

Abstract
Inducible cyclooxygenase (COX-2) and its metabolites have diverse and potent biological actions that are important for both physiological and disease states of lung. The wide variety of prostaglandin (PG) products are influenced by the level of cellular activation, the exact nature of the stimulus, and the specific cell type involved in their production. In turn, the anti-and proinflammatory response of PG is mediated by a blend of specific surface and intracellular receptors that mediate diverse cellular events. The complexity of this system is being at least partially resolved by the generation of specific molecular biological research tools that include cloning and characterization of the enzymes distal to COX-2 and the corresponding receptors to the final cellular products of arachidonic metabolism. The most informative of these approaches have employed genetically modified animals and specific receptor antagonists to determine the exact role of specific COX-2-derived metabolites on specific cell types of the lung in the context of inflammatory models. These data have suggested a number of cell-specific, pathway-specific, and receptor-specific approaches that could lead to effective therapeutic interventions for most inflammatory lung diseases.
Keywords: asthma, acute lung injury, pulmonary fibrosis
Aspirin is an ancient remedy that was first marketed in 1898 and launched more than a century of research that has focused on the involvement of cyclooxygenase (COX) and its enzymatic products in diverse physiological and pathophysiological events. Thirty years after prostaglandins (PG) were identified, Orloff et al. (79a) elucidated their molecular structure and demonstrated that they were derived from arachidonic acid via the COX reaction. This seminal work was greatly advanced when John Vane (102) first demonstrated that aspirin and indomethacin inhibited COX in cell-free homogenated lung tissue from guinea pigs. For their contributions of identifying COX, prostanoids products, and effective pharmacological inhibitors, Bergstrom, Samuelsson, and Vane shared The Noble Prize for Physiology or Medicine in 1982. Armed with these molecular tools and the analytic chemistry necessary to measure prostanoids, many investigators have examined the role of COX-2 and its metabolites in diverse and potent biological action on individual organs and mediators of organ interactions, including work that identified a prominent role in normal physiology and disease state of lung. Although there are several recent general reviews about COX and PG (18, 33, 73, 74, 90), none are specifically focused on information about the role of COX-2 and its metabolites in inflammatory lung diseases. Here, we will mainly focus on recent developments and updated information regarding the role of COX-2 in lung disease with an emphasis on asthma, acute lung injury, and idiopathic pulmonary fibrosis (IPF).
BIOCHEMISTRY OF COX AND PROSTANOIDS
PG are potent biologically active lipid molecules derived from arachidonic acid that are produced by almost every human cell type and act as autocrine, paracrine, and endocrine mediators through an interaction with specific PG receptors. Prostanoids are synthesized de novo from membrane-released arachidonic acid in response to stimulation. PG synthesis starts with the oxidative cyclization of the five carbons at the center of arachidonic acid, which is released by phospholipase A2 from the cell membrane. Free arachidonic acid is presented to the endoplasmic reticulum and nuclear membrane, where membrane-bound COX enzymes catalyze the rate-limiting step for PG synthesis. COX catalyzes the cyclooxygenation reaction through which arachidonic acid is enzymatically cyclized and oxygenated to generate the biocyclic endoperoxide intermediate PGG2. COX reduces a hydroperoxyl in PGG2 to a hydroxyl to form PGH2 via a separate peroxidase active site on the enzyme, resulting in reduction to PGH2. Subsequent isomerases and oxidoreductases catalyze the production of various bioactive PG isomers using PGH2 as the main substrate.
The coupling of PGH2 synthesis to metabolism by down-stream enzymes is intricately orchestrated in a stimulus and cell-specific fashion through the dominance of various distal enzymes. In various cell types of the lung and under different physiological conditions, the downstream metabolism of PGH2 can be dramatically different. PGI synthase (also called prostacyclin synthase) is found in relative abundance in endothelial cells. Two types of PGD synthase, hematogenous and lipocalen PGD synthase, are found in mast cells and macrophages. Microsomal PGE synthase (mPGES) appears in most cell types, but is particularly abundant in airway epithelial cells, and is responsible for PGE2 synthesis. PGF synthase is expressed mostly in the uterus, and thromboxane synthase is present in both platelets and macrophages.
In some cases, COX enzymes and distal PG synthase(s) are coordinately induced. For example, in an inflammatory setting, macrophages increase the expression of both COX-2 and mPGES (57). Also, there is a competition for substrate between COX and lipooxgenases that can contribute to the mix of lipid products, and there are also nonenzymatic mechanisms involved in the transformation of PGH2 into primary PG referred to as isoprostanes, but these are not the focus of this review.
COX-1, the constitutive or noninducible isoform of COX, was first cloned in 1988, and, shortly thereafter, the inducible isoform, COX-2, was discovered. The human gene encoding COX-2 is located on chromosome 1, contains 10 exons, and its RNA transcript is 4.5 kb. COX-2 possesses ~60% amino acid identity with COX-1. Both enzymes are ~600 amino acids in size in most species with an unmodified molecular weight of 68 kDa and ~75–80 kDa after posttranslational modification, which consists mainly of glycosylation. Although there are notable exceptions, in general, COX-1 is the enzyme responsible for basal, constitutive PG synthesis, whereas COX-2 is important in various inflammatory and induced settings. COX-2 gene expression is minimally or not present in most tissues; however, a few hours after stimulation, COX-2 mRNA, protein, and enzymatic activity is dramatically increased followed by a prompt return to basal level over a predictable but relatively short time course. The best-studied inducers of COX-2 are bacterial LPS and proinflammatory cytokines, such as TNF-α and IL-1β. Growth factors and some tumor promoters, such as PMA, also stimulate COX-2 expression in various cell types.
GENE REGULATION OF COX-2
COX-2 gene expression is chiefly regulated at the level of transcription, and, in general, there is an excellent correlation between the time course for COX-2 mRNA expression and COX-2 protein production. The promoter regions of COX-2 genes in mice, rats, and humans have been cloned, sequenced, and mostly characterized. This promoter region contains a canonical TATA box and various putative transcriptional regulatory elements such as cAMP response element (CRE), PU.1, AP2, SP1, GATA box, CCAAT enhancer-binding protein (C/EBP), and NF-κB (8, 26, 44). The mechanisms leading to COX-2 expression involve a combinatorial interaction between the enhancer region and multiple transcription factors that varies in particular cell types and in response to the specific stimulus. The COX-2 promoter contains two putative NF-κB binding sites, and it has been shown that NF-κB regulates COX-2 expression, at least in part, in LPS-stimulated macrophages (10). Stimulation of either protein kinase C (PKC) or Ras signaling enhances mitogen-activated protein kinase (MAPK) activity, which, in turn, activates transcription of COX-2. C/EBP transcriptional factors are also involved in regulating activity of the COX-2 promoter in a cell-specific manner (25, 40). Ets family proteins, such as PU.1, normally contribute to activation of COX-2 transcription, but some members of this same family may repress this process. CRE is involved in COX-2 induction in pulmonary artery smooth muscle cells, and monocytes and mutation of the CRE binding site suppress COX-2 reporter induction (5, 105).
Recently, histone acetyltransferase activity of CBP/p300 coactivator complex has been shown to be important for C/EBPβ and AP-1-mediated induction of COX-2 (44, 96). This enzyme acetylates a key transcription factor, C/EBPβ, which results in increased transcription of the COX-2 gene. Another plausible mechanism for modulating COX-2 expression is the change in chromatin structure adjacent to key DNA binding sequence motif in COX-2 promoter. The transcriptionally inactive chromatin is tightly wrapped around histone proteins and inhibits DNA binding of transcriptional factors. Modifications of histone H3 by phosphorylation potentially relax chromatin structure and increase the exposure of gene promoter elements to various transcription factors, resulting in an increase in the transcriptional activity of COX-2 in macrophages (81).
PG RECEPTORS
PG are released from cells predominantly by facilitated transport through a known PG transporter of the organic anion transporter polypeptide family and potentially by other uncharacterized transporters (86). There are at least nine known PG receptor forms in mouse and man as well as several additional splice variants (73). PG receptors are named by the letter “P” and a prefix of “D,” “E,” “F,” “I,” or “T” to signify preference for PG. To date, four subtypes of EP receptors, EP1–EP4, two receptors for PGD2 (DP1 and DP2), and receptors for PGF2α, PGI2, and thromboxane A2 (TXA2) (FP, IP, and TP, respectively) have been characterized. PG receptors belong to G protein-coupled receptors, which are seven-transmembrane-spanning proteins. The lone exception is the DP2 receptor, which is homologous to a chemoattractant receptor that is expressed on T helper type 2 (Th2) cells (CRTH2). Activation of a given PG receptor by its cognate ligand may elicit varying responses in different cell types and tissues (33). The precise role of PG receptors in pathological settings is determined by many factors, including the receptor expression profile, ligand affinity, differential coupling to signal transduction pathways, and the cellular context in which the receptor is expressed. The intricacy of this system is highlighted by the diverse and often opposing effects of PG within the immune-inflammatory response. The exact role of a specific prostanoid in the inflammatory response is often ambiguous; in certain settings, PG function as proinflammatory mediators, but in others, they appear to have anti-inflammatory properties (33, 73).
In addition to classic prostanoids that act via plasma membrane-derived G protein-coupled receptors, even more distal COX products, such as PGJ2, 15-deoxy-PGJ2 (15d-PGJ2), and PGA2 can activate intracellular nuclear receptor of the peroxisome proliferator-activated receptor-γ (PPARγ) class. The importance of these distal prostanoid products in intracellular signal transduction appears to be an emerging topic of intense investigations.
ALLERGIC AIRWAY INFLAMMATION, ASTHMA, AND COX-2
In asthmatic airways, COX-2 gene expression is increased, which suggests involvement of COX products in the pathogenesis of this disease (95, 97). Additionally, there is an exaggeration of airway eosinophilia, IgE production, and airway hyperresponsiveness in both COX-1- and COX-2-deficient mice (20). As expected from these data, inhibition of COX with the nonselective COX inhibitor indomethacin augments ovalbumin (OVA)-induced allergen airway eosinophilia, Th2 type cytokine production, and airway hyperresponsiveness in a mouse model of allergic asthma (94, 95). All of these data together support the conclusion that endogenous PG play a regulatory role in allergic response with an overall balance favoring suppression of the asthmatic response by COX-2 expression and production of key products. However, on a closer look, the exact role of specific prostanoid products in the pathogenesis of allergic airway disease is ambiguous. Nonselective COX inhibitors have very little effect on airway function in humans with asthma except in a relatively unique cohort of aspirin-sensitive asthmatics (28, 49, 93). This finding suggests a complex involvement of COX in the pathogenesis of asthma because some PG have proallergic inflammatory activity, whereas others have anti-allergic activities, and little is understood about the relationship between pro- and anti-asthmatic prostanoids (Table 1). Most likely, there is a regulated balance between the bronchoconstriction and bronchodilation action of various PG that contributes to bronchial tone, possibly mediated by a balance between PGD2 and PGE2, as discussed in detail (Fig. 1).
Table 1.
Role of PGD2 and PGE2 in the pathogenesis of allergic inflammation
| PGD2 | PGE2 | |
|---|---|---|
| Effect on allergic lung inflammation |
|
|
Fig. 1.
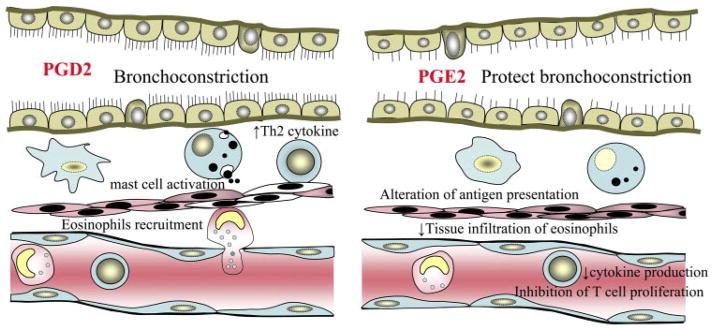
Schematic summary of PGE2 and PGD2 involvement in allergic inflammatory airway diseases. Th2, T helper type 2.
ROLE OF PGD2 IN THE PATHOGENESIS OF ASTHMA
PGD2 is a major eicosanoid, among several other inflammatory mediators, that is released by mast cells and activated macrophages. PGD2 is produced in response to allergic challenge and therefore may have an important role in airway reactivity. In IgE-dependent eicosanoid generation, PGD2 is synthesized in asthma within 2 min of mast cell activation (70) and is known to cause bronchoconstriction in both humans and animals (31). PGD2 is capable of synergistically increasing bronchial reactivity in response to inhaled methacholine (17). Bronchoalveolar lavage fluid from asthmatic patients has a nearly tenfold increased level of PGD2 compared with a nonasthmatic control population (56). The targeted overexpression of PGD2 synthase gene in the lungs of transgenic mice is associated with elevated levels of Th2 cytokines and eosinophil infiltration (15). Although the source of PGD2 production in asthmatic reaction has not been completely defined, it is presumed to be mast cells. In an in vitro experiment, anti-IgE-dependent activation of mast cells resulted in the preferential generation of PGD2, and this is correlated with histamine secretion (34, 54). Whereas mast cells are considered a major source of PGD2 synthesis (54), PGD2 is also produced by other cell types, including Th2 lymphocytes, macrophages, and dendritic cells, during asthmatic attacks that could contribute to an enhanced allergic response (39, 99, 100). Although the cellular target for PGD2 has not been completely defined, it has recently been shown that PGD2 influences Th2 type inflammation through the induction of macrophage-derived chemokine in an allergic airway model (39).
Recent studies have identified that two types of the receptor for PGD2 behave differently in the allergic reaction. One is DP1 and the other is CRTH2/DP2, which is a chemoattractant receptor homolog molecule expressed on Th2. The DP1 receptor belongs to the prostanoid receptor family that consists of eight types and subtypes of receptor, each specific to an individual prostanoid. PGD2 is likely involved in multiple aspects of allergic inflammation through these dual receptor systems, DP1 and CRTH2/DP2. Among the prostanoid receptor, DP1 is the least abundant and in mice is expressed weakly in ileum, lung, stomach, and uterus (37). However, the DP1 receptor is also present on mast cells and eosinophils, and this may mediate production of effecter molecules that contribute to the asthmatic phenotype or predisposition. The DP1 receptor-deficient mice (DP−/−) have a reduced level of Th2 cytokines and less accumulation of lymphocyte in the lung of OVA-induced asthma model, compared with wild-type animals, even though increases in serum IgE concentration are similar to those of wild-type mice. Moreover, DP−/− mice showed decreased infiltration of eosinophils and failed to develop airway hyperreactivity in the OVA-induced model (62).
In addition, a population-based polymorphism study has shown that single nucleotide polymorphism variations in the promoter regions of prostanoid DP receptor (PTGDR) gene, located on chromosome 14q22, correlate to the susceptibility for developing asthma. A person who has a copy of the haplotype with a low transcriptional efficiency has a lower risk of asthma than subjects with no copies of the haplotype. These data, in combination with the animal studies, suggest that less responsiveness to PGD2 by virtue of impaired transcription of PTGDR is protective for developing asthma (78).
In contrast to this, DP1 agonist treatment during the antigen challenge phase decreases eosinophilia and airway hyperresponsiveness in a murine asthma model (92). This suggests that the timing of DP1 activation might be an important issue in this model. Furthermore, it is unknown which cells are responsible for DP1 receptor activity in asthma. Mast cells may be involved because this cell type is a key player in asthma reaction and has abundant DP1 receptors. Interestingly, there is also plentiful expression of the DP1 receptor in bronchiolar and alveolar epithelial cells in the asthmatic airway (15). The airway epithelium is proposed to be a source of proinflammatory cytokines and chemokines in asthma, raising the possibility that PGD2 acting at DP1 in the epithelium contributes to the production and release of these mediators. Further research is necessary to more clearly address this question.
In contrast to DP1, CRTH2/DP2 is preferentially expressed in Th2 cells, eosinophils, and basophils in humans and serves as the novel receptor for PGD2. In human eosinophils, which possess both DP1 and DP2 receptor (67, 92), PGD2 is a potent stimulator of eosinophil chemotaxis, actin polymerization, CD11b expression, and L-selectin shedding through DP2 activation, but it is not DP1 mediated (67, 92). In addition to a role in eosinophil recruitment, DP2 agonist increases the pathology of allergic inflammation. DP2, but not DP1, also mediates PGD2-dependent cell migration of blood eosinophils and basophils (36). Thus DP2 receptor mediates proinflammatory effect of PGD2 in allergic inflammation.
There are little data that address the molecular mechanism of DP signaling. Recently, it has been suggested that the downstream signaling of DP1 receptor is mediated via p38 MAPK and PKC pathways in a cell type-specific manner leading to the activation of NF-κB (58). In response to PGD2, DP2 induces intracellular Ca2+ mobilization and chemotaxis in Th2 cells (36). However, the interaction between DP1 and DP2 in terms of regulating allergic inflammatory signaling has not been addressed.
ROLE OF PGE2 IN THE PATHOGENESIS OF ASTHMA
There is a dispute regarding the role of PGE2 in asthma because of discrepancies between in vitro and in vivo studies. In in vitro studies, PGE2 appears to polarize cellular response toward a Th2 phenotype enhancing IL-4 and IL-5 production (2, 47) and inhibition of macrophage IL-12 production (101). IL-4 and IL-5 are the prototypic Th2 cytokines, and IL-12 is the critical inducer of a polarized Th1 response and plays a role in inhibiting Th2 response (101). In addition, PGE2 also influences Th2-mediated humoral immune response by EP2/4 receptor-driven immunoglobulin class switching to IgE and EP3 receptor-dependent potentiation of mast cell degranulation (14, 76). In spite of these in vitro data, it has been suggested that PGE2 has a bronchoprotective effect in patients with bronchial asthma. PGE2 has been shown to protect against exercise-induced (64), allergen-induced (82), and aspirin-induced bronchoconstriction (13, 59) as well as bronchoconstrictor agents such as methacholine and histamine (59, 103). PGE2 prevent not only allergen-induced bronchoconstriction but also inhibit allergen-induced airway inflammation, including decreased airway eosinophilia in asthma patients (19) and Th2 cytokine production in the OVA-induced murine model (19, 61). COX-1 deficient mice exhibited significantly increased lung inflammation and airway hyperresponsiveness in OVA-induced asthma model, which is correlated with abrogation of PGE2 biosynthesis (20). Within the immune system, PGE2 modulates the function of T cells and macrophages, which are critical for the immune response. PGE2 suppresses proliferation of T cells (24, 65) and inhibits cytokine production of macrophages and alters antigen presentation by inhibiting expression of major histocompatability complex class II proteins (85, 91).
There is little known about interaction between PGE2 and PGD2. Both PG seem to have an opposite role in terms of allergic reaction. It is suggested that PGE2 overrides the pro-asthmatic properties of PGD2. Nebulized PGE2 administered before allergen challenge attenuates the early asthmatic reaction, which may be via its action on downregulation of PGD2 in bronchoalveolar lavage fluid (32).
ROLE OF PGI2 IN THE PATHOGENESIS OF ASTHMA
PGI2 is produced during the allergic reaction in human lung (11) and in murine airway after OVA inhalation (42). PGI2 inhibits allergic mediator release and eosinophil recruitment in experimental animals (6, 52). In OVA-induced asthma model, selective inhibition of COX-2 specifically reduces PGI2 synthesis and results in a marked increase in Th2-mediated lung inflammation. The elevated Th2-mediated inflammatory response elicited by selective COX-2 inhibitors is associated with enhanced airway hyperreactivity and is coincident with a marked increase in the levels of Th2 type of cytokine, including IL-4, IL-5, and IL-13 in the airways (42). In contrast to the proinflammatory effects of prostacyclin, which are important for the generation of edema and pain accompanying inflammation, these findings suggest PGI2 may play a role in inhibiting Th2 inflammatory response.
Interestingly, IP receptor mRNA is upregulated in CD4+ Th2 cells (42), and IP-deficient mice showed the augmentation of allergic inflammation in the airway and skin, associated with the increases in vascular permeability and enhancement of Th2 response (98). Recently, IP receptor has been shown to be involved in airway remodeling in chronic allergen challenge model. IP-deficient mice have more goblet cell hyperplasia and subepithelial fibrosis compared with wild-type mice (72). Even though increased production of IL-10, immunosuppressive cytokine (42), is suggested for a working mechanism, the working mechanism of PGI2 in allergic inflammation is unclear.
ROLE OF PG IN THE PATHOGENESIS OF INFLAMMATION AND ACUTE LUNG INJURY
Role of PG in acute inflammation
It is well known that PG are primarily involved in vasodilatation in the inflammatory process and synergize with other mediators, such as histamine, to cause vascular permeability and edema. Many studies have shown that PGE2 and PGI2 are potent vasodilators that are present at high concentrations at inflammation sites (12). Prostacyclin (PGI2) acts on platelets and blood vessels to inhibit platelet aggregation and to cause vasodilatation and is thought to be important for vascular homeostasis. In inflammation, PGI2 is an important mediator of the edema and pain that accompany acute inflammation. Even in IP receptor-deficient mice that are viable, reproductive, and normotensive, their inflammatory and pain responses are reduced. IP receptor-deficient mice have reduced potentiation of bradykinin-induced microvascular permeability and substantially reduced carrageenan-induced paw edema, suggesting prostacyclin is a mediator of this type of inflammation (71). However, it remains to be shown whether PGI2 and the IP play important roles in other types of inflammations.
The role of PGE2 and EP receptors in inflammation is complicated. The most major cells involved in inflammation including T, B, and dendritic cells have four EP receptors. PGE2 exerts both pro- and anti-inflammatory effects, depending on receptor subtype, cell population, and context of activation. PGE2 enhanced the migration of antigen-stimulated Langerhans cells to lymph nodes and subsequent T cell activation during the contact hypersensitivity response through the EP4 receptor (46). The EP4 receptor also mediates to shift the balance in favor of Th2 response by inhibiting IL-12 production in macrophage and promoting IgE class switching in B cells (14, 75, 101). On the other hand, PGE2 inhibits the production of cytokines like TNF-α in macrophages through the EP4 receptor and T cell proliferation through the EP2 receptor (75). As mentioned before, PGD2 has a proinflammatory role in allergic inflammation and a vasodilatation effect. However, PGD2 may act to inhibit inflammation in other contexts. PGD2 can reduce the migration of lung dendritic cells, which plays a key role in presenting antigens and initiating an adaptive immune response through the selective DP1 activation. After OVA challenge, the DP1 agonist inhibits dendritic cell migration for lung to lymph node, which results in inhibition of T cell proliferation and a lower amount of T cell cytokines (30, 45). These data suggest that PGD2 might contribute to the resolution of allergic inflammation. TXA2 also has been shown to have a similar inhibitory effect on dendritic cells (45).
Role of PG in acute respiratory distress syndrome
Prostanoids have various physiological effects in the lung and are thought to contribute to the pathobiology of acute lung injury. Several studies have shown that COX inhibition before the onset of severe sepsis reduces physiological abnormalities and improves survival in animal models (35, 77). However, treatment with the nonselective COX inhibitor ibuprofen in patients with severe sepsis has no beneficial effect that could not be fully attributed to its antipyretic activity. In large-scale clinical studies, ibuprofen treatment in patients with sepsis did not reduce the incidence or duration of shock or the incidence of acute respiratory distress syndrome (ARDS) and did not significantly improve the rate of survival (1).
Although global inhibition of COX is not an effective prevention for ARDS, there are studies that show that COX-2 and COX metabolites are involved in the development and progression of acute lung injury in animal models. Acute lung injury is associated with increased COX-2 gene expression in murine lung, and selective pharmacological inhibition or gene disruption of COX-2 attenuates lung injury in both an acid-induced mouse model of acute lung injury and in a carrageenan-induced rat pleurisy model (9, 16, 79). In these models, TXA2 and PGI2 are known to be involved in the generation of lung inflammation. In oleic acid lung injury, there is an increase in plasma level of TXB2, a stable metabolite of TXA2, which is correlated with decreased partial oxygen pressure of arterial blood PaO2. Treatment with a TXA2 synthase inhibitor before oleic acid injection prevents the decrease in PaO2 and pulmonary vascular hyperpermeability (41). These data suggest that TXA2 participates in the oleic acid lung injury as an early phase mediator. In acute lung injury, the ventilation/perfusion pattern strongly affects the oxygenation and one of the parameters for disease severity. PGI2 is responsible for perfusion pattern in acute lung injury in an oleic acid-induced lung injury model. Treatment with selective COX-2 inhibitor can prevent decrease of perfusion redistribution by blocking the PGI2 production, which improves the oxygenation in acute lung injury (27).
PGE2 is an important mediator of pulmonary edema in acute lung injury. It has been shown that platelet-activating factor (PAF), triggering edema formation, is at least partly mediated by the release of PGE2 and activation of EP3 receptors (22). In isolated rat lungs, PAF administration stimulated release of PGE2 and increased lung weight as a measure of edema formation. Perfusion with a neutralizing PGE2 antibody attenuated the PAF-induced edema formation. EP3 receptor-deficient mice have an increase in the extravasation of Evans blue dye, a marker of increased permeability, in response to PAF injection (22).
A possible explanation for the lack of efficacy of COX inhibition is that COX-2 products have a protective role in acute lung injury. An early clue of the possible involvement of PG in the resolution of inflammation came from a rat pleurisy model. In this model, PGF2α is increased in resolution but not acute phase of inflammation, suggesting an anti-inflammatory effect of this PG (7). The profiles of PG generated in an inflammatory site change during the course of inflammation and are also dependent on the stimulus and site of inflammation (21). It has been suggested that PG generated in the late phase of inflammation have an anti-inflammatory role. In a rat carrageenin-induced pleurisy model, COX-2 is induced during resolution and is associated with production of PGD2 and cyclopentenone PG (cyPG) 15d-PGJ2. COX-2 inhibitors exacerbate inflammation in this model, whereas replacement with PGD2 and 15d-PGJ2 reversed this effect (21). This study suggests that the late-phase induction of COX-2 may contribute to the resolution of inflammation by producing cyPG, including 15d-PGJ2. 15d-PGJ2 is produced from the COX-2 pathway. 15d-PGJ2 presented in human macrophages and LPS enhanced intracellular accumulation as well as extracellular secretion of 15d-PGJ2 (89). 15d-PGJ2 exerts its anti-inflammatory activity through multiple mechanisms, including the activation of PPARγ (43, 83) and also via the inactivation of the NF-κB pathway by directly inhibiting IKKβ subunit of IKK (84). Recently, NF-E2 related factor 2, a transcriptional factor, was shown to be another downstream regulator of 15d-PGJ2 as anti-inflammatory modulator (66). In a carrageenin-induced acute lung injury model, 15d-PGJ2 protects lung from acute lung injury and hastens resolution through effects on Nrf2 activation and subsequent induction of antioxidant genes such as Prl1 and heme oxygenase-1 in alveolar macrophages (66).
Recently, in studies of acid-induced acute lung injury model, another COX-2-derived lipid mediator, lipoxin, has been shown to enhance resolution of lung inflammation. Lipoxins are a structurally distinct class of eicosanoids that are produced in a variety of tissues including the airways. It carries unique action to promote resolution of cytokine-driven acute inflammation (53). In acid-induced acute lung injury, lipoxin A4 (LXA4) and 15-epi-LXA4 mediate anti-inflammatory actions through the LXA4 receptor (16) (Fig. 2).
Fig. 2.
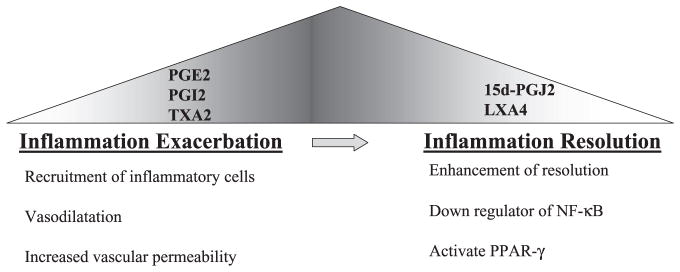
The switching of cyclooxygenase-2 metabolite during the lung inflammatory process. Image illustrates the early phases of inflammatory reaction are leads to early production of prostaglandins, which usually increase the inflammatory reaction by recruiting inflammatory cells and vasodilatation. However, in later stages of the inflammatory reaction, more cyclopentenone prostaglandin 15-deoxy-12,14 PGJ2 (15d-PGJ2) and lipoxin A4 (LXA4) are produced and facilitate the resolution. PPARγ, perxoisome proliferator-activated receptor-γ, TXA2, thromboxane A2.
COX-2 AND PULMONARY FIBROSIS
IPF is a progressive and lethal fibrotic lung disease. Despite some conceptual progress, the pathogenesis of pulmonary fibrosis is incompletely understood. Fibroblast proliferation and collagen synthesis are known to be regulated by a complex interaction between stimulatory and inhibitory mediators. After lung injury there is a disrupted balance between proliferative and suppressive signals that results in fibroblast proliferation recur, which are thought to drive the lung’s response toward fibrosis, rather than normal repair (87).
Although the exact reason for this is not clear, there is quite a large volume of studies that indicate that COX metabolites might be a key mediator of the inhibitory signals on fibroblasts (3, 23, 50, 63, 80). Mice deficient in COX-2 exhibit a fibroproliferative disorder of the kidney even though it has a normal inflammatory response (69). Both genetic disruption and pharmacological blocking of COX enzyme induce an exaggerated fibrotic response in the bleomycin-induced lung fibrosis in a mouse model (38, 48, 68). Among COX metabolites, PGE2 has been the most extensively studied. PGE2 is a major arachidonic metabolite of the lower respiratory tract (80). The PGE2 level in bronchoalveolar lavage fluid from patients with IPF has been shown to be significantly lower than in normal individuals. Furthermore, fibroblasts cultured from patients with IPF failed to induce PGE2 synthesis on stimulation with proinflammatory cytokines or LPS because of aberrant expression of COX-2 (4, 104). PGE2 has been shown to decrease fibroblast proliferation and reduce collagen levels by inhibiting its synthesis and promoting its degradation (3, 50, 63, 104). However, the source of PGE2 in lung remains unclear. Some studies point to epithelial cells because airway epithelial cells express both COX isoforms constitutively, and PGE2 produced by alveolar epithelial cells can suppress the fibroblast proliferation in an in vitro coculture system (51). However, other studies suggested autocrine inhibition of PGE2 on fibroblast proliferation (50, 60). The role of PGD2 and other prostanoids has not been carefully considered or examined in human IPF or animal models of pulmonary fibrosis.
SUMMARY
COX-2 and its metabolites have diverse actions as both pro-and anti-inflammatory mediators in lung injury and inflammation. These molecules must be viewed within the context of a complex milieu of parenchymal and inflammatory cells and an array of other noneicosanoid mediators that result in the overall physiological and pathophysiological status of the host. One possibility is that the temporal sequence of events in acute inflammation is governed by PG profile switching such that PG made during the initial phase are gradually replaced by other PG in the resolution phase (53). Another possibility is that the balance of opposing physiological action of prostanoids determines the inflammatory phenotype. It remains to be seen how this complex system is exactly orchestrated to contribute to the initiation, progression, and resolution phases of various lung diseases. Although global inhibition of COX-2 has not been particularly effective in lung disease, inhibition of distal enzymes or specific PG receptors could have a very beneficial role based on detailed understanding of their role in normal and disease states.
Acknowledgments
GRANTS
This work was supported by the Department of Veterans Affairs and National Heart, Lung, and Blood Institute Grants HL-075557 and HL-66196.
References
- 1.Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB The Ibuprofen in Sepsis Study Group. The effects of ibuprofen on the physiology and survival of patients with sepsis. N Engl J Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 2.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146:108–113. [PubMed] [Google Scholar]
- 3.Bitterman PB, Wewers MD, Rennard SI, Adelberg S, Crystal RG. Modulation of alveolar macrophage-driven fibroblast proliferation by alternative macrophage mediators. J Clin Invest. 1986;77:700–708. doi: 10.1172/JCI112364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI, Crystal RG. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis. 1991;144:1080–1084. doi: 10.1164/ajrccm/144.5.1080. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury DA, Newton R, Zhu YM, El-Haroun H, Corbett L, Knox AJ. Cyclooxygenase-2 induction by bradykinin in human pulmonary artery smooth muscle cells is mediated by the cyclic AMP response element through a novel autocrine loop involving endogenous prostaglandin E2, E-prostanoid 2 (EP2), and EP4 receptors. J Biol Chem. 2003;278:49954–49964. doi: 10.1074/jbc.M307964200. [DOI] [PubMed] [Google Scholar]
- 6.Burka JF, Garland LG. A possible modulatory role for prostacyclin (PGI2) INIgGa-induced release of slow-reacting substance of anaphylaxis in rats. Br J Pharmacol. 1977;61:697–699. doi: 10.1111/j.1476-5381.1977.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capasso F, Dunn CJ, Yamamoto S, Deporter DA, Giroud JP, Willoughby DA. Pharmacological mediators of various immunological and non-immunological inflammatory reactions produced in the pleural cavity. Agents Actions. 1975;5:528–533. doi: 10.1007/BF01972690. [DOI] [PubMed] [Google Scholar]
- 8.Chun KS, Surh YJ. Signal transduction pathways regulating cyclo-oxygenase-2 expression: potential molecular targets for chemoprevention. Biochem Pharmacol. 2004;68:1089–1100. doi: 10.1016/j.bcp.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 9.Cuzzocrea S, Mazzon E, Sautebin L, Dugo L, Serraino I, De Sarro A, Caputi AP. Protective effects of Celecoxib on lung injury and red blood cells modification induced by carrageenan in the rat. Biochem Pharmacol. 2002;63:785–795. doi: 10.1016/s0006-2952(01)00908-x. [DOI] [PubMed] [Google Scholar]
- 10.D’Acquisto F, Iuvone T, Rombola L, Sautebin L, Di Rosa M, Carnuccio R. Involvement of NF-κB in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. FEBS Lett. 1997;418:175–178. doi: 10.1016/s0014-5793(97)01377-x. [DOI] [PubMed] [Google Scholar]
- 11.Dahlen SE, Hansson G, Hedqvist P, Bjorck T, Granstrom E, Dahlen B. Allergen challenge of lung tissue from asthmatics elicits bronchial contraction that correlates with the release of leukotrienes C4, D4, and E4. Proc Natl Acad Sci USA. 1983;80:1712–1716. doi: 10.1073/pnas.80.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies P, Bailey PJ, Goldenberg MM, Ford-Hutchinson AW. The role of arachidonic acid oxygenation products in pain and inflammation. Annu Rev Immunol. 1984;2:335–357. doi: 10.1146/annurev.iy.02.040184.002003. [DOI] [PubMed] [Google Scholar]
- 13.Delamere F, Holland E, Patel S, Bennett J, Pavord I, Knox A. Production of PGE2 by bovine cultured airway smooth muscle cells and its inhibition by cyclooxygenase inhibitors. Br J Pharmacol. 1994;111:983–988. doi: 10.1111/j.1476-5381.1994.tb14840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedyk ER, Phipps RP. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc Natl Acad Sci USA. 1996;93:10978–10983. doi: 10.1073/pnas.93.20.10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujitani Y, Kanaoka Y, Aritake K, Uodome N, Okazaki-Hatake K, Urade Y. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipocalin-type prostaglandin D synthase transgenic mice. J Immunol. 2002;168:443–449. doi: 10.4049/jimmunol.168.1.443. [DOI] [PubMed] [Google Scholar]
- 16.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 17.Fuller RW, Dixon CM, Dollery CT, Barnes PJ. Prostaglandin D2 potentiates airway responsiveness to histamine and methacholine. Am Rev Respir Dis. 1986;133:252–254. doi: 10.1164/arrd.1986.133.2.252. [DOI] [PubMed] [Google Scholar]
- 18.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 19.Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med. 1999;159:31–36. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- 20.Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Langenbach R, Roggli VL, Zeldin DC. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 1999;104:721–732. doi: 10.1172/JCI6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 22.Goggel R, Hoffman S, Nusing R, Narumiya S, Uhlig S. Platelet-activating factor-induced pulmonary edema is partly mediated by prostaglandin E2, E-prostanoid 3-receptors, and potassium channels. Am J Respir Crit Care Med. 2002;166:657–662. doi: 10.1164/rccm.200111-071OC. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein RH, Polgar P. The effect and interaction of bradykinin and prostaglandins on protein and collagen production by lung fibroblasts. J Biol Chem. 1982;257:8630–8633. [PubMed] [Google Scholar]
- 24.Goodwin JS, Bankhurst AD, Messner RP. Suppression of human T-cell mitogenesis by prostaglandin. Existence of a prostaglandin-producing suppressor cell. J Exp Med. 1977;146:1719–1734. doi: 10.1084/jem.146.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorgoni B, Caivano M, Arizmendi C, Poli V. The transcription factor C/EBPβ is essential for inducible expression of the cox-2 gene in macrophages but not in fibroblasts. J Biol Chem. 2001;276:40769–40777. doi: 10.1074/jbc.M106865200. [DOI] [PubMed] [Google Scholar]
- 26.Grall FT, Prall WC, Wei W, Gu X, Cho JY, Choy BK, Zerbini LF, Inan MS, Goldring SR, Gravallese EM, Goldring MB, Oettgen P, Libermann TA. The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2005;272:1676–1687. doi: 10.1111/j.1742-4658.2005.04592.x. [DOI] [PubMed] [Google Scholar]
- 27.Gust R, Kozlowski JK, Stephenson AH, Schuster DP. Role of cyclooxygenase-2 in oleic acid-induced acute lung injury. Am J Respir Crit Care Med. 1999;160:1165–1170. doi: 10.1164/ajrccm.160.4.9811073. [DOI] [PubMed] [Google Scholar]
- 28.Gyllfors P, Bochenek G, Overholt J, Drupka D, Kumlin M, Sheller J, Nizankowska E, Isakson PC, Mejza F, Lefkowith JB, Dahlen SE, Szczeklik A, Murray JJ, Dahlen B. Biochemical and clinical evidence that aspirin-intolerant asthmatic subjects tolerate the cyclooxygenase 2-selective analgetic drug celecoxib. J Allergy Clin Immunol. 2003;111:1116–1121. doi: 10.1067/mai.2003.1450. [DOI] [PubMed] [Google Scholar]
- 29.Hamberg M, Samuelsson B. Detection and isolation of an endoperoxide intermediate in prostaglandin biosynthesis. Proc Natl Acad Sci USA. 1973;70:899–903. doi: 10.1073/pnas.70.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammad H, Jan de Heer H, Soullie T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol. 2003;171:3936–3940. doi: 10.4049/jimmunol.171.8.3936. [DOI] [PubMed] [Google Scholar]
- 31.Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The bronchoconstrictor effect of inhaled prostaglandin D2 in normal and asthmatic men. N Engl J Med. 1984;311:209–213. doi: 10.1056/NEJM198407263110401. [DOI] [PubMed] [Google Scholar]
- 32.Hartert TV, Dworski RT, Mellen BG, Oates JA, Murray JJ, Sheller JR. Prostaglandin E2 decreases allergen-stimulated release of prostaglandin D2 in airways of subjects with asthma. Am J Respir Crit Care Med. 2000;162:637–640. doi: 10.1164/ajrccm.162.2.9904038. [DOI] [PubMed] [Google Scholar]
- 33.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Heavey DJ, Ernst PB, Stevens RL, Befus AD, Bienenstock J, Austen KF. Generation of leukotriene C4, leukotriene B4, and prostaglandin D2 by immunologically activated rat intestinal mucosa mast cells. J Immunol. 1988;140:1953–1957. [PubMed] [Google Scholar]
- 35.Hinshaw LB, Solomon LA, Erdos EG, Reins DA, Gunter BJ. Effects of acetylsalicylic acid on the canine response to endotoxin. J Pharmacol Exp Ther. 1967;157:665–671. [PubMed] [Google Scholar]
- 36.Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–262. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirata M, Kakizuka A, Aizawa M, Ushikubi F, Narumiya S. Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc Natl Acad Sci USA. 1994;91:11192–11196. doi: 10.1073/pnas.91.23.11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodges RJ, Jenkins RG, Wheeler-Jones CPD, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E2 production. Am J Pathol. 2004;165:1663–1676. doi: 10.1016/S0002-9440(10)63423-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honda K, Arima M, Cheng G, Taki S, Hirata H, Eda F, Fukushima F, Yamaguchi B, Hatano M, Tokuhisa T, Fukuda T. Prostaglandin D2 reinforces Th2 type inflammatory responses of airways to low-dose antigen through bronchial expression of macrophage-derived chemokine. J Exp Med. 2003;198:533–543. doi: 10.1084/jem.20022218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. J Biol Chem. 1995;270:24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- 41.Ishitsuka Y, Moriuchi H, Hatamoto K, Yang C, Takase J, Golbidi S, Irikura M, Irie T. Involvement of thromboxane A2 (TXA2) in the early stages of oleic acid-induced lung injury and the preventive effect of ozagrel, a TXA2 synthase inhibitor, in guinea-pigs. J Pharm Pharmacol. 2004;56:513–520. doi: 10.1211/0022357023150. [DOI] [PubMed] [Google Scholar]
- 42.Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol. 2002;169:5997–6004. doi: 10.4049/jimmunol.169.10.5997. [DOI] [PubMed] [Google Scholar]
- 43.Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 44.Joo M, Park GY, Wright JG, Blackwell TS, Atchison ML, Christman JW. Transcriptional regulation of the cyclooxygenase-2 gene in macrophages by PU.1. J Biol Chem. 2004;279:6658–6665. doi: 10.1074/jbc.M306267200. [DOI] [PubMed] [Google Scholar]
- 45.Kabashima K, Murata T, Tanaka H, Matsuoka T, Sakata D, Yoshida N, Katagiri K, Kinashi T, Tanaka T, Miyasaka M, Nagai H, Ushikubi F, Narumiya S. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immun. 2003;4:694–701. doi: 10.1038/ni943. [DOI] [PubMed] [Google Scholar]
- 46.Kabashima K, Sakata D, Nagamachi M, Miyachi Y, Inaba K, Narumiya S. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat Med. 2003;9:744–749. doi: 10.1038/nm872. [DOI] [PubMed] [Google Scholar]
- 47.Katamura K, Shintaku N, Yamauchi Y, Fukui T, Ohshima Y, Mayumi M, Furusho K. Prostaglandin E2 at priming of naive CD4+ T cells inhibits acquisition of ability to produce IFN-γ and IL-2, but not IL-4 and IL-5. J Immunol. 1995;155:4604–4612. [PubMed] [Google Scholar]
- 48.Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-β in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol. 2001;158:1411–1422. doi: 10.1016/s0002-9440(10)64092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirby JG, Hargreave FE, Cockcroft DW, O’Byrne PM. Effect of indomethacin on allergen-induced asthmatic responses. J Appl Physiol. 1989;66:578–583. doi: 10.1152/jappl.1989.66.2.578. [DOI] [PubMed] [Google Scholar]
- 50.Korn JH. Fibroblast prostaglandin E2 synthesis. Persistence of an abnormal phenotype after short-term exposure to mononuclear cell products. J Clin Invest. 1983;71:1240–1246. doi: 10.1172/JCI110873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lama V, Moore BB, Christensen P, Toews GB, Peters-Golden M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol. 2002;27:752–758. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 52.Lellouch-Tubiana A, Lefort J, Simon MT, Pfister A, Vargaftig BB. Eosinophil recruitment into guinea pig lungs after PAF-acether and allergen administration. Modulation by prostacyclin, platelet depletion, and selective antagonists. Am Rev Respir Dis. 1988;137:948–954. doi: 10.1164/ajrccm/137.4.948. [DOI] [PubMed] [Google Scholar]
- 53.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immun. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 54.Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ., II Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982;129:1627–1631. [PubMed] [Google Scholar]
- 56.Liu MC, Bleecker ER, Lichtenstein LM, Kagey-Sobotka A, Niv Y, McLemore TL, Permutt S, Proud D, Hubbard WC. Evidence for elevated levels of histamine, prostaglandin D2, and other bronchoconstricting prostaglandins in the airways of subjects with mild asthma. Am Rev Respir Dis. 1990;142:126–132. doi: 10.1164/ajrccm/142.1.126. [DOI] [PubMed] [Google Scholar]
- 57.Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan CC, Riendeau D. Cloning, expression, and up-regulation of inducible rat prostaglandin E synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276:4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- 58.Mandal AK, Zhang Z, Ray R, Choi MS, Chowdhury B, Pattabiraman N, Mukherjee AB. Uteroglobin represses allergen-induced inflammatory response by blocking PGD2 receptor-mediated functions. J Exp Med. 2004;199:1317–1330. doi: 10.1084/jem.20031666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manning PJ, Lane CG, O’Byrne PM. The effect of oral prostaglandin E1 on airway responsiveness in asthmatic subjects. Pulm Pharmacol. 1989;2:121–124. doi: 10.1016/0952-0600(89)90035-5. [DOI] [PubMed] [Google Scholar]
- 60.Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;289:L391–L399. doi: 10.1152/ajplung.00062.2005. [DOI] [PubMed] [Google Scholar]
- 61.Martin JG, Suzuki M, Maghni K, Pantano R, Ramos-Barbon D, Ihaku D, Nantel F, Denis D, Hamid Q, Powell WS. The immunomodulatory actions of prostaglandin E2 on allergic airway responses in the rat. J Immunol. 2002;169:3963–3969. doi: 10.4049/jimmunol.169.7.3963. [DOI] [PubMed] [Google Scholar]
- 62.Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- 63.McAnulty RJ, Hernandez-Rodriguez NA, Mutsaers SE, Coker RK, Laurent GJ. Indomethacin suppresses the anti-proliferative effects of transforming growth factor-β isoforms on fibroblast cell cultures. Biochem J. 1997;321:639–643. doi: 10.1042/bj3210639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melillo E, Woolley KL, Manning PJ, Watson RM, O’Byrne PM. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med. 1994;149:1138–1141. doi: 10.1164/ajrccm.149.5.8173753. [DOI] [PubMed] [Google Scholar]
- 65.Minakuchi R, Wacholtz MC, Davis LS, Lipsky PE. Delineation of the mechanism of inhibition of human T cell activation by PGE2. J Immunol. 1990;145:2616–2625. [PubMed] [Google Scholar]
- 66.Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxyΔ12,14 prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med. 2005;171:1260–1266. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- 67.Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–1948. doi: 10.1182/blood.v98.6.1942. [DOI] [PubMed] [Google Scholar]
- 68.Moore BB, Coffey MJ, Christensen P, Sitterding S, Ngan R, Wilke CA, McDonald R, Phare SM, Peters-Golden M, Paine R, III, Toews GB. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J Immunol. 2000;165:4032–4039. doi: 10.4049/jimmunol.165.7.4032. [DOI] [PubMed] [Google Scholar]
- 69.Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 70.Murakami M, Austen KF, Arm JP. The immediate phase of c-kit ligand stimulation of mouse bone marrow-derived mast cells elicits rapid leukotriene C4 generation through posttranslational activation of cytosolic phospholipase A2 and 5-lipoxygenase. J Exp Med. 1995;182:197–206. doi: 10.1084/jem.182.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, Ichikawa A, Aze Y, Tanaka T, Yoshida N, Ueno A, Ohishi S, Narumiya S. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 72.Nagao K, Tanaka H, Komai M, Masuda T, Narumiya S, Nagai H. Role of prostaglandin I2 in airway remodeling induced by repeated allergen challenge in mice. Am J Respir Cell Mol Biol. 2003;29:314–320. doi: 10.1165/rcmb.2003-0035OC. [DOI] [PubMed] [Google Scholar]
- 73.Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 75.Nataraj C, Thomas DW, Tilley SL, Nguyen M, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E2 that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen M, Solle M, Audoly LP, Tilley SL, Stock JL, McNeish JD, Coffman TM, Dombrowicz D, Koller BH. Receptors and signaling mechanisms required for prostaglandin E2-mediated regulation of mast cell degranulation and IL-6 production. J Immunol. 2002;169:4586–4593. doi: 10.4049/jimmunol.169.8.4586. [DOI] [PubMed] [Google Scholar]
- 77.Northover BJ, Subramanian G. Analgesic-antipyretic drugs as antagonists of endotoxin shock in dogs. J Pathol Bacteriol. 1962;83:463–468. [PubMed] [Google Scholar]
- 78.Oguma T, Palmer LJ, Birben E, Sonna LA, Asano K, Lilly CM. Role of prostanoid DP receptor variants in susceptibility to asthma. N Engl J Med. 2004;351:1752–1763. doi: 10.1056/NEJMoa031785. [DOI] [PubMed] [Google Scholar]
- 79.Ohara M, Sawa T, Kurahashi K, Wiener-Kronish JP, Doshi V, Kudoh I, Gropper MA. Induction of cyclooxygenase-2 in alveolar macrophages after acid aspiration: selective cyclooxygenase-2 blockade reduces interleukin-6 production. Anesthesiology. 1998;88:1014–1022. doi: 10.1097/00000542-199804000-00022. [DOI] [PubMed] [Google Scholar]
- 79a.Orloff J, Handler JS, Bergstrom S. Effect of PGE-1 on the permeability response of toad bladder to vasopressin, theophylline, and adenosine 3′,5′-monophosphate. Nature. 1965;205:397–398. doi: 10.1038/205397a0. [DOI] [PubMed] [Google Scholar]
- 80.Ozaki T, Rennard SI, Crystal RG. Cyclooxygenase metabolites are compartmentalized in the human lower respiratory tract. J Appl Physiol. 1987;62:219–222. doi: 10.1152/jappl.1987.62.1.219. [DOI] [PubMed] [Google Scholar]
- 81.Park GY, Joo M, Pedchenko T, Blackwell TS, Christman JW. Regulation of macrophage cyclooxygenase-2 gene expression by modifications of histone H3. Am J Physiol Lung Cell Mol Physiol. 2004;286:L956–L962. doi: 10.1152/ajplung.00338.2003. [DOI] [PubMed] [Google Scholar]
- 82.Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148:87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- 83.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 84.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 85.Scales WE, Chensue SW, Otterness I, Kunkel SL. Regulation of monokine gene expression: prostaglandin E2 suppresses tumor necrosis factor but not interleukin-1α or β-mRNA and cell-associated bioactivity. J Leukoc Biol. 1989;45:416–421. [PubMed] [Google Scholar]
- 86.Schuster VL. Molecular mechanisms of prostaglandin transport. Annu Rev Physiol. 1998;60:221–242. doi: 10.1146/annurev.physiol.60.1.221. [DOI] [PubMed] [Google Scholar]
- 87.Selman M, King TE, Jr, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 88.Shi H, Yokoyama A, Kohno N, Hirasawa Y, Kondo K, Sakai K, Hiwada K. Effect of thromboxane A2 inhibitors on allergic pulmonary inflammation in mice. Eur Respir J. 1998;11:624–629. [PubMed] [Google Scholar]
- 89.Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-Deoxy-Δ 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem. 2002;277:10459–10466. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- 90.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 91.Snyder DS, Beller DI, Unanue ER. Prostaglandins modulate macrophage Ia expression. Nature. 1982;299:163–165. doi: 10.1038/299163a0. [DOI] [PubMed] [Google Scholar]
- 92.Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, Trottein F, Dombrowicz D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–3708. doi: 10.4049/jimmunol.174.6.3703. [DOI] [PubMed] [Google Scholar]
- 93.Stevenson DD, Simon RA. Lack of cross-reactivity between rofecoxib and aspirin in aspirin-sensitive patients with asthma. J Allergy Clin Immunol. 2001;108:47–51. doi: 10.1067/mai.2001.116290. [DOI] [PubMed] [Google Scholar]
- 94.Stokes Peebles R, Jr, Dworski R, Collins RD, Jarzecka K, Mitchell DB, Graham BS, Sheller JR. Cyclooxygenase inhibition increases interleukin 5 and interleukin 13 production and airway hyperresponsiveness in allergic mice. Am J Respir Crit Care Med. 2000;162:676– 681. doi: 10.1164/ajrccm.162.2.9911063. [DOI] [PubMed] [Google Scholar]
- 95.Stokes Peebles R, Jr, Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, Christman JW, Kang K-H, Jarzecka K, Furlong J, Mitchell DB, Talati M, Graham BS, Sheller JR. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2002;165:1154–1160. doi: 10.1164/ajrccm.165.8.2106025. [DOI] [PubMed] [Google Scholar]
- 96.Subbaramaiah K, Cole PA, Dannenberg AJ. Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mechanisms. Cancer Res. 2002;62:2522–2530. [PubMed] [Google Scholar]
- 97.Taha R, Olivenstein RON, Utsumi T, Ernst P, Barnes PJ, Rodger IW, Giaid A. Prostaglandin H synthase 2 expression in airway cells from patients with asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:636–640. doi: 10.1164/ajrccm.161.2.9811063. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi Y, Tokuoka S, Masuda T, Hirano Y, Nagao M, Tanaka H, Inagaki N, Narumiya S, Nagai H. Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br J Pharmacol. 2002;137:315–322. doi: 10.1038/sj.bjp.0704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K. Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol. 2000;164:2277–2280. doi: 10.4049/jimmunol.164.5.2277. [DOI] [PubMed] [Google Scholar]
- 100.Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O. The major source of endogenous prostaglandin D2 production is likely antigen-presenting cells. Localization of glutathione-requiring prostaglandin D synthetase in histiocytes, dendritic, and Kupffer cells in various rat tissues. J Immunol. 1989;143:2982–2989. [PubMed] [Google Scholar]
- 101.Van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J, Aarden LA. Prostaglandin E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. 1995;181:775–779. doi: 10.1084/jem.181.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 103.Walters EH, Bevan C, Parrish RW, Davies BH, Smith AP. Time-dependent effect of prostaglandin E2 inhalation on airway responses to bronchoconstrictor agents in normal subjects. Thorax. 1982;37:438–442. doi: 10.1136/thx.37.6.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest. 1995;95:1861–1868. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie W, Fletcher BS, Andersen RD, Herschman HR. V-src induction of the TIS10/PGS2 prostaglandin synthase gene is mediated by an ATF/CRE transcription response element. Mol Cell Biol. 1994;14:6531–6539. doi: 10.1128/mcb.14.10.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]