

Abstract
Extracellular nucleotides can modify the production or drainage of the aqueous humor via activation of P2 receptors and therefore affect the intraocular pressure (IOP). We have synthesized slowly hydrolyzable nucleoside di- and triphosphate analogues, 1, and 8–14. Analogues 8–14 were completely resistant to hydrolysis by alkaline phosphatase over 30 min at 37 °C. In human blood serum, analogues 8–14 exhibited high stability, e.g., analogues 9 and 10–14 were only 15% and 0% degraded after 24 h, respectively. Moreover, analogues 8–14 were highly stable at pH 1.4 (t1/21 h–30 days). Analogues 8–14 were agonists of the P2Y1 receptor (EC50 0.57–9.54μM). Ocular administration of most analogues into rabbits reduced IOP, e.g., analogue 9 reduced IOP by 32% (EC50 95.5 nM). Analogue 9 was more effective at reducing IOP than several common glaucoma drugs and represents a promising alternative to timolol maleate, which cannot be used for the treatment of patients suffering from asthma or cardiac problems.
Introduction
Extracellular nucleotides that activate G protein-coupled P2Y receptors (P2YRs) are attractive pharmaceutical targets due to their ability to modulate various functions in many tissues and organs under normal and pathophysiological conditions.1–5 Extracellular nucleotides and dinucleotides have been shown to play a role in ocular physiology and pathophysiology6 and have been suggested as therapeutic agents for dry eye, retinal detachment, and glaucoma.7
Ocular hypertension, the most common cause of glaucoma, is a target for agents that reduce intraocular pressure (IOPa).8 When topically applied to New Zealand white rabbits, some nucleotides produce an increase in IOP while others decrease IOP. IOP can be reduced by administration of ATP,9 adenosine tetraphosphate (Ap4),10 and diadenosine tetraphosphate (Ap4A).11 In contrast, diadenosine triphosphate (Ap3A) and diadenosine pentaphosphate (Ap5A) can increase IOP.9,11
Receptors for extracellular nucleotides, including P2Y1, P2Y2, and P2Y4, have been identified in trabecular meshwork cells (TM), an area of tissue in the eye that is responsible for draining the aqueous humor.12 Among these P2YR subtypes, activation of the P2Y1R by the selective agonist 2-MeS-ADP reduces aqueous humor outflow in bovines. Other studies have reported the presence of P2Y1 and P2Y2 receptors in bovine TM cells and P2Y1, P2Y4, and P2Y11 receptors in a human TM cell line.13
The therapeutic potential of endogenous nucleotides for the treatment of glaucoma is limited because they are degraded by extracellular enzymes, which reduces their potency, efficacy, and duration of action. In addition, although nucleotides are chemically stable in a pH range of 4–11,14 they are rapidly degraded at more acidic or basic pH. Nucleotides are hydrolyzed enzymatically by the ecto-nucleoside triphosphate diphosphohydrolase family of ectonucleotidases (i.e., e-NTPDase and alkaline phosphatases)15 and ecto-nucleotide pyrophosphatases/phosphodiesterases, (i.e., e-NPPs).16,17 Therefore, there is a need for the identification of enzymatically and chemically stable nucleotide scaffolds that can be used to develop selective and potent P2YR agonists.
A few attempts to improve the stability of nucleotides have been reported,18–26 including the use of phosphate bioisosters of nucleotides such as phosphonate,27,28 phosphoramide,29 and boranophosphate15,20,27,30,31 analogues. Previously, we developed a series of β,γ-methylene-ATP, analogue 1, and β,γ-methylene-ATP(α-borano) derivatives, analogues 2, 3 (Figure 1).27 We showed that replacing a β,γ-bridging oxygen in ATP with a methylene group conferred a significant resistance to enzymatic and chemical (i.e., at pH 1.4 and 37 °C) hydrolysis.27 We also showed that replacing the Pα nonbridging oxygen by a borano isoster decreased enzymatic hydrolysis of the analogues by human blood serum, NTPDase1,2,3,8, NPP1,3, and alkaline phosphatase.27
Figure 1.

Structures of previously tested nucleotides.
Analogues 1–3 also were evaluated as agonists of the P2Y1R.27 The most effective P2Y1R agonist was analogue 1, with an EC50 of 80 nM. Yet, although analogues 2 and 3 were weak P2Y1R agonists (EC50 of 17.2 μM for analogue 3B, whereas analogues 2A, 2B, and 3A were inactive),27 while the structurally related analogue 4A (Figure 1) was a potent P2Y1R agonist with an EC50 of 2.6 nM.15
The decreased activity of analogues 1, 2A, 2B, 3A, and 3B, as compared to analogue 4A, is possibly related to the elevated pKa of the terminal phosphate in analogue 4A, as compared to phosphate (pKa 8.4 vs 6.5, respectively).32,33 Under physiological conditions, i.e., pH 7.4, 91% of ATP is ionized, whereas the β,γ-methylene analogues of ATP are only 9% ionized in solution. Analogues 1–3 are 90% protonated at Pγ at pH 7.4, which likely prevents significant electrostatic interactions with positively charged amino residues in the P2Y1R.34,35
The electronegativity of a dihalogenated methylene group (e.g., CF2, CCl2) lowers the pKa of phosphonate from 8.4 to 6.7–7.0, making it closer to the pKa of phosphate (i.e., 6.5).36,37 Indeed, nucleotide analogues with a β,γ-dihalomethylene group were reported to be promising inhibitors of HIV-1 reverse transcriptase,30,37 agonists of P2X2/3 receptors,38 and potent antagonists of the P2Y12 receptor.39,40
The current study describes the synthesis of several nucleoside-5′-dihalomethylene-di- and triphosphate analogues (i.e., analogues 8–14, Figure 2) and characterizes their activity at the P2Y1R, their chemical stability at acidic pH, and their hydrolytic stability with alkaline phosphatase, NTPDase1,2,3,8, and NPP1,3, and human blood serum. Furthermore, the potency, efficacy, and duration of action of analogues 8–14 have been evaluated for reduction of IOP in vivo. In comparison to several commercially available antiglaucoma drugs, analogues 1 and 9 were found to be promising and novel candidates for the treatment of ocular hypertension and glaucoma through activation of P2 receptors (P2R).
Figure 2.

Structures of nucleotides tested in this study.
Results
Synthesis of Analogues 8–14
ATP analogues in which the β,γ-bridging oxygen is substituted by a methylene group are conventionally prepared via the activation of the 5′-phosphate of nucleoside-5′-monophosphate (NMP) to form a phosphoryl donor, followed by a reaction with methylene bisphosphonate salt (phosphoryl acceptor). Phosphoryl donors were prepared by activation of NMP with carbonyl diimidazole (CDI),41 trifluoroacetic anhydride, and N-methylimidazole42 or dicyclohexylcarbodiimide (DCC),33 followed by condensation with methylene bisphosphonic acid or its salt.
Recently, we reported a facile three-step one-pot synthesis of β,γ-methylene-2-MeS-ATP, analogue 1, at a 35% overall yield.27 Here, we used this synthetic procedure to prepare the β,γ-CF2/CCl2-2-MeS-ATP analogues 8 and 9 (Scheme 1). To ensure the selective phosphorylation of 2-MeS–adenosine at the 5′-OH, we used 2′,3′-methoxy-methylidene-2-MeS–adenosine, analogue 15, as the starting material. Analogue 15 was first treated with POCl3 in trimethylphosphate (TMP) in the presence of proton sponge at 0 °C for 1–3 h, followed by treatment with bis(tributylammonium) dihalomethylene-diphosphonate and tributylamine at 0 °C for 40 min to 1.5 h. Finally, hydrolysis of the cyclic intermediates, analogues 17 and 18, in 0.5 M TEAB and deprotection of the methoxymethylidene groups generated β,γ-CF2-2-MeS-ATP, analogue 8, at a 46% overall yield, and β,γ-CCl2-2-MeS-ATP, analogue 9, at a 10% overall yield, respectively. The formation of analogues 8 and 9 was confirmed by the presence of three typical signals in 31P NMR, as described previously:43 3.24 (m, Pγ), −4.89 (m, Pβ), and −10.61 (d, Pα) ppm for analogue 8, and 8.15 (d, Pγ), 1.04 (dd, Pβ), and −10.22 (d, Pα) ppm for analogue 9.
Scheme 1a.

aReaction conditions for analogue 8: (a) trimethylphosphate, POCl3, proton sponge, 0 °C, 3 h; (b) 0.5 M bis(tributylammonium)difluoromethylene diphosphonate in dry DMF, Bu3N, 0 °C, 1.5 h; (c) 0.5 M TEAB, pH 7, RT, 1 h; (d) (1) 18% HCl, pH 2.3, RT, 3 h, (2) 24% NH4OH, pH 9, RT, 45 min. Reaction conditions for analogue 9: (a) trimethylphosphate, POCl3, proton sponge, 0 °C, 1 h; (b) 1 M bis(tributylammonium)dichloromethylene diphosphonate in dry DMF, Bu3N, 0 °C, 40 min, and then RT, 5 min; (c) 0.5 M TEAB, pH 7, RT, 1 h; (d) as described for analogue 8.
The preparation of α-borano-β,γ-CF2-2-MeS-ATP, analogue 10, α-borano-β,γ-CCl2-ATP, analogue 11, and α-borano-β,γ-CCl2-2-MeS-ATP, analogue 12, required the protection of the 2′- and 3′-hydroxyl groups in analogues 15 and 21 by methoxy-methylidene groups, which remained throughout the entire synthesis until removal in the last step (Scheme 2). In the first synthetic step, we used PCl3, which was rapidly and completely consumed in less than 30 min to form analogues 22 and 23. Next, dihalogenbisphosphonate salt was added and the reactions progressed for 25–60 min at 0 °C, followed by addition of BH3·SMe2 at 0 °C and stirring for 1 h at RT to obtain intermediates 27–29. Finally, hydrolysis of the cyclic intermediates in 0.5 M TEAB produced analogues 30–32. The latter analogues were deprotected to generate analogues 10, 11, and 12 at 21%, 18%, and 7% overall yields, respectively, after LC. The identity and purity of analogues 10–12 were established by 1H and 31P NMR, ESI, or MALDI negative mass spectrometry, and HPLC using two solvent systems. 31P NMR spectra of analogues 10–12 showed a typical Pα signal as a multiplet at about 80 ppm. 1H NMR spectra showed borane hydrogen atoms as a very broad signal at about 0.4 ppm. Because of the chiral center at Pα, each analogue was obtained as a pair of diastereoisomers in a 1:1 ratio. In both the 1H and 31P NMR spectra, there was a slight difference between the chemical shifts for the two diastereoisomers of each analogue. These isomers were well separated by reverse-phase HPLC with about a 1–2 min difference in their retention times with the A isomer eluting before the B isomer.
Scheme 2a.

aReaction conditions for analogue 10: (a) trimethylphosphate, PCl3, proton sponge, 0 °C, 30 min; (b) 0.5 M bis(tributylammonium)dichloromethylene diphosphonate in dry DMF, Bu3N, 0 °C, 1 h; (c) 2 M BH3·SMe in THF, 0 °C, 5 min, and then RT, 60 min; (d) 0.5 M TEAB, pH 7, RT, 1 h; (e) (1) 18% HCl, pH 2.3, RT, 3 h, (2) 24% NH4OH, pH 9, RT, 45 min. Reaction conditions for analogue 11: (a) trimethylphosphate, PCl3, proton sponge, 0 °C, 30 min; (b) 0.5 M bis(tributylammonium)dichloromethylene diphosphonate in dry DMF, Bu3N, 0 °C, 25 min; (c) 2 M BH3·SMe2 in THF, 0 °C, 5 min and then RT, 25 min; (d) 0.5 M TEAB, pH 7, RT, 45 min; (e) as described for analogue 10. Reaction conditions for analogue 12: (a) trimethylphosphate, PCl3, proton sponge, 0 °C, 45 min; (b) 0.5 M bis(tributylammonium)difluoromethylene diphosphonate in dry DMF, Bu3N, 0 °C, 50 min; (c) 2 M BH3·SMe in THF, 0 °C, 5 min then RT, 60 min; (d) and (e) as described for analogue 10.
2-MeS-ADP, 7, is a selective and highly potent P2Y1R agonist. Yet, this agonist suffers from low chemical and enzymatic stability.44 Therefore, we replaced the α,β-bridging oxygen with a dichloro- or difluoromethylene group in an attempt to generate potent P2Y1R agonists with increased chemical and metabolic stabilities (i.e., analogues 13 and 14). The α,β-dihalomethylene-2-MeS-ADP analogues 13 and 14 were prepared as previously reported (Scheme 3).45 Specifically, the 5′-OH of the 2′,3′-protected 2-MeS–adenosine analogues 15 and 33 were activated with tosyl chloride followed by coupling with tris(tetra-n-butylammonium) dihalo diphosphonate salt. Finally, removal of the protecting group provided the α,β-dihalomethylene-2-MeS-ADP derivatives, analogues 13 and 14 at 34% and 18% overall yields, respectively, after LC/MPLC. The relatively high yield of analogue 13 may be due to the higher nucleophility of the difluoro diphosphonate salt, as compared to analogue 14. In addition to analogue 13, a small amount of the dinucleotide, analogue 38, (4% yield) was isolated, due to coupling of analogue 36 with analogue 34. Interestingly, the corresponding dichloro analogue was not detected under the identical reaction conditions.
Scheme 3a.

aReaction conditions for analogue 15: (a) CH2Cl2, DMAP, TsCl, RT, 12 h; (b) tetra-(n-butylammonium)difluoromethylene diphosphonate in dry DMF, RT, 72 h; (c) (1) 18% HCl, pH 2.3, RT, 3 h; (2) 24% NH4OH, pH 9, RT, 45 min. Reaction conditions for analogue 33: (a) CH2Cl2, DMAP, TsCl, RT, 12 h; (b) tetra-(n-butylammonium) dichloromethylene diphosphonate in dry DMF, RT, 72 h; (c) TFA, argon bubbling, RT, 10 min.
Hydrolytic Stability of Analogues 8–14
To explore the potential of analogues 8–14 as drug candidates, we first evaluated their hydrolytic stability. Previously, we investigated the effect of a β,γ-methylene group on the hydrolytic stability of analogues 1–3 at acidic pH, which mimics the acidity of gastric juice (i.e., pH 1.4 at 37 °C).27 2-MeS-β,γ-CH2-ATP, analogue 1, α-B,β,γ-CH2-ATP, analogue 2, and 2-MeS-α-B,β,γ-CH2-ATP, analogue 3, were found to be highly stable at pH 1.4 with t1/2 values for hydrolysis of 65, 19, and 14.5 h, respectively. Although we intend to administer these analogues topically, a higher stability of analogues 8–14 at acidic pH, as compared to analogues 1–3, may suggest a wider range of therapeutic applications.
The hydrolysis of 2-MeS-β,γ-CCl2-ATP, analogue 9, at pH 1.4 and 37 °C was monitored by HPLC over 216 h (Figure 3A), and the hydrolysis rate was determined by integration of the HPLC peaks with time to fit a pseudofirst-order exponential decay rate equation (Figure 3B), yielding a half-life of ∼65 h. Analogue 9 was degraded to 2-MeS-AMP and 2-MeS–adenosine.
Figure 3.

Rates of hydrolysis of analogues 9 and 10B at pH 1.4 at 37 °C, as monitored by HPLC. Hydrolysis of 7.5 mM analogue 9 or 10B in KCl/HCl buffer at pH 1.4 and 37 °C was recorded for 5 days at 7–17 h intervals. (A) HPLC chromatograms of analogue 9 and hydrolysates at t = 6, 52, 216 h. (B) Kinetics of acidic hydrolysis of analogue 9 (t1/2 = 65 h) and time-dependent formation of degradation products. (C) Kinetics of acidic hydrolysis of analogue 10B (t1/2 = 8 h).
The hydrolysis of 2-MeS-α-B-β,γ-CF2-ATP, analogue 10 (B isomer) was similarly determined, yielding a half-life of ∼8 h (Figure 3C). The half-lives of analogues 8, 11 (isomer A), and 12 (isomer B) were 25, 6.7, and 0.7 h, respectively (data not shown). 2-MeS-Rα,β-dichloromethylene-ADP, analogue 14, was highly stable at pH 1.4, with only 15% hydrolysis of the starting material after 6 days. 2-MeS-α,β-difluoromethylene-ADP, analogue 13, was highly stable, with 61% of the starting material remaining after 30 days with the remainder degraded to 2-MeS-adenosine.
Resistance of Analogues 8–14 to Degradation by Alkaline Phosphatase
Alkaline phosphatase is a hydrolase that removes phosphate groups from nucleotides, thereby regulating extracellular nucleotide concentrations in vivo. AP usually catalyzes the hydrolysis of phosphomonoesters yielding Pi and the corresponding alcohol.46
Therefore, we evaluated the effect of α,β/β,γ-dihalomethylene groups in analogues 8–14 on the resistance to hydrolysis by alkaline phosphatase, as compared to ATP, ADP and analogues 1–3. Results indicated that AP degraded 96% of ATP after 3 h (t1/2 = 1.4 h). Likewise, ADP was completely degraded by AP after 3 h, whereas 2-MeS-ADP was 87% degraded after 3 h. Yet, analogues 8–14 were completely resistant to hydrolysis by AP under the same conditions (i.e., 0.2 mg analogue/77.5 μL in the presence of 12.5 units of AP; data not shown).
Resistance of Analogues 8–14 to Hydrolysis in Human Blood Serum
Nucleotides and their analogues undergo dephosphorylation by enzymes in physiological systems.47–50 Blood serum contains such enzymes and, therefore, provides a good model system for assessing the metabolic stability of extracellular nucleotides. Previous studies have used human blood serum51 to demonstrate the metabolic stability of phosphonate modified nucleotide analogues.30,37 Similarly, we have shown that in human blood serum the half-lives of β,γ-CH2-ATP and α-B,β,γ-CH2-ATP, analogues 1–3, were increased by 3.5–20-fold, as compared to ATP.27
Here, we investigated the effect of incorporation of a β,γ-dihalomethylene group in analogues 8–14 on the metabolic stability of these compounds in human blood serum at 37 °C.
As compared to ATP, which is hydrolyzed to ADP and AMP with a half-life of 4.9 h (Figure 4A), analogue 8 was hydrolyzed to the corresponding nucleoside 5′-monophosphate with a half-life of 12.4 h, whereas analogue 9 was only 15% degraded over 24 h (Figure 4B). Furthermore, replacement of the Pα nonbridging oxygen in analogues 10–12 with a BH3 group endowed complete resistance to hydrolysis in human blood serum over 24 h (data not shown). Similarly, replacement of the Pα-Pβ bridging oxygen in 2-MeS-ADP with CF2/CCl2 produced analogues 13 and 14 that were completely resistant to hydrolysis in human blood serum over 24 h, as compared to ADP (t1/2 = 1.4h) or 2-MeS-ADP (t1/2 = 23.5 h) (data not shown).
Figure 4.
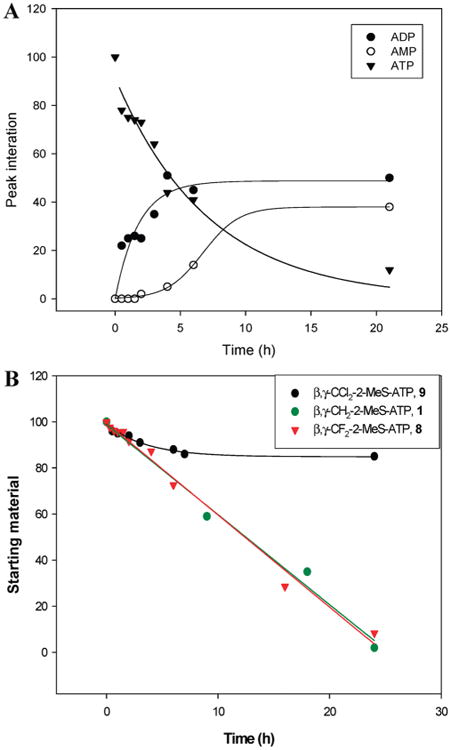
Hydrolysis of ATP and analogues 1, 8, and 9 in human blood serum (180 μL) and RPMI-1640 medium (540 μL) over 24 h at 37 °C, as monitored by HPLC. (A) Hydrolysis of 0.25 mM ATP and production of ADP and AMP. (B) Degradation of analogues 1, 8, and 9 in human blood serum.
Analogues 8–14 are Poor Substrates for ecto-5′-Nucleotidases
NTPDase1,2,3,8 and NPP1,3 are the principal enzymes that metabolize extracellular nucleotides. In comparison to ATP (Table 1), analogues 8–14 were barely hydrolyzed by NTPDase1,2,3,8 (<5%) or NPP1,3 (<10%).
Table 1. Relative Hydrolysis [%] of Analogues 8–14 by Human ecto-Nucleotidasesa.
| analogues | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| human ecto-nucleotidases | 8 | 9 | 10A | 10B | 11A | 11B | 12A | 12B | 13 | 14 |
| NTPDase1 | 2.6 | 0.5 | 1.0 | 0.4 | 5.5 | 3.2 | 0.7 | ND | 1.3 | ND |
| NTPDase2 | 1.1 | 0.5 | 1.8 | 0.9 | 1.3 | 1.4 | 1.5 | ND | 2.4 | 1.7 |
| NTPDase3 | 2.6 | ND | 4.4 | ND | 2.6 | 3.1 | 0.3 | ND | ND | ND |
| NTPDase8 | 5.2 | ND | 1.7 | ND | 4.4 | 4.0 | ND | 1.8 | 0.1 | ND |
| NPP1 | 6.2 | 5.8 | 4.0 | 3.4 | 5.3 | 3.3 | 0.7 | 4.0 | 3.9 | 3.3 |
| NPP3 | 0.5 | 1.9 | 10.0 | 3.2 | ND | ND | 5.0 | ND | ND | 1.8 |
The ATP and ADP analogues 8–14 (100 μM) were used as sub-strates of the ecto-nucleotidases identified in the left column. The relative hydrolysis rates vs 100 μM ATP (set at 100%) were: 403 ± 40, 1006 ± 60, 533 ± 42, 229 ± 20 [nmol Pi·min−1·mg protein−1] for NTPDase1,2,3,8, respectively, and 619 and 245 [nmol nucl·min−1·mg protein−1] for NPP1,3, respectively. ND = not detectable.
Activities of Analogues 8–14 at the P2Y1 Receptor
Earlier, we reported that 2-MeS-β,γ-methylene-ATP was a potent and selective P2Y1 receptor (P2Y1R) agonist.27 Here, we examined whether a β,γ-dihalomethylene substitution in analogues 8–14 would enhance agonist potency at the P2Y1R due to the reduced pKa of the terminal phosphonate (∼7), as compared to the pKa of 8.452 for the corresponding methylene derivatives. Because analogues 8–14 should be 90% ionized under physiological conditions (vs 9% ionization for analogue 1), we expected that negatively charged phosphonates in analogues 8–14 would increase binding to the P2Y1R. Accordingly, the activities of analogues 8–14 were determined at the turkey P2Y1R heterologously expressed in human 1321N1 astrocytoma cells that are devoid of endogenous P2Y receptors.53 P2Y1R activities were evaluated by monitoring increases in the intracellular calcium concentration, [Ca2+]i, induced by the analogues, as compared to 2-MeS-ADP (EC50 0.0025 μM at the P2Y1R).
Surprisingly, analogues 8–14 were found to be weaker agonists of the P2Y1R than 2-MeS-ADP, analogue 7, with EC50 values of 0.57–9.54 μM (Table 2). Replacement of the nonbridging oxygen with BH3 in 2-MeS-α-borano-β,γ-dichloromethylene-ATP, analogue 12A (A isomer), produced the most potent P2Y1R agonist in this series of analogues (EC50 = 0.57μM), whereas the B isomer (analogue 12B) was 2-fold less potent (EC50 = 1.20 μM). Analogues 11A and 11B that lacked a 2-MeS group were inactive at the P2Y1R.
Table 2. Activity of Analogues 8–14 at the P2Y1R, i.e., EC50 (μM) Values for Analogue-Induced Increases in [Ca2+]i.
| analogue no. | EC50 | analogue no. | EC50 |
|---|---|---|---|
| 7 | 0.0025 | 11B | nra |
| 8 | 0.76 | 12A | 0.57 |
| 9 | 9.54 | 12B | 1.20 |
| 10A | nr | 13 | 0.98 |
| 10B | 2.13 | 14 | 3.10 |
| 11A | nr |
nr = no response.
Replacement of the nonbridging oxygen in 2-MeS-β,γ-difluoromethylene-ATP, analogue 8, with BH3 to generate analogue 10B resulted in a 3-fold reduction in P2Y1R activity (EC50 = 0.76 μM for analogue 8 vs EC50 = 2.13 μM for analogue 10B), whereas the A isomer (analogue 10A) had no activity. Although the α,β-dihalomethylene substitution in analogues 13 and 14 decreased the rate of degradation by ecto-nucleotidases (Table 1), as compared to 2-MeS-ADP, analogues 13 and 14 were weak P2Y1R agonists with EC50 values of 0.98 and 3.1 μM, respectively (Table 2). None of the analogues tested (8–14) antagonized the effect of equimolar concentrations of 2-MeS-ADP on P2Y1R activation in 1321N1 cell transfectants (data not shown).
Effect of Nucleotide Analogues on IOP
Nucleotides are present in the aqueous humor,11 although their action has not been fully elucidated due to the multiple P2R subtypes identified in intraocular tissues that are bathed by the aqueous humor.54 Studies suggest that IOP is regulated by G-protein coupled P2Y1Rs in trabecular meshwork cells that control the evacuation of the aqueous humor12 and ligand-gated ion channel P2X2 receptors located on parasympathetic nerve terminals innervating the ciliary bodies.55 P2X receptor activation potentiates the release of acetylcholine, which induces contraction of ciliary muscle to open the trabecular meshwork and reduce IOP.56 In contrast, activation of G-protein coupled P2Y2 receptors increases IOP.56 The current study examined the effect of analogues 8–14, as compared to 2-MeS-β,γ-methylene-ATP (analogue 1), on reduction of IOP in normotensive rabbits to identify novel and potent candidates for the treatment of ocular hypertension.56 For all the single dose experiments, 10 μL of a 100 μM solution of tested compounds was instilled.
Analogues 8 and 11A increased IOP (Figure 5A), whereas analogues 1, 9, 11B, 12A/B, 13, and 14 reduced IOP (Figure 5A-C). Analogues 1 and 9 reduced IOP by ∼32% (at an instilled volume of 10μL of a 100 μM solution) and were more effective than Ap4A and Ap4.10,11 On the basis of concentration response curves, analogues 1, 9, 13, and 14 exhibited the strongest hypotensive effects with pD2 values of 4.5 ± 0.7, 7.0 ± 0.7, 5.1 ± 0.3, and 4.5 ± 0.4, respectively (all assays were performed by instilling 10 μL of 1 nM–100 μM analogue solutions). The rank order of potency for reduction of IOP was 9 > 13 > 1 > 14, with EC50 values of 95.5 nM, and 7.9, 30.2, and 31.6 μM, respectively (Figure 5D). In comparison to other agents shown to reduce IOP, analogues 1 and 9 were more effective than latanoprost (a prostglandin analogue) and dorzolamide hydrochloride (a carbonic anhydrase inhibitor) and equally effective as timolol maleate (a β-blocker) (all commercial drugs were tested at a fixed volume of 40 μL) (Figure 5E). The duration effect of the reduction in IOP induced by the nucleotides was comparable to the commercially available compounds. The reduction in IOP induced by analogues 1 and 9 persisted for 3.5 and 4.5 h, respectively, whereas latanoprost was effective for 5.5 h (Figure 5F).
Figure 5.

(A) Effect of analogues 1, 8–9, and 11–14 (100 μM) on rabbit IOP measured over 8 h vs control: (1) an equal volume of saline was administered to the contralateral eye; (2) an equal volume of saline was administered to other animals. IOP was measured twice before the application of any drug and was nearly identical in all animals. (B) Time course for the effects of analogues 1, 9, 13, and 14 (100 μM) on rabbit IOP over 8 h vs control. Values are the means ± SEM of results from 10 independent experiments. (C) Time-course for the effects of analogues 11B, 12A, and 12B (100 μM) on rabbit IOP over 8 h. Values are the means ± SEM of results from eight independent experiments. (D)Dose–response curves for the maximal effectsof analogues 1, 9, 13, and 14 (100 μM) on rabbit IOP. Values are the means ± SEM of results from eight independent experiments. (E) Maximal effects of analogues 1 and 9 (100 μM, 10 μL) on rabbit IOP, as compared to latanoprost (0.005%), dorzolamide hydrochloride (2%), and timolol maleate (0.5%) applied in 40 μL, as compared to control. (F) Duration of the effects of analogues 1 and 9 (100 μM, 10 μL), as compared to latanoprost (0.005%), dorzolamide hydrochloride (2%), and timolol maleate (0.5%) applied in 40 μL, as compared to control. The duration of the effect, termed mean-time effect, was calculated as the time between 50% maximal decrease in IOP after drug administration and a 50% recovery of the initial IOP value.75
Discussion
Chemical Stability of Analogues 8–14
Previously, we analyzed the chemical stability of analogues 1–3 at gastric juice pH of 1.4 (t1/2 65, 19, and 14.5 h, respectively).27 Here, we determined whether a β,γ-dihalomethylene group increases the chemical stability of these nucleotide analogues as compared to the presence of a β,γ-methylene group.
We found that analogues 8 and 9 exhibited high stability at pH 1.4 with half-lives of 25 and 65 h, respectively, comparable to the t1/2 values of the methylene analogues 1–3. Hydrolytic cleavage of the Pα–O–Pβ bond in both analogues 8 and 9 results in formation of 2-MeS-AMP and β,γ-dihalomethylene diphosphonate. We hypothesize that the lower chemical stability of analogue 8 as compared to 9 may be due to the higher electronegativity of fluorine vs chlorine, resulting in a better leaving group in β,γ-difluoromethylene diphosphonate. Furthermore, we postulate that accessibility of a water molecule for nucleophilic attack on Pα or Pβ is more restricted for analogue 9 than analogue 8 due to the steric hindrance of the larger chlorine atom resulting in a longer t1/2.
Analogues 10–12 were less stable at pH 1.4 (t1/2 = 8.0, 6.7, and 0.7 h, respectively), as compared to analogues 8 and 9, due to the low stability of the P–B bond under acidic conditions.57,58
Analogues 13 and 14 were significantly more stable at pH 1.4, as compared to analogues 8 and 9, respectively. This may be related to differences in the stability of the partial negative charge on the 5′-phosphonate (Pα) and 5′-oxygen atoms of analogues 8/9 and analogues 13/14, respectively, after hydrolysis, i.e., greater stability of a phosphate vs phosphonate group resulting in more extensive hydrolysis and shorter t1/2 values for analogues 8/9 vs 13/14.
Enzymatic Stability of Analogues 8–14
Previously, we reported on the stability of analogue 1 vs ATP in human blood serum (t1/2 = 12.1 vs 3.6 h, respectively),27 whereas here we found that analogue 9 was even more stable, exhibiting only 15% degradation after 24 h in human blood serum vs ATP (t1/2 = 4.9 h). We postulate that the enhanced stability of analogue 9 is probably due to steric effects. The bulky chlorine atoms cause analogue 9 to be a weak substrate for enzymatic dephosphorylation. Likewise, analogues 10–14 were not enzymatically hydrolyzed due to the replacement of the nonbridging oxygen with a borano group.
Previously, we found that 2-MeS-α-borano-ATP (A-isomer), analogue 4, was susceptible to hydrolysis by alkaline phosphatase, resulting in α-borano-2-MeS-AMP and 2-MeS-α-borano-ADP.27 For example, alkaline phosphatase hydrolyzed 60% of 2-MeS-α-borano-ATP (A-isomer) within 12 min at 37 °C, and only traces of 2-MeS-α-borano-ATP remained after 100 min. The substitution of the Pβ, Pγ-bridging CH2 group with a CX2 group as in analogues 10 and 12 rendered these analogues completely resistant to hydrolysis by alkaline phosphatase over 30 min at 37 °C. Furthermore, analogues 8–14 were highly resistant to hydrolysis by NTPDases1,2,3,8, and NPP1,3 (less than 5% hydrolysis, as compared to ATP). In particular, analogue 12B was completely resistant to hydrolysis by NTPDase1,2,3 and NPP3. The extremely low rate of enzymatic hydrolysis of these analogues is related primarily to the presence of a phosphonate moiety at Pβ, Pγ (analogues 8–12) or Pα, Pβ (analogues 13, 14). Bulky Cl atoms at CX2 further increase the enzymatic stability of the analogues, as compared to F atoms (e.g., greater stability for analogue 9 vs analogue 8). The further addition of a borano modification at Pα of analogue 9 yielded analogue 12, the most stable analogue synthesized, indicating that NTPDase and NPP are not tolerant to steric hindrance at Pα.
Activity of Analogues 8–14 at the P2Y1R
Although analogues 8–14 were shown to be resistant to enzymatic hydrolysis, they were less potent agonists of the P2Y1R than the more rapidly hydrolyzed 2-MeS-ADP.
Whereas the activity of analogue 1 at the P2Y1R was 20-fold lower than 2-MeS-ADP (EC50 = 0.0025 μM),27 the activities of the corresponding β,γ-dihalomethylene analogues 8 and 9 were 300- to 3800-fold lower than 2-MeS-ADP, respectively.
Apparently, the presence of a thiomethyl group at the adenine C-2 position is essential for P2Y1R activity because analogues 11A and 11B were inactive. Yet, any halogen substitution at the phosphonate carbon was not tolerated by the P2Y1R.
Considering the rank order of agonist potency obtained at the P2Y1R, analogue 1 > 8 > 9, we believe that the major parameter determining the affinity of this series of P2Y1R agonists is the steric constraints of the ligand binding site of the P2Y1R rather than the pKa values of the analogues' phosphonate moieties.52,59 The smaller size of the β,γ-methylene group presumably allows tighter receptor binding than the larger β,γ-dihalomethylene group, consistent with the EC50 values at the P2Y1R obtained for analogues 13 and 14 (Table 2).
Effect of Nucleotide Analogues on IOP
In the current study, we demonstrate the ability of analogues 1, 9, 11B, 12A, 12B, 13, and 14 to reduce IOP in male New Zealand white rabbits, whereas analogues 8 and 11A increased IOP. The two compounds exhibiting the strongest hypotensive effect (maximal hypotensive effect of about 32% reduction) were analogues 1 and 9 (EC50 values of 30.2 μM and 95.5 nM, respectively), equally or more effective than hypotensive drugs currently on the market (Figure 5E). This is a significant finding because nucleotide analogues activate different receptors than hypotensive drugs commonly used for treatment of glaucoma and may represent alternatives for patients unable to use the currently approved drugs. Side effects are common with current glaucoma medications. Thus, β-blockers (e.g., timolol maleate) can cause bradycardia and hypertension60 and are not suitable for patients with cardiac problems or asthma, obstructive pulmonary disease, and corneal dystrophy.61 Cholinergic agents (e.g., pilocarpine) produce fixed pupils and induce myopia and cataracts,62–64 whereas prostaglandins (e.g., latanoprost) cause eyelash growth, iris pigmentation,65 and muscle and joint pain.60 The development of stable and potent nucleotide analogues should expand the limited repertoire of drugs currently used for the treatment of glaucoma.
We found no correlation between the potency of nucleotide analogues at the P2Y1R and their ability to reduce IOP because analogues 1 and 9 activated the P2Y1R with EC50 values in the nanomolar and micromolar range, respectively, yet analogue 9 was more potent than analogue 1 at reducing IOP. These findings may imply that these analogues reduce IOP not solely via activation of the P2Y1R, as previously described,55 but via activation of other P2 receptors. Interestingly, replacing the β,γ-bridging oxygen with a β,γ-difluoromethylene group increases IOP (analogue 8), whereas substitution with β,γ-dichloromethylene generates an analogue that is strongly hypotensive (analogue 9). Likewise, replacement of the Pα nonbridging oxygen with BH3 results in a significant reduction in IOP (except for analogue 11A), as does a thiomethyl substitution at the C-2 position in the adenine ring.
The enhanced activity of analogue 9 for reduction of IOP, as compared to analogues 11–14, is possibly linked to the significantly enhanced chemical and metabolic stability of analogue 9 vs analogues 11–14.
Summary
We synthesized chemically and enzymatically stable analogues 8–14 based on ADP and ATP scaffolds modified by α,β/β,γ-dihalomethylene groups within the phosphate chain. These analogues were agonists at the P2Y1 R. Analogues 1 and 9, in particular, exhibited the ability to reduce IOP in normotensive rabbits, with EC50 values of 30.2 μM and 95.5 nM, respectively. Moreover, these analogues were more effective at reducing IOP than several commonly prescribed glaucoma drugs (e.g., latanoprost and dorzolamide hydrochloride). In addition, the duration of the hypotensive effect induced by analogue 9 was comparable to latanoprost, dorzolamide hydrochloride, and timolol maleate. We conclude that compound 9 is an attractive drug candidate for the treatment of glaucoma. Furthermore, although slightly less efficacious than timolol maleate, compound 9 represents a promising alternative to timolol maleate, as the latter is a β-blocker and cannot be used for the treatment of patients suffering from asthma, bronchitis or cardiovascular problems.
Experimental Section
General
All air- and moisture-sensitive reactions were carried out in flame-dried, N2-flushed, two-neck flasks sealed with rubber septa, and the reagents were introduced with a syringe. Progress of reactions was monitored by TLC on precoated Merck silica gel plates (60F-254). Visualization of reactants and products was accomplished by UV light. Compounds were characterized by nuclear magnetic resonance using Bruker AC-200, DPX-300, or DMX-600 spectrometers. 1H NMR spectra were measured at 200, 300, or 600 MHz. Nucleotides were characterized also by 31P NMR in D2O, using 85% H3PO4, and 19F NMR using trifluorochloromethane as an external reference on Bruker AC-200 and DMX-600 spectrometers. High resolution mass spectra were recorded on an AutoSpec-E FISION VG mass spectrometer by chemical ionization. Nucleotides were analyzed under ESI (electron spray ionization) conditions on a Q-TOF microinstrument (Waters, UK). Primary purification of the nucleotides was achieved on a LC (Isco UA-6) system using a column of Sephadex DEAE-A25, swollen in 1 M NaHCO3 at 4 °C for 1 day. The resin was washed with deionized water before use. The LC separation was monitored by UV detection at 280 nm. Final purification of the nucleotides and separation of diastereomers were achieved on a HPLC system (Elite Lachrom, Merck-Hitachi) using a semipreparative reverse-phase column (Gemini 5uC-18 110A 250mm × 10.00mm; 5 μm; Phenomenex, Torrance, CA). The purity of the nucleotides was evaluated with an analytical reverse-phase column system (Gemini 5u C-18 110A, 150 mm × 4.60 mm; 5 μm; Phenomenex, Torrance, CA) using two solvent systems: solvent system I, (A) 100 mM triethylammonium acetate (TEAA), pH 7:(B) CH3CN; solvent system II, (A) 0.01 M KH2PO4, pH = 4.5:(B) CH3CN. The details of the solvent system gradients used for the separation of each product are given below. The purity of the nucleotides was generally ≥92%. All commercial reagents were used without further purification unless otherwise noted. All reactants in moisture-sensitive reactions were dried overnight in a vacuum oven. RPMI (Roswell Park Memorial Institute) 1640 medium was obtained from Sigma-Aldrich. 2′,3′-O-Methoxymethylideneadenosine and 2-MeS-adenosine were prepared, as previously described.15,66 2′,3′-O-Methoxymethylidene-2-MeS-adenosine was purified with a MPLC system (Biotage, Kungsgatan, Uppsala, Sweden) using a silica gel (25+M) column and the following gradient scheme: 3 column volumes (CV) of 100:0 (A) CHCl3:(B) EtOH, 5 CV of a gradient from 100:0 to 90:10 A:B, and 4 CV of 90:10 A:B at a flow rate of 12.5 mL/min. pH measurements were performed with an Orion microcombination pH electrode and a Hanna Instruments pH meter. TEAB was prepared according to the literature.67 Bis(tributylammonium)difluoromethylene diphosphonate salt was purchased from RI Chemical Inc. For preparation of human blood serum, whole blood taken from healthy volunteers was obtained from a blood bank (Tel-Hashomer Hospital, Israel). Blood was stored for 12 h at 4 °C and centrifuged in plastic tubes at 1500g for 15 min at RT. The serum was separated and stored at −80 °C.
Preparation of Bis(tributylammonium)dichloromethylene Diphosphonate Salt
A H+ Dowex column was used for ion exchange chromatography. First, 30 mL of Dowex was placed in a column with cotton wool at the bottom, and then the column was washed with 10% NaOH (150 mL) until the pH of the effluent was basic. Then the column was washed with distilled water until the pH of the effluent reached neutral. Then the column was washed with 10% HCl (300 mL), followed by distilled water until the effluent reached acidic and neutral pH, respectively. A flask containing Bu3N (2 equiv) in EtOH was placed in an ice bath under the column and stirred.
The disodium form of dichloromethylene diphosphonate salt was dissolved in distilled water, poured onto the column, and the column was washed with distilled water until the pH of the effluent was neutral. The effluent was dropped into the Bu3N/EtOH solution. The final solution of bis(tributylammonium)dichloromethylene diphosphonate salt was then freeze-dried.
Preparation of 2-MeS-adenosine-5′-O-triphosphate-β,γ-methylene-dihalogen Derivatives, 8 and 9
1,8-Bis(dimethylamino)naphthalene (156 mg, 0.72 mmol, 2 equiv) was added at 0 °C to 2′,3′-O-methoxymethylidene-2-MeS-adenosine, analogue 15, (130 mg, 0.36 mmol) in trimethylphosphate (2.5 mL) in a flame-dried two-neck flask under N2, and the reaction was stirred for 20 min until a clear solution was attained. POCl3 (66 μL, 0.72 mmol, 2 equiv) was added at 0 °C The solution was stirred at 0 °C for 1 h. A 0.5 M solution of bis(tributylammonium)dihalomethylene diphosphonate salt (660 mg, 1.13 mmol, 3 equiv) in dry DMF (1.4 mL) and tributylamine (360 μL, 1.46 mmol, 4 equiv) was added at 0 °C, and the reaction mixture was stirred for 90 min. A 0.5 M solution of TEAB (15 mL) was added at RT, and the reaction mixture was stirred for 60 min and then freeze-dried. The residue was dissolved in water and treated with 18% HCl until the pH was 2.3, and then the mixture was stirred for 3 h at RT. Finally, the mixture was treated with 24% NH4OH solution, and the pH was adjusted to 9. The solution was stirred for 45 min at RT and then freeze-dried. The resulting residue was dissolved in deionized water (100 mL) and extracted with diethyl ether (2 × 50 mL) and then chloroform (50 mL). The aqueous phase was freeze-dried. The resulting residue was separated on an activated Sephadex DEAE-A25 column (0−0.5 M NH4HCO3; total volume 2 L). The relevant fractions were collected, freeze-dried, and excess NH4HCO3 was removed by repeated freeze-drying with deionized water to yield the product as a white powder. The product was purified by LC, yielding analogue 8 at a 46% yield (110 mg) and analogue 9 at a 10% yield (19.6 mg). Finally, the nucleotide triethylammonium counterions were exchanged for Na+ ions by passing analogues 8 and 9 through a Sephadex-CM C-25 Na+-form column. The spectral data for analogues 8 and 9 were consistent with the literature.43
Preparation of Adenosine-5′-O-(α-boranotriphosphate)-β,γ-methylene-dihalogen Derivatives, 10–12
2′,3′-O-Methoxymethylidene adenosine derivatives (analogues 14 or 20; 50 mg, 0.14 mmol) were dissolved in trimethylphosphate (0.7 mL) in a flame-dried two-neck flask under N2. 1,8-Bis(dimethylamino)naphthalene (60 mg, 0.28 mmol, 2 equiv) was added at 0 °C, and the reaction was stirred for 20 min until a clear solution was attained. PCl3 (23 μL, 0.28 mmol, 2 equiv) was added at 0 °C, and a white solid precipitated. The suspension was stirred at 0 °C for 45 min. Then, a 0.5 M solution of bis(tributylammonium) dihalomethylene diphosphonate salt (245 mg, 0.42 mmol, 3 equiv) in dry DMF (0.6 mL) and tributylamine (134 μL, 0.56 mmol, 4 equiv) was added at 0 °C, and the reaction mixture was stirred for 50 min. A 2 M solution of BH3·SMe2 complex in THF (0.7 mL, 2.81 mmol, 10 equiv) was added at 0 °C, and the reaction mixture turned clear. The solution was stirred for 5 min at 0 °C and then for 60 min at RT. Finally, a 0.5 M TEAB solution (15 mL) was added at RT, and the mixture was stirred for 60 min and then freeze-dried. The residue was dissolved in water, treated with 18% HCl until the pH was 2.3, and then the mixture was stirred for 3 h at RT. Finally, the mixture was treated with 24% NH4OH, and the pH was adjusted to 9. The solution was stirred for 45 min at RT and then freeze-dried. The resulting residue was dissolved in deionized water (100 mL) and extracted with diethyl ether (2 × 30 mL) and chloroform (30 mL). The aqueous phase was freeze-dried. The resulting residue was separated on an activated Sephadex DEAE-A25 column (0−0.5 M NH4HCO3; total volume 2 L). The relevant fractions were collected, freeze-dried, and excess NH4HCO3 was removed by repeated freeze-drying cycles with deionized water to obtain the product as a white powder. The product was purified by LC yielding analogue 10 at a 21% yield (13 mg), analogue 11 at a 18% yield (14 mg), and analogue 12 at a 7.2% yield (18.6 mg). The diastereomers of analogues 10, 11, and 12 were separated on a HPLC column, under the conditions described below. Finally, the purified diasteroisomers were passed through a Sephadex-CM C-25 Na+-form column to exchange triethylammonium ions for Na+.
Separation of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CF2 (10A and 10B)
The separation of analogue 10 diastereoisomers, 10A and 10B, was accomplished using a semipreparative reverse-phase Gemini 5u column and isocratic elution with 80:20 (A) 100 mM TEAA, pH 7:(B) MeOH at a flow rate of 5 mL/min. Fractions containing purified isomers [Rt = 12.78 min (10A isomer); 14.75 min (10B isomer)] were collected and freeze-dried. Excess buffer was removed by repeated freezedrying cycles, with the solid residue dissolved each time in deionized water. Diastereoisomers 10A and 10B were obtained at 21% overall yield (18.6 mg) after LC separation.
Characterization of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CF2 (10A)
Retention time on a semipreparative column: 12.78 min. 1H NMR (D2O; 600 MHz): δ 8.43 (s; H-8; 1H), 6.13 (d; J = 5.40 Hz; H-1′; 1H), 4.80 (t; J = 5.40 Hz; H-2′; 1H), 4.65 (m; H-3′; 1H), 4.36 (m; H-4′; 1H), 4.31 (m; H-5′; 1H), 4.13 (m; H-5″; 1H), 2.58 (s; CH3; 3H), and 0.45 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 83.50 (m; Pα-BH3), 4.71 (m; Pγ), and −2.25 (m; Pβ) ppm. 19F NMR (D2O 188 MHz): δ −115.38 (t, J = 79.27) ppm. MS-ESI m/z: 584 (M−). TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.12. Purity data obtained on an analytical column: retention time: 7.82 min (95.3% purity) using solvent system I with a gradient from 85:15 to 50:50 A:B over 15 min at a flow rate of 1 mL/min. Retention time: 4.89 min (92.8% purity) using solvent system II with a gradient from 85:15 to 50:50 A:B over 20 min at a flow rate of 1 mL/min.
Characterization of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CF2 (10B isomer)
Retention time on a semipreparative column: 14.75 min. 1H NMR (D2O; 600 MHz): δ 8.39 (s; H-8; 1H), 6.12 (d; J = 5.40 Hz; H-1′; 1H), 4.80 (t; J = 5.40 Hz; H-2′; 1H), 4.57 (m; H-3′; 1H), 4.37 (m; H-4′; 1H), 4.26 (m; H-5′; 1H), 4.19 (m; H-5″; 1H), 2.58 (s; CH3; 3H), and 0.48 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 84.80 (m; Pα-BH3), 4.73 (m; Pγ), and −2.30 (m; Pβ) ppm. 19F NMR (D2O 188 MHz): δ −115.40 (t, J = 79.27) ppm. MS-ESI m/z: 584 (M−). TLC (NH4-OH:H2O:2-propanol 2:8:11), Rf = 0.12. Purity data obtained on an analytical column: retention time: 8.17 min (87.0% purity) using solvent system I with a gradient from 85:15 to 50:50 A:B over 15 min at a flow rate of 1 mL/min. Retention time: 3.08 min (94.95% purity) using solvent system II with a gradient from 70:30 to 50:50 A:B over 10 min at a flow rate of 1 mL/min.
Separation of Adenosine-5′-O-(a-boranotriphosphate)-β,γ-CCl2 (11)
The separation of diastereomers 11A and 11B was accomplished using a semipreparative reverse-phase Gemini 5u column (C-18 110A; 250 mm × 10.00 mm; 5 μm) and isocratic elution with 91:9 (A) 100 mM TEAA, pH 7:(B) MeOH at a flow rate of 5 mL/min. Fractions containing purified isomers [Rt = 8.37 min (11A isomer); 10.87 min (11B isomer)] were collected and freeze-dried. Excess buffer was removed by repeated freezedrying cycles with the solid residue dissolved each time in deionized water. Diastereoisomers 11A and 11B were obtained at 18% overall yield (14 mg) after LC separation.
Characterization of Adenosine-5′-O-(α-boranotriphosphate)-β,γ-CCl2 (11A)
Retention time on a semipreparative column: 8.37 min. 1H NMR (D2O; 300 MHz): δ 8.51 (s; H-8; 1H), 8.14 (s; H-2; 1H), 6.04 (d; J = 5.7 Hz; H-1′; 1H), 4.78 (H-2′ and H-3′ signals are hidden by the water signal), 4.30 (m; H-5′; 2H), 4.05 (m; H-4′; 1H), and 0.37 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 83.80 (m; Pα-BH3), 9.10 (d; J = 19.14 Hz; Pγ), and 2.02 (m; Pβ) ppm. MS-ESI m/z: 570 (M−). HRMS-FAB (negative) m/z: calculated for C11H18BN5O11Na2P3: 546.0104; found: 546.0104. TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.3. Purity data obtained on an analytical column: retention time: 6.88 min (98% purity) using solvent system I with a gradient from 95:5 to 70:30 A:B over 10 min at a flow rate of 1 mL/min. Retention time: 1.55 min (98% purity) using solvent system II with a gradient from 85:15 to 70:30 A:B over 10 min at a flow rate of 1 mL/min.
Characterization of Adenosine-5′-O-(α-boranotriphosphate)-β,γ-CCl2 (11B)
Retention time on a semipreparative column: 10.87 min. 1H NMR (D2O; 300 MHz): δ 8.49 (s; H-8; 1H), 8.14 (s; H-2; 1H), 6.04 (d; J = 5.7 Hz; H-1′; 1H), 4.78 (H-2′ signal is hidden by the water signal), 4.47 (m; H-3′; 1H), 4.23 (m; H-5′; H-5″; 2H), 4.15 (m; H-4′; 1H), and 0.37 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 84.58 (m; Pα-BH3), 9.14 (d; J = 18.46 Hz; Pγ), and 2.11 (m; Pβ) ppm. MS-ESI m/z: 570 (M−). TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.3. Purity data obtained on an analytical column: retention time: 7.62 min (97% purity) using solvent system I with a gradient from 95:5 to 70:30 A:B over 10 min at a flow rate of 1 mL/min. Retention time: 1.32 min (98% purity) using solvent system II with a gradient from 95:5 to 70:30 A:B over 10 min at a flow rate of 1 mL/min.
Separation of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CCl2 (12A and 12B)
The separation of diastereoisomers 12A and 12B was accomplished using a semipreparative reversephase Gemini 5u column (C-18 110A; 250 mm × 10.00 mm; 5 μm) and isocratic elution with 82:18 (A) 100 mM TEAA, pH 7:(B) MeOH at a flow rate of 5 mL/min. Fractions containing purified isomers [Rt = 9.33 min (12A isomer); 10.61 min (12B isomer)] were collected and freeze-dried. Excess buffer was removed by repeated freeze-drying cycles with the solid residue dissolved each time in deionized water. Diastereoisomers 12A and 12B were obtained at a 7.2% overall yield (13 mg) after LC separation.
Characterization of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CCl2 (12A)
Retention time on a semipreparative column: 9.33 min. 1H NMR (D2O; 300 MHz): δ 8.34 (s; H-8; 1H), 6.04 (d; J = 5.10 Hz; H-1′; 1H), 4.78 (H-2′ and H-3′ signals are hidden by the water signal), 4.30 (m; H-4′; 1H), 4.05 (m; H-5′; H-5″; 2H), 2.49 (s; CH3; 3H), and 0.42 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 83.40 (m; Pα-BH3), 9.28 (d; J = 17.32 Hz; Pγ), and 2.65 (m; Pβ) ppm. MS-ESI + m/z: 548 (M−). TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.15. Purity data obtained on an analytical column: retention time: 7.96 min (90.4% purity) using solvent system I with a gradient from 95:5 to 65:35 A:B over 13 min at a flow rate of 1 mL/min. Retention time: 2.36 min (87.0% purity) using solvent system II with a gradient from 95:5 to 60:40 A:B over 10 min at a flow rate of 1 mL/min.
Characterization of 2-MeS-adenosine-5′-O-(α-boranotriphosphate)-β,γ-CCl2 (12B)
Retention time on a semipreparative column: 10.61 min. 1H NMR (D2O; 300 MHz): δ 8.32 (s; H-8; 1H), 6.02 (d; J = 5.40 Hz; H-1′; 1H), 4.78 (H-2′ and H-3′ signals are hidden by the water signal), 4.28 (m; H-4′; 1H), 4.18 (m; H-5′; H-5″; 2H), 2.48 (s; CH3; 3H), and 0.40 (m; BH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 84.49 (m; Pα-BH3), 9.17 (d; J = 19.16 Hz; Pγ), and 2.35 (m; Pβ) ppm. MS-ESI m/z: 548 (M−). TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.15. Purity data obtained on an analytical column: retention time: 3.87 min (92.0% purity) using solvent system I with a gradient from 85:15 to 60:40 A:B over 10 min at a flow rate of 1 mL/min. Retention time: 4.11 min (91.0% purity) using solvent system II with a gradient from 90:10 to 60:40 A:B over 10 min at a flow rate of 1 mL/min.
Preparation and Characterization of 2′,3′-O-Methoxymethylidene-5′-O-tosyl-2-MeS-adenosine, 34
A solution of 4-dimethylaminopyridine (132 mg, 1.07 mmol, 4 equiv) in CH2Cl2 (1 mL) and a solution of TsCl (130 mg, 0.67 mmol, 2.5 equiv) in CH2Cl2 (1 mL) were added to a suspension of 2′,3′-O-methoxymethylidene-2-MeS-adenosine, analogue 15 (95 mg; 0.27 mmol), in CH2Cl2 (3 mL) in a flame-dried two-neck flask under N2 at RT. The suspension turned clear, and the reaction mixture was stirred for 2 h at RT. CH2Cl2 (30 mL) was added to the reaction mixture, which was then extracted with saturated NaHCO3 solution (3 × 30 mL). The organic phase was treated with Na2SO4 and filtered. The solvent was removed under reduced pressure, and the residue was separated using a MPLC system with a silica (25+M) column and the following gradient scheme: 3 column volumes (CV) of 100:0 (A) CH2Cl2:(B) EtOH, 5 CV of a gradient from 100:0 to 90:10 A:B, and 4 CV of 90:10 A:B at a flow rate of 25 mL/min. The relevant fractions were collected, and the solvent was removed under reduced pressure, yielding analogue 34 at a 85% yield (115 mg) as a white solid. 1H NMR (CDCl3; 200 MHz): δ 7.83 (s; H-8; 2H), 7.69, 7.23 (2 m, 2H), 6.34, 6.29 (2 br s, NH2; 2H), 6.17, 6.05 (2d; J = 2.60 Hz; H-1′; 1H), 6.07, 6.01 (2s, CH-Ome; 1H), 5.56, 5.53 (2dd; J = 2.60 Hz; H-2′; 1H, J = 7.20 Hz; H-2′; 1H), 5.21, 5.06 (2dd, J = 7.20 Hz; H-3′; 1H, J = 3.20 Hz; H-3′; 1H), 4.66, 4.52 (2m, H-4′; 1H), and 4.31 (m, H-5′; H-5″; 4H) ppm. MS-ESI-m/z: 584 (M−). TLC (CHCl3:EtOH, 9:1), Rf = 0.64.
Preparation and Characterization of 2′,3′-O-Isopropylidene-5′-O-tosyl-2-MeS-adenosine, 35
A solution of 4-DMAP (134mg, 1.09 mmol, 4 equiv) in CH2Cl2 (2 mL) and a solution of TsCl (156 mg, 0.82 mmol, 3 equiv) in CH2Cl2 (0.5 mL) were added to a solution of 2′,3′-O-isopropylidene adenosine, analogue 33 (97 mg; 0.27 mmol), in CH2Cl2 (1 mL) in a flame-dried two-neck flask under N2 at RT, and the reaction mixture was stirred for 2 h. CH2Cl2 (50 mL) was added to the reaction mixture, which was then extracted with saturated NaHCO3 solution (3 × 30 mL). The organic phase was treated with Na2SO4 and filtered. The solvent was removed under reduced pressure, and the residue was separated using a MPLC system with a silica gel (25+M) column and the following gradient scheme: 3 column volumes (CV) of 100:0 (A) CH2Cl2:(B) EtOH, 5 CV of a gradient from 100:0 to 90:10 A:B, and 4 CV of 90:10 A:B at a flow rate of 25 mL/min. The relevant fractions were collected, and the solvent was removed under reduced pressure, yielding analogue 35 at a 51% yield (71 mg) as a white solid. 1H NMR (CDCl3; 300 MHz): δ 7.90 (s; H-8; 1H), 7.67 (d, J = 8.40, 2H), 6.05 (s; H-1′; 1H), 5.35 (m; H-2′; 1H), 5.00 (m, H-3′; 1H), 4.50 (m, H-4′; 1H), and 4.25 (m, H-5′; H-5″; 2H). MS-ESI m/z: 508 (MH+). MS-ESI+ m/z: 508 (MH+). TLC(CHCl3: EtOH 95:5), Rf = 0.79.
Preparation and Characterization of 2-MeS-adenosine-5′-O-diphosphate-α,β-difluoromethylene, 13
2′,3′-O-Methoxymethylidene-5′-O-tosyl-2-MeS-adenosine, analogue 34 (90 mg; 0.17 mmol), was dissolved in dry DMF (0.4 mL) in a flame-dried two-neck flask under N2. A solution of tris(tetra-n-butylammonium)difluoro methylenediphosphonate45 (0.28 mmol; 1.5 equiv) in dry DMF (0.3 mL) was added at RT, and the reaction was stirred for 72 h. Deionized water (20 mL) was added, and the reaction was treated with 18% HCl until the pH was 2.3, and then the mixture was stirred for 3 h at RT. Then, the mixture was treated with 24% NH4OH, and the pH was adjusted to 9. The solution was stirred for 45 min at RT and then freeze-dried. The resulting residue was applied to an activated Sephadex DEAEA25 column (0−0.3 M NH4HCO3; total volume of 2 L). The relevant fractions were collected, freeze-dried, and excess NH4HCO3 was removed by repeated freeze-drying with deionized water to yield analogue 13 as a white powder. The residue was separated using a MPLC system with a C-18 (12+M) column and the following gradient scheme: 5 column volumes (CV) of 100:0 (A) TEAA:(B) MeOH, 7.5 CV of a gradient from 100:0 to 60:40 A:B, and 3 CV of 60:40 A:B at a flow rate of 12 mL/min. Finally, triethylammonium ions were exchanged for Na+ by passing the pure analogue 13 through a Sephadex-CM C-25 Na+-form column. Analogue 13 was obtained at a 34% yield (61 mg) after MPLC separation. 1H NMR (D2O; 300 MHz): δ 8.18 (s; H-8; 1H), 5.93 (d; J = 5.40 Hz; H-1′; 1H), 4.78 (H-2′ and H-3′ signals are hidden by the water signal), 4.43 (m; H-4′; 1H), 4.19 (m; H-5′; 1H), 4.15 (m; H-5″; 1H), and 2.37 (s; CH3; 3H) ppm. 31P NMR (D2O; 81 MHz): δ 4.48 (m; Pα, Pβ) ppm. 19F NMR (D2O 188 MHz): δ 119.16 (dd, J1 = 84.72 Hz, J2 = 80.95 Hz) ppm. MS-ESI m/z: 506 (M−). HRMS-FAB (negative) m/z: calculated for C12H15F2N5O9Na3P2S1: 573.9721; found: 573.966. TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.5. Purity data obtained on an analytical column: retention time: 6.85 min (100% purity) using solvent system I with a gradient from 85:15 to 50:50 A:B over 15 min at a flow rate of 1 mL/min. Retention time: 5.17 min (99.94% purity) using solvent system II with a gradient from 85:15 to 50:50 A:B over 18 min at a flow rate of 1 mL/min.
Characterization of 2-MeS-adenosine, 5′,5‴-[P,P′-Difluoromethylene bisphosphonate], 38
1H NMR (D2O; 300 MHz): δ (s; H-8; 1H), 5.93 (d; J = 5.40 Hz; H-1′; 1H), 4.78 (H-2′and H-3′ signals are hidden by the water signal), 4.43 (m; H-4′; 1H), 4.19 (m; H-5′, 1H), 4.15 (m; H-5″; 1H), and 2.37 (s; CH3; 3H) ppm. 31P NMR (D2O; 81 MHz): δ 4.48 (m; Pα, Pβ) ppm. 19F NMR (D2O 188 MHz): δ 119.16 (dd, J1 = 84.72 Hz, J2 = 80.95 Hz) ppm. MS-ESI m/z: 506 (M−). HRMS-FAB (negative) m/z: calculated for C23H28F2N10O12Na3P2S2: 869.0460; found: 869.056. TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.5. Purity data obtained on an analytical column: retention time: 6.85 min (100.0% purity) using solvent system I with a gradient from 85:15 to 50:50 A:B over 15 min at a flow rate of 1 mL/min. Retention time: 5.17 min (99.94% purity) using solvent system II with a gradient from 85:15 to 50:50 A:B over 18 min at a flow rate of 1 mL/min.
Preparation and Characterization of 2-MeS-adenosine, 5′,5‴-[P,P′-Dichloro-methylene bisphosphonate], 35
2′,3′-O-Isopropylidene-5′-O-tosyl-2-MeS-adenosine, analogue 14 (42 mg; 0.08 mmol), was dissolved in dry DMF (0.2 mL) in a flame-dried two-neck flask under N2. A solution of tris(tetra-n-butylammonium)dichloro methylenediphosphonate (0.16 mmol; 2 equiv) in dry DMF (0.3 mL) was added at RT, and the reaction was stirred for 72 h. Neat TFA was added for 10 min under argon bubbling and then evaporated under reduced pressure to yield a yellow solid. The resulting residue was separated on an activated Sephadex DEAE-A25 column (0−0.3 M NH4HCO3; total volume of 1.4 L). The relevant fractions were collected, freezedried, and excess NH4HCO3 was removed by repeated freezedrying with deionized water to yield analogue 14 as a white powder. The residue was separated using a HPLC system with a semipreparative C-18 column and the following gradient scheme (A) 100 mM TEAA:(B) MeOH with gradients of 85:15 to 75:25 over 10 min, 75:25 to 70:30 over 2 min, and 70:30 over 3 min at a flow rate of 5 mL/min. Retention time: 10.96 min. The relevant fractions were collected, freeze-dried, and excess TEAA was removed by repeated freeze-drying with deionized water to yield analogue 14 as a white powder. Finally, the triethylammonium ions were exchanged for Na+ by passing the pure analogue 14 through a Sephadex-CM C-25 Na+-form column. Analogue 14 was obtained at a 18% yield (9 mg) after HPLC separation. Retention time on a semipreparative column: 14.75 min. 1H NMR (D2O; 600 MHz): δ 8.39 (s; H-8; 1H), 6.09 (d; J = 4.80 Hz; H-1′; 1H), 4.83 (m; H-2′; 1H), 4.78 (H-3′ signal is hidden by the water signal), 4.61 (m; H-4′; 1H), 4.38 (m; H-5′; H-5″; 2H), and 2.53 (s; CH3; 3H) ppm. 31P NMR (D2O; 243 MHz): δ 10.88 (d; J = 15.79 Hz) and 8.70 (d; J = 15.79 Hz) ppm. MS-ESI m/z: 538 (M−). TLC (NH4OH:H2O:2-propanol 2:8:11), Rf = 0.26. Purity data obtained on an analytical column: retention time: 7.38 min (100% purity) using solvent system I with a gradient from 85:15 to 50:50 A:B over 10 min at a flow rate of 1 mL/min. Retention time: 4.38 min (99.9% purity) using solvent system II with a gradient from 85:15 to 50:50 A:B over 10 min at a flow rate of 1 mL/min.
Evaluation of the Stability of ADP, ATP, and Analogues 7 –14 in Human Blood Serum
The assay mixture containing 40 mM nucleotide derivative in deionized water (4.5 μL), human blood serum (180 μL), and RPMI-1640 medium (540 μL) was incubated at 37 °C for 0–24 h and samples were removed at 0.5–12 h intervals. Each sample was then heated to 80 °C for 30 min, treated with CM Sephadex (1–2 mg), stirred for 2 h, centrifuged for 6 min (13000 rpm, 17000g), and extracted with chloroform (2 × 500 μL). The aqueous layer was freeze-dried. Each resulting residue was applied to an activated starta X-AW weak anion exchange cartridge and then freeze-dried. The residue was purified using HPLC with a Gemini analytical column (5u, C-18, 110A; 150 mm × 4.60 mm) and gradient elution with solvent system II at 100:0 A:B over 10 min for analogue 5, 100:0 to 70:30 A:B over 3 min for analogue 6, 90:10 to 80:20 A:B over 10 min, and then 80:20 to 60:40 A:B over 5 min for analogue 7, 85:15 A:B over 10 min for analogue 8, 90:10 to 50:50 A:B over 18 min for analogue 9, 80:20 to 30:70 A:B over 10 min for analogue 14, and 80:20 to 30:70 A:B over 10 min for analogue 13 or gradient elution with solvent system I at 90:10 to 60:40 A:B over 12 min for analogues 10A and 10B, 90:10 A:B over 15 min for analogues 11A and 11B, and 80:20 A:B over 15 min for analogues 12A and 12B at a flow rate of 1 mL/min. The hydrolysis rates for the nucleotide analogues in human blood serum were determined by measuring the change in the integration of the respective HPLC peaks with time.
Evaluation of the Stability of Analogues 5 14 with Alkaline Phosphatase
Enzyme activity was determined by the release of p-nitrophenol from p-nitrophenyl phosphate measured with a UV-vis spectrophotometer at 405 nm.68 Relative enzyme activity and resistance of analogues 5–14 to enzymatic hydrolysis were determined at 37 °C using a solution of 0.2 mg of analogue in 77.5 μL of deionized water, 0.1 M Tris-HCl (pH 9.8), and 0.1 M MgCl2 with calf intestine alkaline phosphatase (Fermentas Inc., Glen Burnie, MD; 10unit/μL; 1.25 μL; 12.5u). After 3 h, the reaction was stopped by incubation of the sample at 80 °C for 15 min. Each sample was applied to an activated starta X-AW weak anion exchange cartridge and then freezedried. The residue was purified by HPLC with a Gemini analytical column (5u C-18 110A; 150 mm × 4.60 mm) using the gradient elution system described for the hydrolysis of analogues in human blood serum at a flow rate of 1 mL/min (see above). The hydrolysis rates for analogues 5–14 with alkaline phosphatase were determined by measuring the change in the integration of the respective HPLC peaks with time.
NTPDase1,2,3,8 (EC 3.6.1.5) Assays
Activity was measured, as previously described,69 at 37 °C in 0.2 mL of Tris-Ringers buffer: (in mM) 120 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgSO4, 25 NaHCO3, 5 glucose, 80 Tris, pH 7.4; (Sigma–Aldrich, Oakville, ON, Canada). ecto-Nucleotidases were expressed by transiently transfecting COS-7 cells in 10 cm plates using Lipofectamine (Invitrogen), as previously described.69 For the preparation of protein extracts, transfected cells were washed three times with Tris-saline buffer at 4 °C, collected by scraping in harvesting buffer (95 mM NaCl, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 45 mM Tris at pH 7.5), and washed twice by 300g centrifugation for 10 min at 4 °C. Cells were resuspended in harvesting buffer containing 10 mg/mL aprotinin and sonicated. Nuclei and cellular debris were discarded by centrifugation at 300g for 10 min at 4 °C, and the supernatant (crude protein extract) was aliquoted and stored at −80 °C until used for activity assays. Protein concentration was estimated by the Bradford microplate assay using bovine serum albumin (BSA) as a standard.70 NTPDase protein extracts were added to the reaction mixture and then preincubated at 37 °C for 3 min. The reaction was initiated by addition of ATP (Sigma–Aldrich, Oakville, ON, Canada) or analogues 8–14 at a final concentration of 100 μM, and the reaction was stopped after 20 min with 50 μL of malachite green reagent (Sigma–Aldrich). The released inorganic phosphate (Pi) was measured at 630 nm according to Baykov et al.70 The activity obtained with protein extracts from untransfected cells was subtracted from the activity obtained with extracts from NTPDase-transfected cells. The activity of untransfected cell extracts never exceeded 5% of the activity of extracts from NTPDase-transfected cells.
NPP1,3 (EC 3.1.4.1; EC 3.6.1.9) Assays
Evaluation of the activity of human NPP1 and NPP3 with ATP (Sigma–Aldrich) and analogues 8–14 was performed, as previously described,72 with some modifications. Reactions were carried out at 37 °C in 0.2 mL of the following mixture: (in mM) 1 CaCl2, 140 NaCl, 5 KCl, and 50 Tris, pH 8.5 (Sigma–Aldrich). Human NPP1 or NPP3 extract was added to the reaction mixture and preincubated at 37 °C for 3 min. The reaction was initiated by addition of ATP or analogues 8–14 at a final concentration of 100 μM. The reaction was stopped after 20 min by transferring a 0.1 mL aliquot of the reaction mixture to 0.125 mL of ice-cold 1 M perchloric acid (Fisher Scientific, Ottawa, ON, Canada). The samples were centrifuged for 5 min at 13000g. Supernatants were neutralized with 1 M KOH (Fisher Scientific) at 4 °C and centrifuged for 5 min at 13000g. An aliquot of supernatant (20 μL) was separated by reverse-phase HPLC to evaluate the degradation of ATP and analogues 8–14 using a SUPELCOSIL LC-18-T column (15 cm × 4.6 mm; 3 mm Supelco; Bellefonte, PA) with a mobile phase composed of 25 mM TBA, 5 mM EDTA, 100 mM KH2PO4/K2HPO4, pH 7.0, and 2% methanol at a flow rate of 1 mL/min.
Evaluation of the Chemical Stability of Analogues 8–14
A nucleotide analogue (1.5 mg) was dissolved in 0.2 M HCl/KCl buffer (0.8 mL) and the final pH was adjusted to 1.4 using 0.2 M HCl. Reactions continued at 37 °C for 1–31 days, with samples taken at 1–24 h intervals. The stabilities of the analogues were evaluated by HPLC to monitor degradation products using a Gemini analytical column (5u C-18 110A; 150 mm × 4.60 mm) and the gradient elution system described for the hydrolysis of analogues in human blood serum at a flow rate of 1 mL/min (see above). The hydrolysis rates of analogues 8–14 at pH 1.4 and 37 °C were determined by measuring the change in the integration of the respective HPLC peaks with time.
Intracellular Calcium Measurements
Human 1321N1 astrocytoma cells stably expressing the turkey P2Y1 receptor were grown in Dulbecco's modified Eagle's medium containing 5% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 500 μg/mL Geneticin (G-418; Life Technologies, Inc.). Changes in the intracellular free calcium concentration, [Ca2+]i, were detected by dual-excitation spectrofluorometric analysis of suspensions from cells loaded with fura-2, as previously described.73,74 Cells were treated with the indicated nucleotide analogue at 37 °C in 10 mM Hepesbuffered saline (pH 7.4) containing 1 mM CaCl2 and 1 mM MgCl2, and the maximal increase in [Ca2+]i was determined at different analogue concentrations to calculate the EC50. Concentration–response data were analyzed with the Prism curve fitting program (GraphPAD Software, San Diego, CA). Data were obtained from three experiments performed in triplicate.
Animals
Twenty-four male New Zealand white rabbits (2.5 ± 0.5 kg) were kept in individual cages with free access to food and water on controlled 12 h/12 h light/dark cycles. All the protocols used adhered to the ARVO Statement for the Use of Animals in Ophthalmology and Vision Research and also were in accordance with the European Communities Council Directive (86/609/EEC).
IOP Measurements
IOP was measured by means of a TonoVET rebound tonometer supplied by Tiolat Oy (Helsinki, Finland). The application of this tonometer to animals does not require the use of any anesthetic. For single dose experiments, different analogues were applied unilaterally to the cornea at a concentration of 100 μM and a fixed volume of 10 μL. The contralateral eye received the same volume of saline solution (0.9% NaCl, vehicle). Two IOP measurements were taken before any analogue was instilled. Experiments were performed following a blinded design where no visible indication was given to the experimenter as to the nature of the applied solution. IOP was followed up to 8 h to study the time course of the effect. Some of the analogues were assayed over a range of doses from 1 nM to 100 μM to generate dose–response curves. For these experiments, IOP was measured as the maximal response obtained with each dose of the analogue. Dose–response curves were calculated by plotting the IOP value for a given concentration versus that concentration (from 1 nM to 100 μM). pD2 values were obtained by fitting the values to a dose–response curve equation according to ORIGIN 8.0 software. With the pD2 value, it was possible to calculate the EC50 by multiplying by −1 and then taking the antilogarithm. The obtained value is the EC50 expressed in molar concentration. In all experiments, on any given day, only a single dose was tested on a single animal, which was washed out at least 2 days between doses. The commercial hypotensive agents, Latanoprost (0.005%), dorzolamide hydrochloride (2%), and timolol maleate (0.5%), were assayed by applying a volume of 40 μL.
Statistical Analysis
All data are presented as the mean ± SEM. Significant differences were determined by two-tailed Student's t tests. The plotting and fitting of dose–response curves was carried out with Microcal Origin v.7.0 software (Microcal Software, USA).
Acknowledgments
Part of this work was supported by grants from the Canadian Institutes of Health Research (CIHR) to J.S. In addition, J.S. was a recipient of a New Investigator award from the CIHR and of a junior 2 scholarship from FRSQ.
Footnotes
U.S. Provisional application no. 61/282, 133 was filed on 22 December, 2009.
Abbreviations: IOP, intraocular pressure; P2R, P2 receptor; E-NTPDase, ecto-nucleoside triphosphate diphosphohydrolase; E-NPPs, ecto-nucleotide pyrophosphatases; MPLC, medium pressure liquid chromatography; TEAA, triethylammonium acetate; HRMS-MALDI, high resolution mass spectrometry matrix-assisted laser desorption/ionization; MS-ESI, mass spectrometry-electron spray ionization; CDI, carbonyl diimidazole; DCC, dicyclohexylcarbodiimide; TMP, trimethylphosphate; AP, alkaline phosphatase.
References
- 1.Williams M, Jarvis MF. Purinergic and pyrimidinergic receptors as potential drug targets. Biochem Pharmacol. 2000;59:1173–1185. doi: 10.1016/s0006-2952(99)00341-x. [DOI] [PubMed] [Google Scholar]
- 2.Guile SD, Ince F, Ingall AH, Kindon ND, Meghani P, Mortimore MP. The medicinal chemistry of the P2 receptor family. Prog Med Chem. 2001;38:115–187. doi: 10.1016/s0079-6468(08)70093-6. [DOI] [PubMed] [Google Scholar]
- 3.Fischer B. Therapeutic applications of ATP-(P2)-receptors agonists and antagonists. Expert Opin Ther Pat. 1999;9:385–399. [Google Scholar]
- 4.Hillmann P, Ko GY, Spinrath A, Raulf A, von Kugelgen I, Wolff SC, Nicholas RA, Kostenis E, Holtje HD, Muller CE. Key Determinants of nucleotide-activated G protein-coupled P2Y2 receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem. 2009;52:2762–2775. doi: 10.1021/jm801442p. [DOI] [PubMed] [Google Scholar]
- 5.Burnstock G, Verkhratsky A. Evolutionary origins of the purinergic signalling system. Acta Physiol. 2009;195:415–447. doi: 10.1111/j.1748-1716.2009.01957.x. [DOI] [PubMed] [Google Scholar]
- 6.Crooke A, Guzman-Aranguez A, Peral A, Abdurrahman MKA, Pintor J. Nucleotides in ocular secretions: their role in ocular physiology. Pharmacol Ther. 2008;119:55–73. doi: 10.1016/j.pharmthera.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Guzman-Aranguez A, Crooke A, Peral A, Hoyle CHV, Pintor J. Dinucleoside polyphosphates in the eye: from physiology to therapeutics. Prog Retinal Eye Res. 2007;26:674–687. doi: 10.1016/j.preteyeres.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Pintor J. Adenine nucleotides and dinucleotides as new substances for the treatment of ocular hypertension and glaucoma. Curr Opin Invest Drugs. 2005;6:76–80. [PubMed] [Google Scholar]
- 9.Peral A, Gallar J, Pintor J. Adenine nucleotide effect on intraocular pressure: involvement of the parasympathetic nervous system. Exp Eye Res. 2009;89:63–70. doi: 10.1016/j.exer.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Pintor J, Pelaez T, Peral A. Adenosine tetraphosphate, Ap4, a physiological regulator of intraocular pressure in normotensive rabbit eyes. J Pharmacol Exp Ther. 2004;308:468–473. doi: 10.1124/jpet.103.058669. [DOI] [PubMed] [Google Scholar]
- 11.Pintor J, Peral A, Pelaez T, Martin S, Hoyle CHV. Presence of diadenosine polyphosphates in the aqueous humor: their effect on intraocular pressure. J Pharmacol Exp Ther. 2003;304:342–348. doi: 10.1124/jpet.102.041368. [DOI] [PubMed] [Google Scholar]
- 12.Soto D, Pintor J, Peral A, Gual A, Gasull X. Effects of dinucleoside polyphosphates on trabecular meshwork cells and aqueous humor outflow facility. J Pharmacol Exp Ther. 2005;314:1042–1051. doi: 10.1124/jpet.105.085274. [DOI] [PubMed] [Google Scholar]
- 13.Crosson CE, Yates PW, Bhat AN, Mukhin YV, Husain S. Evidence for multiple P2Y receptors in trabecular meshwork cells. J Pharmacol Exp Ther. 2004;309:484–489. doi: 10.1124/jpet.103.060319. [DOI] [PubMed] [Google Scholar]
- 14.El-Tayeb A, Qi A, Mueller CE. Synthesis and structure–activity relationships of uracil nucleotide derivatives and analogues as agonists at human P2Y2, P2Y4, and P2Y6 receptors. J Med Chem. 2006;49:7076–7087. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 15.Nahum V, Zuendorf G, Levesque SA, Beaudoin AR, Reiser G, Fischer B. Adenosine 5′-O-(1-Boranotriphosphate) derivatives as novel P2Y1 receptor agonists. J Med Chem. 2002;45:5384–5396. doi: 10.1021/jm020251d. [DOI] [PubMed] [Google Scholar]
- 16.Grobben B, Claes P, Roymans D, Esmans EL, Van Onckelen H, Slegers H. Ecto-nucleotide pyrophosphatase modulates the purinoceptor-mediated signal transduction and is inhibited by purinoceptor antagonists. Br J Pharmacol. 2000;130:139–145. doi: 10.1038/sj.bjp.0703289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmermann H. Ectonucleotidases: some recent developments and a note on nomenclature. Drug Dev Res. 2001;52:44–56. [Google Scholar]
- 18.Blackburn GM, Taylor GE, Thatcher GRJ, Prescott M, McLennan AG. Synthesis and resistance to enzymic hydrolysis of stereochemically-defined phosphonate and thiophosphate analogs of P1,P4-bis(5′-adenosyl) tetraphosphate. Nucleic Acids Res. 1987;15:6991–7004. doi: 10.1093/nar/15.17.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cusack NJ, Hourani SMO, Loizou GD, Welford LA. Pharmacological effects of isopolar phosphonate analogs of ATP on P2-purinoceptors in guinea pig tenia coli and urinary bladder. Br J Pharmacol. 1987;90:791–795. doi: 10.1111/j.1476-5381.1987.tb11233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He K, Hasan A, Shaw BR. The synthesis of ribonucleoside 5′-O-(α-P-boranotriphosphates), separation of diastereomers and incorporation by RNA polymerases. Nucleic Acids Symp Ser. 1997;36:159. [Google Scholar]
- 21.Kowalska J, Lewdorowicz M, Darzynkiewicz E, Jemielity J. A simple and rapid synthesis of nucleotide analogues containing a phosphorothioate moiety at the terminal position of the phosphate chain. Tetrahedron Lett. 2007;48:5475–5479. [Google Scholar]
- 22.Lin J, Porter KW, Shaw BR. Synthesis and properties of novel triphosphate analogues: ribonucleoside and deoxyribonucleoside (α-P-borano, α-P-thio)triphosphates. Nucleosides, Nucleotides Nucleic Acids. 2001;20:1019–1023. doi: 10.1081/NCN-100002482. [DOI] [PubMed] [Google Scholar]
- 23.Misiura K, Szymanowicz D, Stec WJ. Synthesis of nucleoside α-thiotriphosphates via an oxathiaphospholane approach. Org Lett. 2005;7:2217–2220. doi: 10.1021/ol050617r. [DOI] [PubMed] [Google Scholar]
- 24.Romaniuk PJ, Eckstein F. Structure of the metal-nucleotide complex in the acetate kinase reaction. A study with γ-32P-phosphorothioate analogs of ATP. J Biol Chem. 1981;256:7322–7328. [PubMed] [Google Scholar]
- 25.Stingelin J, Bolen DW, Kaiser ET. Synthesis, purification, and characterization of the A and B diastereomers of gamma-32P-labeled adenosine 5′-O-(2-thiotriphosphate. J Biol Chem. 1980;255:2022–2025. [PubMed] [Google Scholar]
- 26.Tulapurkar ME, Laubinger W, Nahum V, Fischer B, Reiser G. Subtype specific internalization of P2Y1 and P2Y2 receptors induced by novel adenosine 5′-O-(1-boranotriphosphate) derivatives. Br J Pharmacol. 2004;142:869–878. doi: 10.1038/sj.bjp.0705859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eliahu SE, Camden J, Lecka J, Weisman GA, Sevigny J, Gelinas S, Fischer B. Identification of hydrolytically stable and selective P2Y1 receptor agonists. Eur J Med Chem. 2009;44:1525–1536. doi: 10.1016/j.ejmech.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joseph SM, Pifer MA, Przybylski RJ, Dubyak GR. Methylene ATP analogs as modulators of extracellular ATP metabolism and accumulation. Br J Pharmacol. 2004;142:1002–1014. doi: 10.1038/sj.bjp.0705865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Z, Wang X, Li M, Sohma Y, Zou X, Hwang TC. High affinity ATP/ADP analogues as new tools for studying CFTR gating. J Physiol. 2005;569:447–457. doi: 10.1113/jphysiol.2005.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyle NA, Rajwanshi VK, Prhavc M, Wang G, Fagan P, Chen F, Ewing GJ, Brooks JL, Hurd T, Leeds JM, Bruice TW, Cook PD. Synthesis of 2′,3′-dideoxynucleoside 5′-α-P-borano-β,γ-(difluoromethylene)triphosphates and their inhibition of HIV-1 reverse transcriptase. J Med Chem. 2005;48:2695–2700. doi: 10.1021/jm040101y. [DOI] [PubMed] [Google Scholar]
- 31.Barral K, Priet S, Sire J, Neyts J, Balzarini J, Canard B, Alvarez K. Synthesis, in vitro antiviral evaluation, and stability studies of novel α-borano-nucleotide analogues of 9-[2-(phosphonomethoxy)ethyl]adenine and (R)-9-[2-(phosphonomethoxy)-propyl]adenine. J Med Chem. 2006;49:7799–7806. doi: 10.1021/jm060030y. [DOI] [PubMed] [Google Scholar]
- 32.Bystrom CE, Pettigrew DW, Remington SJ, Branchaud BP. ATP analogs with non-transferable groups in the γ position as inhibitors of glycerol kinase. Bioorg Med Chem Lett. 1997;7:2613–2616. [Google Scholar]
- 33.Myers TC, Nakamura K, Flesher JW. Phosphonic acid analogs of nucleoside phosphates. I. The synthesis of 5′-adenylyl methylenediphosphonate, a phosphonic acid analog of adenosine triphosphate. J Am Chem Soc. 1963;85:3292–3295. [Google Scholar]
- 34.Major DT, Fischer B. Molecular recognition in purinergic receptors. 1. A comprehensive computational study of the h-P2Y1-receptor. J Med Chem. 2004;47:4391–4404. doi: 10.1021/jm049772m. [DOI] [PubMed] [Google Scholar]
- 35.Major DT, Nahum V, Wang Y, Reiser G, Fischer B. Molecular recognition in purinergic receptors. 2. Diastereoselectivity of the h-P2Y1-receptor. J Med Chem. 2004;47:4405–4416. doi: 10.1021/jm049771u. [DOI] [PubMed] [Google Scholar]
- 36.Blackburn GM, Kent DE, Kolkmann F. Three new β,γ-methylene analogs of adenosine triphosphate. J Chem Soc, Chem Commun. 1981:1188–1190. [Google Scholar]
- 37.Wang G, Boyle N, Chen F, Rajappan V, Fagan P, Brooks JL, Hurd T, Leeds JM, Rajwanshi VK, Jin Y, Prhavc M, Bruice TW, Cook PD. Synthesis of AZT 5′-triphosphate mimics and their inhibitory effects on HIV-1 reverse transcriptase. J Med Chem. 2004;47:6902–6913. doi: 10.1021/jm040116w. [DOI] [PubMed] [Google Scholar]
- 38.Spelta V, Mekhalfia A, Rejman D, Thompson M, Blackburn GM, North RA. ATP analogues with modified phosphate chains and their selectivity for rat P2X2 and P2X2/3 receptors. Br J Pharmacol. 2003;140:1027–1034. doi: 10.1038/sj.bjp.0705531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 40.El-Tayeb A, Griessmeier KJ, Mueller CE. Synthesis and preliminary evaluation of [3H]PSB-0413, a selective antagonist radio-ligand for platelet P2Y12 receptors. Bioorg Med Chem Lett. 2005;15:5450–5452. doi: 10.1016/j.bmcl.2005.08.104. [DOI] [PubMed] [Google Scholar]
- 41.Padyukova NS, Dixon HBF, Efimtseva EV, Ermolinsky BS, Mikhailov SN, Karpeisky MY. Synthesis and properties of novel NTP derivatives. Nucleoides, Nucleotides Nucleic Acids. 1999;18:1013–1014. [Google Scholar]
- 42.Mohamady S, Jakeman DL. An improved method for the synthesis of nucleoside triphosphate analogs. J Org Chem. 2005;70:10588–10591. doi: 10.1021/jo0518598. [DOI] [PubMed] [Google Scholar]
- 43.Cusack NJ, Hourani SMO, Loizou GD, Welford LA. Pharmacological effects of isopolar phosphonate analogs of ATP on P2-purinoceptors in guinea pig tenia coli and urinary bladder. Br J Pharmacol. 1987;90:791–795. doi: 10.1111/j.1476-5381.1987.tb11233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. Adenine nucleotide analogues locked in a northern methanocarba conformation: enhanced stability and potency as P2Y1 receptor agonists. J Med Chem. 2002;45:2090–2100. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davisson VJ, Davis DR, Dixit VM, Poulter CD. Synthesis of nucleotide 5′-diphosphates from 5′-O-tosyl nucleosides. J Org Chem. 1987;52:1794–1801. [Google Scholar]
- 46.Hull WE, Halford SE, Gutfreund H, Sykes BD. Phosphorus-31 nuclear magnetic resonance study of alkaline phosphatase: the role of inorganic phosphate in limiting the enzyme turnover rate at alkaline pH. Biochemistry. 1976;15:1547–1561. doi: 10.1021/bi00652a028. [DOI] [PubMed] [Google Scholar]
- 47.Gendaszewska-Darmach E, Maszewska M, Zaklos M, Koziolkiewicz M. Degradation of extracellular nucleotides and their analogs in HeLa and HUVEC cell cultures. Acta Biochim Pol. 2003;50:973–984. [PubMed] [Google Scholar]
- 48.Bonan CD, Schetinger MRC, Battastini AMO, Sarkis JJF. Ectonucleotidases and synaptic plasticity: implications in physiological and pathological conditions. Drug Dev Res. 2001;52:57–65. [Google Scholar]
- 49.Schetinger MRC, Morsch VM, Bonan CD, Wyse ATS. NTPDase and 5′-nucleotidase activities in physiological and disease conditions: new perspectives for human health. BioFactors. 2007;31:77–98. doi: 10.1002/biof.5520310205. [DOI] [PubMed] [Google Scholar]
- 50.Terkeltaub R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signalling. 2006;2:371–377. doi: 10.1007/s11302-005-5304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arzumanov AA, Semizarov DG, Victorova LS, Dyatkina NB, Krayevsky AA. γ-Phosphate-substituted 2′-deoxynucleoside 5′-triphosphates as substrates for DNA polymerases. J Biol Chem. 1996;271:24389–24394. doi: 10.1074/jbc.271.40.24389. [DOI] [PubMed] [Google Scholar]
- 52.Blackburn GM, Kent DE, Kolkmann F. The synthesis and metal binding characteristics of novel, isopolar phosphonate analogs of nucleotides. J Chem Soc, Perkin Trans 1. 1984:1119–1125. [Google Scholar]
- 53.Parr CE, Sullivan DM, Paradiso AM, Lazarowski ER, Burch LH, Olsen JC, Erb L, Weisman GA, Boucher RC, Turner JT. Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proc Natl Acad Sci USA. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pintor J, Sanchez-Nogueiro J, Irazu M, Mediero A, Pelaez T, Peral A. Immunolocalisation of P2Y receptors in the rat eye. Purinergic Signalling. 2004;1:83–90. doi: 10.1007/s11302-004-5072-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Markovskaya A, Crooke A, Guzman-Aranguez AI, Peral A, Ziganshin AU, Pintor J. Hypotensive effect of UDP on intraocular pressure in rabbits. Eur J Pharmacol. 2008;579:93–97. doi: 10.1016/j.ejphar.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 56.Pintor J, Peral A. Therapeutic potential of nucleotides in the eye. Drug Dev Res. 2001;52:190–195. [Google Scholar]
- 57.Nahum V, Fischer B. Boranophosphate salts as an excellent mimic of phosphate salts: Preparation, characterization, and properties. Eur J Inorg Chem. 2004:4124–4131. [Google Scholar]
- 58.Li H, Hardin C, Shaw BR. Hydrolysis of Thymidine Boranomonophosphate and Stepwise Deuterium Substitution of the Borane Hydrogens.31P and11B NMR Studies. J Am Chem Soc. 1996;118:6606–6614. [Google Scholar]
- 59.Bystrom CE, Pettigrew DW, Remington SJ, Branchaud BP. ATP analogs with non-transferable groups in the γ position as inhibitors of glycerol kinase. Bioorg Med Chem Lett. 1997;7:2613–2616. [Google Scholar]
- 60.Higginbotham EJ, Schuman JS, Goldberg I, Gross RL, VanDenburgh AM, Chen K, Whitcup SM, Abelson MB, Baerveldt G, Best S, Branch JD, Brandt JD, Brennan J, Brooks AMV, Butrus S, Cacioppo R, D'Mellow G, DuBiner HB, Duzman E, Forester RJ, Frantz J, Fried WI, Gieser DK, Lewis RA, Logan A, McGovern R, Mundorf T, Nardin GF, Nussdorf J, Rait J, Shields RL, Walters TR, Whitsett JC, Wilensky JT, Williams R, Beck A, Cantor L, Cioffi G, Cohen JS, Cooke D, Crichton A, Dudley DF, Evans R, Greenberg S, Gupta N, Gurevich L, Kasner O, Kellum D, Koby M, Kwedar J, McGarey D, Mikelberg F, Noecker R, Remis LL, Ritch R, Rotberg M, Schenker HI, Sharpe E, Sherwood MB, Sokol J, Solish A, Whiteside-Michel J, Wirostko B. One-year, randomized study comparing bimatoprost and timolol in glaucoma and ocular hypertension. Arch Ophthalmol. 2002;120:1286–1293. doi: 10.1001/archopht.120.10.1286. [DOI] [PubMed] [Google Scholar]
- 61.Kaiserman I, Fendyur A, Vinker S. Topical beta blockers in asthmatic patients–is it safe? Curr Eye Res. 2009;34:517–522. doi: 10.1080/02713680902989337. [DOI] [PubMed] [Google Scholar]
- 62.Quigley HA. Open-angle glaucoma. N Engl J Med. 1993;328:1097–1106. doi: 10.1056/NEJM199304153281507. [DOI] [PubMed] [Google Scholar]
- 63.Hoyng PFJ, Van Beek LM. Pharmacological therapy for glaucoma: a review. Drugs. 2000;59:411–434. doi: 10.2165/00003495-200059030-00003. [DOI] [PubMed] [Google Scholar]
- 64.Alward WLM. Medical management of glaucoma. N Engl J Med. 1998;339:1298–1307. doi: 10.1056/NEJM199810293391808. [DOI] [PubMed] [Google Scholar]
- 65.Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544–547. doi: 10.1016/s0002-9394(14)70870-0. [DOI] [PubMed] [Google Scholar]
- 66.Griffin BE, Jarman M, Reese CB, Sulston JE. The synthesis of oligoribonucleotides. II. Methoxymethylidene derivatives of ribonucleosides and 5′-ribonucleotides. Tetrahedron. 1967;23:2301–2313. doi: 10.1016/0040-4020(67)80067-x. [DOI] [PubMed] [Google Scholar]
- 67.Bachelet M, Guibe M. Preparation of triethylammonium bicarbonate. Bull Soc Chim Fr. 1951:554. [Google Scholar]
- 68.Brandenberger H, Hanson R. Spectrophotometric determination of acid and alkaline phosphatases. Helv Chim Acta. 1953;36:900–906. [Google Scholar]
- 69.Kukulski F, Levesque SA, Lavoie EG, Lecka J, Bigonnesse F, Knowles AF, Robson SC, Kirley TL, Sevigny J. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3, and 8. Purinergic Signalling. 2005;1:193–204. doi: 10.1007/s11302-005-6217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 71.Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 72.Levesque SA, Lavoie EG, Lecka J, Bigonnesse F, Sevigny J. Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol. 2007;152:141–150. doi: 10.1038/sj.bjp.0707361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grynkiewicz G, Poenie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 74.Garrad RC, Otero MA, Erb L, Theiss PM, Clarke LL, Gonzalez FA, Turner JT, Weisman GA. Structural basis of agonist-induced desensitization and sequestration of the P2Y2 nucleotide receptor. Consequences of truncation of the C terminus. J Biol Chem. 1998;273:29437–29444. doi: 10.1074/jbc.273.45.29437. [DOI] [PubMed] [Google Scholar]
- 75.Morales M, Gomez-Cabrero A, Peral A, Gasull X, Pintor J. Hypotensive effect of profilin on rabbit intraocular pressure. Eur J Pharmacol. 2007;567:145–148. doi: 10.1016/j.ejphar.2007.04.020. [DOI] [PubMed] [Google Scholar]