

Abstract
Dinucleoside polyphosphates, NpnN′, exert their physiological effects via P2 receptors (P2Rs). NpnN′ are attractive drug candidates as they offer better stability and specificity compared to nucleotides, the most common P2R ligands. To further improve the agonist properties of NpnN′, we synthesized novel isosters of dinucleoside polyphosphates where N and N′ are A or U and where the Pα or Pβ phosphate groups are replaced by boranophosphate, denoted as Npn(α-B)N′ or Npn(β-B)N′ (n = 3, 4), respectively. The potency of Npn(α/β-B)N′ analogues was evaluated at tP2Y1, hP2Y2, hP2Y4, and rP2Y6 receptors. The most potent P2Y1R and P2Y6R agonists were the Up4(β-B)A (A isomer, EC50 of 0.5 μM vs 0.004 μM for 2-SMe-ADP) and Up3(α-B)U (B isomer, EC50 of 0.3 μM vs 0.2 μM for UDP), respectively. The receptor subtype selectivity is controlled by the position of the borano moiety on the NpnN′ polyphosphate chain and the type of the nucleobase. In addition, Npn(α/β-B)N′ proved ~22-fold more resistant to hydrolysis by e-NPP1, as compared to the corresponding NpnN′ analogues. In summary, Up4(β-B)A and Up3(α-B)U are potent, stable, and highly selective P2Y1 and P2Y6 receptor agonists, respectively.
INTRODUCTION
Naturally occurring dinucleoside polyphosphates, denoted also as dinucleotides and NpnN′ (N and N′ = A, G, U, C; n = 2–6; analogue 1) (Figure 1),1–3 comprise two nucleosides linked by a polyphosphate chain through phosphoester bonds at the 5′-position of the two ribose moieties. Dinucleotides control various physiological functions.4 In recent years, interest in dinucleoside polyphosphates as potent and stable purinergic receptors agonists has been growing.5
Figure 1.

Naturally occurring dinucleotides.
The biological activities of these extracellular dinucleotides include regulation of vascular tone, platelet aggregation, neurotransmission, glycogen breakdown, and renal Na+ excretion and urine flow.6–8
Extracellular effects of dinucleotides involve activation of nucleotide receptors, P2Rs, and possibly dinucleotide receptors.9–12 The members of the P2 receptor superfamily are subdivided into ligand-gated ion channels (P2X receptors, P2XRs) and G-protein-coupled receptor subtypes (P2Y receptors, P2YRs). The principal physiological agonists of the P2Rs are adenosine 5′-triphosphate (ATP), adenosine 5′-diphosphate (ADP), uridine 5′-triphosphate (UTP), uridine 5′-diphosphate (UDP), and UDP-glucose.13 In addition, several dinucleoside polyphosphates9 potently activate various P2YRs.14,15
P2YRs are attractive pharmaceutical targets because they control many physiological functions.16,13 Currently, most P2YR agonists proposed as drugs consist of a nucleotide scaffold,17,16 the stability of which is still unsatisfactory. One of the approaches to overcome the inherent instability of nucleotide-based drug candidates is the use of dinucleotides that are metabolically more stable than the corresponding nucleotides.18 This approach is rather promising, and indeed several dinucleotides have been tested in preclinical trials. For instance, diadenosine tetraphosphate, Ap4A, diuridine tetraphosphate, Up4U, and uridine deoxycytidine tetraphosphate, Up4dC are effective at lowering blood pressure during anesthesia, and in the treatment of dry eye disease, cystic fibrosis, and retinal detachment.18–21 Up4U and Up4dC, known as diquafosol and denufosol, respectively, are currently in phase III of clinical development.16
Although several endogenous dinucleoside polyphosphates have good pharmacological activities, their in vivo half-lives are relatively short.14 Prolongation of the in vivo half-lives of dinucleotides is a prerequisite to their use as potential drugs. In addition, enhancement of affinities and enhancement of selectivities of endogenous NpnN′ analogues for specific P2 receptor subtypes are other important challenges.
Previously, we demonstrated the improved pharmacological properties of a series of dinucleoside poly(borano)phosphate analogues, Npn(β/γ-B)N′, 2–5 (Figure 2) compared to the parent compounds.22 We found that Ap3/5(β/γ-B)A analogues were potent P2Y1R agonists. Specifically, Ap5(γ-B)A was equipotent to 2-MeS-ADP, thus making it one of the most potent P2Y1R agonists currently known. Moreover, Ap5(γ-B)A did not activate the P2Y2R. In contrast, Up3/5(β/γ-B)U analogues were extremely poor agonists at both P2Y1R and P2Y2R. Npn(β/γ-B)N′ analogues exhibited remarkable chemical stability under physiological conditions, including at a pH mimicking gastric juice acidity. The rate of hydrolysis of Ap3(β-B)A by human e-NPP1 and e-NPP3 was decreased by 40% and 59%, respectively, compared to Ap3A. However, Ap5(γ-B)A was hydrolyzed by e-NPP1 at a rate comparable to that of Ap5A. Apparently, the stabilizing effect of the borano modification is pronounced only when in the vicinity of the cleavage site.
Figure 2.
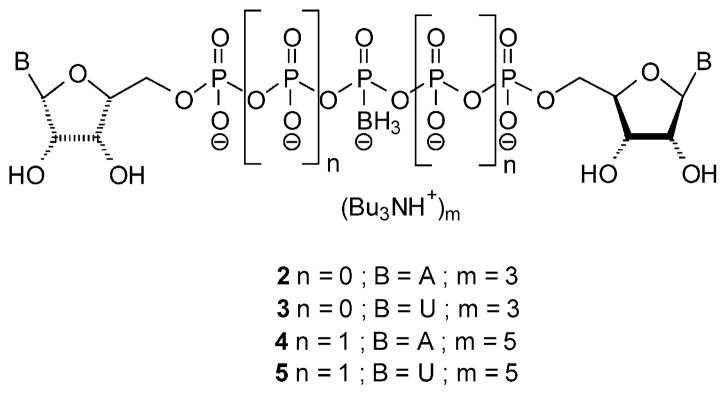
Previously synthesized dinucleoside poly(borano)phosphate analogues.
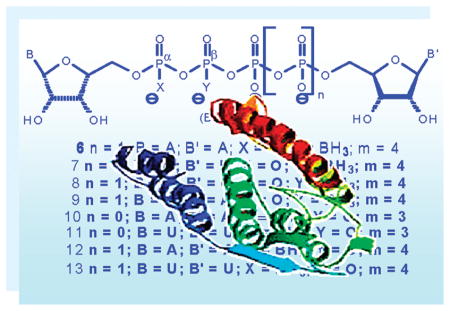
Here, we leveraged the above findings on the beneficial pharmacological properties of the boranophosphate bioisosters for the design of stable, potent, and selective P2Y1 and P2Y6R agonists. Specifically, we report on the synthesis and characterization of dinucleoside tetra- or tri(α/β-borano)phosphate analogues, 6–13 (Figure 3), their resistance to hydrolysis by e-NPP1, and their activity at P2Y1,2,4,6 receptors. In addition we analyze the effect of the position of the boranophosphate moiety, the composition of the nucleobase, and the length of the polyphosphate linker on the pharmacological properties of analogues 6–13. Our findings provide structure–activity relationship (SAR) tools to further design potential drug candidates targeting P2Y1 and P2Y6 receptors.
Figure 3.

Dinucleoside poly(borano)phosphate analogues synthesized and investigated in this study.
RESULTS
Design of Novel Dinucleoside Poly(borano)phosphate Analogues
Recently we reported the synthesis and P2YR activity of diadenosine and diuridine tri- and penta(β/γ-borano)phosphate analogues 2–5.22 The most evident change in receptor affinity was observed for the γ-borano modification of Ap5A, which greatly increased agonist potency for the rP2Y1-GFP receptor, compared to Ap5A. Ap5(γ-B)A (EC50 = 6.3 × 10−8 M), 4, was found to be slightly more potent at the rP2Y1-GFP receptor than 2-MeS-ADP (EC50 = 1.0 × 10−7 M).22 We found that the hydrolytic stability of the Npn(β/γ-B)N′ analogues 2–5 at physiological or gastric pH is dependent on the length of the polyphosphate chain. The dinucleoside triphosphate analogues 2 and 3 were more stable than the pentaphosphate analogues 4 and 5 regarding both chemical and e-NPP1-dependent hydrolysis.
The potent P2Y1R agonist, analogue 4, is not a good drug candidate, since it is hydrolyzed by e-NPP1 at a similar rate as Ap5A. This susceptibility to hydrolysis is probably related to the distance of the borano group from the enzymatic cleavage site between Pα and Pβ.
On the basis of these findings and the enhanced stability of an analogue when the borano modification is in the vicinity of the e-NPP1 cleavage site (Pα-Pβ), we produced here a new series of diadenosine and diuridine tri- and tetra(borano)-phosphate analogues in which the borano modification is in either the Pα or Pβ position, with a view toward developing novel drug candidates targeting certain P2YRs.
Since Ap5(γ-B)A is a potent P2Y1R agonist, whereas Ap3(β-B)A is moderately less potent (EC50 = 6.3 × 10−8 and 9.0 × 10−7 M, respectively), we synthesized Ap4(β-B)A, analogue 6, to evaluate its potency at the P2Y1R. The high potency of Up4U as a P2Y2R agonist (EC50 = 0.06 μM vs 0.015 μM for UTP, an endogenous P2Y2R agonist)14 led us to synthesize Up4(β-B)U, analogue 7, whereas the use of Ap4U as a vasoconstrictive factor23 prompted us to synthesize Ap4(β-B)U, analogue 8, and Up4(β-B)A, analogue 9.
Synthesis of Dinucleoside Poly(β-borano)phosphate Analogues 6–9
Previously, we synthesized dinucleoside poly(borano)phosphate analogues using two nucleoside phosphoroimidazolides24 as P-donors and an inorganic borano-phosphate salt (BPi)25 as a P-acceptor. MgCl2 was added as an activator to overcome the low nucleophilicity of BPi as a P-acceptor.22
Here, we used the same synthetic method to prepare the Np4(β-B)N′ analogues. Briefly, the synthesis of analogues 6–9 involved the activation of a nucleotide bis- and tris-tributylammonium salt, compounds 14 and 15, respectively, with CDI in dry DMF at room temperature for 3 h followed by the addition of BPi and MgCl2 and stirring at room temperature for ~12 h (Scheme 1) to produce analogues 6–9 at up to 40% yield after LC separation.
Scheme 1a.

aReaction conditions: (a) CDI (5 equiv), DMF, room temp 3 h; (b) dry MeOH (5 equiv), room temp 8 min; (c) BPi (1 equiv), DMF, MgCl2 (8 equiv), room temp overnight.
This synthesis involves a concerted reaction of three molecules (two P-donors and one P-acceptor) (Figure 4). Np4(β-B)N′ (~40% yield) (Figure 4), Np3N′ (~ 25% yield) (Figure 5, path a), and Np4N (~10% yield) analogues were the sole products formed. No Npn(B) products, Figure 5 path b, were formed under these conditions.
Figure 4.

Proposed structure for nucleotide-BPi Mg2+ complexes leading to products 6–9.
Figure 5.

Proposed structure for nucleotide-BPi Mg2+ complexes leading to possible byproducts.
The identity and purity of the products were established by 1H and 31P NMR, ESI or FAB mass spectrometry, and HPLC in two solvent systems. 31P NMR spectra showed a typical Pβ signal as a multiplet at about 80 ppm. 1H NMR spectra showed borane hydrogen atoms as a very broad signal at about 0.3 ppm.
Because of the chiral center at Pβ, analogues 6–9 were each obtained as a pair of two diastereoisomers. In both 1H and 31P NMR spectra, there was a slight difference between the chemical shifts for the two diastereoisomers.
These isomers were separated by reverse-phase HPLC, using triethylammonium acetate/MeOH isocratic elution, with about 1 min difference in their retention times. The first eluting isomer was designated as the A isomer, and the other was designated as the B isomer. Surprisingly, the ratio between the diastereoisomers was not always 1:1, as determined by the integration of the respective HPLC peaks. The area under the curve (AUC) for each diastereoisomer was calculated as the average of peak areas from three different HPLC chromatograms. A comparison of the AUC for the Np4(β-B)N′ diastereoisomers showed that the formation of each analogue (6–9) involves a different degree of diastereoselectivity. The diastereoisomer ratio (A/B) was 0.65:0.35 for analogue 6, 0.51:0.49 for analogue 7, 0.52:0.48 for analogue 8, and 0.55:0.45 for analogue 9.
Synthesis of Dinucleoside Poly(α-borano)phosphate Analogues 10–13
The synthesis of Np4(α-B)N′ analogues 12 and 13 is based on the Eckstein procedure for the preparation of triphosphates,26 which was applied by Jones et al.27 to the synthesis of dinucleoside tetra- and pentaphosphates. Specifically, 5′-phosphitylation of 2′,3′-methoxymethylidene nucleoside 14 with 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one (salicylchlorophosphite) followed by reaction with inorganic pyrophosphate yielded the cyclic derivative 16 (Scheme 2). Then, borane–dimethyl sulfide complex was added to yield product 17, which was then treated with either AMP or UMP bis-tributylammonium salt in dry DMF, and MgCl2, to yield the partially protected dinucleotide. ZnCl2 was found to be less effective than MgCl2, yet better than no catalyst. Finally, removal of the 2′,3′-methoxymethylidene group at pH 2.3 and then at pH 9 produced products 12 and 13 at high yields (up to 85% after LC separation).
Scheme 2. a.

aReaction conditions: (a) DMF, pyridine, 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one (1.1 equiv), room temp 10 min; (b) bis(tri-n-butylammonium)pyrophosphate (1.1 equiv), tri-n-butylamine (4 equiv), room temp 10 min; (c) BH3·SMe2, room temp 15 min; (d) NMP, MgCl2, room temp 16 h; (e) pH 2.3, room temp 3 h; pH 9, room temp 40 min.
This method could not be applied to the synthesis of dinucleoside triphosphates 10 and 11. These analogues were synthesized via the activation of the 5′-phosphate of NMP 18 with CDI, followed by coupling with the appropriate NDP(α-B)28 analogue to yield dinucleotide 10 or 11 (Scheme 3). In preliminary experiments to synthesize analogues 10 and 11, we first activated NDP(α-B) with CDI to generate the P-donor and used NMP as the P-acceptor. In a second approach, CDI-activated NMP was the P-donor, and NDP(α-B) was the P-acceptor. The activation of NMP (18) and NDP(α-B) with CDI was monitored by TLC, which showed that NMP completely reacted with CDI after 3 h, whereas the activation of NDP(α-B) was incomplete over this time period, likely because of the lower nucleophilicity of the phosphate in this analogue. Therefore, our subsequent syntheses of analogues 10 and 11 utilized the activation of NMP(Bu3NH)+2 salt with CDI in dry DMF at room temperature for 3 h, followed by the addition of nonactivated NDP(α-B)-(Bu3NH)+2 salt, in the presence of MgCl2 at room temperature for 12 h. Analogue 10 or 11 was formed as the exclusive product at up to 85% after LC separation.
Scheme 3. a.
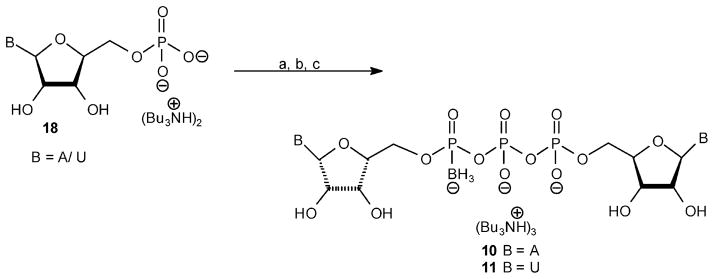
aReaction conditions: (a) CDI (5 equiv), DMF, room temp 3 h; (b) dry MeOH (5 equiv), room temp 8 min; (c) NDP(α-B), MgCl2 (8 equiv), room temp overnight.
Analogues 10–13 were characterized by 31P NMR spectrometry and high resolution mass spectrometry. Because of the chiral center at Pα, analogues 10–13 were each obtained as a pair of diastereoisomers (A and B isomers). Unlike analogues 6–9, the ratio between the diastereoisomers for analogues 10–13 was 1:1. Mg2+ ion serves as a catalyst, and reactions carried out without Mg2+ resulted in yields of <10%.
Activity of Analogues 6–13 at P2Y1/2/4/6-Rs
To study the activity of dinucleotides 6–13 at the P2Y1/2/4/6Rs, we evaluated the magnitude of increase in [Ca2+]i induced by these analogues. These studies were performed in human 1321N1 astrocytoma cells that are devoid of endogenous P2Y receptors but were stably transfected with turkey (t) P2Y1, human (h) P2Y2, human (h) P2Y4, or rat (r) P2Y6 receptors. Concentration–response curves were derived for each dinucleotide and compared with data of agonist for these receptor subtypes (i.e., 2-MeS-ADP for tP2Y1R, UTP for hP2Y2/4Rs, and UDP for rP2Y6R). The results are summarized in Table 1. In control experiments we found that untransfected 1321N1 astrocytoma cells did not respond to any of the analogues or endogenous agonists (data not shown).
Table 1.
EC50 Values for [Ca2+]i Elevation Induced by Analogues 6–13 at tP2Y1, hP2Y2, hP2Y4, and rP2Y6 Receptorsa
| EC50 (μM)
|
||||
|---|---|---|---|---|
| P2Y1 | P2Y2 | P2Y4 | P2Y6 | |
| 6A | 3.0 ± 1.7 | Nr | Nr | Sr |
| 6B | 4.3 ± 1.1 | Nr | Nr | Sr |
| 7A | Sr | Sr | Nr | 1.93 ± 1.11 |
| 7B | Sr | Nr | Nr | 0.98 ± 0.52 |
| 8A | Sr | Nr | Nr | 1.29 ± 0.65 |
| 8B | 6.9 | 64.2 | Nr | 1.92 ± 1.2 |
| 9A | 0.5 ± 0.13 | Sr | Nr | 20.9 ± 4.9 |
| 9B | 0.81 ± 0.55 | Nr | Nr | 11.7 ± 2.9 |
| 10A | 0.76 | Nr | Nr | Nr |
| 10B | 0.8 | Nr | Nr | Nr |
| 11A | 4.4 ± 0.93 | Nr | Nr | 0.72 ± 0.54 |
| 11B | Sr | Nr | Nr | 0.3 ± 0.08 |
| 12A | 1.48 | 25.9 | Nr | Nr |
| 12B | 6.9 | 64.2 | Nr | Nr |
| 13A | Nr | Sr | 9.4 ± 3.8 | 36.1 ± 3.3 |
| 13B | Sr | 24.6 ± 6.5 | 16.8 ± 4.3 | 3.9 ± 1.9 |
| 2-SMe-ADP | 0.004 ± 0.002 | |||
| UTP | 0.64 ± 0.25 | 0.48 ± 0.31 | ||
| UDP | 0.2 ± 0.06 | |||
Sr = slight response at 100 μM. Nr = no response.
Analogues 6–13 had no or weak activity at P2Y2 and P2Y4 receptors, whereas these compounds were relatively selective for either P2Y1 or P2Y6 receptor.
Adenosine containing dinucleotides 6, 10, and 12 were found to be P2Y1-R selective agonists, with EC50 values from 0.5 to 4.3 μM. Uridine containing dinucleotides 7, 11, and 13 were found to be selective P2Y6R agonists, with EC50 values from 0.3 to 3.9 μM. As for the mixed adenosine–uridine compounds, analogue 9 is a selective P2Y1R agonist (EC50 values of 0.5 and 0.81 μM for A and B isomers, respectively), whereas analogue 8 is a selective P2Y6R agonist (EC50 values of 1.29 and 1.92 μM for A and B isomers, respectively).
Analogue 9A is the most potent P2Y1R agonist found here with an EC50 = 0.5 μM, compared to 0.004 μM for 2-MeS-ADP, which is the most potent P2Y1R agonist currently known. Analogue 11B is the most potent P2Y6-R agonist with an EC50 = 0.3 μM, equipotent to UDP (EC50 = 0.2 μM).
None of the analogues antagonized the effect of equimolar concentrations of 2-MeS-ADP on P2Y1R activation, UTP on P2Y2/4R activation, or UDP on P2Y6R activation in 1321N1 cell transfectants (data not shown).
Hydrolysis of Npn(B)N Analogues by Human NPP1
Dinucleoside polyphosphates are substrates of certain members of the ectonucleotide pyrophosphatase/phosphodiesterase (e-NPP) family, namely, e-NPP1, e-NPP2, and e-NPP3.29 These enzymes cleave ApnAs asymmetrically to generate AMP and Apn−1. The degradation products could activate P2R subtypes other than the parent compound. Hence, more stable dinucleotides are desirable, since they result in less degradation products and may serve as drugs with limited side effects. We therefore investigated if analogues 6–13 resist hydrolysis by human e-NPP1.
Since members of the e-NPP family have an alkaline pH optimum, e-NPP1 activity was measured at pH 8.5. Hydrolysis of parent dinucleotides and analogues 6–13 by human e-NPP1 was determined after 20 min at 37 °C. The reaction was started by the addition of a dinucleotide analogue and terminated after 20 min by addition of perchloric acid. Dinucleotides and the nucleotides degradation products were separated and quantified by HPLC. The concentrations of reactants and products were determined from the relative areas for their absorbance maxima peaks.
The acid used to terminate the enzymatic reaction can cause partial degradation of the dinucleotide and nucleotide analogues. Therefore, the percentage of degradation for each analogue was assessed in the absence of enzyme, and this value was subtracted from the percentage of analogue degradation in the presence of enzyme.
Human e-NPP1 hydrolyzed Np4(β-B)N′ analogues 6 and 7 to NMP and either NTP(γ-B) (Scheme 4, path a) or NTP(β-B) (Scheme 4, path b). The time-dependent increase in NMP was monitored by HPLC. The identity of the degradation products was determined by comparing their retention times to those of controls. Good precision was obtained for determinations of NMP concentration, since the peaks of Npn(B)N′ analogues have longer retention times than NMP.
Scheme 4.

Possible Hydrolytic Pathways of Dinucleoside Poly(borano)phosphate Analogues 6 and 7 by hNPP1
The NTP(β-B) degradation product was easily identified, since it appeared as two peaks of the same height in the reverse-phase HPLC chromatogram, corresponding to the two diastereoisomers of this chiral compound. The HPLC chromatogram indicated that most of the degradation product was NTP(β-B) rather than NTP(γ-B) (the NTP(β-B)/NTP(γ-B) ratio was ~95:5), thus making path b of Scheme 4 the major hydrolytic pathway. We concluded that e-NPP1 cleaves dinucleotides 6/7 preferentially away from the boranophosphate moiety.
Np4(β-B)N′ analogues 8 and 9 can be hydrolyzed by e-NPP1 to yield both NMP (Scheme 5, path a) and N′MP (Scheme 5, path b), with N′ being the nucleotide base further from the borane moiety. HPLC analysis of the samples showed that the main hydrolysis product was N′MP (N′MP/NMP was ~94:6). Thus, path b of Scheme 5 is the main hydrolytic pathway. HPLC analysis of the hydroloysis of analogues 8 and 9 in the absence of enzyme showed that most of the NMP generated is due to the acidic medium. Thus, under acidic conditions, path a (Scheme 5) is the main chemical hydrolytic pathway.
Scheme 5.

Possible Hydrolytic Pathways of Dinucleoside Poly(borano)phosphate Analogues 8 and 9 by hNPP1
Npn(α-B)N analogues 10–13 can be hydrolyzed by e-NPP1 to either NMP (Scheme 6, path b) or NMP(α-B) (Scheme 6, path a). HPLC and MS analysis of these samples showed that the major enzymatic cleavage product was NMP. Most of the NMP(α-B) was attributed to acid-induced hydrolysis. Thus, most of the enzymatic degradation of analogues 10–13 is via path b, and most of the acid-induced degradation is via path a.
Scheme 6.

Possible Hydrolytic Pathways of Dinucleoside Poly(borano)phosphate Analogues 12 and 13 by hNPP1
The above data clearly demonstrate the protective effect of the boranophosphate group against e-NPP1-dependent hydrolysis of dinucleotides, in either Pα or Pβ position.
DISCUSSION
Np4(β-B)N′ Analogues, 6–9, Are Obtained via a Metal-Preorganized Concerted Three-Component Reaction
Dinucleoside poly(borano)phosphate products 6–9 are probably formed as a result of a preorganization of a P-acceptor (BPi) and two P-donors (nucleoside phosphoroimidazolides) coordinated with one Mg2+ ion (Figure 4). Although we expected to obtain the Np4(β-B)N′ analogues as exclusive products, Np3N′ (Figure 5, path a) and Np4N byproducts were also isolated. The formation of Np3N′ byproduct is driven by NDP remaining in the reaction mixture due to incomplete reaction with CDI (and not by NMP, as determined by TLC which indicated that over the same time period NMP reacted completely with CDI, whereas NDP did not). Thus, the activated form, NMP-Im, becomes a P-donor, whereas NDP, instead of the less nucleophilic BPi, functions as a P-acceptor. Furthermore, since a phosphate moiety is a better nucleophile than BPi, the Np3N′ byproduct is obtained and not NTP(γ-B) (Figure 5, path b). Since NDP was not completely activated by CDI, nonactivated form and the activated form, NDP-Im, reacted to yield the Np4N byproduct.
The concerted three-component reaction giving rise to analogues 6–9 proved to be diastereoselective in certain cases. Thus, the diastereoisomers ratio (A/B) for Ap4(β-B)A was 65:35 rather than 50:50. Likewise, the ratio for Up4(β-B)A was 55:45. However, the ratio for Up4(β-B)U and Ap4(β-B)U was almost 50:50. It appears that the origin of diastereoselectivity is the nucleobase of the NDP-Im component. When the nucleobase is A rather than U, there is steric hindrance between the borano group and the adenine in the transition state (Figure 6), resulting in the formation of less B-isomer vs A-isomer. Apparently, U does not induce significant steric hindrance, and therefore, analogues 7 and 8 are produced in ~50:50 ratio.
Figure 6.
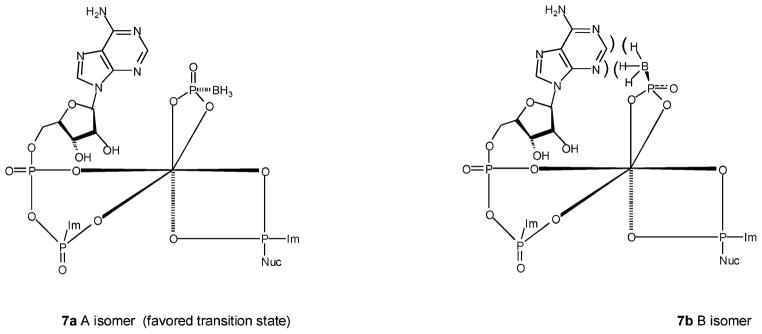
Steric hindrance is the origin of the diastereoselectivity of the reaction leading to products 7A and 7B.
Analogues 6–13 Are Highly Resistant to Hydrolysis by Human e-NPP1
Dinucleoside polyphosphates have considerably longer half-lives in vivo than nucleotides (i.e., NTP, NDP, and NMP), which are rapidly metabolized by members of various enzyme families, including nucleoside triphosphate diphosphohydrolase (NTPDases), ectonucleotide pyrophosphatase/phosphodiesterase (e-NPPs), and alkaline phosphatases.30 Naturally occurring dinucleoside polyphosphates are substrates for only a few e-NPPs.29
Therefore, here, dinucleoside poly(borano)phosphate derivatives 6–13 were tested for their resistance to hydrolysis by human e-NPP1, compared to the corresponding naturally occurring dinucleotides (Figure 7), to evaluate the effect of the borane moiety on the stability of dinucleotides. Naturally occurring dinucleotides (i.e., Up3U, Ap3A, Ap4A, Up4U, and Ap4U) were 17–22% hydrolyzed by human e-NPP1 over 20 min at 37 °C. The identity of the nucleobase (A or U) did not affect stability to enzymatic hydrolysis, since the rate of the hydrolysis was the same for both diadenosine and diuridine analogues with the same length of polyphosphate chain. For example, Ap3A and Up3U were 22% and 21% hydrolyzed over 20 min by e-NPP1, respectively. E-NPP1-dependent hydrolysis is not significantly affected by the length of the NpnN′ polyphosphate chain for either naturally occurring or synthetic analogues. Thus, dinucleoside tri- and tetraphosphates were hydrolyzed at similar rates (18% and 21% hydrolysis over 20 min for Up4U and Up3U, respectively), although dinucleoside triphosphate analogues were found to be slightly less stable, consistent with a previous report.29
Figure 7.

Hydrolysis of dinucleotide poly(borano)phosphate analogues 6–13 by human NPP1. The data represent the percentage of the compound being hydrolyzed over a 20 min period.
Substitution of the nonbridging oxygen atom on either Pα or Pβ with a BH3− group inhibits the hydrolysis of analogues 6–13 by e-NPP1 (Figure 7). All of the borane containing compounds were hydrolyzed by e-NPP1 at a significantly slower rate over 20 min at 37 °C than the corresponding naturally occurring dinucleotides. The presence of BH3− near the cleavage site (i.e., at Pα or Pβ) confers stability to the dinucleoside polyphosphates regarding enzymatic hydrolysis by e-NPP1 compared to a BH3 group at Pγ.22
Within the group of borane containing dinucleotides 6–13, there is a difference in their resistance to hydrolysis by e-NPP1, resulting from a difference in the position of the borane on the polyphosphate chain, i.e., α or β position.
The hydrolysis of dinucleotides is known to be asymmetric and to involve the α,β-pyrophosphate bond.31 We found that when borane is in the β-position, both diastereoisomers exhibit similar hydrolysis rates. Yet, when the borane moiety is in the α-position, there is a considerable difference between the hydrolysis rates of the diastereoisomers, with the A-isomer being more stable. Thus, the nucleophilic attack by the −OH group in water occurs at Pα, rather than Pβ.
Analogues in which the borane moiety is in the β-position relative to adenosine are slightly more stable than those in which the borane moiety is in the β-position to uridine (1.5% vs 3% hydrolysis over 20 min). The most stable analogues with respect to hydrolysis by e-NPP1 are the A-isomers of Ap3(α-B)A, analogue 10, and of Up3(α-B)U, analogue 11, which are completely resistant to hydrolysis by e-NPP1, in contrast to Np4(α-B)N′ (A-isomers), which are ~3% hydrolyzed over 20 min by e-NPP1.
Each dinucleotide has two possible cleavage sites for e-NPP1 in the polyphosphate chain that differ in their proximity to the borane moiety. Analysis of the degradation products showed that the dinucleotides 6–13 were almost exclusively cleaved by e-NPP1 at the site furthest from the borane moiety (Schemes 4–6), probably because the P-BH3 phosphodiester bond is less recognized by the enzyme as a substrate. On the other hand, analysis of the control (acid-induced degradation of analogues 6–13 in the absence of enzyme) showed that acid-induced hydrolysis (used to stop the enzyme activity) occurred preferentially near the borane moiety. This is likely due to a reduction in electron density of the P-BH3 coordinate bond, compared to phosphate, making the P-BH3 more susceptible to nucleophilic attack by a water molecule.
Analogues 6–13 Are Selective P2YR Agonists
The activity of dinucleoside polyphosphates NpnN′ at various purinergic receptors was previously reported.9–11 Adenosine containing dinucleotides have at least minimal activity at P2Y1R, while uridine containing dinucleotides have at least minimal activity at P2Y2R and P2Y6R. The length of the polyphosphate chain determines P2Y subtype specificity, i.e., nucleoside diphosphates have similar specificity to dinucleoside triphosphates, and nucleoside triphosphates are similar to dinucleoside tetraphosphates.14
In the case of dinucleoside poly(borano)phosphates, the presence and position of the borane group in the polyphosphate chain (i.e., Pα, Pβ, or Pγ) also affects P2YR specificity. Although Up4U is a highly potent P2Y2R agonist,14,15 neither Up4(α-B)U nor Up4(β-B)U is an agonist of the P2Y2R subtype (Table 1), implying that the Pα and Pβ oxygen atoms of Up4U are involved in molecular recognition by the P2Y2R. Another interesting finding for the mixed dinucleotides, Ap4(β-B)U and Up4(β-B)A, is that when the borane is closer to the uridine, the analogue is 42-fold more selective for the P2Y1R than the P2Y6R (and inactive at the P2Y2/4Rs). When the borane is closer to the adenosine, the compound is a selective P2Y6R agonist with no activity at P2Y1/2/4Rs. These results led us to define the borane moiety as a “boundary marker”, dividing the molecule into two parts. Thus, in Up4(β-B)A, analogue 9, the boundary marker, divides the molecule into UDP-β-B and ADP. Since ADP is the naturally occurring P2Y1R agonist, Up4(β-B)A acts as a better agonist for the P2Y1R than the P2Y6R. For Ap4(β-B)U, analogue 8, the boundary marker divides the molecule into ADP-β-B and UDP, which is the naturally occurring P2Y6R agonist, and therefore, Ap4(β-B)U is a P2Y6R agonist and inactive at the P2Y1R.
Previously, we observed similar agonist potencies for ATP, ATP-α-B, and Ap3(β-B)A at the P2Y1R, which suggests that these molecules share the same P2Y1R binding site (and binding mode).22 Ap3(β-B)A may occupy the same binding site as ATP if the extra adenosine moiety is oriented parallel to the receptor’s α-helical domain away from the agonist binding site. Similarly for the Npn(α/β-B)N′ analogues, the borane moiety might define the boundary between the part of the dinucleotide that binds to the receptor and the part that is oriented outside the agonist binding binding domain.
When the borane moiety is situated at either Pα or Pβ, each analogue is obtained as two diastereoisomers. At the P2Y1R, the A-isomer is equipotent or slightly more active (up to 4-fold) than the B-isomer. At the P2Y6R, the B-isomer is more potent than the A-isomer (up to 9-fold). These data indicate that the P2Y1 and P2Y6 receptor subtypes have opposite diastereoselectivity. Our previous study on the diastereoselectivity of the P2Y1R showed that for ATP-α-B analogues, A and B isomers possess the Rp and Sp configuration, respectively. The absolute configuration of the two diastereoisomers was determined based on their NMR spectra.22,32 Applying the same considerations to the P2Y1R selective dinucleotides described here resulted in similar conclusions regarding the absolute configuration of the diastereoisomers (i.e., A-isomer is the Rp isomer). Assuming the same elution order of diastereoisomers from the HPLC column, if P2Y1 is a Pα-Rp isomer preferring receptor, then P2Y6 is a Pα-Sp preferring receptor.
CONCLUSION
Np3/4(α-B)N′ and Np4(β-B)N′ (N and N′ = A, U) derivatives are selective P2Y1 or P2Y6 receptor agonists, having no activity at the P2Y2 and P2Y4 receptors. The potency and receptor subtype selectivity can be controlled by the position of the borane moiety in the polyphosphate chain. This is most evident in dinucleotides Ap4(β-B)U and Up4(β-B)A, in which the difference in the position of the borane moiety renders the analogue either a selective P2Y1R or P2Y6R agonist, respectively. Furthermore, a boranophosphate bioisoster at either Pα or Pβ significantly improves metabolic stability with respect to hydrolysis by human e-NPP1. Resistance to e-NPP1-dependent hydrolysis increased up to >20-fold compared to the same compound lacking the borane moiety. Analogues with borane at Pα were more enzymatically stable than analogues with borane at Pβ. When the borane is at Pα, the A isomer is more stable than the B isomer, indicating that e-NPP1-dependent hydrolysis occurs at Pα, whereas when the borane is at Pβ, there is no difference in the rates of hydrolysis for the A and B isomers. The most potent agonists are the P2Y1R-selective A and B isomers of Ap3(α-B)A, analogues 10A/B, and the P2Y6R-selective B isomer of Up3(α-B)U, analogue 11B. Since these analogues are also resistant to e-NPP1-dependent hydrolysis, we propose them as promising scaffolds for the design of future drug candidates targeting P2Y1 and P2Y6 receptors. Improved drug candidates will be reported in due course.
EXPERIMENTAL SECTION
General
All commercial reagents were used without further purification unless otherwise noted. All reactants in moisture-sensitive reactions were dried overnight in a vacuum oven. All air- and moisture-sensitive reactions were carried out in flame-dried, argon-flushed, two-neck flasks sealed with rubber septa, and the reagents were introduced with a syringe. Progress of reactions was monitored by TLC on precoated Merck silica gel plates (60F-254). Visualization of reactants and products was accomplished with UV light. Nucleotides and dinucleoside polyphosphates were characterized by 1H NMR spectrometry at 200 or 300 MHz or by 31P NMR spectrometry in D2O, using 85% H3PO4 as an external reference on a Bruker AC-200 or DPX-300 spectrometer. In certain cases some of the signals of ribose protons were hidden by the very large HOD signal. High resolution mass spectra of nucleotides and dinucleoside polyphosphates were recorded on an AutoSpec-E FISION VG mass spectrometer using ESI (electron spray ionization) on a Q-TOF microinstrument (Waters, U.K.). Primary purification of the dinucleoside polyphosphates was achieved on an LC (Isco UA-6) system using a column of Sephadex DEAE-A25 swollen in 1 M NaHCO3 at 4 °C for 1 day. The resin was washed with deionized water before use. The LC separation was monitored by UV detection at 280 nm. Final purification of the dinucleoside polyphosphates and separation of the diastereomeric pairs were achieved by HPLC (Elite Lachrom, Merck-Hitachi) using a semipreperative reverse-phase column. (Gemini 5 μm C18 110A, 250 mm × 10 mm 5 μm, Phenomenex, Torrance, CA, U.S.). The purity of the dinucleoside polyphosphates was evaluated by analytical reverse-phase column chromatoghraphy (Gemini 5 μm C18 110A, 150 mm × 4.60 mm, 5 μm Phenomenex, Torrance, CA, U.S.) with two solvent systems. Solvent system I consisted of (A) MeOH and (B) 100 mM triethylammonium acetate (TEAA), pH 7. Solvent system II consisted of (A) 0.01 M KH2PO4, pH 4.5, and (B) MeOH. The details of the eluent gradients used for the separation of each product are given below. In addition, novel dinucleotides were characterized by HRMS-FAB (negative). NpnN analogues were prepared according to literature procedures.14 The purity of the dinucleotides was ≥95%. pH measurments were performed with an Orion microcombination pH electrode and a Hanna Instruments pH meter.
2′,3′-O-Methoxymethylideneadenosine was prepared as previously described.33 AMP(Bu3NH+)2 and UMP(Bu3NH+)2 salts were prepared from the corresponding nucleotide free acids and Bu3N (2 equiv) in EtOH. ADP(Bu3NH+)2 and UDP(Bu3NH+)2 were prepared from the corresponding disodium salts. The latter salts were passed through a column of activated Dowex 50WX-8 200 mesh, H+ form. The column eluate was collected in an ice-cooled flask containing Bu3N (2 equiv) and EtOH. The resulting solution was freeze-dried to yield ADP(Bu3NH+)2 or UDP(Bu3NH+)2 as a white solid.
NDP(α-B)(Bu3NH+)2 salt was prepared from the corresponding bisammonium salt. The latter salt was passed through a column of Sephadex Na+-form washed with tetrabutylammonium bromide (4 equiv) and then with deionized water (10 mL). The column eluate was freeze-dried to yield NDP(α-B) (Bu3NH+)2 as a white solid.
Reverse-Phase HPLC Purification and Diastereoisomers Separation
The dinucleotide purification and diasteromers separation were achieved with a semipreparative reverse-phase Elite Lachrom 250-10 column and isocratic elution (MeOH [A]/100 mM triethylammonium acetate (TEAA), pH 7 [B]) at a flow rate of 5 mL/ min. Fractions containing the same compound (similar retention time) were freeze-dried. The excess buffer was removed by repeated freeze-drying cycles until a constant weight was attained, with the solid residue dissolved each time in deionized water. The triethylammonium counterions were exchanged for Na+ by passing the pure dinucleoside poly(borano)phosphate isomer through a Sephadex-CM C-25 (Na+ form) column.
Typical Procedure for the Preparation of Dinucleoside P-β-Tetraboranophosphates (Analogues 6–9)
NMP (Bu3NH+)2 (0.22 mmol) and NDP(Bu3NH+)2 (0.22 mmol) were dissolved in dry DMF (3 mL), followed by addition of CDI (349 mg, 2.15 mmol). The resulting solution was stirred at room temperature for 4 h. Dry MeOH (87 μL, 2.15 mmol) was added. After 8 min BPi(Bu3NH+)2 (100 mg, 0.22 mmol) and MgCl2 (4 equiv) in dry DMF (3 mL) were added. The resulting solution was stirred at room temperature overnight. The semisolid obtained after evaporation of the solvent was chromatographed on a Sepadex DEAE-A25 column by applying a buffer gradient of water (500 mL) to 0.5 M NH4HCO3 (500 mL). The relevant fractions were pooled and freeze-dried to yield a white solid. Product 6 was obtained at 40% yield (80.8 mg). Product 7 was obtained at 38% yield (75.5 mg). Product 8 was obtained at 35% yield (68.9 mg). Product 9 was obtained at 37% yield (72.8 mg).
Synthesis and Separation of P2-Borano-P1,P4-di(5′-adenosine)tetraphosphate (Analogues 6A and 6B)
Analogue 6 was synthesized from AMP(Bu3NH+)2 and ADP(Bu3NH+)2. The separation of diastereoisomers 6A and 6B was accomplished using a semipreparative reverse-phase column and solvent system I, by isocratic elution with 4:96 A/B at a flow rate of 5 mL/min.
P2-Borano-P1,P4-di(5′-adenosine)tetraphosphate (Analogue 6A)
tR = 22.06 min. 31P NMR (D2O, 81 MHz, pH 7): δ 75 (m, Pβ, 1P), −10.5 (d, Pα, 2P), −22.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 200 MHz): δ 8.6 (s, H-8, 1H), 8.3 (s, H-2, 1H), 6.2 (d, H-1′, 1H), 4.5 (m, H-3′, 1H), 4.3 (m, H-4′, 2H), 0–0.97 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C20H30BN10O18P4 833.214, found 833.221. Purity data obtained on an analytical column: retention time of 3.76 min (98% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 1.76 min (97% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P2-Borano-P1,P4-di(5′-adenosine)tetraphosphate (Analogue 6B)
tR = 24.03 min. 31P NMR (D2O, 81 MHz, pH 7): δ 76.0 (m, Pβ, 1P), −11 (d, Pα, 2P), −22.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 200 MHz): δ 8.5 (s, H-8, 1H), 8.2 (s, H-2, 1H), 6.1 (d, H-1′, 1H), 4.4 (m, H-3′, 1H), 4.3 (m, H-4′, 2H), 0–0.97 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 4.49 min (99% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 2.46 min (98.5% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Synthesis and Separation of P2-Borano-P1,P4-di(5′-uridine)-tetraphosphate (Analogues 7A and 7B)
Analogue 7 was synthesized from UMP (Bu3NH+)2 and UDP(Bu3NH+)2. The separation of diastereoisomers 7A and 7B was accomplished using a semi-preparative reverse-phase column and solvent system I, by isocratic elution with 6:94 A/B at a flow rate of 5 mL/min.
P2-Borano-P1,P4-di(5′-uridine)tetraphosphate (Analogue 7A)
tR = 11.06 min. 31P NMR (D2O, 81 MHz, pH 7): δ 77.0 (m, Pβ, 1P), −10.97 (d, Pα, 2P), −23.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 7.98 (d, J = 8.1 Hz H-5, 1H), 5.99 (d, J = 5.1 Hz, H-1′, 1H), 5.97 (d, J = 8.1 Hz, H-6, 1H), 4.44 (m, H-4′, 1H), 4.27 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C18H28BN4O22P4 787.134, found 787.141. Purity data obtained on an analytical column: retention time of 3.5 min (98% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 2.5 min (97.8% purity), using solvent II system and elution with 4:96 A/B at a flow rate of 1 mL/min.
P2-Borano-P1,P4-di(5′-uridine)tetraphosphate (Analogue 7B)
tR = 12.73 min. 31P NMR (D2O, 81 MHz, pH 7): δ 77.0 (m, Pβ, 1P), −10.81 (d, Pα, 2P), −23.0 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 7.97 (d, J = 8.1 Hz, H-5, 1H), 5.99 (d, J = 5.2 Hz, H-1′, 1H), 5.98 (d, J = 8.1 Hz, H-6, 1H), 4.43 (m, H-4′, 1H), 4.26 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 3.76 min (98% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 2.69 min (97.5% purity), using solvent system II and elution with 4:96 A/B at a flow rate of 1 mL/min.
Synthesis and Separation of P3-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogues 8A and 8B)
Analogue 8 was synthesized from AMP (Bu3NH+)2 and UDP (Bu3NH+)2. The separation of diastereoisomers 8A and 8B was accomplished using a semipreparative reverse-phase column and using solvent system I, by isocratic elution with 8:92 A/B at a flow rate of 5 mL/min.
P3-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogue 8A)
tR = 21.47 min. 31P NMR (D2O, 81 MHz, pH 7): δ 77.7 (m, Pβ, 1P), −10.65 (d, Pα, 2P), −21.78 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 8.56 (s, H-8, 1H), 8.26 (s, H-2, 1H), 7.87 (d, J = 8.2 Hz, H-5, 1H), 6.14 (d, H-1′, 1H), 5.95 (d, J = 5.1 Hz, H-1′, 1H), 5.89 (d, J = 8.2 Hz, H-6, 1H), 4.60 (m, H-3′, 1H), 4.38 (m, H-4′, 2H), 4.28 (m, H-5′, 2H), 0–0.97 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C19H29BN7O20P4 810.173, found 810.180. Purity data obtained on an analytical column: retention time of 2.40 min (96% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 1.60 min (95.8% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P3-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogue 8B)
tR = 24.55 min. 31P NMR (D2O, 81 MHz, pH 7): δ 78.5 (m, Pβ, 1P), −10.55 (d, Pα, 2P), −21.30 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 8.57 (s, H-8, 1H), 8.25 (s, H-2, 1H), 7.86 (d, J = 8.2 Hz, H-5, 1H), 6.14 (d, H-1′, 1H), 5.93 (d, J = 5.1 Hz, H-1′, 1H), 5.89 (d, J = 8.2 Hz, H-6, 1H), 4.59 (m, H-3′, 1H), 4.42 (m, H-4′, 2H), 4.27 (m, H-5′, 2H), 0–0.97 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 3.53 min (97.3% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 1.66 min (97% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Synthesis and Separation of P2-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogues 9A and 9B)
Analogue 9 was synthesized from UMP(Bu3NH+)2 and ADP(Bu3NH+)2. The separation of diastereoisomers 9A and 9B, was accomplished using a semipreparative reverse-phase column and solvent system I, by isocratic elution with 7:93 A/B at a flow rate of 5 mL/min.
P2-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogue 9A)
tR = 15.12 min. 31P NMR (D2O, 81 MHz, pH 7): δ 76.5 (m, Pβ, 1P), −11 (d, Pα, 2P), −22.6 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 8.51 (s, H-8, 1H), 8.26 (s, H-2, 1H), 7.87 (d, J = 8.0 Hz, H-5, 1H), 6.11 (d, J = 5.2 Hz, H-1′, 1H), 5.99 (d, H-1′, 1H), 5.98 (d, J = 8.0 Hz, H-6, 1H), 4.57 (m, H-3′, 1H), 4.37 (m, H-4′, 2H), 4.22 (m, H-5′, 2H), 0–0.97 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C19H29BN7O20P4 810.173, found 810.179. Purity data obtained on an analytical column: retention time of 3.36 min (98% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 1.93 min (97.3% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P2-Borano-P1,P4-5′-uridine-5′-adenosinetetraphosphate (Analogue 9B)
tR = 17.74 min. 31P NMR (D2O, 81 MHz, pH 7): δ 76.0 (m, Pβ, 1P), −11 (d, Pα, 2P), −22.6 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 8.51 (s, H-8, 1H), 8.26 (s, H-2, 1H), 7.87 (d, J = 8.1 Hz, H-5, 1H), 6.32 (d, J = 5.2 Hz, H-1′, 1H), 6.14 (d, H-1′, 1H), 5.98 (d, J = 8.1 Hz, H-6, 1H), 4.54 (m, H-3′, 1H), 4.36 (m, H-4′, 2H), 4.22 (m, H-5′, 2H), 0–0.97 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 4.65 min (97% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 2.11 min (96.7% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Typical Procedure for the Preparation of Dinucleoside-P-α-boranotriphosphate (Analogues 10 and 11)
NMP(Bu3NH+)2 (0.2 mmol) was dissolved in dry DMF (2 mL), and CDI (163.7 mg, 1 mmol) was added. The resulting solution was stirred at room temperature for 3 h. Dry MeOH (80 μL, 1 mmol) was added. After 8 min NDP(α-B)(Bu3NH+)2 (0.2 mmol) in dry DMF (2 mL), and MgCl2 (4 equiv) were added. The resulting solution was stirred at room temperature overnight. The semisolid obtained after evaporation of the solvent was chromatographed on a Sephadex DEAE-A25 column. A buffer gradient of water (700 mL) to 0.5 M NH4HCO3 (700 mL) was applied. The relevant fractions were pooled and freeze-dried to yield a white solid. Final purification was achieved on a semipreparative HPLC column. Finally, purified A and B isomers were passed through a Sephadex-CM C-25 (Na+-form) column to exchange triethylammonium counterions for Na+ ions. Product 10 was obtained at 84% yield (50 mg). Product 11 was obtained at 85% yield (60.5 mg).
Synthesis and Separation of P1-Borano-P1,P3-5′-diadenosinetriphosphate (Analogues 10A and 10B)
Analogue 10 was synthesized from AMP(Bu3NH+)2 and ADP(α-B)(Bu3NH+)2. The separation of diastereoisomers 10A and 10B was accomplished using a semipreperative reverse-phase column, using solvent system I, and isocratic elution with 11:89 A/B at a flow rate of 5 mL/min.
P1-Borano-P1,P3-5′-diadenosinetriphosphate (Analogue 10A)
tR = 16.05 min. 31P NMR (D2O, 81 MHz, pH 7): δ 84(m, Pα, 1P), −11.5 (d, Pα, 1P), −23 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 200 MHz): δ 8.45 (s, H-8, 1H), 8.2 (s, H-2, 1H), 6.1 (d, H-1′, 1H), 4.60 (m, H-2′, 1H), 4.30 (m, H-4′, 1H), 4.25 (m, H-5′, 1H), 0.5 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C20H28BN10O15P3 752.223, found 752.227. Purity data obtained on an analytical column: retention time of 3.13 min (96% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 2.55 min (96.5% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P1-Borano-P1,P3-5′-diadenosinetriphosphate (Analogue 10B)
tR = 24.49 min. 31P NMR (D2O, 81 MHz, pH 7): δ 84 (m, Pα, 1P), −11 (d, Pα, 1P), −22.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 200 MHz): δ 8.50 (s, H-8, 1H), 8.1 (s, H-2, 1H), 6.1 (d, H-1′, 1H), 4.60 (m, H-2′, 1H), 4.35 (m, H-4′, 1H), 4.25 (m, H-5′, 1H), 0.7 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 6.68 min (98% purity), using solvent system I and elution with 7:93 A/B at a flow rate of 1 mL/min. Retention time of 3.18 min (98.8% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Synthesis and Separation of P1-Borano-P1,P3-5′-diuridinetriphosphate (Analogues 11A and 11B)
Analogue 11 was synthesized from UMP (Bu3NH+)2 and UDP(α-B)(Bu3NH+)2. The separation of diastereoisomers 11A and 11B was accomplished using a semipreparative reverse-phase column and solvent system I, by isocratic elution with 8:92 A/B at a flow rate of 5 mL/min.
P1-Borano-P1,P3-5′-diuridinetriphosphate (Analogue 11A)
tR = 6.36 min. 31P NMR (D2O, 81 MHz, pH 7): δ 83 (m, Pα, 1P), −11 (d, Pα, 1P), −22.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 8.00 (d, J = 8.1 Hz, H-5, 1H), 5.80 (d, J = 5.2 Hz, H-1′, 1H), 5.75 (d, J = 8.1 Hz, H-6, 1H), 4.40 (m, H-4′, 1H), 4.25 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C18H26BN4O19P3 706.146, found 706.153. Purity data obtained on an analytical column: retention time of 2.12 min (99% purity), using solvent system I and elution with 9:91 A/B at a flow rate of 1 mL/min. Retention time of 2.00 min (98.8% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P1-Borano-P1,P3-5′-diuridinetriphosphate (Analogue 11B)
tR = 9.25 min. 31P NMR (D2O, 81 MHz, pH 7): δ 82.5 (m, Pα, 1P), −11 (d, Pα, 1P), −22.5 (dd, Pβ, 1P) ppm. 1H NMR (D2O, 300 MHz): δ 7.95 (d, J = 8.1 Hz, H-5, 1H), 5.85 (d, J = 5.1 Hz, H-1′, 1H), 5.75 (d, J = 8.1 Hz, H-6, 1H), 4.35 (m, H-4′, 1H), 4.26 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 2.20 min (98% purity), using solvent system I and elution with 9:91 A/B at a flow rate of 1 mL/min. Retention time of 1.81 min (97% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Typical Procedure for the Preparation of Dinucleoside-P-α-boranotetraphosphates (analogues 12 and 13)
To a solution of 2′,3′-O-methoxymethylidene protected nucleoside (0.5 mmol) in anhydrous DMF (2 mL) and pyridine (0.2 mL) was added 2-chloro-4H-1,3,2-benzo-dioxaphosphorin-4-one (111.5 mg, 0.55 mmol) at room temperature. The solution was stirred for 10 min at room temperature. A 0.5 M solution of bis(tri-n-butylammonium)-pyrophosphate in anhydrous DMF (1.5 mL, 0.75 mmol) was vortexed with tri-n-butylamine (0.47 mL, 2 mmol) and immediately added to the reaction mixture. After 10 min, a solution of BH3/SMe2 complex, 2 M in THF (2.5 mL, 5 mmol), was added. After 15 min, a mixture of nucleoside monophosphate monohydrate (2.5 mmol) and magnesium chloride (5 mmol), which had been dried together before by evaporation of DMF, was added and stirring continued for 16 h. The mixture was freeze-dried overnight and chromatographed on a Sephadex DEAE-A25 column. A buffer gradient of 600 mL of water to 600 mL of 0.5 M NH4HCO3 was applied. The relevant fractions were pooled and freeze-dried three times to yield the product as a white solid. Final purification was achieved on a semipreperative C-18 HPLC column. Product 12 was obtained at 82% yield (69.1 mg). Product 13 was obtained at 80% yield (48.3 mg).
Synthesis and Separation of P1-Borano-P1,P3-5′-diadenosinetetraphosphate (Analogues 12A and 12B)
Analogue 12 was synthesized from 2′,3′-O-methoxymethylidene adenosine. The separation of diastereoisomers 12A and 12B was accomplished using a semipreparative reverse-phase column and solvent system I, by isocratic elution with 8:92 A/B at a flow rate of 5 mL/min.
P1-Borano-P1,P3-5′-diadenosinetetraphosphate (Analogue 12A)
tR = 6.62 min. 31P NMR (D2O, 81 MHz, pH 7): δ 83.5 (m, Pα, 1P), −10.5 (d, Pα, 1P), −22.5 (dd, Pβ, 2P) ppm. 1H NMR (D2O, 200 MHz): δ 8.45 (s, H-8, 1H), 8.2 (s, H-2, 1H), 6.25 (d, H-1′, 1H), 4.45 (m, H-2′, 1H), 4.35 (m, H-4′, 1H), 4.25 (m, H-5′, 1H), 0.5 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C20H30BN10O18P4 833.214, found 833.221. Purity data obtained on an analytical column: retention time of 1.94 min (95% purity), using solvent system I and elution with 13:87 A/B at a flow rate of 1 mL/ min. Retention time of 2.15 min (95.3% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P1-Borano-P1,P3-5′-diadenosinetetraphosphate (Analogue 12B)
tR = 8.96 min. 31P NMR (D2O, 81 MHz, pH 7): δ 83.5 (m, Pα, 1P), −10.5 (d, Pα, 1P), −22.0 (dd, Pβ, 2P) ppm. 1H NMR (D2O, 200 MHz): δ 8.45 (s, H-8, 1H), 8.2 (s, H-2, 1H), 6.1 (d, H-1′, 1H), 4.60 (m, H-2′, 1H), 4.35 (m, H-4′, 1H), 4.20 (m, H-5′, 1H), 0.5 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 2.36 min (97% purity), using solvent system I and elution with 13:87 A/B at a flow rate of 1 mL/min. Retention time of 3.21 min (96.5% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Synthesis and Separation of P1-Borano-P1,P4-5′-diuridinetetraphosphate (Analogues 13A and 13B)
Analogue 13 was synthesized from 2′,3′-O-methoxymethylideneuridine. The separation of analogue 13 diastereoisomers 13A and 13B was accomplished using a semipreparative reverse-phase column and solvent system I, by isocratic elution with 10:90 A/B at a flow rate of 5 mL/min.
P1-Borano-P1,P4-5′-diuridinetetraphosphate (Analogue 13A)
tR = 5.55 min. 31P NMR (D2O, 81 MHz, pH 7): δ 83.5 (m, Pα, 1P), −10.8 (d, Pα, 1P), −22.7 (dd, Pβ, 2P) ppm. 1H NMR (D2O, 300 MHz): δ 8.00 (d, J = 8.1 Hz, H-5, 1H), 5.80 (d, J = 5.1 Hz, H-1′, 1H), 5.75 (d, J = 8.1 Hz, H-6, 1H), 4.40 (m, H-4′, 1H), 4.25 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. HR MALDI (negative): calculated for C18H28BN4O22P4 787.134, found 787.139. Purity data obtained on an analytical column: retention time of 2.53 min (96% purity), using solvent system I and elution with 8:92 A/B at a flow rate of 1 mL/min. Retention time of 1.64 min (96.5% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
P1-Borano-P1,-P4-5′-diuridinetetraphosphate (Analogue 13B)
tR = 7.11 min. 31P NMR (D2O, 81 MHz, pH 7): δ 84 (m, Pα, 1P), −10.8 (d, Pα, 1P), −22.9 (dd, Pβ, 2P) ppm. 1H NMR (D2O, 300 MHz): δ 8.00 (d, J = 8.1 Hz, H-5, 1H), 5.80 (d, J = 5.1 Hz, H-1′, 1H), 5.75 (d, J = 8.1 Hz, H-6, 1H), 4.40 (m, H-4′, 1H), 4.25 (m, H-5′, 2H), 0.94–0.99 (m, BH3, 3H) ppm. Purity data obtained on an analytical column: retention time of 2.82 min (95% purity), using solvent system I and elution with 8:92 A/B at a flow rate of 1 mL/min. Retention time of 1.69 min (98% purity), using solvent system II and elution with 5:95 A/B at a flow rate of 1 mL/min.
Intracellular Calcium Measurements
Human 1321N1 astrocytoma cells stably expressing the turkey P2Y1 receptor, the human P2Y2 receptor, the human P2Y4 receptor, or the rat P2Y6 receptor were grown in Dulbecco’s modified Eagle’s medium containing 5% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 500 μg/mL Geneticin (G-418; Life Technologies, Inc.). Changes in the intracellular free calcium concentration, [Ca2+]i, were detected by dual-excitation spectrofluorometric analysis of cell suspensions preloaded with fura-2, as described previously.34 Cells were treated with the indicated nucleotide or analogue at 37 °C in 10 mM Hepes-buffered saline (pH 7.4) containing 1 mM CaCl2 and 1 mM MgCl2, and the maximal increase in [Ca2+]i was determined at various nucleotide or analogue concentrations to calculate the EC50. Concentration–response data were analyzed with the Prism curve fitting program (GraphPAD software, San Diego, CA). Three experiments were conducted on separate days for each P2Y receptor subtype.
Measurement of Enzymatic Hydrolysis of Dinucleoside Poly(borano)phosphate Analogues
e-NPP1 activity was measured at 37 °C in 1 mL of the following incubation medium: (in mM) 1 CaCl2, 200 NaCl, 10 KCl, and 100 Tris, pH 8.5 (Sigma-Aldrich). Then 0.1 mL of human NPP1 enzyme preparation was added to the reaction mixture and the mixture was preincubated for 3 min at 37 °C. The reaction was initiated by addition of 0.5 mL of 0.2 mM substrate. The reaction was stopped after 20 min by transferring a 0.3 mL aliquot of the reaction mixture to 0.375 mL of ice-cold 1 M perchloric acid. The samples were centrifuged for 5 min at 10000g. Supernatants were neutralized with 1 M KOH at 4 °C and centrifuged for 5 min at 10000g. An aliquot of the supernatant (0.2 mL) was analyzed by HPLC to monitor the increase of NMP. The percentage of substrate (dinucleotide) decomposition in 20 min corresponds to the average of two experiments preformed in triplicate.
Quantification of NMP Hydrolysate by HPLC
e-NPP1-dependent degradation products generated from dinucleoside poly-(borano)phosphate analogues 6–13 were isocratically eluted from an analytical reverse-phase Gemini 150-4.60 column with a mobile phase composed of 100 mM triethylammonium acetate (TEAA), pH 7, and 10–18% acetonitrile at a flow rate of 1 mL/min. Nucleotide concentrations were determined from the relative area under the respective HPLC peaks.
ABBREVIATIONS USED
- ADP
adenosine 5′-diphosphate
- ATP
adenosine 5′-triphosphate
- AUC
area under curve
- BPi
boranophosphate
- CDI
carbonyldiimidazole
- GPCR
G-protein-coupled receptor
- HRMS
high resolution mass spectrometry
- LC
liquid chromatography
- NMP
nucleoside monophosphate
- e-NPPs
ectonucleotide pyrophosphatases
- P2R
P2 receptor
- TEAA
triethylammonium acetate
- UDP
uridine 5′-diphosphate
- UTP
uridine 5′-triphosphate
References
- 1.Zamecnik PC, Stephenson ML, Janeway CM, Randerath K. Enzymatic synthesis of diadenosine tetraphosphate and diadenosine triphosphate with a purified lysyl-sRNA synthetase. Biochem Biophys Res Commun. 1966;24:91–97. doi: 10.1016/0006-291x(66)90415-3. [DOI] [PubMed] [Google Scholar]
- 2.Randerath K, Janeway CM, Stephenson ML, Zamecnik PC. Isolation and characterization of dinucleoside tetra- and triphosphates formed in the presence of lysyl-sRNA synthetase. Biochem Biophys Res Commun. 1966;24:98–105. doi: 10.1016/0006-291x(66)90416-5. [DOI] [PubMed] [Google Scholar]
- 3.Pintor J, Rotllan P, Torres M, Miras-Portugal MT. Characterization and quantification of diadenosine hexaphosphate in chromaffin cells: granular storage and secretagogue-induced release. Anal Biochem. 1992;200:296–300. doi: 10.1016/0003-2697(92)90469-n. [DOI] [PubMed] [Google Scholar]
- 4.McLennan AG. Dinucleoside polyphosphates-friend or foe? Pharmacol Ther. 2000;87:73–89. doi: 10.1016/s0163-7258(00)00041-3. [DOI] [PubMed] [Google Scholar]
- 5.Jankowski V, van der Giet M, Mischak H, Morgan M, Zidek W, Jankowski J. Dinucleoside polyphosphates: strong endogenous agonists of the purinergic system. Br J Pharmacol. 2009;157:1142–1153. doi: 10.1111/j.1476-5381.2009.00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flores NA, Stavrou BM, Sheridan DJ. The effects of diadenosine polyphosphates on the cardiovascular system. Cardiovasc Res. 1999;42:15–26. doi: 10.1016/s0008-6363(99)00004-8. [DOI] [PubMed] [Google Scholar]
- 7.Schlueter H, Offers E, Brueggemann G, van der Giet M, Tepel M, Nordhoff E, Karas M, Spieker C, Witzel H, Zidek W. Diadenosine phosphates and the physiological control of blood pressure. Nature (London) 1994;367:186–188. doi: 10.1038/367186a0. [DOI] [PubMed] [Google Scholar]
- 8.van der Giet M, Schmidt S, Tolle M, Jankowski J, Schluter H, Zidek W, Tepel M. Effects of dinucleoside polyphosphates on regulation of coronary vascular tone. Eur J Pharmacol. 2002;448:207–213. doi: 10.1016/s0014-2999(02)01986-6. [DOI] [PubMed] [Google Scholar]
- 9.Hoyle CHV, Hilderman RH, Pintor JJ, Schluter H, King BF. Diadenosine polyphosphates as extracellular signal molecules. Drug Dev Res. 2001;52:260–273. [Google Scholar]
- 10.Flores NA, Stavrou BM, Sheridan DJ. The effects of diadenosine polyphosphates on the cardiovascular system. Cardiovasc Res. 1999;42:15–26. doi: 10.1016/s0008-6363(99)00004-8. [DOI] [PubMed] [Google Scholar]
- 11.Delicado Esmerilda G, Miras-Portugal MT, Carrasquero Luz Maria G, Leon D, Perez-Sen R, Gualix J. Dinucleoside polyphosphates and their interaction with other nucleotide signaling pathways. Pfluegers Arch. 2006;452:563–572. doi: 10.1007/s00424-006-0066-5. [DOI] [PubMed] [Google Scholar]
- 12.Pintor J, Diaz-Hernandez M, Gualix J, Gomez-Villafuertes R, Hernando F, Miras-Portugal MT. Diadenosine polyphosphate receptors from rat and guinea-pig brain to human nervous system. Pharmacol Ther. 2000;87:103–115. doi: 10.1016/s0163-7258(00)00049-8. [DOI] [PubMed] [Google Scholar]
- 13.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR. Structure–activity relationships of dinucleotides: potent and selective agonists of P2Y receptors. Purinergic Signalling. 2005;1:183–191. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pendergast W, Yerxa BR, Douglass JG, Shaver SR, Dougherty RW, Redick CC, Sims IF, Rideout JL. Synthesis and P2Y receptor activity of a series of uridine dinucleoside 5′-polyphosphates. Bioorg Med Chem Lett. 2001;11:157–160. doi: 10.1016/s0960-894x(00)00612-0. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson KA, Boeynaems JM. P2Y nucleotide receptors: promise of therapeutic applications. Drug Discovery Today. 2010;15:570–578. doi: 10.1016/j.drudis.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobson KA, Ivanov AA, de Castro S, Harden TK, Ko H. Development of selective agonists and antagonists of P2Y receptors. Purinergic Signalling. 2009;5:75–89. doi: 10.1007/s11302-008-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC. Pharmacology of INS37217 [P1-(uridine 5′)-P4-(2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- 19.Kikuta Y, Ohiwa E, Okada K, Watanabe A, Haruki S. Clinical application of diadenosine tetraphosphate (Ap4A:F-1500) for controlled hypotension. Acta Anaesthesiol Scand. 1999;43:82–86. doi: 10.1034/j.1399-6576.1999.430117.x. [DOI] [PubMed] [Google Scholar]
- 20.Maminishkis A, Jalickee S, Blaug SA, Rymer J, Yerxa BR, Peterson WM, Miller SS. The P2Y(2) receptor agonist INS37217 stimulates RPE fluid transport in vitro and retinal reattachment in rat. Invest Ophthalmol Visual Sci. 2002;43:3555–3566. [PubMed] [Google Scholar]
- 21.Mundasad MV, Novack GD, Allgood VE, Evans RM, Gorden JC, Yerxa BR. Ocular safety of INS365 ophthalmic solution: a P2Y2 agonist in healthy subjects. J Ocul Pharmacol Ther. 2001;17:173–179. doi: 10.1089/10807680151125519. [DOI] [PubMed] [Google Scholar]
- 22.Nahum V, Tulapurkar M, Levesque SA, Sevigny J, Reiser G, Fischer B. Diadenosine and diuridine poly(borano)phosphate analogs: synthesis, chemical and enzymatic stability, and activity at P2Y1 and P2Y2 receptors. J Med Chem. 2006;49:1980–1990. doi: 10.1021/jm050955y. [DOI] [PubMed] [Google Scholar]
- 23.Jankowski V, Tolle M, Vanholder R, Schonfelder G, van der Giet M, Henning L, Schluter H, Paul M, Zidek W, Jankowski J. Uridine adenosine tetraphosphate: a novel endothelium-derived vasoconstrictive factor. Nat Med (N Y) 2005;11:223–227. doi: 10.1038/nm1188. [DOI] [PubMed] [Google Scholar]
- 24.Hoard DE, Ott DG. Conversion of mono- and oligodeoxyribonucleotides to 5′-triphosphates. J Am Chem Soc. 1965;87:1785–1788. doi: 10.1021/ja01086a031. [DOI] [PubMed] [Google Scholar]
- 25.Nahum V, Fischer B. Boranophosphate salts as an excellent mimic of phosphate salts: preparation, characterization, and properties. Eur J Inorg Chem. 2004:4124–4131. [Google Scholar]
- 26.Ludwig J, Eckstein F. Synthesis of nucleoside 5′-O-(1,3-dithiotriphosphates) and 5′-O-(1,1-dithiotriphosphates) J Org Chem. 1991;56:1777–1783. [Google Scholar]
- 27.Han Q, Gaffney BL, Jones RA. One-flask synthesis of dinucleoside tetra- and penta-phosphates. Org Lett. 2006;8:2075–2077. doi: 10.1021/ol060491d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Xu Z, Liu H, Wennefors CK, Dobrikov MI, Ludwig J, Shaw BR. Synthesis of α-P-modified nucleoside diphosphates with ethylenediamine. J Am Chem Soc. 2005;127:16782–16783. doi: 10.1021/ja055179y. [DOI] [PubMed] [Google Scholar]
- 29.Vollmayer P, Clair T, Goding JW, Sano K, Servos J, Zimmermann H. Hydrolysis of diadenosine polyphosphates by nucleotide pyrophosphatases/phosphodiesterases. Eur J Biochem. 2003;270:2971–2978. doi: 10.1046/j.1432-1033.2003.03674.x. [DOI] [PubMed] [Google Scholar]
- 30.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta, Mol Cell Res. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 31.Guranowski A, Sillero A. In: Ap4A and Other Dinucleoside Polyphosphates. McLennan AG, editor. CRC Press; Boca Raton, FL: 1992. pp. 84–95. [Google Scholar]
- 32.Major DT, Nahum V, Wang Y, Reiser G, Fischer B. Molecular recognition in purinergic receptors. 2. Diastereoselectivity of the h-P2Y1-receptor. J Med Chem. 2004;47:4405–4416. doi: 10.1021/jm049771u. [DOI] [PubMed] [Google Scholar]
- 33.Kikugawa K, Suehiro H, Yanase R, Aoki A. Platelet aggregation inhibitors. IX. Chemical transformation of adenosine into 2-thioadenosine derivatives. Chem Pharm Bull. 1977;25:1959–1969. doi: 10.1248/cpb.25.1959. [DOI] [PubMed] [Google Scholar]
- 34.Garrad RC, Otero MA, Erb L, Theiss PM, Clarke LL, Gonzalez FA, Turner JT, Weisman GA. Structural basis of agonist-induced desensitization and sequestration of the P2Y2 nucleotide receptor. Consequences of truncation of the C terminus. J Biol Chem. 1998;273:29437–29444. doi: 10.1074/jbc.273.45.29437. [DOI] [PubMed] [Google Scholar]