

Abstract
Circulating tumor cells (CTCs) are present at low concentrations in the peripheral blood of patients with solid tumors. It has been proposed that the isolation, ex vivo culture, and characterization of CTCs may provide an opportunity to noninvasively monitor the changing patterns of drug susceptibility in individual patients as their tumors acquire new mutations. In a proof-of-concept study, we established CTC cultures from six patients with estrogen receptor–positive breast cancer. Three of five CTC lines tested were tumorigenic in mice. Genome sequencing of the CTC lines revealed preexisting mutations in the PIK3CA gene and newly acquired mutations in the estrogen receptor gene (ESR1), PIK3CA gene, and fibroblast growth factor receptor gene (FGFR2), among others. Drug sensitivity testing of CTC lines with multiple mutations revealed potential new therapeutic targets. With optimization of CTC culture conditions, this strategy may help identify the best therapies for individual cancer patients over the course of their disease.
Circulating tumor cells (CTCs) are present in the blood of many patients with solid tumors. Most of these cells, which are thought to be involved in metastasis, die in the circulation, presumably due to the loss of matrix-derived survival signals or circulatory shear stress. Nonetheless, if CTCs can be isolated from cancer patients as viable cells that can be genotyped and functionally characterized over the course of therapy, they have the potential to identify treatments that most effectively target the evolving mutational profile of the primary tumor (1). The isolation of viable CTCs is technically challenging: Most methods yield low numbers of partially purified CTCs that are fixed before isolation, damaged during the cell purification process, or irreversibly immobilized on an adherent matrix [see review (2)]. We recently reported a microfluidic technology, the CTC-iChip, which efficiently depletes normal blood cells, leaving behind unmanipulated CTCs (3). The cytological appearance, staining properties, and intact RNA evident within a subset of CTCs isolated by means of this tumor antigen-agnostic CTC isolation platform suggested that the cells may be viable.
To investigate whether the CTCs were in fact viable, we applied the CTC-iChip to blood samples from patients with metastatic estrogen receptor (ER)–positive breast cancer. After testing a range of culture conditions (4–7) (see supplementary methods), we found that CTCs proliferated best as tumor spheres when cultured in serum-free media supplemented with epidermal growth factor (EGF) and basic fibroblast growth factor (FGF) (8) under hypoxic conditions (4% O2) (Fig. 1A). Nonadherent culture conditions were critical, because CTCs senesced after a few cell divisions in adherent monolayer culture (fig. S1). We established long-term oligoclonal CTC cultures (sustained in vitro for >6 months) from CTCs isolated from six patients with metastatic luminal subtype breast cancers (table S1). One or more CTC cell lines were successfully generated from 6 of 36 patients who were either off therapy or progressing on treatment. We were unable to generate CTC cell lines from nine patients who were responding to treatment at the time of attempted CTC culture. For three patients, four additional CTC cell lines were established from blood samples drawn at multiple different time points during therapy (table S1). In these cases, CTCs were successfully cultured only when patients were progressing on treatment (fig. S1).
Fig. 1. Ex vivo expansion of breast cancer CTCs.
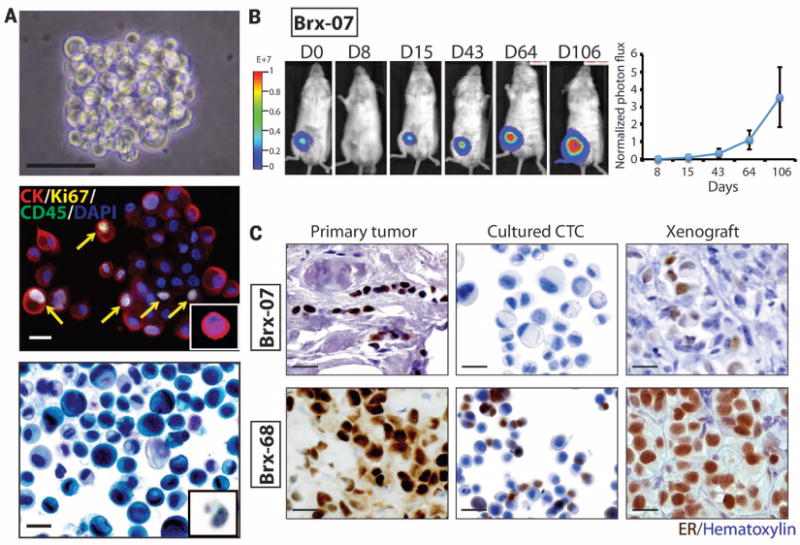
(A) Representative images of nonadherent CTC culture (BRx-07). Top: Phase contrast. Scale bar, 100 μm. Middle: immunofluorescent staining for cytokeratin (CK, red), Ki67 (yellow), CD45 (green), nuclei [4′,6-diamidino-2-phenylindole (DAPI), blue]. Scale bar, 20 μm. Bottom: Light microscopic imaging with Papanicolaou staining. Comparable images for uncultured primary CTCs are shown in the insets. Scale bar, 20 μm. (B) (Left) Bioluminescent images showing growth of NSG mouse xenografts, after implantation of 20,000 cultured CTCs (BRx-07) into the mammary fat pad. (Right) Quantification of bioluminescent signals for BRx-07–derived mouse xenografts (mean ± SD, n = 6). (C) Histology of matched primary breast tumors, cultured CTCs, and CTC-derived mouse xenografts for two CTC lines. All panels show cellular staining with hematoxylin (blue) and immunohistochemical staining for ER expression (brown). Scale bar, 20 μm.
Cultured CTCs shared cytological features with the matched primary CTCs captured on the CTC-iChip (Fig. 1A), and consistent with standard CTC definitions, they stained positive for epithelial cytokeratin (>95% of cells) and negative for the leukocyte marker CD45 (Fig. 1A) (fig. S2). The proliferative index of CTC cultures was ~30%, as defined by Ki67 staining (mean 28.1%, range 24 to 32%), and the initial doubling time of CTC cultures varied from 3 days to 3 weeks (table S1). All six primary tumors were positive for ER expression. Five CTC lines retained ER positivity in culture (>10% of cells), whereas one line (BRx-07) lost ER expression in vitro (Fig. 1C and fig. S2).
We undertook RNA sequencing analysis of each cell line and compared the results with those of 29 uncultured single CTCs from a total of 10 patients, as well as a panel of 13 commonly used established breast cancer cell lines, all using low-template single-cell resolution analysis (fig. S3). CTC cultures clustered with each other, and separately from established breast cancer cell lines or uncultured single CTCs. As expected, both CTC cultures and established breast cancer cell lines had increased proliferative signature, compared with primary uncultured single CTCs (fig. S3). We did not observe increased expression in CTC cultures of defined signaling pathways, including stem cell–related signatures, compared with established breast cancer cell lines.
To test the tumorigenicity of CTC lines, we used lentiviral transduction to label them with both green fluorescent protein (GFP) and luciferase and inoculated 20,000 cells into the mammary fat pad of immunosuppressed non-obese diabetic scid gamma (NSG) female mice implanted with subcutaneous estrogen pellets. Of five CTC lines tested, three (BRx-07, BRx-68, and BRx-61) generated tumors within 3 months at this low inoculum (Fig. 1B and figs. S4 and S5). CTC-derived tumors shared histological and immunohistochemical features with the matched primary patient tumor, including BRx-07, which regained ER expression (Fig. 1C).
All six patients with metastatic breast cancer had received sequential courses of hormonal and other therapies before CTC collection (fig. S6). As part of standard clinical care at Massachusetts General Hospital, a mutational panel [SNaPShot (9)] covering ~140 mutations in 25 genes had been performed on primary tumor specimens (BRx-68 and BRx-42) or on pretreatment biopsies of metastatic lesions (BRx-33, BRx-07, BRx-50 and BRx-61). Point mutations in PIK3CA (H1047R and G1049R), hot-spot mutations in breast cancer, were identified in two cases (BRx-68 and BRx-42), whereas no mutations were found in the four other cases (table S1). The availability of CTC cultures made it possible to undertake more comprehensive mutational analysis from a more abundant and purified tumor cell population. CTC lines were screened for mutations in a panel of 1000 annotated cancer genes, with a hybrid-capture–based next-generation sequencing (NGS) platform. The PIK3CA mutations identified by SNaPShot testing of primary tumors were confirmed by NGS in both CTC cultures (BRx-68 and BRx-42), and multiple additional mutations in other cancer-related genes were identified (Table 1). For all mutations identified in the 1000 cancer gene panel, candidate driver mutations were defined by their absence from matched germline DNA and by their annotation in pan-cancer (10) and COSMIC (Catalogue of somatic mutations in cancer) databases (Table 1), whereas additional mutations in known cancer genes were of uncertain relevance (table S2). To ensure that the candidate driver mutations were not acquired during the in vitro establishment of CTC cell lines, we tested for selected mutations in four additional CTC lines, which had been independently isolated at different time points from each of three patients (BRx-68, BRx-42, and BRx-61). The acquired mutations in ESR1 (BRx-68), TP53 (BRx-68, BRx-61), and KRAS (BRx-42) were universally present in all independent CTC cell lines (Table 1), confirming that they are tumor-derived mutations. In addition, the ESR1 mutation (Y537S) present in multiple BRx-68 CTC lines was also detectable by direct RNA sequencing of uncultured CTCs isolated from this patient (fig. S7).
Table 1.
Mutations detected in cultured CTC lines.
| Case | Gene | DNA | Protein | Allele frequency† | In pretreatment tumor‡ | In multiple CTC lines | Known mutation§ |
|---|---|---|---|---|---|---|---|
| BRx33‖ | ESR1 | A1613G | D538G | 0.24 | – | – | Br,# En |
| NUMA1 | C5501T | S1834L | 0.39 | – | – | Br | |
| BRx07‖ | TP53 | G853A | E285K | 0.99 | No | – | Bl, Br, Co, HN, Lu |
| PIK3CA | A3140T | H1047L | 1 | No | – | Br, Co, GBM, HN, Ki, Lu, Me, Mel, Ov, En | |
| FGFR2 | T1647A | N549K | 0.46 | No | – | Br, En | |
| CDH1 | C790T | Q264* | 1 | Yes | – | Br | |
| APC | G7225A | G2409R | 0.47 | Yes | – | Mel | |
| DGKQ | G2530A | D844N | 0.55 | – | – | Lu | |
| MAML2 | A2569G | M857V | 0.52 | – | – | Lu | |
| BRx68 | TP53 | C1009T | R337C | 0.99 | No | Yes | Br, Co, HN, Hem, Ov |
| ESR1 | A1610C | Y537S | 0.47 | No | Yes | Br#, En | |
| PIK3CA | A3140G | H1047R | 0.7 | Yes | Yes | Br, Co, GBM, HN, Ki, Lu, Me, Mel, Ov, En | |
| MSN | G1153A | E385K | 0.25 | – | – | En | |
| BRx50‖ | ESR1 | T1607C | L536P | 0.06†† | – | – | Br# |
| IKZF1 | G1444T | G482C | 0.09 | – | – | Hem | |
| BRCA2¶ | T6262del | L2039fs | – | – | – | Br (germ line) | |
| BRx42 | PIK3CA | G3145C | G1049R | 0.60 | Yes | Yes | Br, En, Ki |
| PIK3CA | C1097G | P366R | 0.54 | – | – | Br | |
| KRAS | G35T | G12V | 0.99 | No | Yes | Br, Co, Hem, Es, GBM, Lu, Ov, En | |
| IGF1R | G3613A | A1205T | 0.06 | – | – | Hem | |
| BRx61 | TP53 | G610T | E204* | 0.98 | No | Yes | Bl, Br, Ki, Lu, Ov |
Mutant allele frequency within oligoclonal cultured CTC populations was calculated as the ratio of mutant sequence reads to total reads for each gene.
Where sufficient material was available for analysis, matched archival pretreatment tumor specimens were subjected to Sanger sequencing to confirm selected mutations identified in CTC cultures. Insufficient tumor material is marked (–).
List of tumor types reported to harbor the same mutation in pan-cancer (10) or COSMIC databases. Abbreviations: breast (Br), endometrial (En), central nervous system (CNS), bladder (Bl), colorectal (Co), pancreas (Pa), stomach (St), head and neck (HN), lung (Lu), thyroid (Th), glioblastoma (GBM), kidney (Ki), prostate (Pr), medulloblastoma (Me), melanoma (Mel), ovarian (Ov), cervix (Ce), esophageal (Es), hematopoietic and lymphoid tissue (Hem), sarcoma (Sar), cholangiocarcinoma (Ch).
Cases for which DNA from matched normal tissue was not available.
Germline BRCA2 mutation was detected as part of genetic counseling for familial breast cancer. #Mutations reported in recent publications (12–15). ††ESR1 T1607C mutant allele frequency increased to 0.49 after prolonged in vitro culture under low-estrogen conditions (>6 months).
Chain termination codon. fs: frameshift mutation. Abbreviations for amino acid residues: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
Activating mutations in the estrogen receptor (ESR1) were first identified in 1997 and are rare in primary breast cancer (11). While this manuscript was in preparation, multiple research groups reported ESR1 mutations in 18 to 54% of patients treated with aromatase inhibitors (AIs), drugs that suppress estrogen synthesis and thus may favor the emergence of these ligand-independent ER mutants (12–15). We also detected ESR1 mutations in three of six CTC lines (BRx-33, BRx-68, and BRx-50). Each of these patients had received extensive treatment with AIs, and reanalysis of the primary tumor or the pre-AI treatment biopsy of a metastatic lesion showed no evidence of ESR1 mutations (Table 1). Other mutations identified included newly arising mutations in PIK3CA, TP53, KRAS, and fibroblast growth factor receptor–2 (FGFR2) (Table 1). Consistent with its lobular histological subtype, an E-cadherin (CDH1) mutation was detected in one CTC line (BRx-07). Although most mutant allele frequencies indicated heterozygous or homozygous truncal mutations shared by all CTCs, rare mutated alleles consistent with emerging tumor subpopulations were also evident. An ESR1 mutation initially present at 6% allele frequency in BRx-50 increased to 49% allele frequency upon prolonged culture in low-estrogen–containing medium (Table 1), suggesting a proliferative advantage under these conditions. Notably, TP53 mutations, which are thought to be rare in primary luminal breast cancers (16), emerged during tumor progression in three of six cases.
The availability of comprehensive tumor cell genotyping brings with it the challenge of identifying the subset of mutations whose therapeutic targeting is likely to be beneficial to an individual patient. To begin to explore this opportunity, we tested CTC lines for sensitivity to panels of single drug and drug combinations, including standard clinical regimens, as well as experimental agents targeting specific mutations. Conditions were optimized for highly reproducible testing of viability in small numbers of cells (200 cells per well) cultured as aggregates in solution. For each drug, we tested five concentrations (table S3), centered around median inhibitory concentration (IC50) levels established in large-scale cancer cell line screens (17), with relative sensitivity or resistance defined by comparison among the CTC cell lines (Fig. 2 and figs. S8 to S10). Although CTC drug sensitivity testing was blinded to clinical history, and patient treatment selections were not informed by CTC testing, some CTC drug sensitivity measurements were concordant with clinical histories, including sensitivity to paclitaxel (BRx-07) and capecitabine (BRx-68 and BRx-50), and resistance to fulvestrant (BRx-07 and BRx-68), doxorubicin (BRx-07), and olaparib (BRx-50) (fig. S11).
Fig. 2. Drug sensitivity of cultured CTCs.
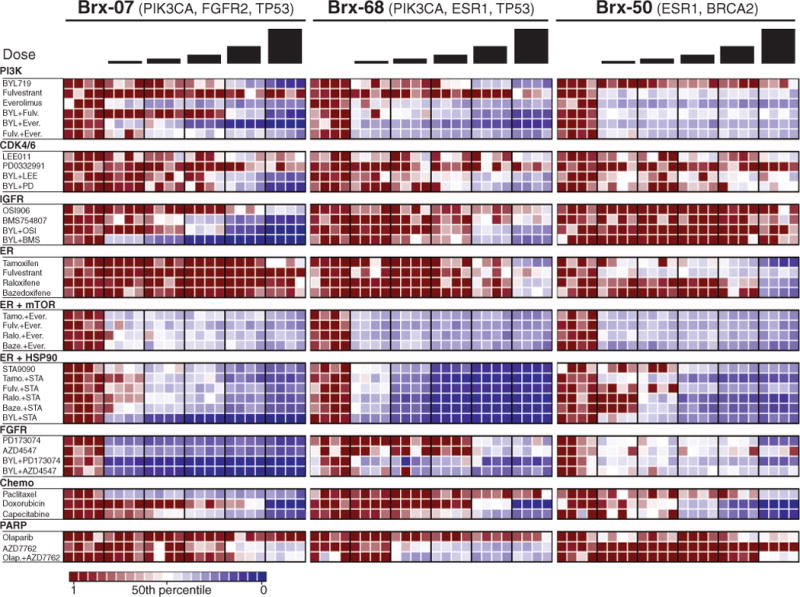
Heatmaps representing cell viability after treatment of BRx-07, BRx-68, and BRx-50 CTC lines with selected anticancer drugs, either alone or in combination. The presumed driving mutation for each CTC line is noted, and drugs are grouped according to therapeutic class and targeted pathway. For each drug, the range of concentrations tested is centered around the IC50 derived from large-scale breast cancer cell line screens (17), and each concentration represents a twofold increase from the previous dose, with each concentration tested in quadruplicate. Drug concentrations are listed in table S3. Signal from viable cells remaining after drug treatment is normalized to corresponding vehicle [dimethyl sulfoxide (DMSO)]–treated controls, with ratios plotted ranging from red (more viable) to blue (less viable). Drug abbreviations: BYL, BYL719; Fulv, fulvestrant; Ever, everolimus; LEE, LEE011; PD, PD0332991; OSI, OSI906; BMS, BMS754807; Tamo, tamoxifen; Ralo, raloxifene; Baze, bazedoxifene; STA, STA9090; Olap, Olaparib.
We selected two mutated drug targets identified in CTCs but not in the primary tumor for more detailed analysis; namely, ESR1 and PIK3CA mutations (additional drug responses in cultured CTCs are shown in fig. S12). To facilitate interpretation of the effect of drug combinations, responses to selected drugs are represented in a 2 × 2 matrix highlighting cooperative drug effects versus independent cytotoxicity (Fig. 3; see quantitation in fig. S13). The three de novo acquired ESR1 mutations affected distinct but adjacent residues within the ER ligand-binding domain and were present at different allele frequencies within the oligoclonal CTC cell lines. The most commonly reported ESR1 mutation, Y537S (12–14), was observed in BRx-68 (47% allele frequency, consistent with a heterozygous mutation in all cells), with two other mutations, D538G and L536P, in BRx-33 and BRx-50 (24 and 6% allele frequencies, respectively). Each mutation arose within the context of distinct additional mutations (Table 1 and table S2). Of note, all ESR1 mutation-positive CTC lines maintained ER expression in culture.
Fig. 3. Combinatorial drug targeting of mutant ESR1 and PIK3CA in CTC lines.
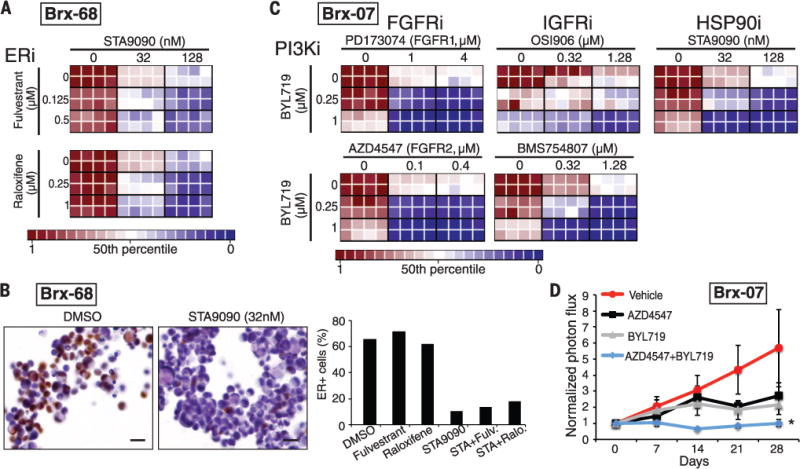
(A) Heatmaps representing cell viability in the BRx-68 CTC line, carrying an ESR1 mutation (allele frequency 47%), treated with HSP90 inhibitor (STA9090) together with the selective estrogen receptor modulator (SERM) tamoxifen or degrader (SERD) fulvestrant. For these drug-combination studies, the concentrations of each drug was varied independently, and results are shown in eight replicates. Cooperative drug interactions are represented by a diagonal gradient, showing increasing cell killing as both drug concentrations increase independently. (B) Down-regulation of ER protein expression measured by immunohistochemical staining (brown) of BRx-68 CTC cultures treated for 24 hours with an HSP90 inhibitor (STA9090) versus vehicle (DMSO). Nuclei are stained with hematoxylin. Scale bar, 20 μm. Bar graph shows quantification of percent ER-positive cells. More than 200 cells were quantified in each condition. (C) Heatmaps representing cell viability in the BRx-07 line harboring mutations in PIK3CA (99% allele frequency) and FGFR2 (46% allele frequency). Drugs targeting the products of these mutated oncogenic drivers were tested, along with compounds inhibiting nonmutated targets (IGFR and HSP90). Drug combinations shown are PI3Ki + FGFRi; PIK3Ki + IGFRi; PIK3Ki + HSP90i. (D) Response of BRx-07 CTC-derived mouse xenografts to the PI3K inhibitor BYL719 (n = 4), the FGFR2 inhibitor AZD4547 (n = 3), the combination of the two inhibitors (BYL719+AZD4547) (n = 4), or diluent control (n = 4). Mean ± SD. In vivo drug administration was initiated after mammary fat pad inoculation with genotyped CTC cultures and establishment of an expanding tumor xenograft, and tumor-derived bioluminescent measurements were normalized to pretreatment levels.
The optimal therapy for breast cancer patients whose ER+ tumor has acquired an ESR1 mutation is unknown; consistent with previous models (12–14, 18), we found that the selective estrogen receptor modulators (SERMs) tamoxifen and raloxifene, and the selective ER degrader (SERD) fulvestrant, were ineffective in BRx-68 cells, either alone or in the clinically approved combination with inhibitors of the phosphatidylinositol 3-kinase–mammalian target of rapamycin (PI3K-mTOR) pathway (everolimus) (19) (Fig. 2). However, the HSP90 inhibitor STA9090 demonstrated cytotoxicity alone and in combination with both raloxifene and fulvestrant (Fig. 3A). ER is a client protein for HSP90, and mutated receptors are highly dependent on this chaperone for their stability (20). Indeed, treatment with a low dose of STA9090 (32 nM) suppressed ER levels in BRx-68 cells but had no effect in MCF7 breast cancer cells with wild-type ER, or in BRx-50 cells, where the low allele frequency of mutant ESR1 is not associated with sensitivity to HSP90 inhibitors (Fig. 3B and figs. S12 to S14). Clinical studies of HSP90 inhibitors, along with novel ER inhibitors, will be required to define the optimal treatment for breast cancer patients whose tumor has acquired an ESR1 mutation.
The BRx-07 cell line is noteworthy because it harbors activating mutations in both PIK3CA and FGFR2, both of which were acquired de novo during the course of therapy. Based on their respective allele frequencies, PIK3CA was homozygously mutated in all cells, whereas the FGFR2 mutation was heterozygous (Table 1). Cultured CTCs were highly sensitive to the PIK3CA inhibitor BYL719 (21) and the FGFR2 inhibitor AZD4547 (22), and moderately responsive to the FGFR1 inhibitor PD173074 (23) (Fig. 2). Combined inhibition of both PIK3CA and FGFR2 showed cooperative effects (Fig. 3C and fig. S13), suggesting that both of these mutations may function as acquired oncogenic drivers in this tumor. Because combinations of PIK3CA and FGFR inhibitors have not been tested in clinical settings, we further quantified responses in a panel of established breast cancer cell lines. Of seven PIK3CA-mutant breast cancer lines, six were responsive to BYL719 (fig. S15). In addition to their characteristic PIK3CA mutation, two lines harbored mutations of unknown importance in FGFR4 (Y367C; MDA-MB-453 cells) and in FGFR2 (K570E; EFM-19 cells). The former showed cooperative cytotoxicity by BYL719 and AZD4547, whereas the latter was insensitive to FGFR inhibition (fig. S15). One of five PIK3CA-mutant breast cancer lines without an FGFR gene mutation showed modest sensitivity to AZD4547 (CAL51), whereas the other four were resistant. Thus, the combination of genotyping and functional testing for drug susceptibility is essential to defining therapeutically relevant driver mutations in both breast cancer cell lines and CTC cultures.
In vitro screening of additional drugs for cooperation with PIK3CA-targeted agents identified inhibitors of the insulin-like growth factor receptor 1 (IGF1R, inhibitors OSI906 and BMS754807) and HSP90 (inhibitor STA9090, Ganetespib) (Fig. 3C). Although neither of these is mutated in BRx-07 cells, IGF1R has been implicated in modulating signaling loops that mitigate sensitivity to PI3K inhibitors (24), and HSP90 is involved in stabilization of mutant kinases (20). To extend drug sensitivity studies to mouse xenografts, we generated BRx-07–derived mammary tumors and treated these with BYL719, AZD4547, the two agents in combination, or diluent control. In vivo tumor suppression was observed after treatment with either drug individually, whereas the combination completely abrogated tumor growth (Fig. 3D).
In this proof-of-concept study, we have shown that the culture of tumor cells circulating in the blood of patients with breast cancer provides an opportunity to study patterns of drug susceptibility, linked to the genetic context that is unique to an individual tumor. In patients with hormone-responsive breast cancer, most of whom have bone metastases that are not readily biopsied, the ability to noninvasively and repeatedly analyze live tumor cells shed into the blood from multiple metastatic lesions may enable monitoring of emerging subclones with altered mutational and drug sensitivity profiles. The successful culture of CTCs stems partly from the application of a microfluidic device capable of effectively depleting leukocytes from a blood specimen while preserving viable tumor cells for ex vivo expansion (3). The proliferation of cultured CTCs as non-adherent spheres differs from that of characteristic epithelial cancer cell cultures and may reflect intrinsic properties of tumor cells that remain viable in the bloodstream after loss of attachment to basement membrane. A recent report documented direct inoculation of the mouse femur with blood-derived cancer cells from a patient who had very high numbers of CTCs, but in vitro culture was not successful (25). Our results differ from the adherent in vitro CTC cultures described by Zhang et al. (26), but these lines appear to share the identical TP53, BRAF, and KRAS genotype of the highly tumorigenic MDA-MB-231 cell line.
Optimization of CTC culture conditions will be needed before this strategy can be incorporated into clinical practice. In addition, further characterization of the nonadherent CTC-derived cell lines described here will be required to define how they differ from cells cultured from primary tumor biopsies or directly implanted into mouse models (4, 14). In the future, strategies such as that described here may be an essential component of “precision medicine” in oncology, where treatment decisions are based on evolving tumor mutational profiles and drug sensitivity patterns in individual patients.
Supplementary Material
Acknowledgments
We are grateful to all the patients who participated in this study. We thank Dr. Lecia Sequist for coordinating the clinical studies, A. McGovern, C. Hart, and the Massachusetts General Hospital (MGH) clinical research coordinators P. Spuhler, A. Shah, J. Ciciliano, and V. Pai for bioengineering technical support; R. Milano, K. Lynch, H. Robinson, and M. Liebers for technical support; L. Collins (Beth Israel Deaconess Medical Center) for providing pathological specimens; and L. Libby for mouse studies. N. Aceto is a fellow of the Human Frontiers Science Program, the Swiss National Science Foundation, and the Swiss Foundation for Grants in Biology and Medicine. This work was supported by grants from the Breast Cancer Research Foundation (D.A.H), Stand Up to Cancer (D.A.H., M.T., S.M.), the Wellcome Trust (D.A.H., C.B.), National Foundation for Cancer Research (D.A.H.), NIH CA129933 (D.A.H.), NIBIB EB008047 (M.T., D.A.H.), Susan G. Komen for the Cure KG09042 (S.M.), National Cancer Institute–MGH Proton Federal Share Program (S.M.), the MGH-Johnson and Johnson Center for Excellence in CTCs (M.T., S.M.), and the Howard Hughes Medical Institute (M.Y., D.A.H.). A.J.I. holds equity in, and is a paid consultant for, Enzymatics, Inc. M.T., D.A.H., and the Massachusetts General Hospital have filed for patent protection for the CTC-iChip technology. RNA-Seq reads have been deposited into Gene Expression Omnibus: uncultured CTCs (accession no. GSE51827); the six cultured CTC lines (accession no. GSE55807).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/345/6193/216/suppl/DC1
Materials and Methods
References (27–30)
REFERENCES AND NOTES
- 1.Haber DA, Gray NS, Baselga J. Cell. 2011;145:19–24. doi: 10.1016/j.cell.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 2.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. J Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozkumur E, et al. Sci Transl Med. 2013;5:179ra47. doi: 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, et al. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato T, Clevers H. Methods Mol Biol. 2013;945:319–328. doi: 10.1007/978-1-62703-125-7_19. [DOI] [PubMed] [Google Scholar]
- 6.Ince TA, et al. Cancer Cell. 2007;12:160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Debnath J, Muthuswamy SK, Brugge JS. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 8.Dontu G, et al. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dias-Santagata D, et al. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence MS, et al. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. Cancer Res. 1997;57:1244–1249. [PubMed] [Google Scholar]
- 12.Robinson DR, et al. Nat Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toy W, et al. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, et al. Cell Reports. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merenbakh-Lamin K, et al. Cancer Res. 2013;73:6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas Network. Nature. 2012;490:61. [Google Scholar]
- 17.Garnett MJ, et al. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Mol Endocrinol. 1996;10:1388–1398. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 19.Baselga J, et al. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Trepel JB, Neckers LM, Giaccone G. Curr Opin Investig Drugs. 2010;11:1466–1476. [PubMed] [Google Scholar]
- 21.Furet P, et al. Bioorg Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Gavine PR, et al. Cancer Res. 2012;72:2045–2056. doi: 10.1158/0008-5472.CAN-11-3034. [DOI] [PubMed] [Google Scholar]
- 23.Mohammadi M, et al. EMBO J. 1998;17:5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollak M. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 25.Baccelli I, et al. Nat Biotechnol. 2013;31:539–544. doi: 10.1038/nbt.2576. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, et al. Sci Transl Med. 2013;5:180ra48. doi: 10.1126/scitranslmed.3005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.