

Abstract
Both purinergic signaling through nucleotides such as ATP (adenosine 5′-triphosphate) and noradrenergic signaling through molecules such as norepinephrine regulate vascular tone and blood pressure. Pannexin1 (Panx1), which forms large-pore, ATP-releasing channels, is present in vascular smooth muscle cells in peripheral blood vessels and participates in noradrenergic responses. Using pharmacological approaches and mice conditionally lacking Panx1 in smooth muscle cells, we found that Panx1 contributed to vaso-constriction mediated by the α1 adrenoreceptor (α1AR), whereas vasoconstriction in response to serotonin or endothelin-1 was independent of Panx1. Analysis of the Panx1-deficient mice showed that Panx1 contributed to blood pressure regulation especially during the night cycle when sympathetic nervous activity is highest. Using mimetic peptides and site-directed mutagenesis, we identified a specific amino acid sequence in the Panx1 intracellular loop that is essential for activation by α1AR signaling. Collectively, these data describe a specific link between noradrenergic and purinergic signaling in blood pressure homeostasis.
INTRODUCTION
Purinergic signaling is central in the regulation of vascular tone, which can be mediated by adenosine 5′-triphosphate (ATP) and its metabolic breakdown products (1). ATP can act as either a vasoconstrictor or a vasodilator (2). In the vascular wall, there are multiple sources for ATP; for example, ATP can be released from perivascular nerves and endothelial cells, as well as from circulating erythrocytes (3). Previously, we showed that cultured smooth muscle cells (SMCs) isolated from the vasculature release ATP in response to phenylephrine, an α1 adrenoreceptor (α1AR) agonist (4), and that ATP, purinergic receptors, and the ATP-release channel formed by pannexin1 (Panx1) are synergistically involved in phenylephrine-mediated vasoconstriction (4).
The pannexins comprise a family of membrane channels similar to innexins, the gap junction–forming proteins in invertebrates (5). Pannexins share topological similarities but no sequence homology with the gap junction–forming connexin proteins in vertebrates; thus, pannexins represent a distinct class of channel-forming proteins (6–8). Besides Panx1, two other isoforms have been described, Panx2 and Panx3. Panx1 is the most widely distributed in vertebrate tissues, whereas the presence of Panx2 and Panx3 is restricted to specific tissues (9, 10). In the systemic vasculature, Panx1 is found in all endothelial cells, but only in some SMCs; the protein is absent in SMCs of conduit arteries and becomes more abundant as the resistance of the arteries increases (11). Functionally, in apoptotic cells, Panx1 channels are activated for cell clearance (12, 13) to support the innate immune response (14), and in neurons, Panx1 channels are activated in response to cerebral ischemia (15) or to decreases in circulating oxygen (16). Because Panx1 forms large-pore channels, allowing the release of ATP and other intracellular ions and metabolites, channel activity is regulated by various receptors to avoid loss of cellular electrochemical and metabolic homeostasis, which would result in rapid cell death (17–19). For example, Panx1-dependent ATP release occurs in response to activation of thrombin receptors (20), N-methyl-D-aspartate (NMDA) receptors (21), histamine receptors (22), and purinergic receptors (23, 24).
Here, we examined Panx1 opening in response to α1AR activation in intact arteries using pharmacological blockers and an inducible SMC-specific Panx1 knockout mouse. We investigated the mechanisms downstream of α1AR stimulation leading to activation of Panx1 channels using a heterologous system expressing both Panx1 channels and the α1D subtype of AR (α1DAR). Using peptides analogous to different intracellular regions of Panx1 amino acid sequence, as well as specific Panx1 mutants, we identified a region required for Panx1 activation by α1AR stimulation. These findings suggest that Panx1 channels are opened downstream of α1AR, potentiating vasoconstriction by a mechanism dependent on a discrete intracellular loop region of the channel. Our data provide new insights into the molecular mechanisms that control vascular tone and blood pressure and physiological Panx1 opening.
RESULTS
Pharmacological or molecular inhibition of Panx1 reduces α1AR-dependent responses in isolated arteries
We have previously demonstrated that Panx1 is localized in SMCs of several resistance arteries, including thoracodorsal arteries (TDAs) and mesenteric and cremasteric arterioles (4, 11, 17, 18). Using pressure arteriography on TDAs dissected from C57BL/6 mice [as described (25) and aortic ring contraction as described (26)], we measured the effect of pannexin inhibitors, probenecid (22, 27) and 10Panx1 (21, 28), on the contractile response upon stimulation with different vasoconstrictors: phenylephrine (Fig. 1A), noradrenaline (also called norepinephrine) (Fig. 1B), serotonin [also known as 5-hydroxytryptamine (5-HT)] (Fig. 1C), and endothelin-1 (Fig. 1D). Both pannexin inhibitors significantly inhibited phenylephrine- and noradrenaline-mediated contraction of TDAs (Fig. 1, A and B), which was associated with a decrease in the maximum effect of both agonists (EMAX), without affecting the EC50 (median effective concentration) values (Table 1). In contrast, 10Panx1 and probenecid had no effect on contraction of TDAs measured in response to cumulative concentrations of serotonin or endothelin-1 (Fig. 1, C and D, and Table 1).
Fig. 1. Pharmacological inhibition of Panx1 reduces vasoconstriction and ATP release selectively upon activation of α1AR.
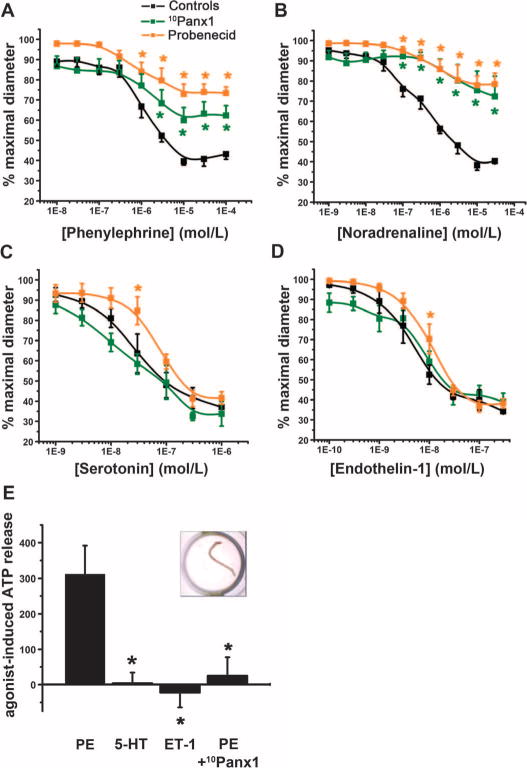
(A to D) Effect of 10Panx1 (300 μM) and probenecid (2 mM) on contractile response of pressurized TDAs stimulated with the indicated concentrations of agonists. n = 5to7. *P < 0.05 compared to untreated response (black curves) using two-way analysis of variance (ANOVA). (E) Relative ATP released from intact TDAs in response to phenylephrine (PE) in the presence or absence of 10Panx1 (300 μM), serotonin (5-HT), or endothelin-1 (ET-1). Data are presented as a percent increase in ATP concentration from unstimulated conditions. The insert shows an image of a TDA in a well of a 96-well dish. n = 5 to 11. *P < 0.05 compared to phenylephrine using a Kruskal-Wallis test.
Table 1. Effect of probenecid and 10Panx1 on TDA constriction in response to phenylephrine, noradrenaline, serotonin, or endothelin-1.
EC50 and EMAX were calculated using the cumulative concentration response curves shown in Fig. 1 (A to D). EC50 represents the concentration needed to produce 50% of the maximum effect (EMAX). EMAX is expressed as the percentage of maximal diameter. Data are presented as means ±SEM.
| Phenylephrine | Noradrenaline | |||||
|---|---|---|---|---|---|---|
| Control | Probenecid | 10Panx1 | Control | Probenecid | 10Panx1 | |
| EC50 (μM) | 1.21 ± 0.27 | 2.71 ± 1.36 | 2.38 ± 0.53 | 0.56 ± 0.25 | 1.30 ± 0.65 | 1.61 ± 0.49 |
| EMAX | 41.1 ± 2.21 | 72.6 ± 3.51* | 60.6 ± 5.80* | 33.3 ± 1.04 | 74.9 ± 3.04* | 73.4 ± 10.6* |
| Serotonin | Endothelin-1 | |||||
| Control | Probenecid | 10Panx1 | Control | Probenecid | 10Panx1 | |
| EC50 (nM) | 50.8 ± 18.9 | 83.3 ± 8.42 | 47.9 ± 23.1 | 4.57 ± 0.88 | 12.8 ± 4.05 | 6.53 ± 1.36 |
| EMAX | 34.8 ± 2.66 | 39.5 ± 3.29 | 30.9 ± 4.92 | 41.9 ± 4.82 | 34.6 ± 4.50 | 41.5 ± 3.91 |
P<0.05 compared to control using a Kruskal-Wallis test (n = 5 to 7).
To determine whether the TDA response involved release of ATP, we dissected TDAs from C57BL/6 mice and measured the accumulation of extracellular ATP in response to various agonists in the medium (Fig. 1E, inset). To prevent ATP degradation, we exposed the TDAs to the ectonucleotidase inhibitor ARL67156 for 30 min before exposure to the contractile agonist. The amount of ATP in the medium surrounding the vessel was then determined with an ATP bioluminescence assay. Phenylephrine stimulated ATP release from TDAs, and this effect was significantly reduced when Panx1 channels were blocked with 10Panx1 before stimulation (Fig. 1E). In contrast to phenylephrine, neither serotonin nor endothelin-1 promoted ATP release from intact TDAs (Fig. 1E). Together, these pharmacological studies suggested a link between α1AR stimulation and Panx1 activation, which indicated the involvement of a purinergic component through the release of cellular ATP in adrenergic-stimulated vasoconstriction.
To further test the role of Panx1 in α1AR-mediated vasoconstriction, we generated a tamoxifen-inducible, SMC-specific Panx1 knockout mouse model. We confirmed cell type–specific deletion of Panx1 by immunofluorescence analysis of TDA cross sections. Although we detected Panx1 in both endothelial cells and SMCs of arteries from Cre−/Panx1WT, Cre−/Panx1Fl, and Cre+/Panx1WT mice (littermate controls; all injected with tamoxifen), Panx1 immunoreactivity was absent in the SMCs of Cre+/Panx1Fl mice injected with tamoxifen (Fig. 2A and fig. S1A). Western blotting of TDA lysates showed reduced amounts of Panx1, and we assume that the residual Panx1 was from endothelial cells (fig. S1B). Although knockdown of a pannexin isoform may result in compensation by other pannexin isoforms (18), we did not detect an increase in the abundance of Panx2 and Panx3 in the tamoxifen-treated Cre+/Panx1Fl mouse TDAs (fig. S1A). We also confirmed that the knockdown of Panx1 in SMCs did not alter total intracellular ATP content (fig. S1C) or α1DAR abundance (fig. S1B) in intact arteries.
Fig. 2. Inducible SMC deletion of Panx1 selectively inhibits vasoconstriction and ATP release upon α1AR stimulation.
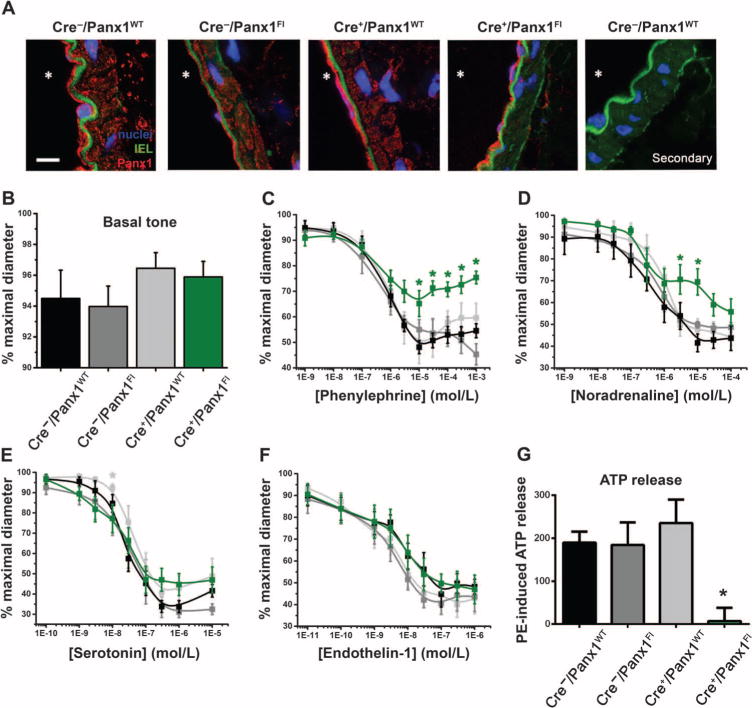
(A) Representative immunofluorescence micrographs showing Panx1 labeling (red) on cross sections of TDAs isolated from mice of the indicated genotypes. All mice had been injected with tamoxifen for 10 days. The far right panel shows a negative control (secondary antibody only) on a cross section of a TDA isolated from Cre−/Panx1WT mice. The autofluorescence of the internal elastic lamina (IEL) appears in green, and the nuclei were labeled with DAPI (4′,6-diamidino-2-phenylindole) (blue). * indicates the lumen. Scale bar, 10 μm. (B) Basal tone exhibited by TDAs from each genotype (exposed to tamoxifen for 10 days). (C to F) Contraction of pressurized TDAs isolated from Cre−/Panx1WT mice (black curves), Cre−/Panx1Fl mice (dark gray curves), Cre+/Panx1WT mice (light gray curves), and Cre+/Panx1Fl (green curves) all injected with tamoxifen for 10 days and stimulated with cumulative concentrations of phenylephrine (n = 6 to 16), noradrenaline (n = 4 to 8), serotonin (n = 5 to 10), or endothelin-1 (n = 4 to 8). *P < 0.05 compared to Cre−/Panx1WT using a two-way ANOVA. (G) Histogram showing the phenylephrine (PE)–induced ATP release from intact TDAs isolated from mice of the indicated genotypes, all injected with tamoxifen for 10 days. Data are presented as a percent increase in ATP concentration from unstimulated conditions. *P < 0.05 compared to Cre−/Panx1WT using a Kruskal-Wallis test. n = 4 to 8.
Initial functional tests performed on TDAs from the SMC-specific Panx1 knockout mice and littermate controls revealed no difference in basal vascular tone (Fig. 2B). However, in TDAs from the SMC-specific Panx1 knockout mice, the contractile responses to phenylephrine or noradrenaline were significantly reduced (Fig. 2, C and D). The EMAX for phenylephrine was reduced to 69.1 ± 3.06% of maximal diameter compared to an EMAX of about 50% of maximal diameter for TDAs isolated from the three control genotypes; the EC50 values were not different among the different genotypes (Table 2). To confirm that this effect was specific to Panx1 deletion and not to a deficient α1AR response in these mice, we measured phenylephrine-induced vasoconstriction in abdominal aortic rings, which are devoid of Panx1 in the SMC layer (11), isolated from tamoxifen-treated or control Cre+/Panx1Fl mice. Abdominal aortic rings from tamoxifen-treated and control mice showed the same EMAX and EC50 for phenylephrine-induced vasoconstriction (fig. S1D and table S1). The contractile responses of TDAs isolated from the SMC-specific Panx1 knockout mice to serotonin or endothelin-1 were not significantly different from those of TDAs isolated from Cre−/Panx1WT, Cre−/Panx1Fl, or Cre+/Panx1WT mice (Fig. 2, E and F, and Table 2). Consistent with α1AR activating Panx1 to promote ATP release, SMC-specific Panx1 knockout attenuated phenylephrine-induced ATP release from TDAs (Fig. 2G). The specificity of Cre in targeting knockout in SMCs was further demonstrated using TdTomato mice (fig. S1E).
Table 2. Contractile properties of TDAs isolated from Cre−/Panx1WT, Cre−/Panx1Fl, Cre+/Panx1WT, or Cre+/Panx1Fl mice.
The EC50 and EMAX are calculated from the data presented in Fig. 2 (C to F). EC50 represents the concentration needed to produce 50% of the maximum effect (EMAX). EMAX is expressed as the percentage of maximal diameter. Data are presented as means ± SEM.
| Phenylephrine | ||||
|---|---|---|---|---|
| Cre−/Panx1WT | Cre−/Panx1FL | Cre+/Panx1WT | Cre+/Panx1FL | |
| EC50 (μM) | 0.66 ± 0.13 | 0.64 ± 0.37 | 1.79 ± 1.13 | 7.84 ± 5.60 |
| EMAX | 51.8 ± 2.66 | 46.3 ± 5.94 | 53.5 ± 4.91 | 69.1 ± 3.06* |
| Noradrenaline | ||||
| Cre−/Panx1WT | Cre/Panx1FL | Cre+/Panx1WT | Cre+/Panx1FL | |
| EC50 (μM) | 6.28 ± 1.45 | 10.3 ±1.3 | 6.78 ± 3.20 | 50.5 ± 24.8 |
| EMAX | 41.4 ± 5.10 | 45.6 ± 5.40 | 48.2 ± 1.82 | 56.4 ± 5.61# |
| Serotonin | ||||
| Cre−/Panx1WT | Cre−/Panx1FL | Cre+/Panx1WT | Cre+/Panx1FL | |
| EC50 (nM) | 23.8 ± 5.07 | 65.2 ± 40.9 | 40.7 ± 12.2 | 14.2 ± 5.53 |
| EMAX | 36.1 ± 2.68 | 30.4 ± 1.78 | 40.1 ± 4.59 | 38.2 ± 5.60 |
| Endothelin-1 | ||||
| Cre−/Panx1WT | Cre−/Panx1FL | Cre+/Panx1WT | Cre+/Panx1FL | |
| EC50 (nM) | 18.4 ± 10.9 | 3.77 ± 0.56 | 5.64 ± 2.94 | 30.1 ± 14.6 |
| EMAX | 41.8 ± 4.95 | 42.2 ± 6.50 | 42.5 ± 4.82 | 38.9 ± 6.90 |
P < 0.05
P < 0.07 compared to Cre−/Panx1WT using a Kruskal-Wallis test (n = 4 to 16).
SMC-specific Panx1 knockout impairs blood pressure regulation
On the basis of the central role of α1AR signaling in blood pressure homeostasis (29), we evaluated the effect of Panx1 deletion on systemic blood pressure. Using radiotelemetry, we monitored real-time blood pressure parameters in conscious Cre+/Panx1Fl mice and littermate controls before and after tamoxifen injections. Administration of tamoxifen significantly decreased the mean arterial blood pressure in Cre+/Panx1Fl mice by 4.6 ± 2.1 mmHg, with no evident changes observed in the mice that retained endogenous amount of Panx1 (Cre−/Panx1WT, Cre−/Panx1Fl, and Cre+/Panx1WT) (Fig. 3A). This effect appeared independent of heart rate, which was unchanged after tamoxifen injection in Cre+/Panx1Fl mice (569 ± 4 beats/min before tamoxifen versus 549 ± 7 beats/min after tamoxifen). The mean arterial blood pressure measured during the day was unchanged (Fig. 3B), whereas during the night cycle (the active period for rodents during which sympathetic nerve activity is greatest), the blood pressure was significantly decreased in the SMC-specific Panx1 knockout mice (Fig. 3C). Thus, both pharmacological and molecular evidence from intact arteries and live animals suggested a functional link in SMCs between the sympathetic signaling through α1AR and Panx1-mediated ATP release in controlling vascular tone and systemic blood pressure.
Fig. 3. Inducible SMC deletion of Panx1 reduces blood pressure in freely moving mice.
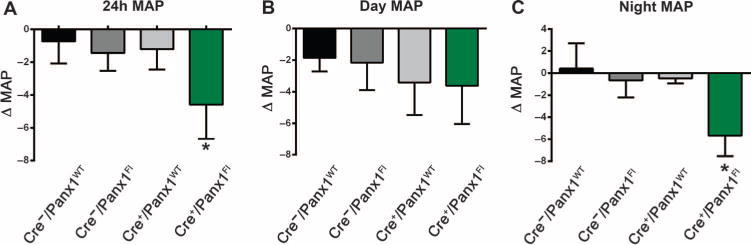
(A) Difference in the 24-hour mean arterial pressure (MAP) of mice of the indicated genotypes before and after tamoxifen injections. (B) Difference in the MAP during the day cycle (12-hour light: 6:00 a.m. to 6:00 p.m.) of mice of the indicated genotypes before and after tamoxifen injections. (C) Difference in the MAP during the night cycle (12-hour no light: 6:00 p.m. to 6:00 a.m.) of mice of the indicated genotypes before and after tamoxifen injections. *P < 0.05 comparing the MAP before and after tamoxifen injection using a nonparametric paired t test (Wilcoxon). n = 4 to 7.
α1AR-stimulated vasoconstriction involves the intracellular loop of Panx1
To determine the region of Panx1 necessary for channel activation in response to α1AR stimulation, we produced short peptides derived from the intracellular regions of the Panx1 channel. We generated two peptides mimicking unique mouse Panx1 (mPanx1) sequences in the first intracellular loop (IL1 and IL2) and two peptides mimicking unique mPanx1 sequences in the C-terminal region, one in the second intracellular loop (CT1) and one in the C-terminal tail (CT2) (Fig. 4A and Table 3). All peptides also contained a TAT sequence to facilitate entry into cells (30, 31), and we verified the intracellular localization of each peptide by immunofluorescence detection of the TAT sequence in TDAs preincubated with each peptide (fig. S2A). We tested the effect of each peptide on phenylephrine-induced contractile responses in pressurized TDAs from C57BL/6 mice (Fig. 4, B to E). The CT1, CT2, and IL1 peptides did not affect phenylephrine-induced constriction (Fig. 4, B to D, and Table 4). In contrast, the IL2 peptide significantly decreased phenylephrine-induced constriction (Fig. 4E and Table 4). Note that these differences in effects of phenylephrine were similar in magnitude to those observed in arteries from SMC-specific Panx1 knockout mice and littermate controls (Fig. 2C and Table 2). A scrambled version of the IL2 peptide, as well as the TAT sequence alone, had no significant effect on the phenylephrine response (fig. S2B and table S2).
Fig. 4. A peptide analog to a Panx1 intracellular loop sequence inhibits vasoconstriction and ATP release upon α1AR stimulation and reduces blood pressure.
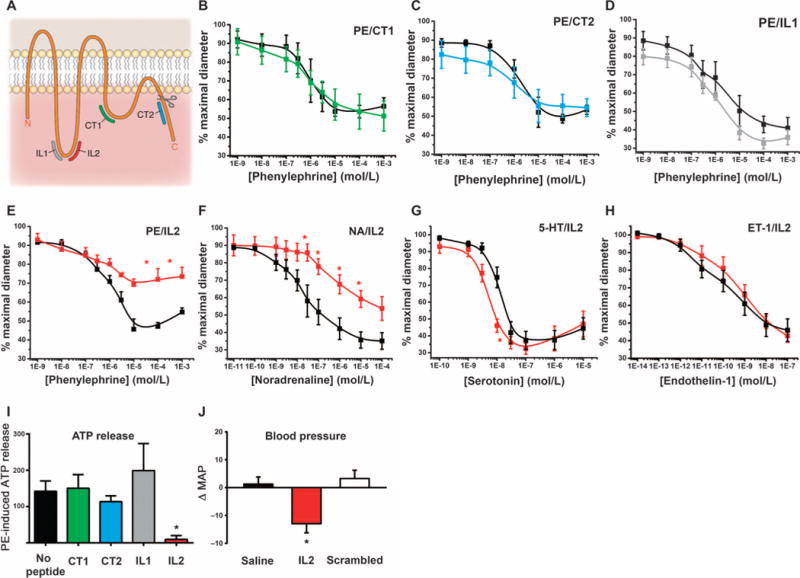
(A) Diagram showing the position of each of the four peptides on mPanx1. The scissors indicate a caspase cleavage site. (B to H) Effects of the indicated peptide inhibitor on phenylephrine-induced constriction of pressurized TDAs (B to E), and effect of IL2 peptide on constriction of pressurized TDAs induced by the indicated concentrations of agonists (E to H). The black curves represent constriction in the absence of peptide. n = 4 to 7. *P< 0.05 compared to constriction in the absence of peptide using a two-way ANOVA. (I) Effect of the indicated peptide on phenylephrine (PE)–induced ATP release. Data are presented as a percent increase in ATP concentration from unstimulated conditions. n = 3 to 8. *P< 0.05 compared to no peptide using a Kruskal-Wallis test. (J) Difference between the MAPs (ΔMAP) measured before and after injection of saline, IL2 peptide, or its scrambled IL2 peptide in C57BL/6 mice. *P< 0.05 comparing before and after vehicle and peptide injections using a nonparametric paired t test (Wilcoxon). n = 7 to 9.
Table 3. Amino acid sequences of intracellular loop and C-terminal region peptides.
All listed peptides were linked with a TAT sequence (YGRKKQRRR). The numbers indicate the peptide location in the mPanx1 amino acid sequence. The CT1 peptide has previously been published to inhibit NMDA-mediated Panx1 opening (21).
| Amino acid sequence (mPanx1) | |
|---|---|
| IL1 | 178-VGQSLWEISE-187 |
| IL2 | 191-KYPIVEQYLK-200 |
| CT1 | 305-RRLKVYEILPTFDVLH-318 |
| CT2 | 381-IPTSLQTKGE-390 |
| Scrambled IL2 | IYLYVEQKPY |
Table 4. Effect of four Panx1 mimetic peptides on constriction of TDAs in response to phenylephrine, noradrenaline, serotonin, or endothelin-1.
The EC50 and EMAX are calculated from the data presented in Fig. 4 (B to H). EC50 represents the concentration needed to produce 50% of the maximum effect (EMAX). EMAX is expressed as the percentage of maximal diameter. Data are presented as means ± SEM.
| Phenylephrine | ||||||
|---|---|---|---|---|---|---|
| No peptide | CT1 | No peptide | CT2 | |||
| EC50 (μM) | 1.67 ± 1.13 | 1.43 ± 0.59 | 1.06 ± 0.32 | 4.78 ± 3.93 | ||
| EMAX | 52.5 ± 3.30 | 50.7 ± 9.08 | 49.0 ± 2.49 | 52.3 ± 4.86 | ||
| No peptide | IL1 | No peptide | IL2 | |||
| EC50 (μM) | 3.26 ± 1.53 | 1.14 ± 0.43 | 1.15 ± 0.35 | 0.57 ± 0.22 | ||
| EMAX | 40.1 ± 5.10 | 32.3 ± 3.81 | 47.70 ± 1.6 | 68.6 ± 5.11* | ||
| Noradrenaline | Serotonin | Endothelin-1 | ||||
| No peptide | IL2 | No peptide | IL2 | No peptide | IL2 | |
| EC50 (μM) | 0.49 ± 0.42 | 2.00 ± 1.90 | 11.7 ± 1.65 | 5.03 ± 0.82* | 6.01 ± 3.74 | 0.88 ± 0.39 |
| EMAX | 27.1 ± 4.69 | 53.4 ± 7.00* | 38.6 ± 5.70 | 36.7 ± 3.52 | 38.2 ± 6.67 | 41.9 ± 3.73 |
P < 0.05 compared to no peptide using a Mann-Whitney test (n = 4 to 7).
Because Panx1 activity appeared selectively involved in α1AR-mediated responses, we tested the effect of the IL2 peptide on the response to noradrenaline (Fig. 4F), serotonin (Fig. 4G), and endothelin-1 (Fig. 4H). As expected, whereas the IL2 peptide inhibited noradrenaline-induced constriction, vasoconstriction mediated by serotonin or endothelin-1 was similar in the presence or absence of the IL2 peptide (Table 4).
Additionally, phenylephrine-induced ATP release was significantly decreased only by the IL2 peptide (Fig. 4I) with the scrambled version of IL2 and the TAT sequence alone having no effect (fig. S2C). Continuous recording of blood pressure in conscious C57BL/6 mice revealed that acute injection of the IL2 peptide significantly reduced the mean arterial blood pressure by 13.0 ± 3.3 mmHg within 1.5 hours, whereas its scrambled version had no significant effect (Fig. 4J).
α1AR-stimulated activation of the Panx1 channel involves the intracellular loop of Panx1
To test whether α1AR stimulation triggered opening of the Panx1 channel and the importance of the region represented by the IL2 peptide, we cotransfected mPanx1 and α1DAR into human embryonic kidney (HEK) 293 cells and monitored whole-cell currents by patch clamp electrophysiology. Whole-cell currents recorded from cotransfected HEK293 cells displayed characteristics typical of Panx1 currents (21, 27, 28, 32–34): the cells showed an outwardly rectifying current-voltage relationship and inhibition of this current by carbenoxolone (CBX) (Fig. 5A). In cells expressing both mPanx1 and α1DAR, phenylephrine increased this current, and CBX abolished the current (Fig. 5A, right). This phenylephrine-induced current activation required cotransfection of Panx1 and was significantly smaller without cotransfection of α1DAR (Fig. 5B, top). Similarly, only cells cotransfected with Panx1 and α1DAR exhibited phenylephrine-induced ATP release (Fig. 5B, bottom).
Fig. 5. Characterization of α1DAR-mediated ATP release by Panx1 using a heterologous system.
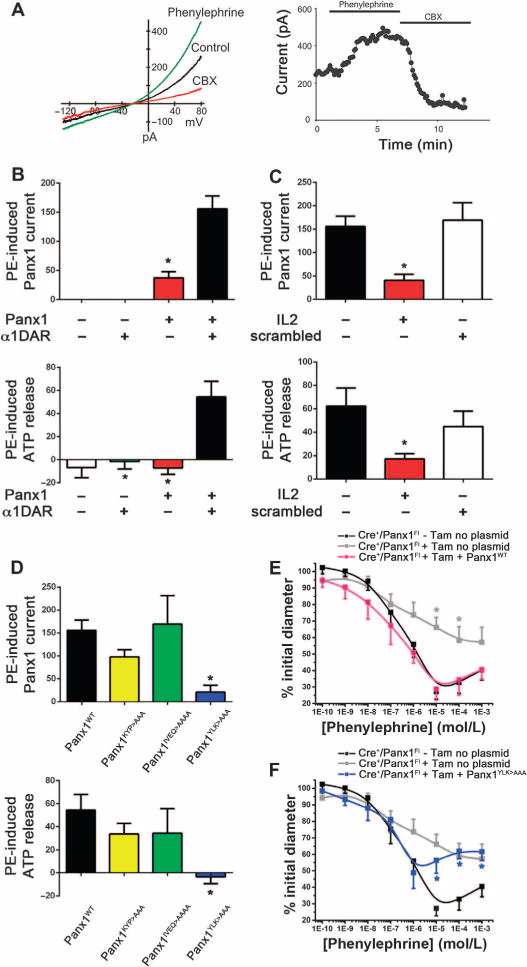
(A) Left panel: Representative current-voltage (I–V) curve obtained from whole-cell patch clamp recording of HEK293 cells cotransfected with Panx1 and α1DAR before (control, black curve) and after stimulation with phenylephrine (green curve), and upon application of CBX (red curve). Right panel: Representative time course of whole-cell current recorded from cotransfected HEK293 cell showing the effect of phenylephrine and CBX. (B) Phenylephrine-induced Panx1 current (top panel) and phenylephrine-induced ATP release (bottom panel) in untransfected HEK293 cells or HEK293 cells transfected with the indicated constructs. (C) Effect of the IL2 peptide and scrambled IL2 peptide on phenylephrine-induced Panx1 current (top panel) and phenylephrine-induced ATP release (bottom panel). (D) Phenylephrine-induced Panx1 current (top panel) and phenylephrine-induced ATP release (bottom panel) in HEK293 cells cotransfected with α1DAR and the indicated Panx1 construct. Panx1 current data [(B) to (D), top panels] are presented as a percent increase of CBX-sensitive current at +80 mV, or as a percent increase of ATP concentration from unstimulated conditions [(B) to (D), bottom panels]. (B to D) *P < 0.05 compared to cotransfected conditions (B), no peptide (C), or wild-type Panx1 (Panx1WT) (D) using a Kruskal-Wallis test. (E) Phenylephrine-induced contraction of pressurized TDAs isolated from Cre+/Panx1Fl mice not injected with tamoxifen and electroporated without plasmid (black curve), Cre+/Panx1Fl mice after injection with tamoxifen for 10 days and then electroporated without plasmid (gray curve), or Cre+/Panx1Fl mice after injection with tamoxifen for 10 days and then electroporated with Panx1WT (pink curve). (F) Phenylephrine-induced contraction of pressurized TDAs from control mice as indicated in (E) (black and gray curves) and Cre+/Panx1Fl mice after injection with tamoxifen for 10 days and then electroporated with Panx1YLK>AAA (blue curve). (E and F) *P < 0.05 compared to control Cre+/Panx1Fl (black curve) using a two-way ANOVA. n = 6 mice.
When cotransfected cells were incubated with the IL2 peptide, both phenylephrine-induced Panx1 current and ATP release were significantly decreased, whereas the scrambled version of the IL2 peptide had no effect (Fig. 5C). To better define the activating region within the Panx1 intracellular loop, we generated Panx1 mutants with alanine substitutions for selected amino acids corresponding to the IL2 sequence (amino acids 191 to 200): Panx1KYP>AAA, Panx1IVEQ>AAAA, and Panx1YLK>AAA. We individually cotransfected each mutated construct with α1DAR into HEK293 cells and monitored phenylephrine-induced Panx1 activation. Phenylephrine stimulation of cells expressing α1DAR and either Panx1KYP>AAA or Panx1IVEQ>AAAA resulted in similar increases in Panx1 current and ATP release as were observed with the control cells expressing α1DAR and Panx1WT (Fig. 5D). However, HEK293 cells expressing α1DAR and Panx1YLK>AAA failed to increase Panx1 current and ATP release upon phenylephrine stimulation (Fig. 5D), identifying these residues as important for α1AR-dependent Panx1 activation. Importantly, the absence of phenylephrine-induced channel activation was not due to an inability of the channel to traffic to the plasma membrane or to disrupted channel function independently of phenylephrine-induced activation, because the Panx1YLK>AAA mutant channel produced basal CBX-sensitive currents that were not different from those detected in cells expressing the wild-type channel (fig. S3).
We further tested the importance of the YLK sequence within the intracellular loop of Panx1 on phenylephrine-induced vasoconstriction using rescue experiments with intact arteries from mice lacking endogenous Panx1. We reintroduced Panx1 into the SMCs of TDAs from Cre+/Panx1Fl mice that had received tamoxifen by selectively transfecting SMCs with a plasmid encoding Panx1WT. After transfection of Panx1WT into these arteries, the phenylephrine-induced vasoconstriction was similar to that in control, sham-transfected arteries from Cre+/Panx1Fl mice that were not injected with tamoxifen (Fig. 5E and Table 5). Consistent with our in vitro analysis, transfection of the Panx1YLK>AAA mutant into SMCs of TDAs from tamoxifen-injected Cre+/Panx1Fl mice failed to rescue phenylephrine-induced constriction, and the response plateaued at a similar level of constriction as seen in sham-transfected TDAs from tamoxifen-injected Cre+/Panx1Fl mice (Fig. 4F and Table 5). These studies identified a molecular signature within the Panx1 intracellular loop that is essential for coordination of α1AR-dependent Panx1 channel activation in vascular SMCs and, in turn, vascular constriction.
Table 5. Effect of transfection of TDAs with Panx1WT or Panx1YLK>AAA on phenylephrine-induced constriction.
The EC50 and EMAX are calculated from the data presented in Fig. 4 (E and F). EC50 represents the concentration needed to produce 50% of the maximum effect (EMAX). EMAX is expressed as the percentage of maximal diameter. Data are presented as means ± SEM.
| Phenylephrine | ||||
|---|---|---|---|---|
| Cre+/Panx1FL No tamoxifen | Cre+/Panx1FL With tamoxifen | Cre+/Panx1FL + Panx1WT | Cre+/Panx1FL + Panx1YLK>AAA | |
| EC50 (μM) | 0.64 ± 0.23 | 1.13 ± 0.77 | 0.45 ± 0.33 | 0.63 ± 0.53 |
| EMAX | 32.9 ± 4.85 | 57.5 ± 9.20*# | 34.2 ± 6.15 | 56.9 ± 5.84*# |
P < 0.05 in comparison to no tamoxifen
P < 0.05 in comparison to Cre+/Panx1FL + Panx1WT using a Kruskal-Wallis test.
DISCUSSION
In the resistance vasculature, sympathetic vasoconstriction contributes to the overall control of systemic blood pressure by regulating peripheral vascular resistance. This process occurs, in part, after the release of noradrenaline from perivascular sympathetic nerves and binding to ARs located on the adjacent SMCs. This constriction occurs mainly through activation of the α1DAR isoform on SMCs (4, 29, 35, 36). Recent work from our laboratory has implicated Panx1 channels in the control of adrenergic vasoconstriction in the peripheral vasculature; however, less is known about the molecular mechanisms controlling this event, and whether Panx1 channels are involved in response to other contractile agonists. Here, we showed, using multiple in vitro, ex vivo, and in vivo models, that a functional interaction exists between α1AR and Panx1, but not between Panx1 and other vasoconstricting receptors, in vascular SMCs.
The first line of evidence used a pharmacological approach with two independent, well-described Panx1 inhibitors on pressurized arteries subjected to cumulative doses of contractile agonists that are all G protein (G protein)–coupled receptors (GPCRs). These results demonstrated a specific functional interaction between AR activation by phenylephrine or noradrenaline and Panx1 channels: Panx1 channel inhibition blunted agonist-induced vasoconstriction without affecting vasoconstriction elicited by serotonin and endothelin-1. In support of a purinergic role for Panx1 activity, isolated intact arteries stimulated with phenylephrine released ATP into the extracellular milieu, an effect also ablated with Panx1 inhibitors and absent in response to serotonin and endothelin-1.
In the vasculature, serotonin vasoconstriction is mostly mediated by 5-HT2A receptors and, in some vascular beds, by 5-HT1B or 5-HT1D receptors (37–39), and endothelin-1 induces SMC contraction upon binding to ETA receptors (40, 41). The α1AR, 5-HT2A, and ETA are all Gq protein–coupled receptors, leading to activation of phospholipase C and subsequent calcium release from the endoplasmic reticulum. Several studies have reported activation of Panx1 by Gq-coupled receptors in other cell types. For example, bradykinin is linked to Panx1-mediated ATP release from human subcutaneous fibroblasts (42). Histamine induces an increase in ATP release through Panx1 channels from human subcutaneous fibro-blasts (22) and from endothelial cells (20). Additionally, the stimulation of protease-activated receptor (PAR)–1 by thrombin is linked to ATP release through Panx1 channels from endothelial cells (20), and a PAR-3–dependent pathway stimulates ATP release through Panx1 channels from lung epithelial cells (43, 44). These reports indicate that Panx1 may provide additional secondary signaling activation (an increase in intracellular Ca2+ concentration) through ATP release and subsequent purinergic receptor activation, which would ultimately enhance the response to metabotropic GPCR signaling (17). However, our results here suggested that there is specificity to this GPCR-mediated activation of Panx1, because only agonists of α1AR and not those of serotonin or endothelin receptors stimulated Panx1 channel activity, ATP release, and vasoconstriction in SMCs.
Although α1AR, 5-HT2A, and ETA are all Gq-coupled receptors, other downstream signaling molecules can contribute to the cellular response to their activation. For example, calcium sensitization of the contractile apparatus through Rho kinases has been described upon stimulation of various vascular beds with phenylephrine, serotonin, or endothelin-1 (26, 45–49), and Panx1-mediated ATP release has been associated to Rho kinase activity in lung epithelial cells (43, 44). The Rho kinase pathway is therefore a possible target in this pathway; however, the specificity for α1AR would need elucidation. Furthermore, phenylephrine-mediated constriction has also been linked to activation of a Gi–cAMP (adenosine 3′,5′-monophosphate)–dependent signaling pathway in rat mesenteric arteries (50, 51) and swine renal arteries (52), and pharmacological evidence has demonstrated a possible interaction between α1AR and the Gi pathway in promoting cardiomyocyte contraction (53). Although our work shows that purinergic signaling is a key component to the α1AR vasoconstriction pathway, the mechanism linking α1AR stimulation and Panx1 channel opening requires more detailed analysis of the signaling pathways downstream of α1AR, 5-HT2A, 5-HT1B/1D, and ETA.
Although our pharmacological assessment of Panx1 involvement in adrenergic vasoconstriction suggested a prominent role for the channel in this process, there are inherent limitations to the current pharmacological tools available for inhibition of pannexin channels with several blockers showing cross-inhibition with connexin hemichannels (54). Because of this potential confounding issue, we used a genetic approach by creating an inducible, SMC-specific Panx1 knockout mouse model (Cre+/Panx1Fl). Because global deletion of Panx1 from birth can induce a compensatory increase in the Panx3 isoform in SMCs in the arterial circulation in the adult mouse (18), we used an inducible knockout model that enabled spatial and temporal control of Panx1 expression in the adult mouse. Analysis of Panx2 and Panx3 abundance in the vasculature of these mice revealed no compensatory increases in either isoform with complete deletion of Panx1 specifically from the SMC layer in adult mice. Conditional deletion of Panx1 in SMCs significantly reduced the constriction to α1AR agonists, providing further support for a central role of Panx1 channels in adrenergic vasoconstriction. We noted that Panx1 deletion was more effective at reducing vasoconstriction to phenylephrine than to noradrenaline. Several studies have reported the involvement of other AR isoforms in noradrenaline-mediated responses in arteries. In particular, the α2AR and the β2AR, respectively coupled to Gi and Gs, are found in both SMC and endothelial cells depending on the vascular bed and the species (36, 55–58). On the basis of these observations, we predict that noradrenaline signaling through one of the other AR isoforms, likely α2AR, is responsible for the reduced effect of Panx1 knockout on the vasoconstriction to noradrenaline compared to that produced by the more selective α1AR agonist phenylephrine. Although the postjunctional receptors involved in the noradrenergic response in TDAs are unknown, the effect of SMC Panx1 knockout not only reduced phenylephrine- and noradrenaline-mediated vasoconstriction but also resulted in a decrease of MAP in freely moving mice. Our radiotelemetry data on Cre+/Panx1Fl mice demonstrated a significant hypotension, which was exaggerated at night during the period of greatest sympathetic activity. These data indicate a potentially key role of SMC Panx1 channels in noradrenergic vasoconstriction and regulation of systemic blood pressure in the live animal.
Similar to pharmacological studies, genetic deletion of Panx1 from SMCs prevented ATP release in response to α1AR stimulation, which is consistent with our previous work reporting a functional role for ATP release in arterial constriction to phenylephrine (4). Previously, we reported that degradation of extracellular ATP with apyrase and inhibition of SMC P2Y purinergic receptors with Reactive Blue 2 reduced phenylephrine-induced vasoconstriction. Although other cells composing the vascular wall, including nerves and endothelial cells, as well as circulating erythrocytes, can provide releasable pools of ATP (3), our pharmacological, molecular, and genetic data described here provide evidence that SMCs can also release ATP from the intact arterial wall. In addition, other nonvascular SMCs release ATP, including SMCs from the colon (59) and SMCs from the bladder (60). Although the mechanisms of ATP liberation from those cells are still under investigation, pannexins are present in these nonvascular SMCs (61, 62).
Using molecular techniques, we disrupted the α1AR-Panx1 functional interaction by screening interfering peptides that were based on Panx1 intracellular amino acid sequences. One peptide mimicking a sequence in the Panx1 intracellular loop (IL2) inhibited α1AR-dependent vasoconstriction and ATP release from intact arteries. Although consistent with the pharmacological inhibition and genetic deletion of Panx1, the pharmacological inhibitors 10Panx1 and probenecid (Fig. 1) were more effective in reducing the phenylephrine-induced constriction compared to the IL2 peptide (Fig. 4) or the knockdown of Panx1 in our conditional knockout mouse model (Fig. 2). This difference is most likely due to inherent nonspecific effect of 10Panx1 and probenecid (54). Intraperitoneal injection of the IL2 peptide acutely reduced MAP in C57BL/6 mice, consistent with our inducible conditional knockout model.
To further define the α1AR-Panx1 interaction, we turned to a heterologous cell culture system expressing α1DAR and Panx1. With this system, we observed enhanced ATP release and CBX-sensitive Panx1 currents after phenylephrine application; these effects were inhibited with the same IL2 peptide that blunted phenylephrine-induced vasoconstriction and ATP release from intact arteries. Because the IL2 peptide mimics the endogenous Panx1 sequence in positions 191 to 200, we progressively substituted amino acids in this region of full-length Panx1 with alanine residues to more precisely define the motif required for channel activation by α1AR stimulation. Using our heterologous system, we identified a three–amino acid stretch (198YLK200) in the Panx1 intracellular loop that, when mutated, renders the channel insensitive to α1AR-dependent activation. Last, using in situ transfection, we rescued the α1AR-dependent vasoconstriction in vessels by heterologous expression of wild-type Panx1 in the SMC of intact arteries from Cre+/Panx1Fl mice; by contrast, the Panx1YLK>AAA mutant did not rescue α1AR-dependent vasoconstriction.
Although the exact mechanism by which the α1AR functionally interacts with the Panx1 YLK motif remains unclear, sequence analysis may provide initial insight. The region of Panx1 containing the amino acids 191 to 200 has a high propensity to form α helix structure, which are known to constitute a fundamental recognition element in many protein-protein interactions (7, 63). α Helix–mediated protein-protein interactions are practical targets for chemical design of small molecular inhibitors (64). Additionally, there is a region containing several proline residues proximal to the IL2 sequence in the Panx1 intracellular loop (65). This may prove to be important because proline-rich regions can introduce hinge points in the tertiary structure of proteins, creating flexibility and providing hallmark locations where protein-protein interactions occur (66). In agreement, connexins, which have a similar topology to Panx1, contain a proline-rich region in their C-terminal tail, which is the main site of interaction with protein partners (67). Another intriguing aspect of the YLK motif is the presence of a tyrosine, which could be a potential regulatory phosphorylation site. Recent evidence in support of Panx1 regulation by kinases has been reported in several cell types, including kinases of the Src family in hippocampal neurons and in macrophages (21, 23). However, direct evidence of Panx1 phosphorylation has only been provided in skeletal myocytes, in which increased amounts of Panx1 phosphoserine and phosphothreonine are associated with Panx1 activity in electrically stimulated rat skeletal muscles (68). Several kinases are activated in the α1AR signaling pathway, including kinases from the Rho kinase family, PKA (cAMP-dependent protein kinase), PKC (protein kinase C), and Src family of protein tyrosine kinases (45–49, 69). It is thus tempting to speculate that Panx1 may be phosphorylated at Tyr198 by a kinase activated upon α1AR stimulation.
Our data demonstrated that α1AR and Panx1 participate in vasoconstriction through a unique functional interaction in vascular SMCs that could be important for adrenergic control of blood pressure. Targeting this signaling mechanism may therefore provide an approach to intervene in blood pressure disorders. To this end, several Panx1 inhibitors have been successfully used in vivo in animal models to target Panx1-mediated signaling processes, including the Food and Drug Administration (FDA)–approved gout remedy probenecid (70). In addition, another Panx1 channel inhibitor, mefloquine, induces hypotension when injected into anesthetized guinea pigs (71, 72). Similar to probenecid, mefloquine is another FDA-approved drug to prevent and treat malaria, and it is noteworthy that this drug has a listed side effect of hypotension. Trovafloxacin, an FDA-approved antibiotic that was later removed from the market due to its side effects, can also inhibit Panx1 channels (13). Our work suggested that it may be possible to target the YLK sequence of Panx1, either with peptides (such as our IL2 peptide) or through a small-molecule approach and targeting this sequence, which may provide a highly specific mechanism for therapeutically regulating vasoconstriction and blood pressure. Collectively, this work provides new insight into the basis of α1AR-mediated vasoconstriction by indicating that noradrenergic signaling activates Panx1 to promote purinergic signaling and that this signaling mechanism may have a potentially key role in blood pressure homeostasis.
MATERIALS AND METHODS
Animals
Wild-type C57BL/6 and tdTomato male mice were purchased from Taconic and Jackson, respectively, and were used at 8 to 12 weeks of age. Panx1Fl/Fl mice were generated as previously described (13). Briefly, Panx1-targeted embryonic stem cells were obtained from the Knockout Mouse Project (KOMP) Repository and injected into blastocysts of C57BL/6J mice. Smooth muscle myosin heavy chain Cre modified estrogen receptor–binding domain (SMMHC-CreERT2) mice were a gift from S. Offermanns (73). In these mice, Cre is on the Y chromosome, is only expressed in SMCs, and is inducible by tamoxifen (73). We verified SMC expression by breeding SMMHC-CreERT2 mice with tdTomato mice, which express the tdTomato gene with a loxP-flanked STOP cassette. Panx1Fl/Fl mice were bred with C57BL/6 mice to obtain Panx1Fl/WT mice, which were further bred together to produce Panx1Fl/Fl mice (Cre−/Panx1Fl) and Panx1WT/WT mice (Cre−/Panx1WT). Male SMMHC-CreERT2+ were bred with female Panx1Fl/Fl mice, producing SMMHC-CreERT2+/Panx1Fl/WT male and SMMHC-CreERT2−/Panx1Fl/WT females. This progeny was later crossed together and resulted in SMMHC-CreERT2+/Panx1Fl/Fl mice (Cre+/Panx1Fl) and SMMHC-CreERT2+/Panx1WT/WT mice (Cre+/Panx1WT). The four different genotypes (Cre−/Panx1Fl, Cre−/Panx1WT, Cre+/Panx1WT, and Cre+/Panx1Fl) were injected intraperitoneally with tamoxifen (1 mg/kg per day) for 10 days. All mice were housed and used in accordance with the University of Virginia Animal Care and Use Committee guidelines.
Chemicals and reagents
Serotonin (5-HT), bovine serum albumin (BSA), and CBX were purchased from Fisher. ARL67156 was purchased from Tocris, and 10Panx1 was produced by GenScript. All other reagents were obtained from Sigma. Proben-ecid was prepared by dissolving in 1 M NaOH, and pH was adjusted to pH7.4.
Peptides
Amino acid sequences of the four peptides used in the study are indicated in Table 3. We have produced two peptide analogs to the mPanx1 intracellular loop, designated IL1 and IL2, and one peptide corresponding to the C-tail of Panx1 protein (CT2; Fig. 1A). The IL1, IL2, and CT2 peptides were produced by AnaSpec and attached to a TAT sequence (YGRKKQRRR) from the HIV tat transactivation protein (30, 31). The CT1 peptide has been shown to prevent NMDA-induced Panx1 activation in neurons (21). All peptides were diluted in water.
Plasmids
The α1DAR plasmid was purchased from OriGene (NM_013460), contained a C-terminal Myc-DDK tag (FLAG), and was expressed in a pCMV6-Entry vector. mPanx1–hemagglutinin (HA) in pEBB was obtained from K. Ravichandran at the University of Virginia. Mutations of Panx1 at the region of amino acids 191 to 200 (KYPIVEQYLK) were performed using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) using the primers in table S3. All mutations were confirmed by plasmid sequencing.
Cell culture and transfection
HEK293T cells were cultured as previously described (33), and were used until passage 20. For ATP measurements, 50,000 cells per well were seeded in 24-well plates precoated with 0.01% poly-L-lysine and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Briefly, 0.4 μg of α1DAR and 0.4 μg of Panx1WT, Panx1KYP>AAA, Panx1IVEQ>AAA, or Panx1YLK>AAA plasmids were added to each well along with 2 μl of Lipofectamine 2000. For electrophysiology measurements, 4 μg of α1DAR plasmid; 2 μg of Panx1WT, Panx1KYP>AAA, Panx1IVEQ>AAA, or Panx1YLK>AAA plasmids; and 0.5 μg of GFP (green fluorescent protein) plasmid were mixed with 10 μl of Lipofectamine 2000 and added to a well of a six-well plate seeded with ~70% confluent HEK cells. Cells were split the next morning and plated onto a glass coverslip for recording. ATP measurements and whole-cell recordings were performed 24 hours after transfection.
Pressure myography
The contractile responses of pressurized TDAs were measured as previously described (25). Briefly, TDAs were isolated and placed in cold Krebs-Hepes until cannulation in a pressure arteriograph (Danish Myo Technology). After a 30-min equilibration period, cumulative concentrations of contractile agonists were applied to the TDAs pressurized at 80 mmHg, and the vessel was visualized with an Olympus IX71 microscope attached to a Hamamatsu EM-CCD (electron multiplier charge-coupled device) camera coupled to SlideBook imaging software. At the end of the dose response, the viability of the endothelium was verified by applying 1 μM acetylcholine, and only TDAs exhibiting relaxation reaching 80% of the maximal diameter were analyzed. Last, maximal diameter was measured at the end of each experiment in calcium-free Krebs-Hepes supplemented with EGTA (1 mM) and sodium nitroprusside (10 μM). In cases where the vessels were not constricting to the contractile agonist, their viability was assessed by applying 40 mM KCl, and only TDAs reaching 30% of the maximal diameter were considered for analysis. The contractile responses, as well as the basal tone, are expressed as a percentage of the maximal diameter.
Vessel transfection was performed as previously described by us (4). Briefly, after isolation, TDAs were placed in cold, sterile RPMI supplemented with penicillin (2 mM)/streptomycin (50 U/ml) (Gibco), CaCl2 (2 mM), and 1% BSA. Vessels were transferred to a cuvette containing 100 μl of Nucleofector solution (Lonza) containing 5 μg of plasmid, and subjected to electroporation. Arteries were then placed in supplemented RPMI medium in an incubator for 14 to 18 hours until cannulation in the pressure arteriograph as described above.
Wire myography
Abdominal aorta were isolated from Cre+/Panx1Fl injected with peanut oil (vehicle control) or with tamoxifen, cut into 2-mm rings, and mounted on a myograph (Danish Myo Technology) as previously described (26). Each ring was bathed in Krebs solution containing 115.2 mM NaCl, 22.14 mM NaHCO3, 7.88 mM D-glucose, 4.7 mM KCl, 1.18 mM KH2PO4, 1.16 mM MgSO4, 1.80mMCaCl2, 0.114 mM ascorbic acid, and 0.027 mM Na2EDTA and continuously bubbled with 95% O2, 5% CO2. Rings were stretched at 1.2× resting length and allowed to equilibrate for 30 min at 37°C before application of 154 mM K+. Next, rings were washed in Krebs solution and subjected to cumulative concentrations of phenylephrine. The tension induced by the different doses of phenylephrine was expressed as the percentage of the tension induced by 154 mM K+.
Electrophysiology
Whole-cell recordings were obtained at room temperature from transiently transfected HEK293T cells using Axopatch 200B amplifier (Molecular Devices) in a bath solution composed of 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, and 10 mM glucose (pH 7.3). Patch pipettes (3 to 5 megohms) were filled with a Cs-/TEA-based internal solution, as previously described (33). Ramp voltage commands with 7-s intervals were applied using pCLAMP software and Digidata1322A digitizer (Molecular Devices). Peak whole-cell currents were determined at +80 mV and normalized to cell capacitance. Current mediated by Panx1 was defined by its sensitivity to CBX. In this system, no CBX-sensitive current is observed in HEK293T cells without Panx1 (32). Basal current was recorded for about 2 min, and phenylephrine (20 μM) was applied to the bath solution, which was followed by addition of CBX (50 μM). When indicated, cells were preincubated with bath solution containing IL2 peptide or its scrambled version at room temperature for 40 to 60 min before the experiment. The effect of phenylephrine on Panx1 current was quantified relative to CBX-sensitive basal current, and data were expressed as percent increase in CBX-sensitive current. All experiments were performed on 8 to 22 cells for each condition tested. Data were analyzed using a Kruskal-Wallis test (nonparametric one-way ANOVA) followed by Dunn’s post hoc test.
ATP assay
Mouse TDAs of equal length were isolated and washed thoroughly to eliminate red blood cells. Vessels were placed in a well of a 96-well plate containing Krebs-Hepes solution supplemented with 1% BSA at 37°C for 30 min to allow for degradation of any ATP released as a result of mechanical stimulation during manipulation of the vessel. For measurements performed on HEK cells: 24 hours after transfection, cells cultured in 24-well plates were cautiously washed twice with warm Krebs-Hepes-BSA and 300 μl of Krebs-Hepes-BSAwas added to each well after the last wash as previously described (33). The plate was then kept in the incubator at 37°C with 5% CO2 for 30 min to allow for degradation of ATP eventually released during the washes.
The ectonucleotidase inhibitor ARL67156 (300 μM) was applied to each well. When indicated, 10Panx1 (300 μM) or the peptide analogs to intracellular regions of Panx1 (3 μM) were added along with the ARL67156. After 30 min of incubation at 37°C with the inhibitor(s), phenylephrine (100 μM) or equivalent volume of Krebs-Hepes-BSAwas added, and the plate was kept at 37°C. After 5 min of phenylephrine stimulation, the medium surrounding the TDAs or the cells was transferred to an Eppendorf tube and centrifuged for 5 min at 5000g to eliminate eventual cell debris. Fifty microliters of the supernatant was transferred to a white-wall 96-well plate and placed in a FLUOstar Omega luminometer. The ATP concentration was measured by adding 50 μl of luciferin:luciferase reagent (ATP Bioluminescence Assay Kit HSII, Roche), which was injected into each well, and the luminescence was immediately recorded. Data are expressed as a percent increase in ATP concentration compared to unstimulated condition. Each stimulated condition was compared to unstimulated condition performed on the same day, and all experiments were performed on at least 15 wells of HEK cells or three TDAs. Data were analyzed using a Kruskal-Wallis test (nonparametric one-way ANOVA) followed by Dunn’s post hoc test.
Total ATP, or ATP content, was measured using the ATP Bioluminescence Assay Kit HSII (Roche) according to the manufacturer’s instructions. Briefly, each TDA was homogenized in 500 μl of cell lysis reagent using a douncer on ice. The homogenates were further centrifuged for 5 min at 5000g and 50 μl of the supernatant was transferred to a white-wall 96-well plate, and the ATP concentration was measured as described above.
Immunofluorescence
Experiments on isolated arteries were performed as previously described (4). Briefly, mice were deeply anesthetized and transcardially perfused with 5 ml of heparinized phosphate-buffered saline (PBS) followed by 5 ml of 4% paraformaldehyde in PBS. TDAs were isolated and placed in 4% paraformaldehyde for 1 hour before paraffin embedding. Cross sections of TDAs were subjected to paraffin removal, washed, and blocked for 30 min. Next, sections were incubated overnight with primary antibodies directed against Panx1, Panx2, and Panx3 as described in (11). All antibodies were produced in rabbit and were detected using an anti-rabbit secondary antibody coupled to Alexa Fluor 594 (11). Sections were observed using an Olympus FluoView 1000 as previously described (25).
To verify the intracellular localization of the different TAT-coupled peptides, TDAs were isolated from C57BL/6 and placed in Krebs-Hepes in the presence of the different peptides for 30 min. The vessels were next placed in 4% paraformaldehyde for further paraffin embedding. After paraffin removal, cross sections were incubated with a primary antibody directed against the TAT sequence.
Western blot
TDAs were isolated from mice and blood was thoroughly washed to avoid contamination of Panx1 present in erythrocytes. Two arteries from one mouse were homogenized on ice using a douncer containing 150 μl of lysis buffer [RIPA (radioimmunoprecipitation assay) buffer supplemented with protease cocktail inhibitor, Sigma]. After sonication, protein concentration was measured using a BCA assay (Pierce), and samples were mixed with Laemmli buffer and boiled. Protein lysates were subjected to electrophoresis on 4 to 12% bis-tris gel (Invitrogen), transferred to a nitrocellulose membrane, and blocked for 30 min using PBS–0.05% Tween (PBS-T) containing 3% BSA. Membranes were incubated overnight at 4°C with the primary antibody and washed twice in PBS-T before adding the corresponding LI-COR secondary antibody for an hour at room temperature. Membranes were washed, visualized, and quantitated using LI-COR Odyssey software as previously described (11).
Blood pressure measurements
Blood pressure was measured using radiotelemetry units [Data Sciences International (DSI)] implanted in C57BL/6, Cre−/Panx1Fl, Cre−/Panx1WT, Cre+/Panx1WT, and Cre+/Panx1Fl mice. The catheter of a radiotelemetry unit (TA11PA-C10, DSI) was implanted in the mouse left carotid artery under isoflurane anesthesia, and the catheter was tunneled trough the radio-transmitter placed in a subcutaneous pouch along the right flank of the mouse, as previously described (74). After implantation, mice were allowed to recover for 7 days.
For experiments on the conditional knockout mouse model: blood pressure was measured using Dataquest A.R.T. 20 software (DSI) for 5 days before starting tamoxifen injections at 1 mg/kg for 10 days. Blood pressure was recorded for another 5 days starting 24 hours after the last tamoxifen injection. The change in MAP (ΔMAP) was calculated by subtracting the average MAP measured for 5 days before tamoxifen injections to the MAP measured for 5 days after the tamoxifen injections. Day MAP was measured during inactivity of the mice [the light cycle (6:00 a.m. to 6:00 p.m.)], and night MAP was measured when the mice were most active [the nocturnal cycle (6:00 p.m. to 6:00 a.m.)]. MAPs before and after tamoxifen injections were compared with a Wilcoxon test (nonparametric paired t test).
For experiments on C57BL/6: basal blood pressure was measured continuously for 30 min before intraperitoneal injection of saline solution or peptide (20 mg/kg in a volume not exceeding 100 μl). Blood pressure was recorded for another 1.5 hours, and the MAP data from 1 to 1.5 hours after injection were averaged and compared to the basal blood pressure. The ΔMAP was calculated by subtracting the average MAP measured for 30 min before injection to the MAP measured for 30 min 1 hour after injection. MAPs before and after peptide or saline injections were compared with a Wilcoxon test (nonparametric paired t test).
Data analysis
All data were analyzed using GraphPad Prism and Origin software and are presented as means ± SEM. All vasoreactivity experiments were analyzed using a two-way ANOVA.
Supplementary Material
Fig. S1. Effect of SMC deletion of Panx1 on Panx2 and Panx3 abundance, ATP content, α1DAR abundance, and phenylephrine-induced contraction of aortic rings.
Fig. S2. Effect of IL2 scrambled peptide and TAT peptide on phenylephrine-mediated constriction and ATP release.
Fig. S3. Effect of mutations in the Panx1 IL2 region on the channel function.
Table S1. Contractile properties of aortic rings isolated from control Cre+/Panx1Fl mice and Cre+/Panx1Fl injected with tamoxifen for 10 days.
Table S2. Effect of scrambled IL2 peptide and TAT peptide on constriction of TDAs in response to phenylephrine.
Table S3. Primers used for the generation of Panx1KYP>AAA, Panx1IVEQ>AAAA, and Panx1YLK>AAA plasmids.
Acknowledgments
We thank the Histology Core at the University of Virginia, D. Hess and K. Karpe for technical help, and T. J. Johnson and L. Biwer for critical feedback. We also thank D. Laird and S. Penuela from the University of Western Ontario for providing Panx1 (both C-tail and extracellular loop), Panx2, and Panx3 antibodies.
Funding: This work was supported by NIH grants HL088554 (B.E.I.), P01 HL120840 (B.E.I., D.A.B., and K.S.R.), GM107848 (D.A.B. and K.S.R.), DK094907 (T.H.L.), GM086457 (A.V.S.), and R01DK088905 (A.V.S.). The American Heart Association provided postdoctoral (M.B.) and predoctoral (A.W.L. and J.K.S.) fellowships that supported this work. A.W.L., J.T.B., and L.J.D. were supported by NIH training grants (HL007284).
Footnotes
Author contributions: M.B. collected and analyzed the majority of the data, and prepared the figures and manuscript. Y.-H.C. created and characterized the heterologous system, and Y.-H.C. and J.K.S. performed electrophysiology recordings. A.W.L. performed Western blots and helped with ATP measurements on intact arteries. T.H.L. assisted with interpretation of blood pressure data. T.P. assisted in vasoreactivity and immunofluorescence experiments on the conditional knockout mice. J.T.B. performed peptide injections in C57BL/6 mice. J.T.B. and S.M.M. helped with vasoreactivity experiments involving the peptides. L.J.D. and A.K.B. helped with cell maintenance. M.V.A. performed wire myography on aortic rings from conditional knockout mouse model. R.J.T. provided CT2 peptide. K.S.R. shared (previously unpublished) Panx1Fl mice. D.A.B., K.S.R., T.H.L., R.J.T., and A.V.S. helped with experimental design and data interpretation. B.E.I. directed and supported the work through each steps. All the authors discussed the results and participated in manuscript preparation.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Burnstock G, Ralevic V. Purinergic signaling and blood vessels in health and disease. Pharmacol Rev. 2014;66:102–192. doi: 10.1124/pr.113.008029. [DOI] [PubMed] [Google Scholar]
- 2.Burnstock G. Dual control of vascular tone and remodelling by ATP released from nerves and endothelial cells. Pharmacol Rep. 2008;60:12–20. [PubMed] [Google Scholar]
- 3.Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. 2012;95:269–280. doi: 10.1093/cvr/cvs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billaud M, Lohman AW, Straub AC, Looft-Wilson R, Johnstone SR, Araj CA, Best AK, Chekeni FB, Ravichandran KS, Penuela S, Laird DW, Isakson BE. Pannexin1 regulates α1-adrenergic receptor-mediated vasoconstriction. Circ Res. 2011;109:80–85. doi: 10.1161/CIRCRESAHA.110.237594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panchin Y, Kelmanson I, Matz M, Lukyanov K, Usman N, Lukyanov S. A ubiquitous family of putative gap junction molecules. Curr Biol. 2000;10:R473–R474. doi: 10.1016/s0960-9822(00)00576-5. [DOI] [PubMed] [Google Scholar]
- 6.Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, Naus CC, Penuela S, Scemes E, Spray DC, Thompson RJ, Zhao HB, Dahl G. Pannexin channels are not gap junction hemichannels. Channels. 2011;5:193–197. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spagnol G, Sorgen PL, Spray DC. Structural order in Pannexin 1 cytoplasmic domains. Channels. 2014;8:157–166. doi: 10.4161/chan.28854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yen MR, Saier MH., Jr Gap junctional proteins of animals: The innexin/pannexin superfamily. Prog Biophys Mol Biol. 2007;94:5–14. doi: 10.1016/j.pbiomolbio.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bond SR, Naus CC. The pannexins: Past and present. Front Physiol. 2014;5:58. doi: 10.3389/fphys.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penuela S, Bhalla R, Gong XQ, Cowan KN, Celetti SJ, Cowan BJ, Bai D, Shao Q, Laird DW. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J Cell Sci. 2007;120:3772–3783. doi: 10.1242/jcs.009514. [DOI] [PubMed] [Google Scholar]
- 11.Lohman AW, Billaud M, Straub AC, Johnstone SR, Best AK, Lee M, Barr K, Penuela S, Laird DW, Isakson BE. Expression of pannexin isoforms in the systemic murine arterial network. J Vasc Res. 2012;49:405–416. doi: 10.1159/000338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poon IK, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature. 2014;507:329–334. doi: 10.1038/nature13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adamson SE, Leitinger N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett. 2014;588:1416–1422. doi: 10.1016/j.febslet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weilinger NL, Maslieieva V, Bialecki J, Sridharan SS, Tang PL, Thompson RJ. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol Sin. 2013;34:39–48. doi: 10.1038/aps.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol. 2010;299:H1146–H1152. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isakson BE, Thompson RJ. Pannexin-1 as a potentiator of ligand-gated receptor signaling. Channels. 2014;8:118–123. doi: 10.4161/chan.27978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohman AW, Isakson BE. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 2014;588:1379–1388. doi: 10.1016/j.febslet.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandilos JK, Bayliss DA. Physiological mechanisms for the modulation of pannexin 1 channel activity. J Physiol. 2012;590:6257–6266. doi: 10.1113/jphysiol.2012.240911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gödecke S, Roderigo C, Rose CR, Rauch BH, Gödecke A, Schrader J. Thrombin-induced ATP release from human umbilical vein endothelial cells. Am J Physiol Cell Physiol. 2012;302:C915–C923. doi: 10.1152/ajpcell.00283.2010. [DOI] [PubMed] [Google Scholar]
- 21.Weilinger NL, Tang PL, Thompson RJ. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J Neurosci. 2012;32:12579–12588. doi: 10.1523/JNEUROSCI.1267-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinheiro AR, Paramos-de-Carvalho D, Certal M, Costa MA, Costa C, Magalhães-Cardoso MT, Ferreirinha F, Sévigny J, Correia-de-Sá P. Histamine induces ATP release from human subcutaneous fibroblasts, via pannexin-1 hemichannels, leading to Ca2+ mobilization and cell proliferation. J Biol Chem. 2013;288:27571–27583. doi: 10.1074/jbc.M113.460865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, Scemes E. P2X7 receptor-Pannexin1 complex: Pharmacology and signaling. Am J Physiol Cell Physiol. 2008;295:C752–C760. doi: 10.1152/ajpcell.00228.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Piskuric NA, Vollmer C, Nurse CA. P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: Potential role in amplifying the neurotransmitter ATP. J Physiol. 2012;590:4335–4350. doi: 10.1113/jphysiol.2012.236265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Billaud M, Lohman AW, Straub AC, Parpaite T, Johnstone SR, Isakson BE. Characterization of the thoracodorsal artery: Morphology and reactivity. Microcirculation. 2012;19:360–372. doi: 10.1111/j.1549-8719.2012.00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Artamonov MV, Momotani K, Stevenson A, Trentham DR, Derewenda U, Derewenda ZS, Read PW, Gutkind JS, Somlyo AV. Agonist-induced Ca2+ sensitization in smooth muscle: Redundancy of Rho guanine nucleotide exchange factors (RhoGEFs) and response kinetics, a caged compound study. J Biol Chem. 2013;288:34030–34040. doi: 10.1074/jbc.M113.514596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008;295:C761–C767. doi: 10.1152/ajpcell.00227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, Sunada S, Takeo S, Tsujimoto G. The α1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–775. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivès E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 31.Howl J, Nicholl ID, Jones S. The many futures for cell-penetrating peptides: How soon is now? Biochem Soc Trans. 2007;35:767–769. doi: 10.1042/BST0350767. [DOI] [PubMed] [Google Scholar]
- 32.Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11311. doi: 10.1074/jbc.M111.323378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohman AW, Weaver JL, Billaud M, Sandilos JK, Griffiths R, Straub AC, Penuela S, Leitinger N, Laird DW, Bayliss DA, Isakson BE. S-nitrosylation inhibits pannexin 1 channel function. J Biol Chem. 2012;287:39602–39612. doi: 10.1074/jbc.M112.397976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma W, Hui H, Pelegrin P, Surprenant A. Pharmacological characterization of pannexin-1 currents expressed in mammalian cells. J Pharmacol Exp Ther. 2009;328:409–418. doi: 10.1124/jpet.108.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson WF, Boerman EM, Lange EJ, Lundback SS, Cohen KD. Smooth muscle α1D-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. Br J Pharmacol. 2008;155:514–524. doi: 10.1038/bjp.2008.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westcott EB, Segal SS. Ageing alters perivascular nerve function of mouse mesenteric arteries in vivo. J Physiol. 2013;591:1251–1263. doi: 10.1113/jphysiol.2012.244483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watts SW. Serotonin-induced contraction in mesenteric resistance arteries: Signaling and changes in deoxycorticosterone acetate–salt hypertension. Hypertension. 2002;39:825–829. doi: 10.1161/hy0302.104668. [DOI] [PubMed] [Google Scholar]
- 38.Watts SW, Davis RP. 5-Hydroxtryptamine receptors in systemic hypertension: An arterial focus. Cardiovasc Ther. 2011;29:54–67. doi: 10.1111/j.1755-5922.2010.00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill PB, Dora KA, Hughes AD, Garland CJ. The involvement of intracellular Ca2+ in 5-HT1B/1D receptor-mediated contraction of the rabbit isolated renal artery. Br J Pharmacol. 2000;130:835–842. doi: 10.1038/sj.bjp.0703387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierre LN, Davenport AP. Endothelin receptor subtypes and their functional relevance in human small coronary arteries. Br J Pharmacol. 1998;124:499–506. doi: 10.1038/sj.bjp.0701865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rizzoni D, Porteri E, Piccoli A, Castellano M, Bettoni G, Pasini G, Agabiti-Rosei E. The vasoconstriction induced by endothelin-1 is mediated only by ETA receptors in mesenteric small resistance arteries of spontaneously hypertensive rats and Wistar Kyoto rats. J Hypertens. 1997;15:1653–1657. doi: 10.1097/00004872-199715120-00068. [DOI] [PubMed] [Google Scholar]
- 42.Pinheiro AR, Paramos-de-Carvalho D, Certal M, Costa C, Magalhães-Cardoso MT, Ferreirinha F, Costa MA, Correia-de-Sá P. Bradykinin-induced Ca2+ signaling in human subcutaneous fibroblasts involves ATP release via hemichannels leading to P2Y12 receptors activation. Cell Commun Signal. 2013;11:70. doi: 10.1186/1478-811X-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seminario-Vidal L, Kreda S, Jones L, O’Neal W, Trejo J, Boucher RC, Lazarowski ER. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J Biol Chem. 2009;284:20638–20648. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, Jones LC, O’Neal WK, Penuela S, Laird DW, Boucher RC, Lazarowski ER. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem. 2011;286:26277–26286. doi: 10.1074/jbc.M111.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitazawa T, Kitazawa K. Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol. 2012;590:5401–5423. doi: 10.1113/jphysiol.2012.241315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Momotani K, Artamonov MV, Utepbergenov D, Derewenda U, Derewenda ZS, Somlyo AV. p63RhoGEF couples Gαq/11-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ Res. 2011;109:993–1002. doi: 10.1161/CIRCRESAHA.111.248898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai MH, Jiang MJ. Rho-kinase-mediated regulation of receptor-agonist-stimulated smooth muscle contraction. Pflugers Arch. 2006;453:223–232. doi: 10.1007/s00424-006-0133-y. [DOI] [PubMed] [Google Scholar]
- 48.Budzyn K, Paull M, Marley PD, Sobey CG. Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. J Pharmacol Exp Ther. 2006;317:791–796. doi: 10.1124/jpet.105.100040. [DOI] [PubMed] [Google Scholar]
- 49.Loirand G, Sauzeau V, Pacaud P. Small G proteins in the cardiovascular system: Physiological and pathological aspects. Physiol Rev. 2013;93:1659–1720. doi: 10.1152/physrev.00021.2012. [DOI] [PubMed] [Google Scholar]
- 50.Boonen HC, De Mey JG. G-proteins are involved in contractile responses of isolated mesenteric resistance arteries to agonists. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:462–468. doi: 10.1007/BF00169465. [DOI] [PubMed] [Google Scholar]
- 51.Heesen BJ, De Mey JG. Effects of cyclic AMP-affecting agents on contractile reactivity of isolated mesenteric and renal resistance arteries of the rat. Br J Pharmacol. 1990;101:859–864. doi: 10.1111/j.1476-5381.1990.tb14171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karsten AJ, Derouet H, Ziegler M, Eckert RE. Involvement of cyclic nucleotides in renal artery smooth muscle relaxation. Urol Res. 2003;30:367–373. doi: 10.1007/s00240-002-0281-2. [DOI] [PubMed] [Google Scholar]
- 53.Vettel C, Wittig K, Vogt A, Wuertz CM, El-Armouche A, Lutz S, Wieland T. A novel player in cellular hypertrophy: Giβγ/PI3K-dependent activation of the RacGEF TIAM-1 is required for α1-adrenoceptor induced hypertrophy in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2012;53:165–175. doi: 10.1016/j.yjmcc.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 54.Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev. 2014;66:513–569. doi: 10.1124/pr.112.007351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moore AW, Jackson WF, Segal SS. Regional heterogeneity of α-adrenoreceptor subtypes in arteriolar networks of mouse skeletal muscle. J Physiol. 2010;588:4261–4274. doi: 10.1113/jphysiol.2010.194993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Post-junctional α-adrenoceptors and basal limb vascular tone in healthy men. J Physiol. 2002;540:1103–1110. doi: 10.1113/jphysiol.2001.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohyanagi M, Faber JE, Nishigaki K. Differential activation of α1- and α2-adrenoceptors on microvascular smooth muscle during sympathetic nerve stimulation. Circ Res. 1991;68:232–244. doi: 10.1161/01.res.68.1.232. [DOI] [PubMed] [Google Scholar]
- 58.Guimarães S, Moura D. Vascular adrenoceptors: An update. Pharmacol Rev. 2001;53:319–356. [PubMed] [Google Scholar]
- 59.Katsuragi T, Tamesue S, Sato C, Sato Y, Furukawa T. ATP release by angiotensin II from segments and cultured smooth muscle cells of guinea-pig taenia coli. Naunyn Schmiedebergs Arch Pharmacol. 1996;354:796–799. doi: 10.1007/BF00166908. [DOI] [PubMed] [Google Scholar]
- 60.Cheng Y, Mansfield KJ, Sandow SL, Sadananda P, Burcher E, Moore KH. Porcine bladder urothelial, myofibroblast, and detrusor muscle cells: Characterization and ATP release. Front Pharmacol. 2011;2:27. doi: 10.3389/fphar.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diezmos EF, Sandow SL, Markus I, Perera D Shevy, Lubowski DZ, King DW, Bertrand PP, Liu L. Expression and localization of pannexin-1 hemichannels in human colon in health and disease. Neurogastroenterol Motil. 2013;25:e395–e405. doi: 10.1111/nmo.12130. [DOI] [PubMed] [Google Scholar]
- 62.Timóteo MA, Carneiro I, Silva I, Noronha-Matos JB, Ferreirinha F, Silva-Ramos M, Correia-de-Sá P. ATP released via pannexin-1 hemichannels mediates bladder overactivity triggered by urothelial P2Y6 receptors. Biochem Pharmacol. 2014;87:371–379. doi: 10.1016/j.bcp.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 63.Edwards TA, Wilson AJ. Helix-mediated protein–protein interactions as targets for intervention using foldamers. Amino Acids. 2011;41:743–754. doi: 10.1007/s00726-011-0880-8. [DOI] [PubMed] [Google Scholar]
- 64.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Dahl G. SCAM analysis of Panx1 suggests a peculiar pore structure. J Gen Physiol. 2010;136:515–527. doi: 10.1085/jgp.201010440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kay BK, Williamson MP, Sudol M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 67.Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 68.Riquelme MA, Cea LA, Vega JL, Boric MP, Monyer H, Bennett MV, Frank M, Willecke K, Sáez JC. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology. 2013;75:594–603. doi: 10.1016/j.neuropharm.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 69.Robertson TP, Moore JN, Noschka E, Lewis TH, Lewis SJ, Peroni JF. Effects of Rho-kinase and Src protein tyrosine kinase inhibition on agonist-induced vasoconstriction of arteries and veins of the equine laminar dermis. Am J Vet Res. 2007;68:886–894. doi: 10.2460/ajvr.68.8.886. [DOI] [PubMed] [Google Scholar]
- 70.Xiong XX, Gu LJ, Shen J, Kang XH, Zheng YY, Yue SB, Zhu SM. Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem Res. 2014;39:216–224. doi: 10.1007/s11064-013-1212-z. [DOI] [PubMed] [Google Scholar]
- 71.Coker SJ, Batey AJ, Lightbown ID, Díaz ME, Eisner DA. Effects of me-floquine on cardiac contractility and electrical activity in vivo, in isolated cardiac preparations, and in single ventricular myocytes. Br J Pharmacol. 2000;129:323–330. doi: 10.1038/sj.bjp.0703060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iglesias R, Spray DC, Scemes E. Mefloquine blockade of Pannexin1 currents: Resolution of a conflict. Cell Commun Adhes. 2009;16:131–137. doi: 10.3109/15419061003642618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13–LARG–mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 74.Cechova S, Zeng Q, Billaud M, Mutchler S, Rudy CK, Straub AC, Chi L, Chan FR, Hu J, Griffiths R, Howell NL, Madsen K, Jensen BL, Palmer LA, Carey RM, Sung SS, Malakauskas SM, Isakson BE, Le TH. Loss of collectrin, an angiotensin-converting enzyme 2 homolog, uncouples endothelial nitric oxide synthase and causes hypertension and vascular dysfunction. Circulation. 2013;128:1770–1780. doi: 10.1161/CIRCULATIONAHA.113.003301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of SMC deletion of Panx1 on Panx2 and Panx3 abundance, ATP content, α1DAR abundance, and phenylephrine-induced contraction of aortic rings.
Fig. S2. Effect of IL2 scrambled peptide and TAT peptide on phenylephrine-mediated constriction and ATP release.
Fig. S3. Effect of mutations in the Panx1 IL2 region on the channel function.
Table S1. Contractile properties of aortic rings isolated from control Cre+/Panx1Fl mice and Cre+/Panx1Fl injected with tamoxifen for 10 days.
Table S2. Effect of scrambled IL2 peptide and TAT peptide on constriction of TDAs in response to phenylephrine.
Table S3. Primers used for the generation of Panx1KYP>AAA, Panx1IVEQ>AAAA, and Panx1YLK>AAA plasmids.