

Abstract
We herein report the application of the phosphorodiamidate phosphate prodrug approach to a series of thirteen nucleoside analogs with antiviral or anticancer activity. Twenty-five symmetrical phosphorodiamidates were synthesized, bearing esterified l-Alanine (and in one case d-Alanine) in the prodrug moiety, each as single stereoisomer. The presence of an achiral phosphorus represents a potential advantage over the phosphoramidate ProTide approach, where diastereoisomeric mixtures are routinely obtained, and different biological profiles may be expected from the diastereoisomers. Optimization of the synthetic pathway allowed us to identify two general methods depending on the particular nucleoside analogs. All the compounds were biologically evaluated in antiviral and anticancer assays and several showed improvement of activity compared to their parent nucleosides, as in the case of ddA, d4T, abacavir and acyclovir against HIV-1 and/or HIV-2. The biological results were supported by metabolism studies with carboxypeptidase Y monitored by 31P NMR to investigate their bioactivation. This work further validates the phosphorodiamidate approach as a monophosphate prodrug motif with broad application in the antiviral and anticancer fields.
Keywords: Nucleoside analogs, Antiviral, Anticancer, Phosphorodiamidates, Prodrugs
Graphical abstract
Highlights
-
•
Application of diamidate approach to nucleoside analogs as potential antiviral and anticancer agents.
-
•
A markedly improved antiviral activity was observed in some cases, whilst the parent nucleosides were inactive.
-
•
Inhibitory effect on the proliferation of tumor cell lines was also demonstrated.
1. Introduction
Nucleoside analogs (NAs) play a pivotal role in antiviral and anticancer therapy [1], [2]. They are structurally related to the natural nucleosides bearing modifications at the base and/or at the sugar moieties, which, in most of the cases, confer selectivity versus the desired targets [2]. NAs are prodrugs and they need to be converted into their active species, which usually consists of their 5′-triphosphate form [3]. Their bioactivation pathway often involves three consecutive phosphorylation steps starting from the parent nucleoside, which is converted to its mono-, di- and finally triphosphate form. Some NAs, such as abacavir or famciclovir for instance, require also additional bioactivation steps in order to display their biological activity [4], [5]. The first step of phosphorylation is usually considered to be the rate-limiting step in the bioactivation of NAs, with few exceptions, such as zidovudine for which the second phosphorylation may be rate limiting [6]. After long-term treatment with NAs the activity of the nucleoside kinases involved in the first step of bioactivation may be decreased, therefore leading to drug resistance onset [7]. Several monophosphate prodrug strategies are currently under investigations to overcome these issues [8], including the phosphoramidate ProTide approach developed in our group [9], [10], [11]. More recently the phosphorodiamidate technology applied to 6-O-alkyl-2′-C-methylguanosine was reported by us as a new promising approach for the delivery of monophosphates inside the cell. Several stability studies on the 6-O-alkyl-2′-C-methylguanosine diamidates have shown a good stability profile under different conditions such as acid (pH = 2) and mild basic buffer (pH = 8.5–11), and in human serum [12]. This novel prodrug approach has been validated both in vitro and in vivo and some compounds are already under consideration for clinical studies [12]. Similarly, several acyclic nucleoside phosphonate diamidate prodrugs showed a better biological profile compared to the parent compounds [13]. In this approach two amino acid esters are introduced on the monophosphate moiety in order to mask the negative charges. As also in the case of the phosphoramidate diester approach of Wagner and colleagues [14], the phosphorus in the symmetrical diamidate prodrug is achiral, thus avoiding the presence of diastereoisomeric mixtures as in the case of the phosphoramidate ProTide derivatives. In fact, it has been reported how two diastereoisomers may interact differently with the enzymes involved in the bioactivation pathway, thus leading to different biological profiles [15]. Moreover, the diamidate motif bears non-toxic and natural promoieties and obviate the need for a phenyl or naphthyl moiety. The putative bioactivation pathway of diamidate prodrugs, depicted in Scheme 1 , is similar to the one reported for ProTides. The first step (a) may be mediated by an esterase or a carboxypeptidase-type enzyme, which is responsible for the cleavage of one of the two esters. This mechanism has been already described and supported by enzymatic experiments using 31P NMR [12]. The second step (b) involves an intramolecular attack of the carboxylate anion to the phosphorus with elimination of the second amino acid and formation of a five-membered ring (mixed anhydride intermediate). Spontaneous hydrolysis (c) of the cycle then leads to the formation of an intermediate bearing two negative charges. Finally, for the last step (d), a phosphoramidase-type enzyme cleaves the P–N bond to form the NA monophosphate.
Scheme 1.
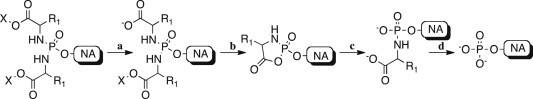
Putative bioactivation pathway of diamidate prodrugs: a) enzyme-mediated ester hydrolysis; b) spontaneous intracellular displacement; c) spontaneous hydrolysis; d) enzyme-mediated P–N bond cleavage.
We were keen to probe the scope of this new diamidate prodrug motif across several therapeutic arenas and for a broad range of NAs. In this context, we herein applied the diamidate approach to NAs with either antiviral or anticancer activity, and the novel prodrug compounds were evaluated for their biological activities. The NAs considered for this study are: 6-O-ethyl-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine (1), stavudine (d4T, 2), 2′,3′-dideoxyadenosine (ddA, 3), zidovudine (AZT, 4), lamivudine (3TC, 5), N-acetyl-lamivudine (N-acetyl-3TC, 6), 4′-azidouridine (4′-AzU, 7), 4′-azidocytidine (4′-AzC, 8), ribavirin (RBV, 9), acyclovir (ACV, 10), abacavir (ABC, 11), the bicyclic nucleoside analog 12 (BCNA, also known as Cf1743) and acadesine (AICA, 13) (Fig. 1 ).
Fig. 1.
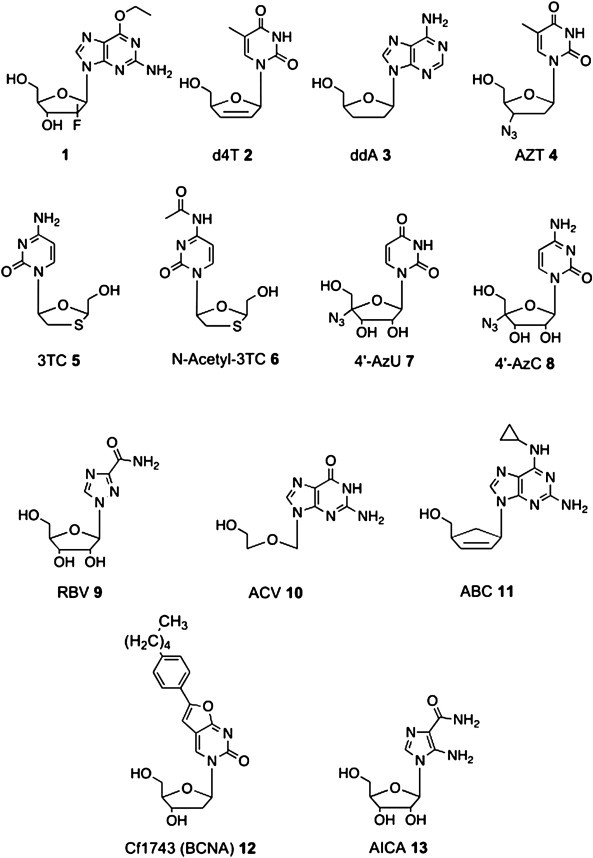
NAs considered for this study.
Different synthetic conditions were necessary depending on solubility and reactivity issues of the parent nucleosides, and a total of twenty-five diamidates were synthesized. Based on the previous work published on ProTides and diamidates, we selected l-alanine (l-Ala) as the amino acid of choice with benzyl and 2,2-dimethylpropyl as preferred ester moieties. For some derivatives, methyl and cyclohexyl esters were considered and, in one case, d-alanine (d-Ala) was used as the amino acid moiety.
2. Results and discussion
2.1. Chemistry
At first, we applied our previously reported successful methodology for the synthesis of anti-HCV 6-O-alkyl-2′-C-methylguanosine 5′-phosphorodiamidates [12] to 6-O-ethyl-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine 1, d4T 2, ddA 3, AZT 4, and 3TC 5. This procedure, called method A in this paper, is represented in Scheme 2 .
Scheme 2.
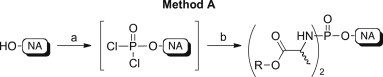
Synthetic method A to phosphorodiamidates 14–21. Reagents and conditions: (a) anhydrous Et3N (1.0–1.2 mol/eq), anhydrous THF, room temperature, 30 min; then POCl3 (1.0–1.2 mol/eq), −78 °C, 30 min; (b) amino acid ester p-TSA salt (3.0–5.0 mol/eq) in anhydrous CH2Cl2, room temperature; then anhydrous Et3N (5.0–10.0 mol/eq), −78 °C, then room temperature, 16–20 h.
In this strategy, the unprotected nucleoside dissolved in THF was treated with phosphorus oxychloride (1 equivalent) in the presence of triethylamine (1 equivalent) to generate a phosphorodichloridate intermediate (31P NMR signal at ∼7–8 ppm), which was not isolated. Then, an excess of the appropriate amino acid ester as p-toluene sulfonate (p-TSA) salt (usually 5 equivalents) and triethylamine (5–10 equivalents) were added, leading after stirring at room temperature for 18–20 h to the desired phosphorodiamidates 14–21 (31P NMR signals at δ P ∼ 11–14 ppm).
Interestingly, in the case of 3TC (5) both 5′-OH and 4-NH2 functional groups reacted yielding the tetradiamidate derivative 22, and the N-4-diamidate derivative 23 (Scheme 3).
Scheme 3.
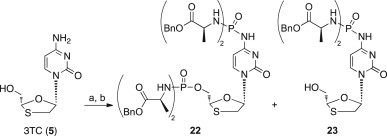
Synthesis of 3TC phosphorodiamidates 22 and 23 with method A. Reagents and conditions: (a) anhydrous Et3N (1.1 eq), POCl3 (1.1 eq), anhydrous THF, −78 °C to rt, 5 h; (b) l-alanine benzyl ester p-TSA salt (5 eq), anhydrous Et3N (10 eq), anhydrous CH2Cl2, −78 °C to rt, 20 h.
However, when applied to the other NAs reported in this work, this method was not successful, probably due to a lack of solubility in the organic solvent used. Based on these findings, a modification of a synthetic approach reported by Yoshikawa et al. for the synthesis of monophosphate species was used [16]. This same method was previously reported for the synthesis of some phosphorodiamidates [17].
This second strategy, named method B, was used for the synthesis of diamidates of 3TC (5), N-acetyl 3TC (6), 4′-AzU (7), 4′-AzC (8), RBV (9), ACV (10), ABC (11), Cf1743 (12), and AICA (13) (Scheme 4 ).
Scheme 4.
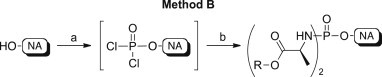
Synthetic method B to phosphorodiamidates 24–40. Reagents and conditions: (a) POCl3 (1.0 mol/eq), trimethylphosphate or triethylphosphate, −5 °C, 4–5 h, or 5 °C, 16 h; (b) amino acid ester p-TSA or HCl salt (5.0 mol/eq), anhydrous CH2Cl2, anhydrous Et3N (10.0 mol/eq) or anhydrous DIPEA (5.0–10.0 mol/eq), −78 °C, 30 min, then room temperature, 16–45 h.
The reaction was carried out using either trimethylphosphate or triethylphosphate and POCl3 to afford the corresponding intermediate dichlorophosphates following its formation by 31P NMR (δ P = ∼7). The addition of an excess of the appropriate amino acid ester salts in the presence of DIPEA or TEA yielded the desired phosphorodiamidates 24–40.
2.2. Biological results
The arylphosphoramidate ProTide technology as a monophosphate prodrug approach has proven to be a powerful tool in terms of enhancing the biological activity, by-passing certain mechanisms of resistance, as well as extending the biological profile of several NAs. Moreover, the increased lipophilicity of these prodrugs may also lead to a better delivery inside the cells by passive diffusion, thus favoring a greater uptake of the drug. For instance, it has been reported how the application of the ProTide approach greatly enhanced the antiviral activity of the anti-hepatitis C virus (HCV) agent 2-amino-6-methoxy-9-(2-C-methyl-β-d-ribofuranosyl) purine, [11] overcame the mechanisms of resistance for 5-fluoro-2′-deoxyuridine [18] and extended the biological profile of BVdU and ACV from antiherpetic to anticancer and anti-human immunodeficiency virus (HIV) agents, respectively [19], [20]. In a similar context, the phosphorodiamidates reported here were tested versus different cancer cell lines as well as versus a range of viruses with the aim to investigate the full potential of this phosphate prodrug moiety. To probe the delivery of the monophosphate inside the cells, thus by-passing the first step of phosphorylation, thymidine kinase-deficient (TK−) mutant cancer cells and herpes virus strains were also used.
2.2.1. Anticancer activity
Table 1 reports the anticancer activity for d4T (2), ddA (3), 3TC (5), N-acetyl-3TC (6), ABC (11), AICA (13) and their respective phosphorodiamidates versus mouse lymphocytic leukemia cells (L1210), human T-lymphocyte cells (CEM), human cervical carcinoma cells (HeLa), for which TK− mutant strains were also included (L1210-TK−, CEM-TK−, and HeLa-TK−), colorectal adenocarcinoma cells (Caco-2) and human colon carcinoma cells (Colo-320). As expected, d4T itself did not show any antiproliferative activity versus all the human cancer cell lines considered in this study. In the case of murine L1210, d4T showed cytostatic activity at ∼9 μM, while it was found ineffective (≥250 μM) versus L1210/TK−, which confirms its expected dependence on TK for phosphorylation for eventual biological activity. To the contrary, its phosphorodiamidates 16–18 showed antiproliferative activity versus all the cancer cell lines in a range between 8.9 and 47 μM for L1210, 73–96 μM for CEM, 96–116 μM for HeLa, 30–133 μM for Colo-320, while being poorly effective versus Caco-2 cell (135–241 μM). More importantly, all the compounds showed retention of activity against all the TK− strains in a range between 7.4 and 25 μM versus L1210-TK−, 37–54 μM vs CEM-TK−, and 29–53 μM vs HeLa/TK− strains. These results are strongly suggestive for the successful delivery of d4T monophosphate inside the cells, thus heading to their independence from TK activation. As an alternative hypothesis, it could have been assumed that the similar antiproliferative activity observed for the d4T prodrugs against the L1210 TK− cells versus wild-type L1210 cells could have been due to a direct effect of the prodrug or prodrug moieties such as the released benzyl part of the molecule. However, this is highly unlikely because in such a case, these prodrugs would have expected to display similar toxicities for the different prodrug molecules (i.e. 16, 22, 24, 25) and this had not been the case.
Table 1.
Inhibitory effects on the proliferation of tumor cell lines in cell culture. IC50 = 50% inhibitory concentration.
| Compd | Nucleoside | AA | R | IC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| L1210 | L1210/TK− | CEM | CEM/TK− | HeLa | HeLa/TK− | Caco-2 | Colo-320 | ||||
| 2 | d4T | – | – | 8.9 ± 3.2c | ≥250 | ≥250 | >250 | >250 | >250 | >250 | 159 ± 36 |
| 16 | d4T | l-Ala | Bn | 8.9 ± 0.4c | 7.4 ± 4.6 | 92 ± 0 | 46 ± 21 | 116 ± 54 | 29 ± 7 | 241 ± 13 | 133±18 |
| 17 | d4T | l-Ala | cHex | 29 ± 5 | 25 ± 3 | 73 ± 9 | 54 ± 17 | 96 ± 0 | 36 ± 9 | 135 ± 7 | 56 ± 24 |
| 18 | d4T | l-Ala | CH2tBu | 47 ± 4 | 23 ± 6 | 96 ± 3 | 37 ± 2 | 110 ± 35 | 53 ± 23 | 174 ± 72 | 30 ± 16 |
| 3 | ddA | – | – | >250 | – | >250 | – | >250 | – | – | – |
| 19 | ddA | l-Ala | CH2tBu | 76 ± 3 | – | 36 ± 4 | – | 35 ± 5 | – | – | – |
| 5 | 3TC | – | – | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 |
| 6 | N-Acetyl-3TC | – | – | >250 | >250 | >250 | >250 | 242 ± 11 | >250 | >250 | ≥250 |
| 22a | 3TC | l-Ala | Bn | >250 | >250 | >250 | 155 ± 38 | ≥250 | >250 | >250 | >250 |
| 23b | 3TC | l-Ala | Bn | ≥250 | 150 ± 18 | 214 ± 50 | 139 ± 67 | ≥250 | 256 ± 21 | >250 | >250 |
| 24 | N-Acetyl-3TC | l-Ala | Bn | 121 ± 6 | – | 120 ± 2 | – | 155 ± 61 | – | ≥250 | >250 |
| 25 | 3TC | l-Ala | Bn | 86 ± 23 | 57 ± 6 | 116 ± 9 | 106 ± 18 | 93 ± 16 | 38 ± 7 | 87 ± 44 | 85 ± 50 |
| 11 | ABC | – | – | >250 | – | >250 | – | 170 ± 1 | – | – | – |
| 34 | ABC | l-Ala | Bn | 54 ± 2 | – | 19 ± 0 | – | 23 ± 6 | – | – | – |
| 35 | ABC | l-Ala | CH2tBu | 48 ± 5 | – | 27 ± 2 | – | 36 ± 17 | – | – | – |
| 13 | AICA | – | – | ≥250 | – | 210 ± 56 | – | 165 ± 18 | ≥250 | – | – |
| 38 | AICA | l-Ala | Bn | >250 | – | >250 | – | >250 | >250 | – | – |
| 39 | AICA | l-Ala | Me | ≥250 | – | >250 | – | >250 | >250 | – | – |
| 40 | AICA | l-Ala | CH2tBu | >250 | – | >250 | – | >250 | >250 | – | – |
Data represent the mean ± SD of at least 2 to 3 independent experiments.
Phosphorotetraamidate.
N-Phosphorodiamidate.
No dose response.
A similar trend was observed for 3TC (5) and its phosphorodiamidate 25; in fact while the parent nucleoside was found inactive versus all the cancer cell lines (>250 μM) compound 25 showed a certain antiproliferative activity in a range between 38 and 116 μM, against all tested tumor cell lines including the TK− mutant strains. As expected, the phosphorotetramidate 22 and the N-phosphorodiamidate 23 did not show any activity of particular interest.
In the case of ddA (3) and ABC (11) and their prodrugs, while the parent nucleosides did not show any potent antiproliferative activity (>250 μM vs L1210, CEM, and HeLa for ddA, >250 μM vs L1210 and CEM, and 170 μM vs HeLa for ABC), their prodrugs showed a great enhancement of the activity showing IC50 values in between 35 and 76 μM for the ddA derivative 19, and in the range of 19–54 μM for ABC phosphate prodrugs 34 and 35. Neither AICA (13) nor its prodrugs 38, 39 and 40 showed any activity, which may indicate a poor conversion of these prodrugs into the free monophosphate as it will be discussed.
2.2.2. Antiviral activity
One of the advantages of the monophosphate prodrug approach is the by-pass of the first phosphorylation step mediated by human and/or viral kinase enzymes. The monophosphate prodrugs here reported were tested for their antiviral activity versus different viruses.
2.2.2.1. Anti-HIV activity
d4T (2), ddA (3), AZT (4), 3TC (5), ACV (10), and ABC (11), and their respective phosphorodiamidates were tested against HIV-1 and HIV-2 in CEM or MT4 cells (Table 2 ). Compounds 16–18 showed a boost of their activity of ∼2–4 fold vs HIV-1 and ∼2–6 fold vs HIV-2 compared to their parent derivative d4T.
Table 2.
Anti-HIV-1 and HIV-2 activity (EC50) in human T-lymphocyte CEM cells.
| Compd | Nucleoside | AA | R | EC50 (μM) |
CC50 (μM) | |
|---|---|---|---|---|---|---|
| HIV-1 | HIV-2 | |||||
| 2 | d4T | – | – | 0.39 ± 0.29 | 0.58 ± 0.33 | ≥250 |
| 16 | d4T | l-Ala | Bn | 0.14 ± 0.028 | 0.096 ± 0.063 | 92 ± 0 |
| 17 | d4T | l-Ala | cHex | 0.15 ± 0.16 | 0.38 ± 0.42 | 73 ± 9 |
| 18 | d4T | l-Ala | CH2tBu | 0.090 ± 0.084 | 0.14 ± 0.042 | 96 ± 3 |
| 3 | ddA | – | – | 7.2 ± 2.5 | 2.8 ± 1.8 | >250 |
| 19 | ddA | l-Ala | CH2tBu | 0.10 ± 0.069 | 0.049 ± 0.028 | 35 ± 5.0 |
| 4 | AZT | – | – | 0.012 ± 0.006 | 0.067 ± 0.018 | >250 |
| 20 | AZT | l-Ala | Bn | 0.0092 ± 0.00035 | 0.030 ± 0.0092 | 30 ± 4 |
| 21 | AZT | l-Ala | CH2tBu | 0.0083 ± 0.00014 | 0.013 ± 0.0042 | 75 ± 40 |
| 5 | 3TC | – | – | 0.099 ± 0.086 | 0.18 ± 0.13 | >250 |
| 22a | 3TC | l-Ala | Bn | 16 ± 5.3 | 58 ± 44 | >250 |
| 23b | 3TC | l-Ala | Bn | 0.28 ± 0.17 | 0.40 ± 0.33 | 214 ± 60 |
| 25 | 3TC | l-Ala | Bn | >50 | >50 | 116 ± 9 |
| 11 | ABC | – | – | 23 ± 5.0 | 18 ± 8.9 | >250 |
| 34 | ABC | l-Ala | Bn | 0.56 ± 0.23 | 1.1 ± 0.82 | 19 ± 0 |
| 35 | ABC | l-Ala | CH2tBu | 1.5 ± 0.47 | 1.2 ± 0.66 | 27 ± 2 |
| 10 | ACV | – | – | >250c | – | >250c |
| 32 | ACV | l-Ala | Bn | 2.01 ± 1.16c | – | 34.93 ± 1.45c |
| 33 | ACV | l-Ala | CH2tBu | 4.07 ± 1.06c | – | 82.76 ± 4.54c |
Data represent the mean ± SD of at least 2 to 3 independent experiments.
Phosphorotetraamidate.
N-Phosphorodiamidate.
MT-4 cell.
In the case of ddA a markedly greater boost in activity (72× vs HIV-1 and 57× vs HIV-2) was observed for its prodrug 19. AZT derivatives 20 and 21 were equipotent to the parent, while 3TC derivatives were significantly less effective, with the exception of N-phosphorodiamidate 23, which was surprisingly only two fold less active. In the case of ABC, compounds 34 and 35 showed an increased potency (15–41 fold) compared to the parent nucleoside.
In the case of ACV, a great improvement of its biological profile was observed. In fact, while ACV is not able to inhibit HIV-1 (EC50 > 250 μM), both phosphorodiamidates 32 and 33 showed an inhibitory activity in the low micromolar range (2–4 μM), thus leading to a >60 fold boost in activity.
The positive results obtained may be due to a combination of both successful delivery of the monophosphate form of the parent nucleosides inside the cells as well as to an enhanced cellular uptake due to an increased lipophilicity. However, these positive features of the monophosphate prodrugs also led, in some cases, to a slightly increased cytotoxicity compared to the parent compounds. In the case of 3TC phosphoramidate 25, the poor anti-HIV activity may be due to a poor metabolic bioactivation of the phosphate prodrug moieties with the subsequent poor release of the free monophosphate form. In fact, the uptake and metabolic activation of the prodrugs is a multistep process for which it is currently not clear which enzymes are contributing to the eventual activity of the prodrugs. Not only the different type of enzymes involved in the drug conversion pathways have yet to be clarified but also their differential specificities related to the nature of the nucleoside and the prodrug part and their activity levels in the different cell systems are not exactly known. Without any doubt, such subtle differences are playing a role in the eventual biological activity and properties of these compounds.
2.2.2.2. Anti-HCV activity
The phosphorodiamidate approach prodrug applied to 6-O-alkyl-2′-C-methylguanosine showed outstanding results versus HCV both in vitro and in vivo [12]. Driven by these encouraging results, a series of diamidates of compounds of interests versus HCV were prepared, including 6-O-ethyl-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine (1), 4′-AzU (7), 4′-AzC (8), RBV (9), and ACV (10) (Table 3 ). Unfortunately, the majority of the compounds showed a poor biological profile, with the exception of compounds 14 and 15 derived from compound 1. In fact, both compounds showed an inhibitory activity against HCV in the sub-micromolar range showing a great boost of activity compared to the parent guanosine nucleoside (EC90 = 69.2 μM) [25]. Notably, the l-alanine derivative 14 gave a 4 fold improved activity compared to 15, which bears the non-natural amino acid d-alanine. Based on previous study, this finding may be the result of a lower conversion of the d-alanine based phosphate prodrug to the monophosphate, as a consequence of its lower affinity with the enzymes involved in the bioactivation pathway.
Table 3.
HCV replicon assays (type 1b) in Huh 7 cells.
| Compd | Nucleoside | AA | R | EC50 (μM) | CC50 (μM) |
|---|---|---|---|---|---|
| 14 | 1 | l-Ala | CH2tBu | 0.05 | 73 |
| 15 | 1 | d-Ala | CH2tBu | 0.23 | >100 |
| 7 | 4′-AzU | – | – | >100, Ref. [21] | >100, Ref. [21] |
| 26 | 4′-AzU | l-Ala | Bn | >40 | >100 |
| 27 | 4′-AzU | l-Ala | CH2tBu | 22 ± 7 | >100 |
| 8 | 4′-AzC | – | – | 7.13, Ref. [22] | >100, Ref. [22] |
| 28 | 4′-AzC | l-Ala | Bn | >40 | >100 |
| 29 | 4′-AzC | l-Ala | CH2tBu | >40 | 72 |
| 9 | RBV | – | – | 87, Ref. [23] | >100, Ref. [23] |
| 30 | RBV | l-Ala | Bn | >100 | >100 |
| 31 | RBV | l-Ala | CH2tBu | >100 | >100 |
| 10 | ACV | – | – | 30, Ref. [24] | >30, Ref. [24] |
| 32 | ACV | l-Ala | Bn | >100 | – |
| 33 | ACV | l-Ala | CH2tBu | >100 | – |
2.2.2.3. Anti-HSV-1 and 2 activity
AICA (13), ACV (10), Cf1743 (12) and their diamidates were then evaluated against human herpes simplex virus (HSV) type 1 and 2 and feline herpes virus as reported in Table 4 .
Table 4.
Antiviral activity of test compounds against herpes simplex virus type 1 and 2 and feline herpes virus and cytotoxicity (MCC) in HEL and CrFK cell cultures.
| Compd | Nucleoside | AA | R | MCC (μM) | EC50 (μM) HEL cell |
|||
|---|---|---|---|---|---|---|---|---|
| Herpes simplex virus 1(KOS) | Herpes simplex virus 2 (G) | Herpes simplex virus 1TK−(KOS)ACVr | Feline herpes virus | |||||
| 13 | AICA | – | – | >100 | >100 | >100 | >100 | >100 |
| 38 | AICA | l-Ala | Bn | >100 | >100 | >100 | >100 | >100 |
| 39 | AICA | l-Ala | Me | >100 | >100 | >100 | >100 | >100 |
| 40 | AICA | l-Ala | CH2tBu | >100 | >100 | >100 | >100 | >100 |
| 10 | ACV | – | – | >100 | 0.23 ± 0.20 | 0.2 ± 0 | 23 ± 5 | >100 |
| 32 | ACV | l-Ala | Bn | >100 | 18 ± 14 | 16 ± 19 | 25 ± 22 | >100 |
| 33 | ACV | l-Ala | CH2tBu | >100 | 33 ± 18 | 40 ± 8 | 20 ± 0 | >100 |
| 12 | Cf1743 | – | – | ≥20 | >20 | >20 | >20 | >100 |
| 36 | Cf1743 | l-Ala | Bn | >100 | >100 | >100 | >100 | >100 |
AICA (13), Cf1743 (12), and their respective phosphorodiamidate prodrugs 36, 38–40 were devoided of activity against human and feline herpes viruses (EC50 > 100 μM) (Table 4). In the case of ACV, while the parent itself showed a submicromolar activity versus HSV-1 and 2, prodrugs 32 and 33 showed a loss (80–200 fold) of antiviral activity. However, when tested against HSV-1 TK−, while ACV was 100 fold less effective (EC50 = 23 μM), its diamidates retained their activity and were found to be equipotent to the parent compound thus indicating their TK independence. Indeed, since ACV obligatorily needs to be converted (activated) to its monophosphate derivative by HSV-1-encoded TK, it has markedly lowered antiviral activity against a HSV-1 TK− strain whereas its prodrugs have not. It has been ascertained that the prodrugs have no direct activity against purified herpetic DNA polymerase, excluding the possibility that the prodrugs might have activity as such against HSV-1 replication.
2.2.2.4. Other antiviral activity
Some of the phosphorodiamidate prodrugs were also examined for their inhibitory activity against a variety of other viruses, including vaccinia virus, vesicular stomatitis virus, influenza virus A (H1N1 and H3N2) and B, feline corona virus (FIPV), parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, Punta Toro virus, respiratory syncytial virus and Coxsackie virus. None of them showed inhibitory activity nor cytotoxicity at 100 μM.
2.3. Enzymatic studies
As depicted in Scheme 1, the putative bioactivation pathway for the phosphorodiamidate phosphate moiety involves two enzymatic steps, mediated by an esterase or carboxypeptidase type enzyme (first step) and a phosphoramidase type enzyme (last step). We have reported the investigation of the first step of the bioactivation pathway for both ProTide [26] and diamidate [12] by incubating the desired compound with carboypeptidase Y enzyme in d 6-acetone and Trizma buffer (pH = 7.6) and following the progress of the process through 31P NMR. All the prodrugs tested, proved to be chemically robust in d 6-acetone and Trizma buffer (pH = 7.6) environment and in absence of carboxypeptidase Y. Fig. 2, Fig. 3 report the first enzymatic cleavage for an ACV prodrug 32 (active compound) and for an AICA prodrug 40 (inactive compound) respectively. Compound 32 (δ P = 13.66) showed a fast metabolism to the first intermediate lacking one ester moiety. The cleavage of only one ester was supported by the presence of two peaks at the 31P NMR (δ P = 14.30 and 14.42), thus indicating the chirality at the phosphorus. After 30 min from the addition of the enzyme a new peak (δ P = 7.16) appeared, which corresponds to the metabolite lacking one amino acid and one ester, in agreement with our previously published results [11], [26]. Notably, compound 32 was fully converted in 7.5 h into its metabolite indicating that the first step in the bioactivation pathway proceeds well for ACV derivative. To the contrary, a poor metabolism was observed for compound 40. In fact, under the same conditions used for compound 32, only a partial conversion into the desired metabolite (δ P = 6.71) was observed and after 24 h the starting material (δ P = 14.20) was still the predominant species.
Fig. 2.
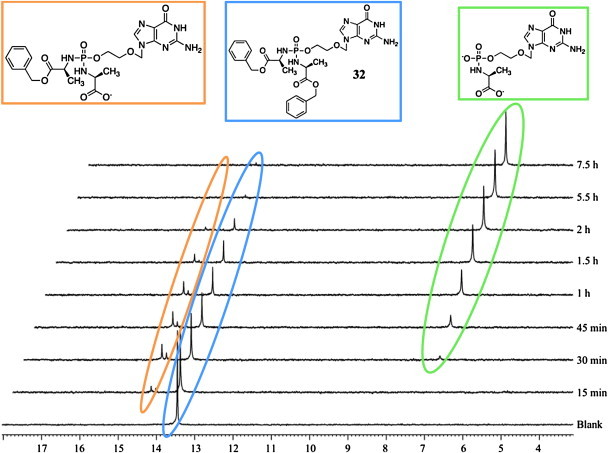
Carboxypeptidase-mediated cleavage of compound 32, monitored by 31P NMR.
Fig. 3.
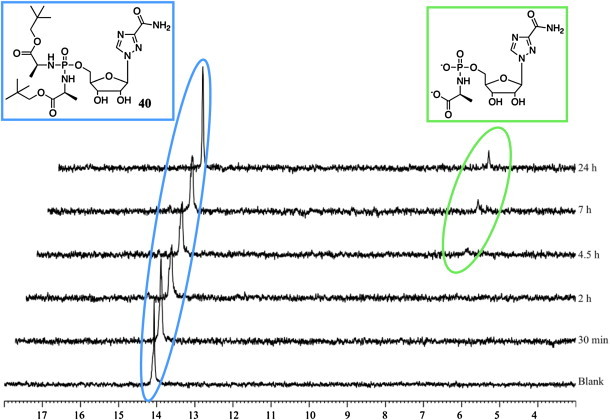
Carboxypeptidase-mediated cleavage of compound 40, monitored by 31P NMR.
From the two examples reported here, we can correlate the biological activity found for 32 and 40 and their bioactivation efficiency, thus supporting the need of an ester cleavage in the bioactivation pathway for this class of compounds. However intracellular activation of these prodrugs still needs to be confirmed.
3. Conclusion
In conclusion, we report the synthesis and biological evaluation of twenty-five phosphorodiamidate prodrugs of known NAs. The key advantage of this nucleoside monophosphate prodrug approach is to overcome the chirality at the phosphorus center present in other monophosphate prodrug approaches, thus allowing the formation of single stereoisomers instead of diastereoisomeric mixtures. A broad anticancer and antiviral evaluation has been performed. Many of the compounds showed a good improvement and/or extension of their biological profile. A great improvement of activity was observed in the case of ddA, ABC and ACV phosphorodiamidates against HIV-1 and 2, while d4T prodrugs showed a good inhibitory activity versus different cancer cell lines. To the contrary, AICA derivatives showed neither anticancer nor antiviral activity. A metabolic assays using carboxypeptidase Y was performed to support the importance of bioactivation of these compounds to exert their activity. In fact, when the compounds were slowly activated in our assays either a low or no activity was observed in the testing, whereas a rapid bioactivation resulted in an improved biological profile. The phosphorodiamidate approach we report shows considerable promise across the biological spectrum where NAs are active and we believe significantly augment the field of nucleotide prodrugs for use in drug discovery.
4. Experimental protocols
4.1. Chemistry
All anhydrous solvents were purchased from Sigma–Aldrich and amino acid esters from Novabiochem. All reagents commercially available were used without further purification. Thin Layer Chromatography (TLC): precoated aluminum backed plates (60 F254, 0.2 mm thickness, Merck) were visualized under both short and long wave UV light (254 and 366 nm). Flash column chromatography was carried out using silica gel supplied by Fisher (60A, 35–70 μm). Analytical High Performance Liquid Chromatography (HPLC) analysis was performed using either a ThermoScientific or a Varian Prostar system. 1H NMR (500 MHz), 13C NMR (125 MHz), 31P NMR (202 MHz) and 19F NMR (470 MHz) spectra were recorded on a Bruker Avance 500 MHz spectrometer at 25 °C. Chemical shifts (δ) are quoted in parts per million (ppm) relative to internal MeOD (δ 3.34 1H NMR, δ 49.86 13C NMR) and CDCl3 (δ 7.26 1H NMR, δ 77.36 13C NMR) or external 85% H3PO4 (δ 0.00 31P NMR). Coupling constants (J) are given in Hertz. The following abbreviations are used in the assignment of NMR signals: s (singlet), d (doublet), t (triplet), q (quartet), qn (quintet), m (multiplet), bs (broad singlet), dd (doublet of doublet), dt (doublet of triplet). Low resolution mass spectrometry was performed on a Bruker Daltonics microTof-LC system, as a service by the School of Chemistry at Cardiff University.
4.1.1. Standard procedure A: synthesis of phosphorodiamidates
To a stirring solution/suspension of the desired nucleoside (1.00 mol/eq) in anhydrous THF, anhydrous Et3N (1.00–1.20 mol/eq) was added under an argon atmosphere. After stirring at room temperature for 30 min, POCl3 (1.00–1.20 mol/eq) was added dropwise at −78 °C. The reaction mixture was stirred at −78 °C for 30 min and then allowed to warm to room temperature. Anhydrous CH2Cl2 and the appropriate amino acid ester salt (3.00–5.00 mol/eq) were added, followed dropwise addition of anhydrous Et3N (5.00–10.00 mol/eq) at −78 °C. The reaction mixture was then stirred at room temperature. After this period, H2O was added and the aqueous phase was extracted with CH2Cl2. The organic phase was washed with brine, dried over anhydrous Na2SO4 or MgSO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography using different eluent systems.
4.1.2. Standard procedure B: synthesis of phosphorodiamidates
To a stirring solution of the desired nucleoside (1.00 mol/eq) in trimethylphosphate or triethylphosphate, POCl3 (1.00 mol/eq) was added dropwise at −5 °C under an argon atmosphere. The reaction mixture was stirred at −5 °C for 4–5 h, or at 5 °C for 16 h. Anhydrous CH2Cl2 and the appropriate amino acid ester salt (5.0 mol/eq) were added, followed by dropwise addition of anhydrous Et3N (10.0 mol/eq) at 0 °C, or anhydrous DIPEA (10.0 mol/eq) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min and then at room temperature for 16–45 h. After this period, H2O was added and the aqueous phase was extracted with CH2Cl2. The organic phase was washed with brine, dried over anhydrous Na2SO4 or MgSO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography using different eluent systems.
4.1.3. Synthesis of 4-acetylamino-1-[(2′R,5′S)-2′-(hydroxymethyl)-1,3-oxathiolan-5′-yl]-1,2-dihydropyrimidin-2-one (6)
Anhydrous DMF (20 mL) was added to (5) (5.0 g, 21.81 mmol) and the mixture was stirred under nitrogen atmosphere for 5 min at room temperature. Acetic anhydride (2.26 mL, 23.99 mmol) was added dropwise, and the mixture was stirred at room temperature overnight. Then, the solvent was removed under reduced pressure, and the residue was azeotroped with toluene. The crude solid was washed with diethyl ether (3 × 20 mL) to give the desired compound (6) as a white solid (82%, 4.84 g). 1H NMR (500 MHz, MeOD) δ H 8.59 (d, J = 7.5 Hz, 1H, H-6), 7.43 (d, J = 7.5 Hz, 1H, H-5), 6.33 (dd, J = 5.3, 3.0 Hz, 1H, H-1′), 5.36 (t, J = 3.6 Hz, 1H, H-4′), 4.05 (dd, J = 12.6, 3.4 Hz, 1H, H-5′), 3.95 (dd, J = 12.6, 3.9 Hz, 1H, H-5′), 3.65 (dd, J = 12.4, 5.4 Hz, 1H, H-2′), 3.27 (dd, J = 12.4, 3.0 Hz, 1H, H-2′), 2.20 (s, 3H, COCH 3).
4.1.4. Synthesis of (2R,3R,4R,5R)-5-(2-amino-6-ethoxy-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)-4-methyltetrahydrofuran-3-ol-5′-O-bis-(2,2-dimethylpropoxy-l-alaninyl)-phosphate(14)
Prepared according to standard procedure A, using (1) (0.15 g, 0.46 mmol) in anhydrous THF (3 mL), anhydrous Et3N (0.06 mL, 0.46 mmol), and POCl3 (0.04 mL, 0.46 mmol). Anhydrous CH2Cl2 (3 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alanine (0.76 g, 2.29 mmol) were added, followed by dropwise addition of anhydrous Et3N (0.64 mL, 4.58 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–3%) in CHCl3 to give the product (14) as an off white solid (9%, 0.03 g). 1H NMR (500 MHz, MeOD) δ H 7.97 (s, 1H, H-8), 6.17 (d, J H–F = 19.0 Hz, 1H, H-1′), 4.57–4.53 (m, 3H, OCH 2CH3 and H-3′), 4.44–4.36 (m, 2H, H-5′), 4.23–4.19 (m, 1H, H-4′), 3.99–3.95 (m, 2H, 2× CHCH3), 3.84, 3.82, 3.72, 3.66 (2AB, J AB = 10.5 Hz, 4H, 2× OCH 2C(CH3)3), 1.45 (t, J = 7.3 Hz, 3H, OCH2CH 3), 1.39 (d, J = 7.1 Hz, 3H, CHCH 3), 1.35 (d, J = 7.1 Hz, 3H, CHCH 3), 1.23 (d, J H–F = 20.0 Hz, 3H, CCH 3), 0.94 (s, 9H, OCH2C(CH 3)3), 0.92 (s, 9H, OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 175.65, 175.60 (2d, J C–P = 6.3 Hz, CO), 162.46 (C-6), 161.99 (C-2), 154.45 (C-4), 139.42 (C-8), 115.71 (C-5), 102.01 (d, J C–F = 180.0 Hz, C-2′), 90.83 (d, J C–F = 39.0 Hz, C-1′), 81.92 (d, J C–P = 7.5 Hz, C-4′), 75.38, 75.36 (OCH2C(CH3)3), 74.04 (d, J C–F = 18.0 Hz, C-3′), 66.15 (d, J C–P = 3.8 Hz, C-5′), 63.58 (OCH2CH3), 51.11 (CHCH3), 32.32, 32.28 (OCH2 C(CH3)3), 26.80, 26.77 (OCH2C(CH3)3), 21.11, 20.98 (CHCH3), 16.89 (d, J C–F = 25.0 Hz, 2′CCH3), 14.88 (OCH2 CH3). 31P NMR (202 MHz, MeOD) δ P 13.98. 19F NMR (470 MHz, MeOD) δ F −162.26. MS (ES+) m/z: 712.31 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 20.63 min.
4.1.5. Synthesis of (2R,3R,4R,5R)-5-(2-amino-6-ethoxy-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)-4-methyltetrahydrofuran-3-ol-5′-O-bis-(2,2-dimethylpropoxy-d-alaninyl)-phosphate (15)
Prepared according to standard procedure A, using (1) (0.15 g, 0.46 mmol) in anhydrous THF (2.4 mL), anhydrous Et3N (0.08 mL, 0.55 mmol), and POCl3 (0.05 mL, 0.55 mmol). Anhydrous CH2Cl2 (3.5 mL) and tosylate salt of 2,2-dimethylpropoxy-d-alanine (0.76 g, 2.30 mmol) were added, followed by dropwise addition of anhydrous Et3N (0.64 mL, 4.60 mmol). The reaction mixture was stirred at room temperature overnight. After work-up, the crude residue was purified by silica gel column chromatography eluting with CHCl3/MeOH (97.5/2.5) to give the product (15) as an off white solid (7%, 0.02 g). 1H NMR (500 MHz, CDCl3) δ H 7.76 (s, 1H, H-8), 6.04 (d, 1H, J H–F = 18.7 Hz, H-1′), 5.31 (bs, 1H, NH), 4.90 (bs, 1H, NH), 4.82–4.72 (m, 1H, H-3′), 4.65–4.61 (m, 1H, H-5′), 4.56 (q, 1H, J = 6.6 Hz, OCH 2CH3), 4.35–4.31 (m, 1H, H-5′), 4.19–4.18 (m, 1H, H-4′), 4.07–3.98 (m, 2H, 2× CHCH3), 3.90, 3.88 (AB, 2H, J AB = 10.4 Hz, OCH 2C(CH3)3), 3.73, 3.71 (AB, 2H, J AB = 10.4 Hz, OCH 2C(CH3)3), 1.47 (t, 3H, J = 7.1 Hz, OCH2CH 3), 1.43 (t, 6H, J = 6.5 Hz, 2× CHCH 3), 1.27 (d, 3H, J H–F = 22.7 Hz, 2′CCH 3), 0.94 (s, 9H, OCH2C(CH 3)3), 0.92 (s, 9H, OCH2C(CH 3)3). 13C NMR (125 MHz, CDCl3) δ C 175.15 (d, J C–P = 6.5 Hz, CO), 174.28 (d, J C–P = 6.8 Hz, CO), 161.32 (C-6), 159.59 (C-2), 153.20 (C-4), 137.37 (C-8), 115.71 (C-5), 101.92, 100.48 (d, J C–F = 180.0 Hz, C-2′), 89.23 (d, J C–F = 40.3 Hz, C-1′), 79.97 (d, J C–P = 7.6 Hz, C-4′), 74.82, 74.55 (OCH2C(CH3)3), 71.93 (C-3′), 63.03 (OCH2CH3), 62.71, 62.57 (2d, J C–P = 17.6 Hz, C-5′), 49.78, 49.43 (CHCH3), 31.36, 31.23 (OCH2 C(CH3)3), 26.33, 26.25, 26.14 (OCH2C(CH3)3), 21.37 (d, J C–P = 6.1 Hz, CHCH3), 21.02 (d, J C–P = 8.1 Hz, CHCH3), 16.59 (d, J C–F = 25.2 Hz, 2′CCH3), 14.47 (OCH2 CH3). 31P NMR (202 MHz, CDCl3) δ P 13.86. 19F NMR (470 MHz, CDCl3) δ F −162.43. MS (ES) m/z: 690.34 (M + H+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 10/90 in 30 min, flow = 1 mL/min, λ = 275 nm, t R = 20.24 min.
4.1.6. Synthesis of d4T-5′-O-bis(benzoxy-l-alaninyl)-phosphate (16)
Prepared according to standard procedure A, using (2) (0.50 g, 2.23 mmol) in anhydrous THF (5 mL), anhydrous Et3N (0.31 mL, 2.23 mmol) POCl3 (0.21 mL, 2.23 mmol). Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (2.35 g, 6.69 mmol) were added, followed by dropwise addition of anhydrous Et3N (3.10 mL, 22.30 mmol). The reaction mixture was stirred at room temperature overnight. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to yield product (16) as a white solid (11% 0.10 g). 1H NMR (500 MHz, MeOD) δ H 7.40–7.30 (m, 11H, OCH2 Ph, H-5), 6.96–6.94 (m, 1H, H-1′) 6.34–6.32 (m, 1H, H-3′), 5.90–5.93 (m, 1H, H-2′), 5.17–5.10 (m, 4H, 2× OCH 2Ph), 4.91 (bs, 1H, H-4′), 4.11–4.09 (m, 2H, H-5′), 3.97–3.89 (m, 2H, 2× CHCH3), 1.88 (s, 3H, CH 3), 1.35 (d, J = 7.0 Hz, 3H, CHCH 3), 1.33 (d, J = 7.0 Hz, 3H, CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.37 (d, J C–P = 5.0 Hz, CO), 175.22 (d, J C–P = 5.0 Hz, CO), 166.42 (C-4), 152.82 (C-2), 137.96 (C-6), 137.35 (ipso OCH2 Ph), 137.33 (ipso OCH2 Ph), 134.99 (C-3′), 129.67, 129.41 (OCH2 Ph), 127.83 (C-2′), 112.17 (C-5), 91.20 (C-1′), 86.48 (d, J C–P = 8.7 Hz, C-4′), 67.97 (OCH2Ph), 66.44 (d, J C–P = 8.7 Hz, C-5′), 51.17 (d, J C–P = 1.9 Hz, CHCH3), 50.99 (d, J C–P = 1.9 Hz, CHCH3), 20.84 (d, J C–P = 5.5 Hz, CHCH3), 20.66 (d, J C–P = 5.5 Hz, CHCH3), 12.73 (5-CH3). 31P NMR (202 MHz, MeOD) δ P 13.63. MS (ES+) m/z: 649.20 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min; flow = 1 mL/min, λ = 280 nm, t R = 18.61 min.
4.1.7. Synthesis of d4T-5′-O-bis-(cyclohexoxy-l-alaninyl)-phosphate (17)
Prepared according to standard procedure A, using (2) (0.53 g, 2.36 mmol) in anhydrous THF (5 mL), anhydrous Et3N (0.33 mL, 2.36 mmol), and POCl3 (0.22 mL, 2.36 mmol). Anhydrous CH2Cl2 (5 mL) and tosylate salt of cyclohexoxy-l-alanine (4.06 g, 11.82 mmol) were added, followed by dropwise addition of anhydrous Et3N (3.30 mL, 23.63 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to afford the product (17) as a solid (46%, 0.66 g). 1H NMR (500 MHz, MeOD) δ H 7.44 (s, 1H, H-6), 6.99 (m, 1H, H-1′), 6.45 (dd, J = 6.0, 1.2 Hz, 1H, H-3′), 5.99 (dd, J = 6.0, 1.2 Hz, 1H, H-2′), 5.02 (bs, 1H, H-4′), 4.78–4.74 (m, 2H, 2× OCH–cHx), 4.21–4.14 (m, 2H, H-5′), 3.90–3.84 (m, 2H, 2× CHCH3), 1.92 (s, 3H, CH 3), 1.90–1.80 (m, 4H, cHx), 1.80–1.70 (m, 4H, cHx), 1.63–1.55 (m, 2H, cHx), 1.54–1.29 (m, 10H, cHx), 1.36 (d, J = 7.0 Hz, 3H, CHCH 3), 1.32 (d, J = 7.0 Hz, 3H, CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.05 (d, J C–P = 5.0 Hz, CO), 174.89 (d, J C–P = 5.0 Hz, CO), 166.37 (C-4), 152.81 (C-2), 137.97 (C-6), 134.98 (C-3′), 127.95 (C-2′), 112.14 (C-5), 91.21 (C-1′), 86.57 (d, J C–P = 8.8 Hz, C-4′), 74.88, 74.86 (2× OCHcHx), 67.44 (d, J C–P = 5.0 Hz, C-5′), 51.22 (d, J C–P = 1.1 Hz, CHCH3), 50.99 (d, J C–P = 1.1 Hz, CHCH3), 32.57, 32.56, 32.50, 32.48 (CH2–cHx), 26.48, 24.75, 24.72, 24.68 (CH2–cHx), 21.15 (d, J C–P = 5.0 Hz, CHCH3), 20.95 (d, J C–P = 5.0 Hz, CHCH3), 12.73 (5-CH3). 31P NMR (202 MHz, MeOD) δ P 12.58. MS (ES+) m/z: 633.25 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 18.48 min.
4.1.8. Synthesis of d4T-5′-O-bis-(2,2-dimethylpropoxy-l-alaninyl)-phosphate (18)
Prepared according to standard procedure A, using (2) (0.50 g, 2.23 mmol) in anhydrous THF (5 mL), anhydrous Et3N (0.31 mL, 2.23 mmol), and POCl3 (0.21 mL, 2.23 mmol). Anhydrous CH2Cl2 (5 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alanine (2.22 g, 6.69 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.52 mL, 11.15 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to afford the product (18) as a white solid (12%, 0.15 g). 1H NMR (500 MHz, MeOD) δ H 7.44 (s, 1H, H-6), 7.01–6.90 (m, 1H, H-1′), 6.46–6.40 (m, 1H, H-3′), 6.00–5.90 (m, 1H, H-2′), 5.03 (bs, 1H, H-4′), 4.21–4.14 (m, 2H, H-5′), 3.98–3.92 (m, 2H, 2× CHCH3), 3.91, 3.89, 3.78, 3.76 (2AB, J AB = 10.0 Hz, 4H, 2× OCH 2C(CH3)3), 1.92 (s, 3H, CH3), 1.40 (d, J = 7.0 Hz, 6H, 2× CHCH 3), 0.97 (s, 9H, OCH2C(CH 3)3), 0.97 (s, 9H, OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 175.64 (d, J C–P = 5.0 Hz, CO), 175.47 (d, J C–P = 5.0 Hz, CO), 166.41 (C-4), 152.85 (C-2), 137.97 (C-6), 134.98 (C-3′), 127.93 (C-2′), 112.14 (C-5), 91.22 (C-1′), 86.57 (d, J C–P = 8.7 Hz, C-4′), 75.45 (OCH2C(CH3)3), 67.54 (d, J C–P = 5.0 Hz, C-5′), 51.19 (d, J C–P = 1.1 Hz, CHCH3), 50.82 (d, J C–P = 1.1 Hz, CHCH3), 32.36 (OCH2 C(CH3)3), 26.87, 26.60 (OCH2C(CH3)3), 21.19 (d, J C–P = 5.0 Hz, CHCH3), 21.01 (d, J C–P = 5.0 Hz, CHCH3), 12.71 (5-CH3). 31P NMR (202 MHz, MeOD) δ P 13.73. MS (ES+) m/z: 609.27 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 17.80 min.
4.1.9. Synthesis of ddA-5′-O-bis-(2,2-dimethylpropoxy-l-alaninyl)-phosphate (19)
Prepared according to standard procedure A, using (3) (0.10 g, 0.42 mmol) in anhydrous THF (2 mL), anhydrous Et3N (0.06 mL, 0.42 mmol), and POCl3 (0.39 mL, 0.42 mmol). Anhydrous CH2Cl2 (3 mL) and tosylate salt of 2,2-dimethylpropoxy (0.70 g, 2.10 mmol) were added, followed by dropwise addition of anhydrous Et3N (0.29 mL, 2.10 mmol). The reaction mixture was stirred at room temperature for 18 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to give the product (19) as a white solid (9%, 0.02 g). 1H NMR (500 MHz; MeOD) δ H 8.55 (bs, 1H, H-8), 8.39 (bs, 1H, H-2), 6.38 (dd, J = 6.5, 3.3 Hz, 1H, H-1′), 4.44–4.39 (m, 1H, H-4′), 4.23–4.19 (m, 1H, H-5′), 4.14–4.10 (m, 1H, H-5′), 3.94–3.87 (m, 2H, 2× CHCH3), 3.84, 3.72 (2AB, J AB = 10.4 Hz, 4H, OCH 2C(CH3)3), 2.67–2.55 (m, 2H, H-2′), 2.21 (m, 2H, H-3′), 1.35 (d, J = 7.1 Hz, 6H, 2× CHCH 3), 0.93 (s, 18H, 2× OCH2C(CH 3)3); 31P NMR (202 MHz; MeOD) δ P 13.98. Reverse-phase HPLC, eluting with H2O/MeOH from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 25.91 min.
4.1.10. Synthesis of 3′-azido-3′-deoxythymidine-5′-O-bis(benzoxy-l-alaninyl)-phosphate (20)
Prepared according to standard procedure A, using (4) (0.20 g, 0.75 mmol) in anhydrous THF (5 mL), anhydrous Et3N (0.10 mL, 0.75 mmol), and POCl3 (0.07 mL, 0.75 mmol). Anhydrous CH2Cl2 (10 mL) and tosylate salt of benzoxy-l-alaninyl (1.31 g, 3.74 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.04 mL, 7.48 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–3%) in CHCl3 to give the product (20) as an off white solid (20%, 0.10 g). 1H NMR (500 MHz, MeOD) δ H 7.51 (s, 1H, H-6), 6.16 (t, J = 6.5 Hz, 1H, H-1′), 5.17–5.10 (m, 4H, 2× OCH 2Ph), 4.38–4.35 (m, 1H, H-3′), 4.19–4.10 (m, 2H, H-5′), 4.02–3.95 (m, 3H, H-4′, 2× CHCH3), 2.36 (m, 2H, H-2′), 1.89 (s, 3H, 5-CH 3), 1.39 (d, 3H, J = 7.0 Hz, CHCH 3), 1.36 (d, 3H, J = 7.0 Hz, CHCH 3). 13C (125 MHz, MeOD) δ C 175.42 (d, J C–P = 3.8 Hz, CO), 175.35 (d, J C–P = 6.3 Hz, CO), 166.25 (C-4), 152.22 (C-2), 137.69 (C-6), 137.29, 137.27 (ipso OCH2 Ph), 129.96, 129.68, 129.66, 129.44, 129.42, 129.38, 129.35, 129.12 (OCH2 Ph), 112.16 (C-5), 86.24 (C-1′), 83.85 (d, J C–P = 8.8 Hz, C-4′), 68.03, 68.00 (OCH2Ph), 66.04 (d, J C–P = 5.0 Hz, C-5′), 61.85 (C-3′), 51.21, 51.16 (CHCH3), 37.68 (C-2′), 20.85, 20.73 (2d, J C–P = 6.3 Hz, CHCH3), 12.71 (5-CH3). 31P NMR (202 MHz, MeOD) δ P 13.69. MS (ES+) m/z: 692.22 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 20.83 min.
4.1.11. Synthesis of 3′-azido-3′-deoxythymidine-5′-O-bis(2,2-dimethylpropoxy)-phosphate (21)
Prepared according to standard procedure A, using (4) (0.20 g, 0.75 mmol) in anhydrous THF (5 mL), anhydrous Et3N (0.10 mL, 0.75 mmol), and POCl3 (0.07 mL, 0.75 mmol). Anhydrous CH2Cl2 (10 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alaninyl (1.24 g, 3.74 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.04 mL, 7.48 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient (0%–3%) of MeOH in CHCl3 to give the product (21) as an off white solid (25%, 0.10 g). 1H NMR (500 MHz, MeOD) δ H 7.57 (s, 1H, H-6), 6.21 (t, J = 7.0 Hz, 1H, H-1′), 4.48–4.45 (m, 1H, H-3′), 4.24–4.21 (m, 2H, H-5′), 4.08–4.06 (m, 1H, H-4′), 4.03–3.96 (m, 2H, 2× CHCH3), 3.91, 3.90, 3.79, 3.77 (2AB, 4H, J AB = 10.5 Hz, 2× OCH 2C(CH3)3), 2.48–2.44 (m, 2H, H-2′), 1.94 (s, 3H, 5-CH 3), 1.45 (d, J = 7.0 Hz, 3H, 2× CHCH 3), 1.44 (d, J = 7.0 Hz, 6H, 2× CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.66 (d, J C–P = 3.8 Hz, CO), 175.62 (d, J C–P = 6.3 Hz, CO), 166.25 (C-4), 152.25 (C-2), 137.79 (C-6), 112.15 (C-5), 86.27 (C-1′), 83.90 (d, J C–P = 7.5 Hz, C-4′), 75.47, 75.44 (OCH2C(CH3)3), 66.09 (d, J C–P = 5.0 Hz, C-5′), 61.90 (C-3′), 51.21, 51.12 (CHCH3), 37.69 (C-2′), 30.78 (OCH2 C(CH3)3), 26.85, 26.70 (OCH2C(CH3)3), 21.16 (d, J C–P = 5.0 Hz, CHCH3), 21.04 (d, J C–P = 6.3 Hz, CHCH3), 12.70 (5-CH3). 31P NMR (202 MHz, MeOD) δ P 13.79. MS (ES+) m/z: 652.59 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 19.93 min.
4.1.12. Synthesis of 3TC(−)-5′-O-bis(benzoxy-l-alaninyl)-N-bis(benzoxy-l-alaninyl)-diphosphate (22) and 3TC(−)-N-bis(benzoxy-l-alaninyl)-phosphate (23)
Prepared according to standard procedure A, using (5) (2.29 g, 10.00 mmol) in anhydrous THF (25 mL), anhydrous Et3N (1.39 mL, 10.00 mmol), and POCl3 (0.93 mL, 10.00 mmol). Anhydrous CH2Cl2 (25 mL) and tosylate salt of l-alanine benzyl ester (17.57 g, 50.00 mmol) were added, followed by dropwise addition of anhydrous Et3N (14.0 mL, 100.00 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (1%–10%) in CH2Cl2, followed by a second purification eluting with CH2Cl2/MeOH 95/5 to give compound (22) as an off-white solid (3%, 0.18 g). 1H NMR (500 MHz; MeOD) δ H 8.13 (d J = 7.6 Hz, 1H, H-6), 7.39–7.28 (m, 20H, 4× OCH2 Ph), 6.29–6.22 (m, 2H, H-5, H-1′), 5.31–5.29 (m, 1H, H-4′), 5.19–5.11 (m, 6H, 3× CH 2OPh), 5.08 (s, 2H, CH 2OPh), 4.28–4.22 (m, 2H, H-5′), 4.10–4.08 (m, 2H, 2× CHCH3), 4.04–3.81 (m, 2H, 2× CHCH3), 3.48 (dd, J = 12.6, 4.4 Hz, 1H, H-2′), 3.01 (dd, J = 12.6, 4.4 Hz, 1H, H-2′), 1.38–1.41 (m, 12H, 4× CHCH 3). 13C NMR (125 MHz; MeOD) δ C 175.49, 175.12 (CO), 163.75 (C-4), 156.36 (C-2), 143.72 (C-6), 137.36, 137.34, 137.32, 137.30 (ipso OCH2 Ph), 129.66, 129.65, 129.62, 129.42, 129.38, 129.36, 129.26, 129.05 (OCH2 Ph), 98.06 (C-1′), 89.15 (C-5), 85.53 (d, J C–P = 8.7 Hz, C-4′), 68.01, 67.97, 67.96, 67.77 (OCH2Ph), 66.94 (d, J C–P = 5.0 Hz, C-5′), 51.25, 51.13, 51.00, 50.94 (CHCH3), 38.40 (C-2′), 20.83, 20.79, 20.76, 20.70 (CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.56, 7.39. MS (ES+) m/z: 1056.31 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 16.34 min.
Further elution with CH2Cl2/MeOH 95/5 afforded (23) off-white solid (2%, 0.09 g).
1H NMR (500 MHz; MeOD) δ H 8.29 (d, J = 7.3 Hz, 1H, H-6), 7.39–7.29 (m, 10H, 2× OCH2 Ph), 6.29 (dd, J = 5.3, 3.1 Hz, 1H, H-1′), 6.20 (d, J = 7.3 Hz, 1H, H-5), 5.32–5.30 (m, 1H, H-4′), 5.17 (s, 2H, CH 2OPh), 5.08 (s, 2H, CH 2OPh), 4.08 (m, 2H, 2× CHCH3), 4.05 (dd, J = 12.2, 4.2 Hz, 1H, H-5′), 3.92 (dd, J = 12.3, 4.2 Hz, 1H, H-5′), 3.59 (dd, J = 12.6, 4.3 Hz, 1H, H-2′), 3.12 (dd, J = 12.6, 4.3 Hz, 1H, H-2′), 1.42–1.38 (m, 6H, 2× CHCH 3). 13C NMR (125 MHz; MeOD) δ C 175.23 (d, J C–P = 6.2 Hz, CO), 166.79 (C-4), 157.76 (C-2), 144.15 (C-6), 137.34 (ipso CH2OPh), 129.60, 129.34, 129.32, 129.25, 129.07 (CH2OPh), 97.26 (C-5), 89.15 (C-4′), 89.04 (C-1′), 67.96, 67.77 (CH2OPh), 63.59 (C-5′), 51.01, 50.91 (CHCH3), 39.08 (C-2′), 20.69 (d, J C–P = 6.2 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 7.40. MS (ES+) m/z: 654.18 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 14.34 min.
4.1.13. Synthesis of N-acetyl-3TC-5′-O-bis(benzoxy-l-alaninyl)-phosphate (24)
Prepared according to standard procedure B, using (6) (0.50 g, 1.84 mmol) in trimethylphosphate (5 mL), and POCl3 (0.18 mL, 1.84 mmol), the reaction mixture was stirred at 0 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (3.24 g, 9.22 mmol) were added, followed by dropwise addition of anhydrous DIPEA (3.21 mL, 18.43 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to give the product (24) as a white solid (1%, 0.01 g). 1H NMR (500 MHz, MeOD) δ H 8.26 (d, J = 7.5 Hz, 1H, H-6), 7.46 (d, J = 7.6 Hz, 1H, H-5), 7.35–7.33 (m, 10H, OCH2 Ph), 6.27 (dd, J = 5.3, 3.8 Hz, 1H, H-1′), 5.36–5.35 (m, 1H, H-4′), 5.17–5.11 (m, 4H, 2× CH 2OPh), 4.33–4.25 (m, 2H, H-5′), 4.03–3.98 (m, 2H, 2× CHCH3), 3.59 (dd, J = 12.4, 4.6 Hz, 1H, H-2′), 3.17 (dd, J = 12.4, 4.6 Hz, 1H, H-2′), 2.19 (s, 3H, COCH 3), 1.41 (dd, J = 7.2, 0.8 Hz, 3H, CHCH 3), 1.38 (dd, J = 7.2, 0.6 Hz, 3H, CHCH 3). 31P NMR (202 MHz, MeOD) δ P 13.75. MS (ES+) m/z: 674.20 (M + H+, 100%).
4.1.14. Synthesis of 3TC-5′-O-bis(benzoxy-l-alaninyl)-phosphate (25)
Prepared according to standard procedure B, using (5) (0.42 g, 1.84 mmol) in triethylphosphate (5 mL), and POCl3 (0.18 mL, 1.84 mmol), the reaction mixture was stirred at 5 °C for 16 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (3.24 g, 9.20 mmol) were added, followed by dropwise addition of anhydrous DIPEA (3.21 mL, 18.43 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–7%) in CH2Cl2 to give the product (25) as an off white solid (6%, 0.07 g). 1H NMR (500 MHz, MeOD) δ H 7.72 (d, J = 7.6 Hz, 1H, H-6), 7.41–7.31 (m, 10H, OCH2 Ph), 6.41 (dd, J = 5.6, 2.9 Hz, 1H, H-1′), 5.85 (d, J = 6.3 Hz, 1H, H-5), 5.18–5.13 (m, 4H, 2× OCH 2Ph), 5.06–5.09 (m, 1H, H-4′), 4.09–3.92 (m, 3H, H-5′, CHCH3), 2.72–2.60 (m, 2H, H-2′), 1.41–1.33 (m, 6H, 2× CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.31 (d, J C–P = 6.2 Hz, CO), 167.59 (C-4), 157.15 (C-2), 142.08 (C-6), 137.32 (ipso OCH2 Ph), 129.64, 129.54, 129.36, 129.30, 129.24 (OCH2 Ph), 96.52 (C-1′), 98.80 (d, J C–P = 7.7 Hz, C-4′), 96.32 (C-5), 67.95, 67.88 (OCH2Ph), 58.40 (d, J C–P = 5.0 Hz, C-5′), 54.84 (CHCH3), 30.13 (C-2′), 20.83 (d, J C–P = 6.2 Hz, CHCH3).31P NMR (202 MHz, MeOD) δ P 11.24. MS (ES+) m/z: 654.17 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 14.64 min.
4.1.15. Synthesis of 4′-azidouridine-5′-O-bis(benzoxy-l-alaninyl)-phosphate (26)
Prepared according to standard procedure B, using (7) (0.25 g, 0.88 mmol) in triethylphosphate (1 mL), and POCl3 (0.08 mL, 0.88 mmol), the reaction mixture was stirred at 5 °C for 16 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (1.54 g, 4.38 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.22 mL, 8.77 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–3%) in CHCl3 to give the product (26) as an off white solid (20%, 0.10 g). 1H NMR (500 MHz, MeOD) δ H 7.51 (d, J = 8.0 Hz, 1H, H-5), 7.38–7.30 (m, 10 H, 2× OCH2 Ph), 6.14 (d, J = 4.0 Hz, 1H, H-1′), 5.80 (d, J = 8.0 Hz, 1H, H-5), 5.18–5.09 (m, 4H, 2× OCH 2Ph), 4.44–4.38 (m, 2H, H-3′, H-2′), 4.10–4.02 (m, 2H, H-5′), 4.02–3.95 (m, 2H, 2× CHCH3), 1.39 (d, 3H, J = 7.0 Hz, CHCH 3), 1.36 (d, 3H, J = 7.0 Hz, CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.40 (d, J C–P = 3.8 Hz, CO), 175.36 (d, J C–P = 6.3 Hz, CO), 165.89 (C-4), 152.27 (C-2), 142.89 (C-6), 137.27, 137.25 (ipso OCH2 Ph), 129.67, 129.65, 129.40, 129.37, 129.33, 129.12 (OCH2 Ph), 103.83 (C-5), 98.94 (d, J C–P = 8.8 Hz, C-4′), 92.58 (C-1′), 73.76, 73.58 (C-3′, C-2′), 68.07, 68.04 (OCH2Ph), 67.60 (d, J C–P = 3.8 Hz, C-5′), 51.16, 51.12 (CHCH3), 20.85 (d, J C–P = 6.3 Hz, CHCH3), 20.65 (d, J C–P = 6.3 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.57. MS (ES+) m/z: 710.18 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 15.97 min.
4.1.16. Synthesis of 4′-azidouridine-5′-O-bis(2,2-dimethylpropoxy-l-alaninyl)-phosphate (27)
Prepared according to standard procedure B, using (7) (0.25 g, 0.88 mmol) in triethylphosphate (1 mL), and POCl3 (0.08 mL, 0.88 mmol), the reaction mixture was stirred at 5 °C for 16 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alanine (1.45 g, 4.38 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.22 mL, 8.77 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–5%) in CHCl3 to give the product (27) as an off white solid (17%, 0.10 g). 1H NMR (500 MHz, MeOD) δ H 7.55 (d, J = 8.0 Hz, 1H, H-5), 6.15 (d, J = 3.5 Hz, 1H, H-1′), 5.83 (d, J = 8.0 Hz, 1H, H-5), 4.45–4.41 (m, 2H, H-3′, H-2′), 4.13–4.06 (m, 2H, H-5′), 4.03–3.96 (m, 2H, 2× CHCH3), 3.91, 3.79 (AB, 4H, J AB = 10.5 Hz, 2× OCH 2C(CH3)3), 1.44, (d, J = 7.0 Hz, 3H, CHCH 3), 1.43 (d, J = 7.0 Hz, 3H, CHCH 3), 0.98 (s, 18H, 2× OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 175.65 (d, J C–P = 5.0 Hz, CO), 175.61 (d, J C–P = 6.3 Hz, CO), 165.89 (C-4), 152.29 (C-2), 142.97 (C-6), 103.85 (C-5), 98.93 (d, J C–P = 10.0 Hz, C-4′), 92.56 (C-1′), 75.53, 75.49 (OCH2C(CH3)3), 73.78, 73.63 (C-3′, C-2′), 67.70 (d, J C–P = 3.8 Hz, C-5′), 51.16, 51.09 (CHCH3), 32.37, 32.36 (OCH2 C(CH3)3), 26.84 (OCH2C(CH3)3), 21.17 (d, J C–P = 5.0 Hz, CHCH3), 20.98 (d, J C–P = 7.5 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.57. MS (ES+) m/z: 670.25 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 17.55 min.
4.1.17. Synthesis of 4′-azidocytidine-5′-O-bis(benzoxy-l-alaninyl)-phosphate (28)
Prepared according to standard procedure B, using (8) (0.25 g, 0.75 mmol) in trimethylphosphate (1 mL), and POCl3 (0.07 mL, 0.75 mmol), the reaction mixture was stirred at 5 °C for 16 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (1.32 g, 3.75 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.04 mL, 7.50 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–5%) in CHCl3 to give the product (28) as an off white solid (16%, 0.08 g). 1H NMR (500 MHz, MeOD) δ H 7.60 (d, J = 7.5 Hz, 1H, H-5), 7.27–7.19 (m, 10H, 2× OCH2 Ph), 6.00 (d, 1H, J = 4.5 Hz, H-1′), 5.85 (d, 1H, J = 7.5 Hz, H-5), 5.07–4.99 (m, 4H, 2× OCH 2Ph), 4.28 (d, J = 6.0 Hz, 1H, H-3′), 4.25 (dd, J = 6.0, 4.0 Hz, 1H, H-2′), 4.00–3.93 (m, 2H, H-5′), 3.90–3.84 (m, 2H, 2× CHCH3), 1.26 (d, J = 7.0 Hz, 3H, CHCH 3), 1.24 (d, J = 7.0 Hz, 3H, CHCH 3).13C NMR (125 MHz, MeOD) δ C 175.83 (d, J C–P = 5.0 Hz, CO), 175.37 (d, J C–P = 6.3 Hz, CO), 167.71 (C-4), 158.22 (C-2), 143.38 (C-6), 137.24, 137.22 (ipso OCH2 Ph), 129.71, 129.68, 129.45, 129.40, 129.37, 129.12 (OCH2 Ph), 97.98 (d, J C–P = 9.0 Hz, C-4′), 96.98 (C-5), 93.84 (C-1′), 74.52 (C-2′), 73.33 (C-3′), 67.61 (d, J C–P = 5.0 Hz, C-5′), 51.16, 51.12 (CHCH3), 20.84 (d, J C–P = 5.0 Hz, CHCH3), 20.73 (d, J C–P = 5.0 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.53. MS (ES+) m/z: 709.20 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 15.11 min.
4.1.18. Synthesis of 4′-azidocytidine-5′-O-bis(2,2-dimethylpropoxy-l-alaninyl)-phosphate (29)
Prepared according to standard procedure B, using (8) (0.25 g, 0.75 mmol) in trimethylphosphate (1 mL), and POCl3 (0.07 mL, 0.75 mmol), the reaction mixture was stirred at 5 °C for 16 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alanine (1.24 g, 3.75 mmol) were added, followed by dropwise addition of anhydrous Et3N (1.04 mL, 7.50 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (0%–5%) in CHCl3 to give the product (29) as an off white solid (21%, 0.10 g). 1H NMR (500 MHz, MeOD): δ H 7.76 (d, 1H, J = 7.5 Hz, H-5), 6.15 (d, 1H, J = 4.0 Hz, H-1′), 6.00 (d, 1H, J = 7.5 Hz, H-5), 4.41 (d, 1H, J = 6.0 Hz, H-3′), 4.36 (dd, 1H, J = 6.0, 4.5 Hz, H-2′), 4.16–4.08 (m, 2H, H-5′), 4.05–3.96 (m, 2H, 2× CHCH3), 3.91, 3.90, 3.79, 3.78 (2AB, 4H, J AB = 10.5 Hz, 2× OCH 2C(CH3)3), 1.44 (d, 6H, J = 7.0 Hz, 2× CHCH 3), 0.98 (s, 18H, 2× OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 176.74 (d, J C–P = 3.8 Hz, CO), 175.63 (d, J C–P = 5.0 Hz, CO), 167.72 (C-4), 158.18 (C-2), 143.38 (C-6), 98.90 (d, J C–P = 9.0 Hz, C-4′), 96.98 (C-5), 93.96 (C-1′), 75.47, 75.46 (OCH2C(CH3)3), 74.39 (C-2′), 73.32 (C-3′), 67.69 (d, J C–P = 5.0 Hz, C-5′), 51.16, 51.12 (CHCH3), 32.34, 32.33 (OCH2 C(CH3)3), 26.79 (OCH2C(CH3)3), 21.07 (d, J C–P = 5.0 Hz, CHCH3), 20.94 (d, J C–P = 7.5 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.59. MS (ES+) m/z: 669.27 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, flow = 1 mL/min, λ = 254, t R = 16.59.
4.1.19. Synthesis of ribavirin-5′-O-bis(benzoxy-l-alaninyl)-phoshate (30)
Prepared according to standard procedure B, using (9) (0.30 g, 1.23 mmol) in trimethylphosphate (5 mL), and POCl3 (0.11 mL, 1.23 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (5 mL), and a suspension of tosylate salt of benzoxy-l-alanine (2.16 g, 6.15 mmol) in anhydrous CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (2.16 mL, 12.30 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (4%–10%) in CH2Cl2 to give the product (30) as an off white solid (25%, 0.20 g). 1H NMR (500 MHz, MeOD) δ H 8.69 (s, 1H, H-5), 7.36–7.31 (m, 10H, 2× OCH2 Ph), 5.95–5.94 (m, 1H, H-1′), 5.17–5.08 (m, 4H, 2× OCH 2Ph), 4.55–4.54 (m, 1H, H-2′), 4.45–4.43 (m, 1H, H-3′), 4.22–4.07 (m, 3H, H-4′, H-5′), 3.98–3.91 (m, 2H, 2× CHCH3), 1.34 (d, J = 7.2 Hz, 3H, CHCH 3), 1.29 (d, J = 7.0 Hz, 3H, CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.40, 175.36 (CO), 163.24 (CONH2), 158.71 (C-3), 146.88 (C-5), 137.33, 137.35 (ipso OCH2 Ph), 129.59, 129.35, 129.32, 129.31 (OCH2 Ph), 93.67 (C-1′), 84.93 (d, J C–P = 7.7 Hz, C-4′), 76.50 (C-2′), 71.83 (C-3′), 67.94, 67.91 (OCH2Ph), 66.36 (d, J C–P = 5.1 Hz, C-5′), 51.09, 51.08 (CHCH3), 20.74 (d, J C–P = 6.1 Hz, CHCH3), 20.59 (d, J C–P = 6.4 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.75. MS (ES+) m/z: 669.20 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/MeOH from 90/100 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 13.88 min.
4.1.20. Synthesis of ribavirin-5′-O-bis(2,2-dimethylpropoxy-l-alaninyl)-phoshate (31)
Prepared according to standard procedure B, using (9) (0.30 g, 1.23 mmol) in trimethylphosphate (5 mL), and POCl3 (0.11 mL, 1.23 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of 2,2-dimethylpropoxy-l-alanine (2.04 g, 6.15 mmol) in anhydrous CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (2.16 mL, 12.30 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient (4%–10%) of MeOH in CH2Cl2 to give the product (31) as an off white solid (21%, 0.16 g). 1H NMR (500 MHz, MeOD) δ H 8.73 (s, 1H, H-5), 5.97 (m, 1H, H-1′), 4.56–4.55 (m, 1H, H-2′), 4.48–4.46 (m, 1H, H-3′), 4.28–4.13 (m, 3H, H-4′, H-5′), 4.00–3.92 (m, 2H, 2× CHCH3), 3.89–3.75 (m, 4H, 2× OCH 2C(CH3)3), 1.40–1.37 (6H, m, 2× CHCH 3), 0.96 (s, 9H, 2× OCH2C(CH 3)3), 0.95 (s, 9H, 2× OCH2C(CH 3)3).13C NMR (125 MHz, MeOD) δ C 174.30 (d, J C–P = 4.1 Hz, CO), 174.20 (d, J C–P = 3.2 Hz, CO), 161.80 (CONH2), 157.30 (C-3), 145.47 (C-5), 92.29 (C-1′), 83.53 (d, J C–P = 7.4 Hz, C-4′), 74.81 (C-2′), 73.99, 73.96 (OCH2C(CH3)3), 70.47 (C-3′), 65.11 (d, J C–P = 5.3 Hz, C-5′), 49.68 (CHCH3), 30.91, 30.90 (OCH2 C(CH3)3), 23.38 (OCH2C(CH3)3), 19.66 (d, J C–P = 5.6 Hz, CHCH3), 19.54 (d, J C–P = 5.9 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.84. MS (ES+) m/z: 607.28 (M + H+, 100%). Reverse-phase HPLC, eluting with H2O/MeOH from 90/100 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 21.27 min.
4.1.21. Synthesis of acyclovir-5′-O-bis[(benzoxy-l-alaninyl)]-phosphate (32)
Prepared according to standard procedure B, using (10) (0.30 g, 1.33 mmol) in trimethylphosphate (5 mL), and POCl3 (0.12 mL, 1.33 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of benzoxy-l-alanine (2.34 g, 6.65 mmol) in anhydrous CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (2.32 mL, 13.30 mmol). The reaction mixture was stirred at room temperature for 45 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with gradient (2%–10%) of MeOH in CH2Cl2 to give a white solid, which was dissolved in CH2Cl2 and washed with 0.05 N HCl (1 × 5 mL), brine (1 × 5 mL), 5% NaHCO3 (1 × 5 mL), brine (1 × 5 mL), dried over MgSO4, filtered and concentrated to give the product (32) as a white solid (26%, 0.22 g).
1H NMR (500 MHz, MeOD) δ H 7.81 (s, 1H, H-8), 7.34–7.26 (m, 10H, 2× OCH2 Ph), 5.41 (s, 2H, H-1′), 5.12–5.10 (m, 4H, 2× OCH 2Ph), 4.02–3.93 (m, 4H, H-5′, 2× CHCH3), 3.64 (t, J = 4.2 Hz, 2H, H-4′), 1.35 (d, J = 7.1 Hz, 3H, CHCH 3), 1.32 (d, J = 7.1 Hz, 3H, CHCH 3). 13C NMR (125 MHz, MeOD) δ C 174.13 (d, J C–P = 5.1 Hz, CO), 158.14 (C-6), 154.34 (C-2), 151.90 (C-4), 138.35 (C-8), 135.87, 135.85 (ipso OCH2 Ph), 128.57, 128.24, 128.21, 127.99, 127.95, 127.92 (OCH2 Ph), 116.26 (C-5), 72.36 (C-1′), 68.16 (d, J C–P = 7.3 Hz, C-4′), 66.60, 66.57 (OCH2Ph), 64.29 (d, J C–P = 5.3 Hz, C-5′), 49.76, 49.74 (CHCH3), 19.50 (d, J C–P = 6.2 Hz, CHCH3), 19.40 (d, J C–P = 6.3 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.53. MS (ES+) m/z: 650.20 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 16.65 min.
4.1.22. Synthesis of acyclovir-5′-O-bis[(2,2-dimethylpropoxy-l-alaninyl)]-phosphate (33)
Prepared according to standard procedure B, using (10) (0.30 g, 1.33 mmol) in trimethylphosphate (5 mL), and POCl3 (0.12 mL, 1.33 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of 2,2-dimethylpropoxy-l-alanine (2.20 g, 6.65 mmol) in anhydrous CH2Cl2 (7 mL) were added, followed by dropwise addition of anhydrous DIPEA (2.32 mL, 13.30 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient (3%–10%) of MeOH in CH2Cl2. The product was dissolved in CH2Cl2 and washed with 0.05 N HCl (1 × 5 mL), brine (1 × 5 mL), 5% NaHCO3 (1 × 5 mL), brine (1 × 5 mL), dried over MgSO4, filtered and concentrated to give the product (33) as a white solid (22%, 0.17 g). 1H NMR (500 MHz, MeOD) δ H 7.92 (s, 1H, H-8), 5.52 (s, 2H, H-1′), 4.12–4.10 (m, 2H, H-5′), 3.98–3.95 (m, 2H, 2× CHCH3), 3.89–3.86 (m, 3H, H-4′, OCH 2C(CH3)3), 3.79–3.76 (m, 3H, H-4′, OCH 2C(CH3)3), 1.42–1.39 (m, 6H, 2× CHCH 3), 0.96 (s, 9H, OCH2C(CH 3)3), 0.95 (s, 9H, OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 174.77 (d, J C–P = 5.9 Hz, CO), 174.72 (d, J C–P = 5.2 Hz, CO), 159.49 (C-6), 155.76 (C-2), 153.54 (C-4), 139.92 (C-8), 117.38 (C-5), 75.39 (d, J C–P = 5.7 Hz, C-4′), 73.81 (C-1′), 69.72 (d, J C–P = 7.0 Hz, OCH2C(CH3)3), 65.76 (d, J C–P = 4.6 Hz, C-5′), 49.95 (CHCH3), 32.37 (OCH2 C(CH3)3), 26.86 (OCH2C(CH3)3), 21.14 (d, J C–P = 6.0 Hz, CHCH3), 21.06 (d, J C–P = 6.1 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.69. MS (ES+) m/z: 610.26 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 18.01 min.
4.1.23. Synthesis of abacavir-5′-O-bis(benzoxy-l-alaninyl)-phosphate (34)
Prepared according to standard procedure B, using (11) (0.20 g, 0.69 mmol) in trimethylphosphate (5 mL), and POCl3 (0.06 mL, 0.69 mmol), the reaction mixture was stirred at −5 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of benzoxy-l-alanine (1.23 g, 3.49 mmol) in CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (1.22 mL, 6.98 mmol). The reaction mixture was stirred at room temperature for 15 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with CHCl3/MeOH (95/5) to afford the product (34) as a white foam (47%, 0.23 g). 1H NMR (500 MHz, MeOD) δ H 7.67 (s, 1H, H-8), 7.32–7.26 (m, 10H, 2× OCH2 Ph), 6.08 (dt, J = 5.7, 2.1 Hz, 1H, H-2′), 5.90 (dt, J = 5.6, 2.2 Hz, 1H, H-3′), 5.49–5.46 (m, 1H, H-1′), 5.11–5.02 (m, 4H, 2× OCH 2Ph), 3.96–3.86 (m, 4H, H-5′, 2× CHCH3), 3.03–3.00 (m, 1H, H-4′), 2.90–2.87 (m, 1H, CH–cPr), 2.71 (dt, J = 13.9, 8.5 Hz, 1H, H-6′), 1.59 (dt, J = 13.8, 6.2 Hz, 1H, H-6′), 1.35–1.31 (m, 6H, 2× CHCH 3), 0.84–0.80 (m, 2H, CH 2–cPr), 0.58–0.55 (m, 2H, CH 2–cPr). 13C NMR (125 MHz, MeOD): δ C 175.50, 175.40 (CO), 161.90, 157.50 (C-6, C-2, C-4), 138.20 (C-2′), 137.37, 137.30 (ipso OCH2 Ph), 137.10 (C-8), 131.50 (C-3′), 129.62, 129.60, 129.38, 129.35, 129.32 (OCH2 Ph), 114.90 (C-5), 69.00 (d, J C–P = 5.4 Hz, C-5′), 67.80 (OCH2Ph), 60.50 (C-1′), 51.10 (CHCH3), 47.10 (d, J C–P = 8.3 Hz, C-4′), 35.90 (C-6′), 24.30 (CH–cPr), 20.70 (d, J C–P = 6.0 Hz, CHCH3), 20.60 (d, J C–P = 5.9 Hz, CHCH3), 7.67 (CH2–cPr). 31P NMR (202 MHz, MeOD) δ P 13.52. MS (ES+) m/z: 689.30 (M + H+, 100%). Reverse-phase HPLC eluting with H2O/MeOH from 90/10 to 0/100 in 25 min, flow = 1 mL/min, λ = 254 nm t R = 22.15 min.
4.1.24. Synthesis of abacavir-5′-O-bis-(2,2-dimethylpropoxy-l-alaninyl)-phosphate (35)
Prepared according to standard procedure B, using (11) (0.20 g, 0.69 mmol) in trimethylphosphate (5 mL), and POCl3 (0.07 mL, 0.60 mmol), the reaction mixture was stirred at −5 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of 2,2-dimethylpropoxy-l-alanine (1.16 g, 3.49 mmol) in CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (1.22 mL, 6.98 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with CHCl3/MeOH (95:5) to afford the product (35) as a colorless oil (44%, 0.20 g). 1H NMR (500 MHz, MeOD) δ H 7.70 (s, 1H, H-8), 6.16 (dt, J = 5.5, 2.0 Hz, 1H, H-2′), 5.95 (dt, J = 5.5, 2.0 Hz, 1H, H-3′), 5.55–5.51 (m, 1H, H-1′), 4.05–3.92 (m, 4H, H-5′, 2× CHCH3), 3.85, 3.84, 3.74, 3.70 (2AB, J AB = 10.5 Hz, 4H, 2× OCH 2C(CH3)3), 3.15–3.12 (m, 1H, H-4′), 2.93–2.91 (m, 1H, CH–cPr), 2.80 (dt, J = 13.8, 8.6 Hz, 1H, H-6′), 1.69 (dt, J = 13.5, 6.6 Hz, 1H, H-6′), 1.38–1.41 (m, 6H, 2× CHCH 3), 0.96 (s, 18H, 2× OCH2C(CH 3)3), 0.86–0.82 (m, 2H, CH 2–cPr), 0.62–0.59 (m, 2H, CH 2–cPr). 13C NMR (125 MHz, MeOD) δ C 175.8 (d, J C–P = 5.0 Hz, CO), 175.7 (d, J C–P = 4.8 Hz, CO), 161.9, 157.5 (C-2, C-6, C-4), 138.1 (C-2′), 137.1 (C-8), 131.7 (C-3′), 114.9 (C-5), 75.38, 75.34 (OCH2C(CH3)3), 69.1 (d, J C–P = 5.5 Hz, C-5′), 60.5 (C-1′), 51.1 (CHCH3), 47.2 (d, J C–P = 8.3 Hz, C-4′), 36.0 (C-6′), 32.35, 32.33 (OCH2 C(CH3)3), 26.80, 26.79 (OCH2C(CH3)3), 24.3 (CH–cPr), 21.06 (d, J C–P = 6.1 Hz, CHCH3), 21.02 (d, J C–P = 5.8 Hz, CHCH3), 7.67, 7.64 (CH2–cPr). 31P NMR (202 MHz, MeOD) δ P 13.65. MS (ES+) m/z: 649.36 (M + H+, 100%). Reverse-phase HPLC eluting with H2O/MeOH 90/10 to 0/100 in 25 min, flow = 1 mL/min, λ = 254 nm, t R = 23.99 min.
4.1.25. Synthesis of 3-(2′-deoxy-β-d-ribofuranosyl)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one-5′-O-bis-(benzoxy-l-alaninyl)-phosphate (36)
Prepared according to standard procedure B, using (12) (0.26 g, 0.65 mmol) in trimethylphosphate (5 mL), and POCl3 (0.06 mL, 0.65 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (5 mL) and a suspension of tosylate salt of benzoxy-l-alanine (1.14 g, 3.25 mmol) in anhydrous CH2Cl2 (5 mL) were added, followed by dropwise addition of anhydrous DIPEA (1.13 mL, 6.50 mmol). The reaction mixture was stirred at room temperature for 20 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with gradient of MeOH (2%–10%) in CH2Cl2 to give the product (36) as a yellow solid (19%, 0.10 g). 1H NMR (500 MHz, MeOD) δ H 8.70 (s, 1H, H-4), 7.64 (d, J = 8.3 Hz, 2H, Ph), 7.36–7.21 (m, 12H, Ph, OCH2 Ph), 7.08 (s, 1H, H-5), 6.30 (t, J = 6.0 Hz, 1H, H-1′), 5.17–5.07 (m, 4H, 2× OCH 2Ph), 4.44–4.41 (m, 1H, H-3′), 4.31–4.28 (m, 1H, H-4′), 4.20–4.16 (m, 2H, 2× CHCH3), 4.05–3.95 (m, 2H, H-5′), 2.68–2.63 (m, 1H, H-2′), 2.60 (t, J = 7.8 Hz, 2H, α-CH 2), 2.24–2.19 (m, 1H, H-2′), 1.61 (qn, J = 7.5 Hz, 2H, β-CH 2), 1.41 (d, J = 7.2 Hz, 3H, CHCH 3), 1.37–1.28 (m, 7H, γ-CH 2, δ-CH 2, CHCH 3), 0.90 (t, J = 6.9 Hz, 3H, CH2CH 3). 13C NMR (125 MHz, MeOD) δ C 175.45 (d, J C–P = 5.6 Hz, CO), 175.39 (d, J C–P = 4.5 Hz, CO), 172.91 (C-7a), 157.08 (C-2), 156.65 (C-6), 146.19 (para-C), 138.63 (C-4), 137.43, 137.21, 137.19, 130.16, 129.95, 129.63, 129.57, 129.37, 129.33, 129.30, 129.26, 129.20, 129.10, 128.28, 128.02, 127.33, 125.94 (OCH2 Ph, Ph), 110.17 (C-4a), 99.45 (C-5), 90.00 (C-1′), 87.60 (d, J C–P = 8.2 Hz, C-4′), 71.30 (C-3′), 67.97, 67.92 (OCH2Ph), 65.64 (d, J C–P = 4.8 Hz, C-5′), 51.18 (CHCH3), 42.63 (C-2′), 36.77, 32.62, 32.14, 23.61 ((CH2)4), 20.92 (d, J C–P = 5.7 Hz, CHCH3), 20.82 (d, J C–P = 6.4 Hz, CHCH3), 14.48 (CH3). 31P NMR (202 MHz, MeOD) δ P 14.03. MS (ES+) m/z: 823.29 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 26.51 min.
4.1.26. Synthesis of 3-(2′-deoxy-β-d-ribofuranosyl)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one-5′-O-bis-(2,2-dimethylpropoxy-l-alaninyl)-phosphate (37)
Prepared according to standard procedure B, using (12) (0.30 g, 0.75 mmol) in trimethylphosphate (4 mL), and POCl3 (0.07 mL, 0.75 mmol), the reaction mixture was stirred at −5 °C for 4 h. Anhydrous CH2Cl2 (6 mL) and a suspension of tosylate salt of 2,2-dimethylpropoxy-l-alanine (1.25 g, 3.75 mmol) in anhydrous CH2Cl2 (6 mL) were added, followed by dropwise addition of anhydrous DIPEA (1.31 mL, 7.50 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with gradient of MeOH (3%–5%) in CH2Cl2 to give a pale yellow solid. The product was dissolved in CH2Cl2 and washed with 0.05 N HCl (2 × 5 mL), brine (1 × 5 mL), 5% NaHCO3 (2 × 5 mL), brine (1 × 5 mL), dried over MgSO4, filtered and concentrated to give the product (37) as a pale yellow solid (10%, 0.06 g). 1H NMR (500 MHz, MeOD) δ H 8.79 (s, 1H, H-4), 7.72 (d, 2H, J = 8.3 Hz, Ph), 7.28 (d, 2H, J = 8.3 Hz, Ph), 7.19 (s, 1H, H-5), 6.35 (t, J = 6.0 Hz, 1H, H-1′), 4.50–4.48 (m, 1H, H-3′), 4.38–4.34 (m, 1H, H-4′), 4.29–4.23 (m, 2H, H-5′), 4.05–3.94 (m, 2H, 2× CHCH3), 3.91–3.85 (m, 4H, 2× OCH 2C(CH3)3), 2.70–2.64 (m, 3H, H-2′, α-CH 2), 2.32–2.26 (m, 1H, H-2′), 1.65 (qn, J = 7.5 Hz, 2H, β-CH 2), 1.46 (d, J = 7.2 Hz, 3H, CHCH 3), 1.41 (d, J = 7.2 Hz, 3H, CHCH 3), 1.38–1.31 (m, 4H, γ-CH 2, δ-CH 2), 0.87 (t, J = 6.9 Hz, 3H, CH 3). 13C NMR (125 MHz, MeOD): δ C 175.72 (d, J C–P = 6.4 Hz, CO), 175.58 (d, J C–P = 4.9 Hz, CO), 173.02 (C-7a), 157.20 (C-2), 156.73 (C-6), 146.29 (para-C), 138.72 (C-4), 130.17, 127.39, 125.96 (Ph), 110.28 (C-4a), 99.51 (C-5), 89.94 (C-1′), 87.24 (d, J C–P = 8.2 Hz, C-4′), 75.51, 75.40 (OCH2C(CH3)3), 71.16 (C-3′), 65.67 (d, J C–P = 4.8 Hz, C-5′), 51.16 (CHCH3), 51.08 (d, J C–P = 2.0 Hz, CHCH3), 42.61 (C-2′), 36.77, 32.57, 32.34, 32.30, 32.15, 23.59 (OCH2 C(CH3)3, (CH2)4), 26.82, 26.77 (OCH2C(CH3)3), 21.22 (d, J C–P = 5.5 Hz, CHCH3), 21.13 (d, J C–P = 6.3 Hz, CHCH3), 14.45 (CH3). 31P NMR (202 MHz, MeOD) δ P 14.18. MS (ES+) m/z: 783.35 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 0/100 in 30 min, flow = 1 mL/min, λ = 254 nm, t R = 29.08 min.
4.1.27. Synthesis of acadesine-5′-O-bis(benzoxy-l-alaninyl)-phosphate (38)
Prepared according to standard procedure B, using (13) (0.30 g, 1.16 mmol) in trimethylphosphate (4 mL), and POCl3 (0.11 mL, 1.16 mmol), the reaction mixture was stirred at −5 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of benzoxy-l-alanine (2.04 g, 5.80 mmol) were added, followed by dropwise addition of DIPEA (2.00 mL, 11.61 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–5%) in CH2Cl2 to afford the product (38) as a colorless oil (2%, 0.02 g). 1H NMR (500 MHz, MeOD) δ H 7.40–7.31 (m, 11H, OCH2 Ph and H-5), 5.56 (d, J = 6.0 Hz, 1H, H-1′), 5.18–5.10 (m, 4H, 2× OCH 2Ph), 4.41 (dd, J = 6.0, 6.1 Hz, 1H, H-2′), 4.23 (dd, J = 5.0, 3.5 Hz, 1H, H-3′), 4.16–4.09 (m, 3H, H-4′, H-5′), 3.97–3.89 (m, 2H, 2× CHCH3), 1.40–1.31 (m, 6H, 2× CHCH 3). 13C NMR (125 MHz, MeOD) δ C 175.46 (d, J C–P = 5.0 Hz, CO), 175.40 (d, J C–P = 5.1 Hz, CO), 169.25 (CONH2), 145.44 (C-2), 130.21 (C-5), 137.29 (ipso OCH2 Ph), 129.63, 129.62, 129.39, 129.36, 129.32, 129.24 (OCH2 Ph), 113.82 (C-3), 89.54 (C-1′), 85.00 (d, J C–P = 7.1 Hz, C-4′), 74.71 (C-2′), 71.42 (C-3′), 68.01 (OCH2Ph), 66.27 (d, J C–P = 5.0 Hz, C-5′), 51.17 (CHCH3), 20.75 (d, J C–P = 6.2 Hz, CHCH3), 20.58 (d, J C–P = 7.2 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.73. MS (ES+) m/z: 683.21 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 90/10 to 70/30 in 10 min; 70/30 to 40/60 in 20 min; 40/60 to 0/100 in 5 min; flow = 1 mL/min, λ = 265 nm, t R = 18.72 min.
4.1.28. Synthesis of acadesine-5′-O-bis(methoxy-l-alaninyl)-phosphate (39)
Prepared according to standard procedure B, using (13) (0.20 g, 0.77 mmol) in trimethylphosphate (3.5 mL), and POCl3 (0.18 mL, 1.77 mmol), the reaction mixture was stirred at −5 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and chloridate salt of methoxy-l-alanine (0.54 g, 3.87 mmol) were added, followed by dropwise addition of anhydrous DIPEA (1.30 mL, 7.74 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–20%) in CH2Cl2 to afford the product (39) as a colorless oil (3%, 0.01 g). 1H NMR (500 MHz, MeOD) δ H 7.41 (s, 1H, H-5), 5.56 (d, 1H, J = 6.0 Hz, H-1′), 4.45 (dd, J = 6.2, 6.0 Hz, 1H, H-2′), 4.26 (dd, 1H, J = 5.5, 3.0 Hz, H-3′), 4.21–4.19 (m, 3H, H-4′, H-5′), 3.88–3.84 (m, 2H, 2× CHCH3), 3.73 (s, 3H, OCH 3), 3.72 (s, 3H, OCH 3), 1.39–1.34 (m, 6H, 2× CHCH 3). 13C NMR (125 MHz, MeOD) δ C 176.16 (d, J C–P = 4.6 Hz, CO), 176.09 (d, J C–P = 5.5 Hz, CO), 169.29 (CONH2), 145.46 (C-2), 130.19 (C-5), 113.78 (C-3), 89.57 (C-1′), 85.07 (d, J C–P = 7.5 Hz, C-4′), 74.68 (C-2′), 71.53 (C-3′), 66.22 (d, J C–P = 4.6 Hz, C-5′), 52.80 (OCH3), 51.00 (d, J C–P = 1.7 Hz, CHCH3), 51.99 (d, J C–P = 1.7 Hz, CHCH3), 20.83 (d, J C–P = 6.4 Hz, CHCH3), 20.65 (d, J C–P = 6.4 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.88. MS (ES+) m/z: 531.15 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 80/20 in 10 min; 80/20 to 70/30 in 10 min; 70/30 to 0/100 in 10 min; flow = 1 mL/min, λ = 265 nm, t R = 10.43 min.
4.1.29. Synthesis of acadesine-5′-O-bis(2,2-dimethylpentoxy-l-alaninyl)-phosphate (40)
Prepared according to standard procedure B, using (13) (0.30 g, 1.16 mmol) in trimethylphosphate (4 mL), and POCl3 (0.11 mL, 1.16 mmol), the reaction mixture was stirred at −5 °C for 5 h. Anhydrous CH2Cl2 (5 mL) and tosylate salt of 2,2-dimethylpropoxy-l-alanine (1.92 g, 5.80 mmol) were added, followed by dropwise addition of anhydrous DIPEA (2.00 mL, 11.61 mmol). The reaction mixture was stirred at room temperature for 16 h. After work-up, the crude residue was purified by silica gel column chromatography eluting with a gradient of MeOH (2%–10%) in CH2Cl2 to give the product (40) as a colorless oil (1%, 0.01 g). 1H NMR (500 MHz, MeOD) δ H 7.41 (s, 1H, H-5), 5.56 (d, J = 6.0 Hz, 1H, H-1′), 4.42 (dd, J = 5.6, 5.5 Hz, 1H, H-2′), 4.25 (dd, J = 5.5, 3.0 Hz, 1H, H-3′), 4.22–4.17 (m, 3H, H-4′, H-5′), 4.00–3.94 (m, 2H, 2× CHCH3), 3.90, 3.78 (2AB, J AB = 11.0 Hz, 4H, 2× OCH 2(CH3)3), 1.42 (d, J = 7.0 Hz, 3H, CHCH 3), 1.40 (d, J = 7.0 Hz, 3H, CHCH 3), 0.97 (s, 9H, OCH2C(CH 3)3), 0.96 (s, 9 H, OCH2C(CH 3)3). 13C NMR (125 MHz, MeOD) δ C 175.70 (d, J C–P = 2.6 Hz, CO), 175.67 (d, J C–P = 2.6 Hz, CO), 169.25 (CONH2), 145.44 (C-2), 130.21 (C-5), 113.82 (C-3), 89.58 (C-1′), 85.02 (d, J C–P = 7.5 Hz, C-4′), 75.46 (OCH2(CH3)3), 75.42 (OCH2(CH3)3), 74.68 (C-2′), 71.48 (C-3′), 66.22 (d, J C–P = 4.5 Hz, C-5′), 51.00 (d, J C–P = 1.9 Hz, CHCH3), 32.35 (OCH2 C(CH3)3), 26.79 (OCH2C(CH3)3), 21.07 (d, J C–P = 6.4 Hz, CHCH3), 20.93 (d, J C–P = 6.4 Hz, CHCH3). 31P NMR (202 MHz, MeOD) δ P 13.94. MS (ES+) m/z: 643.29 (M + Na+, 100%). Reverse-phase HPLC, eluting with H2O/CH3CN from 100/0 to 80/20 in 10 min; 80/20 to 50/50 in 20 min; 50/50 to 0/100 in 5 min; flow = 1 mL/min, λ = 265 nm, t R = 27.32 min.
4.2. Cytostatic activity assays
The tumor cells were seeded in 96-well microtiter plates and exposed to different concentrations of the test compounds. After 2 days (L1210, L1210/TK−) and 3 days (CEM, CEM/TK−, HeLa, HeLa TK−, Caco-2, Colo-320), cell number was determined using a Particle counter (Coulter Z-1, Analis, Ghent, Belgium). The IC50 represents the compound concentration required to inhibit tumor cell proliferation by 50%.
4.3. Antiviral assays
The antiviral assays [except anti-human immunodeficiency virus (HIV) assays] were based on inhibition of virus-induced cytopathicity in HEL [herpes simplex virus type 1 (HSV-1), HSV-2 (G), vaccinia virus and vesicular stomatitis virus], Vero (parainfluenza-3, reovirus-1, Sindbis, Coxsackie B4, and Punta Toro virus), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus), CrFK (feline herpes virus), feline corona virus (FIPV) and MDCK (influenza virus A (H1N1, H3N2) and B) cell cultures. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 cell culture inhibitory dose-50 (CCID50) of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures). After a 1 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (200, 40, 8,… μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV-1(IIIB) or HIV-2(ROD)/mL and seeded in 200 μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically. MT-4 cells (1_104 cells per mL) were suspended in fresh culture medium and infected with 10_L (0.7 ng of p24) of X4LAI.04 viral stock per mL of cell suspension. Infected cell suspensions were then transferred to microplate wells, mixed with 1 mL of medium containing the test compound at an appropriate dilution and further incubated at 37 °C. After 3 days, p24 production was measured in the MT-4 cell culture supernatants. The results are given as the mean ± standard error of the mean of the concentration required to suppress viral replication by 50% (median effective concentration [EC50]). The value of EC50 was calculated by fitting the data points to a sigmoidal dose–response curve, with Prism software, (version 4.0; GraphPad).
Viability assays. Viability assays were performed in the MT-4 cell cultures with the Nucleocounter automated cell counting system (ChemoMetec). Total number of cells and number of dead cells in the cultures untreated and treated with ACV ProTides were enumerated using a propidium iodide-based assay according to the manufacturers' protocol. Data were collected and analyzed using Nucleoview software (Chemometec, Denmark).
4.4. Enzymatic assays
Compound 32 (5.0 mg) or 40 (4.8 mg) were dissolved in d 6-acetone (0.15 mL) and Trizma buffer (pH = 7.6) (0.30 mL) and a 31P NMR was recorded (blank). Then a solution of carboxypeptidase Y (0.1 mg) in Trizma buffer (0.15 mL) was added and a 31P NMR experiment was performed recording the experiment every 5 min, for 24 h at room temperature.
Acknowledgment
This work was supported by Inhibitex, and by a grant from the KU Leuven to JB (GOA 10/014). The authors thank Ms. Helen Murphy for excellent secretarial assistance and Mrs. Leentje Persoons, Mrs. Frieda De Meyer, Mrs. Leen Ingels and Mrs. Lizette van Berckelaer for excellent technical assistance.
References
- 1.Robins R.K. The potential of nucleoside analogs as inhibitors of retroviruses and tumors. Pharm. Res. 1984:11–18. doi: 10.1023/A:1016370407633. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Wagner C.R., Iyer V.V., McIntee E.J. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000;20:417–451. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 4.Faletto M.B., Miller W.H., Garvey E.P., Clair M.H.St., Daluge S.M., Good S. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother. 1997;41:1099–1107. doi: 10.1128/aac.41.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rashidi M.R., Smith J.A., Clarke S.E., Beedham C. In vitro oxidation of famciclovir and 6-deoxypenciclovir by aldehyde oxidase from human, guinea pig, rabbit, and rat liver. Drug Metab. Dispos. 1997;25:805–813. [PubMed] [Google Scholar]
- 6.Furman P.A., Fyfe J.A., St. Clair M.H., Weinhold K., Rideout J.L., Freeman G.A., Nusinoff Lehrman S., Bolognesi D.P., Broder S., Mitsuya H., Barry D.W. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1986;83:8333–8337. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert C., Bestman-Smith J., Boivin G. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updates. 2002;5:88–114. doi: 10.1016/s1368-7646(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 8.Hecker S.J., Erion M.D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 9.Cahard D., McGuigan C., Balzarini J. Aryloxy phosphoramidate trimesters as Pro-Tides. Mini-Rev. Med. Chem. 2004;4:371–482. doi: 10.2174/1389557043403936. [DOI] [PubMed] [Google Scholar]
- 10.Mehellou Y., Balzarini J., McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. Chem. Med. Chem. 2009;11:1779–1791. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 11.McGuigan C., Madela K., Aljarah M., Gilles A., Brancale A., Zonta N., Chamberlain S., Vernachio J., Hutchins J., Hall A., Ames B., Ghorovits E., Ganguly B., Kolykhalov A., Wang J., Muhammad J., Patti J.M., Henson G. Design, Synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg. Med. Chem. Lett. 2010;20:4850–4854. doi: 10.1016/j.bmcl.2010.06.094. [DOI] [PubMed] [Google Scholar]
- 12.McGuigan C., Madela K., Aljarah M., Bourdin C., Arrica M., Barrett E., Jones S., Kolykhalov A., Bleiman B., Bryant K.D., Ganguly B., Gorovits E., Henson G., Hunley D., Hutchins J., Muhammad J., Obikhod A., Patti J., Walters C.R., Wang J., Vernachio J., Ramamurty C.V.S., Battina S.K., Chamberlain S. Phosphorodiamidates as a promising new phosphate prodrug motif for antiviral drug discovery: application to anti-HCV agents. J. Med. Chem. 2011;54:8632–8645. doi: 10.1021/jm2011673. [DOI] [PubMed] [Google Scholar]
- 13.Jansa P., Baszczyňski O., Dračínský M., Votruba I., Zídek Z., Bahador G., Stepan G., Cihlar T., Mackman R., Holý A., Janeba Z. A novel and efficient one-pot synthesis of symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities. Eur. J. Med. Chem. 2011;46:3748–3754. doi: 10.1016/j.ejmech.2011.05.040. [DOI] [PubMed] [Google Scholar]
- 14.Drontle D.P., Wagner C.R. Designing a pronucleotide stratagem: lessons from amino acid phosphoramidates of anticancer and antiviral pyrimidines. Mini-Rev. Med. Chem. 2004;4:409–419. doi: 10.2174/1389557043403945. [DOI] [PubMed] [Google Scholar]
- 15.Sofia M.J., Bao D., Chang W., Du J., Nagarathnam D., Rachakonda S., Reddy P.G., Ross B.S., Wang P., Zhang H.-R., Bansal S., Espiritu C., Keilman M., Lam A.M., Micolochick Steuer H.M., Niu C., Otto M.J., Furman P.A. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010;53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 16.Yoshikawa M., Kato T., Takenishi T. Studies of phosphorylation. Iii. Selective phosphorylation of unprotected nucleosides. Bull. Chem. Soc. Jpn. 1969;42:3505–3508. [Google Scholar]
- 17.Jones B.C.N.M., McGuigan C., O'Connor T.J., Jeffries D.J., Kinchington D. Synthesis and anti-HIV activity of some novel phosphorodiamidate derivatives of 3′-azido-3′-deoxythymidine (AZT) Antivir. Chem. Chemother. 1991;2:35–39. [Google Scholar]
- 18.McGuigan C., Murziani P., Slusarczyk M., Gonczy B., Voorde J.V., Liekens S., Balzarini J. Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem. 2011;54:7247–7258. doi: 10.1021/jm200815w. [DOI] [PubMed] [Google Scholar]
- 19.McGuigan C., Thiery J.-C., Daverio F., Jiang W.G., Davies G., Mason M. Anti-cancer ProTides: tuning the activity of BVDU phosphoramidates related to thymectacin. Bioorg. Med. Chem. 2005;13:3219–3227. doi: 10.1016/j.bmc.2005.02.041. [DOI] [PubMed] [Google Scholar]
- 20.Lisco A., Vanpouille C., Tchesnokov E.P., Grivel J.-C., Biancotto A., Brichacek B., Elliott J., Fromentin E., Shattock R., Anton P., Gorelick R., Balzarini J., McGuigan C., Derudas M., Gotte M., Schinazi R.F., Margolis L. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe. 2008;4:260–270. doi: 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrone P., Luoni G.M., Kelleher M.R., Daverio F., Angell A., Mulready S., Congiatu C., Rajyaguru S., Martin J.A., Levêque V., Le Pogam S., Najera I., Klumpp K., Smith D.B., McGuigan C. Application of the phosphoramidate ProTide approach to 4′-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J. Med. Chem. 2007;50:1840–1849. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 22.McGuigan C., Kelleher M.R., Perrone P., Mulready S., Luoni G., Daverio F., Rajyaguru S., Le Pogam S., Najera I., Martin J.A., Klumpp K., Smith D.B. The application of phosphoramidate ProTide technology to the potent anti-HCV compound 4′-azidocytidine (R1479) Bioorg. Med. Chem. Lett. 2009;19:4250–4254. doi: 10.1016/j.bmcl.2009.05.099. [DOI] [PubMed] [Google Scholar]
- 23.Derudas M., Brancale A., Naesens L., Neyts J., Balzarini J., McGuigan C. Application of the phosphoramidate ProTide approach to the antiviral drug ribavirin. Bioorg. Med. Chem. 2010;18:2748–2755. doi: 10.1016/j.bmc.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.M. Derudas, C. Vanpouille, A. Lisco, L. Margolis, J. Balzarini, C. McGuigan, Acyclic nucleoside ProTides: from antiherpetic to broad spectrum antiviral compounds, 241st ACS National Meeting, Anaheim, CA, USA, March 27th–31st 2011, MEDI-1.
- 25.Reddy P.G., Bao D., Chang W., Chun B.-K., Du J., Nagarathnam D., Rachakonda S., Ross B.S., Zhang H.-R., Bansal S., Espiritu C.L., Keilman M., Lam A.M., Niu C., Micolochick Steuer H., Furman P.A., Otto M.J., Sofia M.J. 2′-Deoxy-2′-α-fluoro-2′-β-C-methyl 3′,5′-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: discovery of PSI-352938. Bioorg. Med. Chem. Lett. 2010;20:7376–7380. doi: 10.1016/j.bmcl.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 26.Derudas M., Carta D., Brancale A., Vanpouille C., Lisco A., Margolis L., Balzarini J., McGuigan C. The application of phosphoramidate protide technology to acyclovir confers anti-HIV inhibition. J. Med. Chem. 2009;52:5520–5530. doi: 10.1021/jm9007856. [DOI] [PMC free article] [PubMed] [Google Scholar]