cellular or immune components involved in replication and persistence may impact susceptibility to the three different viruses.
The Bayesian phylogenetic signal estimator has a number of advantages over other approaches. Whereas many implementations of alternative estimators are restricted to single traits, our extension of general-purpose phylogenetic diffusion models can be used to determine the phylogenetic association of multivariate traits of any dimension. As noted by Freckleton et al. (2002), simultaneously estimating the precision matrix and λ allows quantifying the correlation between pairs of traits that is optimal under a common random effects Brownian process. In addition, our framework is also equipped with ancestral trait reconstruction, which is also naturally achieved under the appropriate degree for statistical dependence. The phylogenetic signal estimator can also be connected to model extensions that relax the Brownian motion assumptions. We have previously presented a relaxed random walk model that accommodates diffusion rate heterogeneity by rescaling the precision matrix in a branch-specific manner (Lemey et al., 2010). Future studies will therefore be able to examine how phylogenetic signal estimates are affected by violations of the constant variance assumption in Brownian processes. Most of the examples we study also demonstrate that the there is a need to relax the zero-mean displacement assumption and incorporate some degree of drift. The Ornstein-Uhlenbeck (OU) process has been proposed as a ‘mean reverting’ extension of Brownian motion (Hansen, 1997; Butler & King, 2004; Blomberg et al., 2003), but perhaps more natural generalizations may be developed through stochastic modeling in our Bayesian framework. While it is important to model trait evolutionary processes more realistically, it may also be useful to accommodate heterogeneity in phylogenetic signal throughout evolutionary history because different lineages in a phylogeny may exhibit different degrees of phylogenetic signal (Münkemüller et al., 2012).
Bayesian inference is a natural framework to accommodate different sources uncertainty. In addition to phylogenetic error and uncertainty in the sequence and trait evolutionary process, we also take into account measurement error for our phylogenetic signal estimates when multiple measurements are available for the tip traits. One drawback of adequately accommodating uncertainty through simultaneous estimation is the computation time that may need to be invested, in particular when attempting to average over all plausible evolutionary histories. While we show here that this is still feasible for data sets including more than 600 taxa, we abandoned random trees when comparing model fit using marginal likelihood estimation. We use a stepping stone sampling approach for marginal likelihood estimation, which has proven to provide a relatively accurate measure of model fit, but at the price of considerable computational burden (Baele et al., 2012). Therefore, future studies may need to pursue more efficient testing procedures, for example by allowing λ to shrink to zero with some prior probability in our inference and estimating Bayes factors through comparison of prior and posterior odds (see, e.g., Suchard et al. (2005)).
In summary, many questions in evolutionary biology need to be addressed using a comparative phylogenetic approach. Although we have focused on phylogenetic signal in this study, we hope to have demonstrated that Bayesian phylogenetic diffusion models offer a flexible framework for evolutionary hypothesis testing. We also hope that future advances will open up more opportunities for unraveling trait evolutionary processes and their underlying genetic determinants, for viral pathogens as well as other organisms in general.
Supplementary Material
Table 1.
Comparison of bias, mean squared error (MSE) and coverage for different phylo-genetic signal estimators across a range of heritability values.
| Estimator | bias
|
MSE | coverage | ||
|---|---|---|---|---|---|
| total | h2 < 0.5 | h2 > 0.5 | |||
| Blomberg’s K | 0.031 | −0.101 | 0.120 | 0.048 | NA |
| Pagel’s λML, MLE | 0.073 | 0.130 | −0.024 | 0.045 | 0.830 |
| λB, β(1.0, 1.0) | 0.018 | 0.009 | −0.004 | 0.020 | 0.820 |
Acknowledgments
The research leading to these results has received funding from the European Union Seventh Framework Programme [FP7/2007-2013] under Grant Agreement no. 278433-PREDEMICS and ERC Grant agreement no. 260864. This work was also supported by National Institutes of Health grants R01 AI107034 and R01 HG006139 and National Science Foundation grants DMS 1264153 and IIS 1251151. This research was supported by the Fonds door Wetenschappelijk Onderzoek Vlaanderen (FWO) (krediet no. 1.5.252.12N).
The SHCS is financed in the framework of the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation (SNF grant #33CS30-134277 and -148522). Furthermore, the SHCS drug resistance database was financed by the following funding sources: the SHCS projects #470, 528, 569, the SHCS Research Foundation, the Swiss National Science Foundation (grant #324730-130865 (to HFG), the European Union Seventh Framework Program (Grant FP7/2007-2013), under the Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN; grant 223131, to HFG), the Yvonne-Jacob Foundation (to HFG), and by a further research grant of the Union Bank of Switzerland, in the name of an anonymous donor to HFG, an unrestricted research grant from Gilead, Switzerland to the SHCS research foundation, and by the University of Zurichs Clinical research Priority Program (CRPP) Viral infectious diseases: Zurich Primary HIV Infection Study (to HFG).
We thank Samuel Alizon for sharing data and R scripts, Tanja Stadler for sharing python scripts, Hanneke Schuitemaker & Zelda Euler for sharing sequence data, and Christophe Fraser & George Shirreff for sharing insights on their preliminary work.
We thank the patients participating in the SHCS and the members of the SHCS: Aubert V, Battegay M, Bernasconi E, Böni J, Bucher HC, Burton-Jeangros C, Calmy A, Cavassini M, Egger M, Elzi L, Fehr J, Fellay J, Francioli P, Furrer H (Chairman of the Clinical and Laboratory Committee), Fux CA, Gorgievski M, Günthard H (President of the SHCS), Haerry D (deputy of ”Positive Council”), Hasse B, Hirsch HH, Hirschel B, Hösli I, Kahlert C, Kaiser L, Keiser O, Kind C, Klimkait T, Kovari H, Ledergerber B, Martinetti G, Martinez de Tejada B, Metzner K, Müller N, Nadal D, Pantaleo G, Rauch A (Chairman of the Scientific Board), Regenass S, Rickenbach M (Head of Data Center), Rudin C (Chairman of the Mother & Child Substudy), Schmid P, Schultze D, Schöni-Affolter F, Schüpbach J, Speck R, Taffe P, Tarr P, Telenti A, Trkola A, Vernazza P, Weber R, Yerly S.
References
- Abouheif E. A method for testing the assumption of phylogenetic independence in comparative data. Evolutionary Ecology Research. 1999;1:895–909. [Google Scholar]
- Alizon S, von Wyl V, Stadler T, Kouyos RD, Yerly S, Hirschel B, Böni J, Shah C, Klimkait T, Furrer H, Rauch A, Vernazza PL, Bernasconi E, Battegay M, Bürgisser P, Telenti A, Günthard HF, Bonhoeffer S Swiss HIV Cohort Study. Phylogenetic approach reveals that virus genotype largely determines hiv set-point viral load. PLoS Pathogens. 2010;6:e1001123. doi: 10.1371/journal.ppat.1001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Molecular Biology and Evolution. 2012;29:2157–67. doi: 10.1093/molbev/mss084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D, Meade A, Pagel M. Constrained models of evolution lead to improved prediction of functional linkage from correlated gain and loss of genes. Bioinformatics. 2007;23:14–20. doi: 10.1093/bioinformatics/btl558. [DOI] [PubMed] [Google Scholar]
- Bedford T, Suchard MA, Lemey P, Dudas G, Gregory V, Hay AJ, McCauley JW, Russell CA, Smith DJ, Rambaut A. Integrating influenza antigenic dynamics with molecular evolution. Elife. 2014;3:e01914. doi: 10.7554/eLife.01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N, Däumer M, Oette M, Korn K, Hoffmann D, Kaiser R, Lengauer T, Selbig J, Walter H. Geno2pheno: Estimating phenotypic drug resistance from hiv-1 genotypes. Nucleic Acids Research. 2003;31:3850–5. doi: 10.1093/nar/gkg575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F, Rambaut A, Suchard MA, Lemey P. Spread: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg SP, Garland T, Jr, Ives AR. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution. 2003;57:717–45. doi: 10.1111/j.0014-3820.2003.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Boltz S, Debreuve E, Barlaud M. Image Analysis for Multimedia Interactive Services. 2007. knn-based high-dimensional kullback-leibler distance for tracking; p. 16. [Google Scholar]
- Bouvin-Pley M, Morgand M, Moreau A, Jestin P, Simonnet C, Tran L, Goujard C, Meyer L, Barin F, Braibant M. Evidence for a continuous drift of the hiv-1 species towards higher resistance to neutralizing antibodies over the course of the epidemic. PLoS Pathogens. 2013;9:e1003477. doi: 10.1371/journal.ppat.1003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, Euler Z, Welkers MRA, Boeser-Nunnink BDM, Grijsen ML, Prins JM, Schuitemaker H. Adaptation of hiv-1 envelope gp120 to humoral immunity at a population level. Nature Medicine. 2010;16:995–7. doi: 10.1038/nm.2203. [DOI] [PubMed] [Google Scholar]
- Butler M, King A. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. American Naturalist. 2004;164:683–695. doi: 10.1086/426002. [DOI] [PubMed] [Google Scholar]
- Dawid AP. Some matrix-variate distribution theory: notational considerations and a bayesian application. Biometrika. 1981;68:265–274. [Google Scholar]
- de Villemereuil P, Wells JA, Edwards RD, Blomberg SP. Bayesian models for comparative analysis integrating phylogenetic uncertainty. BMC Evolutionary Biology. 2012;12:102. doi: 10.1186/1471-2148-12-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with beauti and the beast 1.7. Molecular Biology and Evolution. 2012;29:1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler Z, Bunnik EM, Burger JA, Boeser-Nunnink BDM, Grijsen ML, Prins JM, Schuitemaker H. Activity of broadly neutralizing antibodies, including pg9, pg16, and vrc01, against recently transmitted subtype b hiv-1 variants from early and late in the epidemic. Journal of Virology. 2011;85:7236–45. doi: 10.1128/JVI.00196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. American Naturalist. 1985;125:1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- Fraser C, Lythgoe K, Leventhal GE, Shirreff G, Hollingsworth TD, Alizon S, Bonhoeffer S. Virulence and pathogenesis of hiv-1 infection: an evolutionary perspective. Science. 2014;343:1243727. doi: 10.1126/science.1243727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freckleton RP, Harvey PH, Pagel M. Phylogenetic analysis and comparative data: a test and review of evidence. American Naturalist. 2002;160:712–26. doi: 10.1086/343873. [DOI] [PubMed] [Google Scholar]
- Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, Mumford JA, Holmes EC. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–32. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Guo H, Weiss RE, Gu X, Suchard MA. Time squared: repeated measures on phylogenies. Molecular biology and evolution. 2007;24:352–362. doi: 10.1093/molbev/msl165. [DOI] [PubMed] [Google Scholar]
- Hansen T. Stabilizing selection and the comparative analysis of adaptation. Evolution. 1997;52:1341–1351. doi: 10.1111/j.1558-5646.1997.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W. Geiger: investigating evolutionary radiations. Bioinformatics. 2008;24:129–31. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
- Hartfield M, Murall CL, Alizon S. Clinical applications of pathogen phylogenies. Trends Mol Med. 2014;20:394–404. doi: 10.1016/j.molmed.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Harvey PH, Purvis A. Comparative methods for explaining adaptations. Nature. 1991;351:619–24. doi: 10.1038/351619a0. [DOI] [PubMed] [Google Scholar]
- Heitjan DF, Basu S. Distinguishing “missing at random” and “missing completely at random”. The American Statistician. 1996;50:207–213. [Google Scholar]
- Hodcroft E, Hadfield JD, Fearnhill E, Phillips A, Dunn D, O’Shea S, Pillay D, Leigh Brown AJ UK HIV Drug Resistance Database & UK CHIC Study. The contribution of viral genotype to plasma viral set-point in hiv infection. PLoS Pathog. 2014;10:e1004112. doi: 10.1371/journal.ppat.1004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth TD, Laeyendecker O, Shirreff G, Donnelly CA, Serwadda D, Wawer MJ, Kiwanuka N, Nalugoda F, Collinson-Streng A, Ssempijja V, Hanage WP, Quinn TC, Gray RH, Fraser C. Hiv-1 transmitting couples have similar viral load set-points in rakai, uganda. PLoS Pathogens. 2010;6:e1000876. doi: 10.1371/journal.ppat.1000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool A, Leventhal GE, Bonhoeffer S. Virus-induced target cell activation reconciles set-point viral load heritability and within-host evolution. Epidemics. 2013;5:174–80. doi: 10.1016/j.epidem.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Housworth EA, Martins EP, Lynch M. The phylogenetic mixed model. Am Nat. 2004;163:84–96. doi: 10.1086/380570. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung’u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. Adaptation of hiv-1 to human leukocyte antigen class i. Nature. 2009;458:641–5. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koel Björn F, Burke David F, Bestebroer Theo M, van der Vliet Stefan, Zondag Gerben C M, Vervaet Gaby, Skepner Eugene, Lewis Nicola S, Spronken Monique I J, Russell Colin A, Eropkin Mikhail Y, Hurt Aeron C, Barr Ian G, de Jong Jan C, Rimmelzwaan Guus F, Osterhaus Albert D M E, Fouchier Ron A M, Smith Derek J. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342(6161):976–9. doi: 10.1126/science.1244730. [DOI] [PubMed] [Google Scholar]
- Kouyos RD, von Wyl V, Yerly S, Böni J, Taffé P, Shah C, Bürgisser P, Klimkait T, Weber R, Hirschel B, Cavassini M, Furrer H, Battegay M, Vernazza PL, Bernasconi E, Rickenbach M, Ledergerber B, Bonhoeffer S, Günthard HF. Molecular epidemiology reveals long-term changes in hiv type 1 subtype b transmission in switzerland. J Infect Dis. 2010;201:1488–97. doi: 10.1086/651951. [DOI] [PubMed] [Google Scholar]
- Langford SE, Ananworanich J, Cooper DA. Predictors of disease progression in hiv infection: a review. AIDS research and therapy. 2007;4:11. doi: 10.1186/1742-6405-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin SR, Karasov WH, Ives AR, Middleton KM, Garland T., Jr Morphometrics of the avian small intestine compared with that of nonflying mammals: a phylogenetic approach. Physiological and Biochemical Zoology. 2008;81:526–50. doi: 10.1086/590395. [DOI] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Drummond A, Suchard M. Bayesian phylogeography finds its root. PLoS Computational Biology. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Bedford T, Faria N, Bielejec F, Baele G, Russell CA, Smith DJ, Pybus OG, Brockmann D, Suchard MA. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza h3n2. PLoS Pathogens. 2014;10:e1003932. doi: 10.1371/journal.ppat.1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Pybus OG. Hiv evolutionary dynamics within and among hosts. AIDS Reviews. 2006;8:125–40. [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Molecular Biology and Evolution. 2010;27:1877–85. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. r package version 0.6-4 edition. 2012. FNN: Fast Nearest Neighbor Search Algorithms and Applications. [Google Scholar]
- Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. Antiretroviral-drug resistance among patients recently infected with hiv. New England Journal of Medicine. 2002;347:385–94. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathogens. 2011;7:e1002260. doi: 10.1371/journal.ppat.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB. Phylogenetic niche conservatism, phylogenetic signal and the relationship between phylogenetic relatedness and ecological similarity among species. Ecology Letters. 2008;11:995–1003. doi: 10.1111/j.1461-0248.2008.01229.x. [DOI] [PubMed] [Google Scholar]
- Lynch M. Methods for the analysis of comparative data in evolutionary biology. Evolution. 1991;45:1065–1080. doi: 10.1111/j.1558-5646.1991.tb04375.x. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Rinaldo CR, Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in hiv-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–70. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Margolick JB, Phair JP, Rinaldo CR, Detels R, Jacobson LP, Muñoz A. Prognostic value of hiv-1 rna, cd4 cell count, and cd4 cell count slope for progression to aids and death in untreated hiv-1 infection. Journal of the American Medical Association. 2007;297:2349–50. doi: 10.1001/jama.297.21.2349. [DOI] [PubMed] [Google Scholar]
- Minin V, Bloomquist E, Suchard M. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Molecular Biology and Evolution. 2008;25:1459–71. doi: 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran P. Notes on continuous stochastic phenomena. Biometrika. 1950;37:17–23. [PubMed] [Google Scholar]
- Münkemüller T, Lavergne S, Bzeznik B, Dray S, Jombart T, Schiffers K, Thuiller W. How to measure and test phylogenetic signal. Methods in Ecology and Evolution. 2012;3:743–756. [Google Scholar]
- O’Meara BC, Ané C, Sanderson MJ, Wainwright PC. Testing for different rates of continuous trait evolution using likelihood. Evolution. 2006;60:922–33. [PubMed] [Google Scholar]
- Orme D, Freckleton R, Thomas G, Petzoldt T, Fritz S, Isaac N, Pearse W. R package version 0.5.2. 2013. caper: Comparative Analyses of Phylogenetics and Evolution in R. [Google Scholar]
- Orne D, Freckleton R, Thomas G, Petzoldt T, Fritz S, Issac N, Pearse W. Caper: comparative analyses of phylogenetics and evolution in R. 2013 http://cran.r-project.org/web/packages/caper/index.html.
- Pagel M. Inferring the historical patterns of biological evolution. Nature. 1999;401:877–84. doi: 10.1038/44766. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Pinheiro J, Bates D, DebRoy S, Team RC. nlme: Linear and Nonlinear Mixed Effects Models. 2013. [Google Scholar]
- Pybus OG, Suchard MA, Lemey P, Bernardin FJ, Rambaut A, Crawford FW, Gray RR, Arinaminpathy N, Stramer SL, Busch MP, Delwart EL. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15066–71. doi: 10.1073/pnas.1206598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell LJ. phytools: an r package for phylogenetic comparative biology (and other things) Methods in Ecology and Evolution. 2012;3:217–223. [Google Scholar]
- Revell LJ, Collar DC. Phylogenetic analysis of the evolutionary correlation using likelihood. Evolution. 2009;63:1090–100. doi: 10.1111/j.1558-5646.2009.00616.x. [DOI] [PubMed] [Google Scholar]
- Revell LJ, Graham Reynolds R. A new bayesian method for fitting evolutionary models to comparative data with intraspecific variation. Evolution. 2012;66:2697–707. doi: 10.1111/j.1558-5646.2012.01645.x. [DOI] [PubMed] [Google Scholar]
- Revell LJ, Mahler DL, Peres-Neto PR, Redelings BD. A new phylogenetic method for identifying exceptional phenotypic diversification. Evolution. 2012;66:135–46. doi: 10.1111/j.1558-5646.2011.01435.x. [DOI] [PubMed] [Google Scholar]
- Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC, de Jong JC, Kelso A, Klimov AI, Kageyama T, Komadina N, Lapedes AS, Lin YP, Mosterin A, Obuchi M, Odagiri T, Osterhaus ADME, Rimmelzwaan GF, Shaw MW, Skepner E, Stohr K, Tashiro M, Fouchier RAM, Smith DJ. The global circulation of seasonal influenza a (H3N2) viruses. Science. 2008;320:340–346. doi: 10.1126/science.1154137. [DOI] [PubMed] [Google Scholar]
- Shapiro B, Ho SYW, Drummond AJ, Suchard MA, Pybus OG, Rambaut A. A bayesian phylogenetic method to estimate unknown sequence ages. Molecular Biology and Evolution. 2011;28:879–87. doi: 10.1093/molbev/msq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirreff G, Alizon S, Cori A, Günthard HF, Laeyendecker O, van Sighem A, Bezemer D, Fraser C. How effectively can hiv phylogenies be used to measure heritability? Evolution, Medicine and Public Health. 2013;2013:209–24. doi: 10.1093/emph/eot019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–6. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Suchard M, Weiss R, Sinsheimer J. Bayesian selection of continuous-time Markov chain evolutionary models. Molecular Biology and Evolution. 2001;18:1001–1013. doi: 10.1093/oxfordjournals.molbev.a003872. [DOI] [PubMed] [Google Scholar]
- Suchard M, Weiss R, Sinsheimer J. Models for estimating Bayes factors with applications in phylogeny and tests of monophyly. Biometrics. 2005;61:665–673. doi: 10.1111/j.1541-0420.2005.00352.x. [DOI] [PubMed] [Google Scholar]
- Swiss HIV Cohort Study. Schoeni-Affolter F, Ledergerber B, Rickenbach M, Rudin C, Günthard HF, Telenti A, Furrer H, Yerly S, Francioli P. Cohort profile: the swiss hiv cohort study. International Journal of Epidemiology. 2010;39:1179–89. doi: 10.1093/ije/dyp321. [DOI] [PubMed] [Google Scholar]
- Tang J, Tang S, Lobashevsky E, Zulu I, Aldrovandi G, Allen S, Kaslow RA Zambia-UAB HIV Research Project. Hla allele sharing and hiv type 1 viremia in seroconverting zambians with known transmitting partners. AIDS Research and Human Retroviruses. 2004;20:19–25. doi: 10.1089/088922204322749468. [DOI] [PubMed] [Google Scholar]
- van der Kuyl AC, Jurriaans S, Pollakis G, Bakker M, Cornelissen M. Hiv rna levels in transmission sources only weakly predict plasma viral load in recipients. AIDS. 2010;24:1607–8. doi: 10.1097/QAD.0b013e32833b318f. [DOI] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR Protocol G Principal Investigators. Broad and potent neutralizing antibodies from an african donor reveal a new hiv-1 vaccine target. Science. 2009;326:285–9. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. Structural basis for broad and potent neutralization of hiv-1 by antibody vrc01. Science. 2010;329:811–7. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.