

Abstract
Purpose
O2-(2,4-dinitrophenyl)1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] or JS-K is a nitric oxide-producing prodrug of the arylated diazeniumdiolate class with promising anti-tumor activity. JS-K has challenging solubility and stability properties. We aimed to characterize and compare Pluronic® P123-formulated JS-K (P123/JS-K) with free JS-K.
Methods
We determined micelle size, shape, and critical micelle concentration of Pluronic® P123. Efficacy was evaluated in vitro using HL-60 and U937 cells and in vivo in a xenog raft in NOD/SCID IL2Rγnull mice using HL-60 cells. We compared JS-K and P123/JS-K stability in different media. We also compared plasma protein binding of JS-K and P123/JS-K. We determined the binding and Stern Volmer constants, and thermodynamic parameters.
Results
Spherical P123/JS-K micelles were smaller than blank P123. P123/JS-K formulation was more stable in buffered saline, whole blood, plasma and RPMI media as compared to free JS-K. P123 affected the protein binding properties of JS-K. In vitro it was as efficacious as JS-K alone when tested in HL-60 and U937 cells and in vivo greater tumor regression was observed for P123/JS-K treated NOD/SCID IL2Rγnull mice when compared to free JS-K-treated NOD/SCID IL2Rγnull mice.
Conclusions
Pluronic® P123 solubilizes, stabilizes and affects the protein binding characteristics of JS-K. P123/JS-K showed more in vivo anti-tumor activity than free JS-K.
Keywords: O2-(2,4-dinitrophenyl)1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate]; JS-K; nitric oxide; acute myeloid leukemia (AML); Pluronic® P123; glutathione
1. Introduction
Acute myeloid leukemia (AML) is the most common type of adult acute leukemia. The The American Cancer Society estimates that 18,860 cases of AML will be diagnosed in the United States in 2014 with 10,460 AML-related deaths (1). The success of conventional chemotherapeutic agents in the treatment of AML is frequently limited by the development of drug resistance and severe toxicity (2).
Nitric oxide (NO), a biologically occurring molecule, is involved in a plethora of physiological, biological and pathological functions (3). NO has shown promising therapeutic activity against multiple diseases including cardiovascular and pulmonary diseases (4). NO has also shown in vitro tumoricidal activity (5). It can lead to apoptosis by targeting various cellular sites and bringing about post-translational modifications such as S-nitrosylation, S-glutathionylation, DNA nitration or deamination and ADP ribosylation (6). NO induces differentiation in acute myeloid leukemia (AML) cells (7). Due to instability of the NO radical and its hypotensive effects, NO cannot be administered directly or in high doses. NO donors of the diazeniumdiolates class generate NO spontaneously. They induce apoptosis and differentiation in AML cells (8). However, these compounds cannot be used clinically for the treatment of malignant diseases because of the pleiotropic effects of NO, and particularly NO-induced vasodilatation through activation of the soluble guanylate cyclase/cGMP pathway (9). Alternatively, arylated diazeniumdiolates react with glutathione (GSH) to release NO. Although the reaction can occur spontaneously, it is catalyzed by the glutathione S-transferases (GST). GST's are upregulated in malignant cells (10). O2-(2,4-dinitrophenyl)1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] or JS-K (Figure 1), an arylated diazeniumdiolate has potent anti-leukemic activity in vitro and in vivo (10). In in vivo murine models, JS-K was also found to be effective against prostate cancer (10), hepatoma (11), multiple myeloma (12) and non-small cell lung cancer (13). JS-K also possesses anti-angiogenic activity both in vitro and in vivo (14). In multiple myeloma and breast cancer studies, JS-K did not affect the growth of normal human peripheral blood mononuclear cells (12) and normal mammary epithelial cells, respectively (15).
Figure 1.
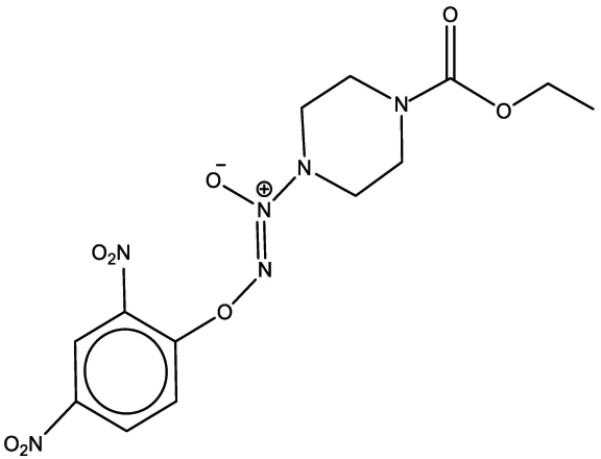
Structure of JS-K.
Pluronic® block copolymers are used in multiple different applications. They are amphiphilic molecules arranged as A-B-A blocks of hydrophilic poly(ethylene oxide) (PEO or A) and hydrophobic poly(propylene oxide) (PPO or B). The block copolymers possess different hydrophilic-lipophilic balance (HLB) due to varying concentrations of ethylene oxide and propylene oxide units. HLB is important for the critical micelle concentration (CMC) i.e. the concentration above which these copolymers self assemble into micelles in an aqueous solution (16).
Plasma protein binding is an important parameter of the drug development process that needs to be established in order to predict drug distribution characteristics. Drug-binding proteins can act as a reservoir or drug depot. Only free drug molecules are available at the target site (17). Therefore, in preclinical settings, it is important to determine the plasma protein binding characteristics of an active pharmaceutical ingredient (API) to assess safety, efficacy and bioavailability (18). Two major plasma proteins involved in drug binding are human serum albumin (HSA) and alpha1-acid glycoprotein (AGP) (19). Plasma protein binding could be analyzed using the conventional equilibrium dialysis approach or the comparatively new technique of fluorescence quenching.
In the present study, a Pluronic® P123 micelle formulation of JS-K was developed and pre-clinical studies were carried out. We performed in vitro cytotoxicity analysis and also an in vivo tumor regression study in a mouse model. The Pluronic® micelles were characterized for their size, shape and critical micelle concentration. Finally, we performed serum-binding analysis using two different techniques, namely equilibrium dialysis and fluorescence quenching. While both techniques provided valuable information in terms of binding characteristics, we were able to assess thermodynamic parameters associated with drug-protein binding using fluorescence quenching.
2. Materials and Methods
2.1 Materials
JS-K was synthesized as previously described (20) and provided by Dr. Joseph Saavedra (Leidos Biomedical Research, Inc, Frederick, MD). JS-K stocks were prepared in amber colored vials to prevent any photo-degradation. Pluronic® polymers were obtained from BASF (Florham Park, NJ). Human serum albumin (HSA) and alpha 1 acid glycoprotein (AGP) used for protein binding studies were from Sigma (St. Louis, MO). Low binding SpectraPor dialysis membranes [MWCO: 12,000-14,000] were from Spectrum Laboratories Inc (Los Angeles, CA). All other chemicals were from Sigma (St. Louis, MO) unless otherwise noted.
2.2 Cell culture
Human myeloid leukemia HL-60 cells and human monocytic leukemia U937 cells (ATCC, Manassas, VA) were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin and mycozap (Lonza, Allendale, NJ). Cells were cultured at 37°C in a 5% CO2 humidified atmosphere. Five micromolar JS-K stocks in dimethyl sulfoxide (DMSO) were serially diluted in phosphate buffered saline (PBS) before addition to the cultures. The final concentration of DMSO added to the cultures was 0.1% or less. For each experiment, JS-K was added at the time of culture initiation, cells were harvested at the indicated time points, washed in PBS and assays conducted.
2.3 Preparation of JS-K-loaded P123 micelles
An 11.25% P123 micelle stock solution was prepared in deionized water. The solution was further diluted to 2.25% in phosphate-buffered saline (PBS - pH 6.5). One mM micelle JS-K (P123/JS-K) was prepared by heating 980 μL of 2.25% P123 at 50°C and adding 20 μL of a 50 mM JS-K stock in DMSO. The weight loading of drug in P123 micelles was 1.7%. Further dilutions were made in PBS (pH 6.5). The formulated drug was used immediately in order to avoid photo-degradation.
2.4 Comparison of different Pluronic® polymers
JS-K was loaded in P123, P105 and F127 Pluronic® polymers. The proportion of JS-K to Pluronic® polymer in solution was 3.84% by weight. The JS-K micelle preparations were dialyzed for 5.5 hours or 17 hours. The different Pluronic® formulations (before and after dialysis) were incubated with HL-60 cells for three days and MTS cytoxicity analysis was performed (see below).
2.5 In vitro cytotoxic activity
The in vitro cytotoxic activity of JS-K, P123/JS-K micelles and blank P123 micelles was assessed using the MTS assay (Promega, Madison, WI) in HL-60 and U-937 cells. Briefly, cells were seeded in 96-well plate at a density of 105 cells per mL. Cells were treated with 0, 0.09, 0.2, 0.4, 0.6 and 0.8 μM of free JS-K or P123/JS-K or an equivalent volume of blank P123 in triplicates. After incubation for 72 hours, 20 μL of MTS reagent were added to each well and incubated further for 1.5 hours. Absorbance was read at 490 nm in a microplate reader (Modulus Microplate, Turner Biosystems, Sunnyvale, CA). A media blank was used to correct for background absorbance. Results were expressed as a growth percent of untreated control and IC50 values were derived from growth vs. drug concentration curves.
2.6 P123 Pluronic® micelle formulation of JS-K
JS-K stocks (50 mM) in DMSO were mixed with stocks of Pluronic® P123 polymers prepared in PBS. Micellization was allowed to occur spontaneously with gentle heating (50°C). The proportion of JS-K to P123 in solution was 1.7% by weight. For experiments with free JS-K, DMSO stocks of JS-K (50 mM) were diluted to a final concentration of JS-K of 1 mM in PBS/80% DMSO in stability experiments or PBS/40% DMSO for protein binding experiments, or PBS/20% DMSO for in vivo experiments, or only PBS for other experiments.
2.7 P123 JS-K formulation characterization
a) Particle size measurement
Particle mean size measurements were performed using the dynamic light scattering method using a Malvern Nano zeta sizer (Westborough, MA). The size analysis was performed on blank P123 micelles (2.25%) in PBS and JS-K-loaded P123 micelles (final loading 1.7%).
b) Transmission electron microscopy (TEM) analysis
The morphology of P123 micelles was studied by TEM after negative staining with a phosphotungstic acid solution (2% w/v). A 1 mM solution of JS-K formulated in 2.25% P123 micelles was prepared. TEM images were obtained only with P123/JS-K micelles as the empty micelles were found to be comparatively less stable without drug.
c) Critical micelle concentration (CMC)
CMC of Pluronic® P123 micelles was analyzed using the Iodine UV-absorption spectra method with iodine as a hydrophobic probe as previously reported (21). Briefly, a KI/I2 solution was prepared by dissolving 0.5 g Iodine (I2) and 1 g Potassium Iodide (KI) in 50 mL water for injection (WFI). P123 concentrations ranging from 0.000005% to 0.1% were prepared in WFI. To each 5 mL solution, 25 μL of KI/I2 solution was added. The vials were covered with foil to avoid photo-degradation. The samples were incubated for nearly 12 hours at room temperature. Measurements were performed at 366 nm using a UV-vis spectrometer (Ultrospec 2000, Pharmacia Biotech, Piscataway, NJ). Triplicate readings were performed for each sample. CMC value was analyzed by plotting absorbance vs. log% Pluronic® weight and was interpreted at the point where a sharp increase in absorbance was observed. The data were analyzed by piece-wise linear model with a single change in slope. A grid search method was used to determine the optimal change point. The statistical analysis was performed using the R statistical computing software version 2.15.0 (Vienna, Austria).
2.8 P123/JS-K formulation stability in different media
The stability of JS-K formulated in DMSO was compared to JS-K formulated in P123 Pluronic® micelles in different media. JS-K recovery with either formulation was tested in 4 mM GSH (dissolved in PBS; pH 7.4); RPMI-1640 media, human plasma and human whole blood. At the indicated time points, aliquots from the relevant test solution were collected in 95% Acetonitrile/Formate (ACN/Formate) in water in polypropylene vials. The extracts were spin filtered (0.2 μm, low binding nylon filters) for two minutes. JS-K levels were measured by HPLC using an isocratic mobile phase consisting of 25% 0.025 M ammonium formate (pH 4.2), 25% ACN and 50% methanol at a flow rate of 1 mL/min for 6-8 minutes.
2.9 P123/JS-K protein binding studies
The two most common proteins in the blood are human serum albumin (HSA) and alpha 1 acid glycoprotein (AGP). While HSA preferentially binds with acidic drugs, AGP binds mainly with basic or neutral drugs (22). We studied interaction of free drug and micelle-formulated drug with these two proteins by fluorescence quenching of tryptophan present in these proteins. Free or P123-formulated JS-K was prepared at concentrations 20 μM-700 μM or 100μM-700μM, respectively for temperature controlled fluorometric studies.
In preliminary experiments, we compared the equilibrium dialysis technique with fluorescence quenching. Both preliminary experiments were carried out at room temperature. The purpose of these experiments was to test the sensitivity and closeness of results obtained from either technique. Also, with the dialysis technique, since higher drug concentrations were used for extended hours, free JS-K precipitation was observed and therefore saturation/equilibrium was not attained. Dialysis was carried out using Spectra/Por dialysis membranes (MWCO:12,000-14,000) which were activated for 30 minutes in HPLC grade water and rinsed before use. The membranes were held in a 20 mL glass vial. The glass vial was covered with aluminum foil to prevent photodegradation of JS-K. The drug preparation (JS-K in DMSO or JS-K in P123) with 4% final HSA was added in the dialysis bag and a similar sink (i.e. HSA and DMSO or P123) was created in the glass vial. The whole apparatus was kept at constant stirring position and the samples were collected from both sides of the membrane after two, four, six and eight hours. We found that for P123/JS-K, equilibrium was achieved after two hours. For free JS-K, we could not achieve equilibrium due to possible degradation of the drug over time. Therefore, two hours of equilibrium dialysis was selected for both formulations. The samples were then soaked for 30 minutes in 95% acetonitrile/5% ammonium formate in water (pH~3) and then spin filtered (0.2 micron, low binding nylon) for two minutes. JS-K levels in the bag and the glass vial were measured by HPLC or UPLC. For quantitation, AUC values were calculated. Percent recovery from each fraction was calculated based on the total initial amount of JS-K added. The association and dissociation constants were calculated using the SigmaPlot software (Systat, Chicago, IL) with the built-in equation for Ligand Binding.
For the fluorometric analysis, the formulation was prepared as described above. Final protein concentration was 4% for HSA and 0.09% for AGP. Drug and protein were added in 96 well black opaque plates to avoid the inner filter effect. Fluorescence was read at an excitation of 280 nm and emission of 360 nm. Corresponding blank fluorescence was read and subtracted as background from the fluorescence of interest. For the temperature-controlled studies, plates were incubated at the desired temperature in a PCR thermal controller (PTC-100™, MJ Research Inc, Watertown, MA) for 30 minutes and read within ten minutes on a fluorescence reader (Synergy 4, BioTek, Winooski, VT) previously adjusted to the same temperature. Fluorometric analysis was carried out using the Stern Volmer equation. For HSA, Ksv and Ka were obtained at 298, 303 and 310 K. Thermodynamic parameters such as enthalpy, entropy and free energy were calculated to assess the nature of binding involved.
2.10 In vivo studies of JS-K and P123/JS-K
To study the in vivo antineoplastic activity of JS-K and P123/JS-K, NOD/SCID IL2Rγnull mice were injected subcutaneously with 5 × 106 HL-60 cells. When tumors became palpable, treatment with JS-K (4 μmol/kg) or P123/JS-K (4 μmol/kg or 5 μmol/kg) or P123 only (volume equivalent to a P123/JS-K dose of 5 μmol/kg) was started. Dose selection was made based on prior experience. We have shown that a dose of 4 μmol/kg of free JS-K could be administered without producing significant hypotension (10). We were able to escalate the dose with P123/JS-K to 5 μmol/kg without observing significant hypotension in mice (unpublished data). Treatments were administered intravenously every other day. The no-treatment control group did not receive any treatments. Mouse weight and tumor size were measured every other day using a Vernier caliper. Tumor volume was calculated using the formula: width × length × [(width + length)/2] × 0.5236. Animals were sacrificed after 8 treatments using isofluorane inhalation. The protocol was approved by the University of Utah Institutional Animal Care and Use Committee.
2.11 High Performance Liquid Chromatography (HPLC) and Ultra Performance Liquid Chromatography (UPLC)
HPLC to measure JS-K levels was conducted on a Water's Alliance HPLC system controlled by Empower software. We used an isocratic mobile phase consisting of A = 30% 0.02 M ammonium formate (pH 3), 40% acetonitrile (ACN) and 30% methanol and D = 100% methanol. Flow rate was kept constant at 1 mL/min for 6-8 minutes. During chromatographic separation, JS-K was monitored by absorbance at 300 nm. The method results in a standard detection curve over a linear range of 6.1 – 49.1 ng with a lower limit of quantitation of 6.4 ng. Very low concentrations of JS-K were measured using a Water's UPLC with Empower 2 software. The samples were run on a gradient of 0.02 M formic acid, pH ~2.7 and 100% acetonitrile at a flow rate of 0.6 mL/min.
2.12 Statistical Analysis
The omnibus null hypothesis of no difference in mean tumor volume between any of the groups was tested using one-way analysis of variance. Tukey's Honest Significant Difference test was used to adjust the p-values for the individual differences for multiple comparisons. Statistical analysis was performed using R statistical computing software version 2.13.0 (Vienna, Austria). At specific time points, individual tumor volumes were compared using the Student's t-test assuming a 2-tailed distribution with unequal variance. Differences were considered significant for P values below 0.05.
We also performed statistical analysis using the trapezoid method. For each mouse treatment group, the cumulative area under the curve (AUC) of the tumor volume was calculated starting from the first treatment to the last (eighth) treatment. Cumulative AUC (for the last treatment) of each group was used for running one-way analysis of variance. Differences were considered significant for P values below 0.05.
3. Results
3.1 P123 JS-K drug retention and loading
We performed preliminary experiments to analyze the efficacy of JS-K loading and cytotoxicity using different Pluronics®. JS-K was loaded in P123, P105 and F127 as explained in the methods section. We tested the cytoxicity of the obtained three different micelle formulations. The three micelles tested have different HLB values (F127>P105>P123) (23). The micelles were dialyzed and tested for cytotoxicity as explained in the materials and methods section. The order of efficacy obtained against HL-60 cells after dialysis was P123>P105>F127 (results not shown). To determine the extent of JS-K loading and retention in micelles, JS-K was loaded in P123. The proportion of JS-K to Pluronic® polymer in solution was 10% by weight. The percent of JS-K retained after two hours of dialysis was determined by HPLC. Percent retention of JS-K in P123 was 75 ± 10%. These preliminary experiments showed that JS-K in a micellar formulation is retained after dialysis.
3.2 P123 micelle JS-K in vitro cytotoxicity
P123 micelle JS-K (1.7% JS-K/P123 by weight) in vitro cytotoxicity was analyzed using two different acute myeloid leukemia cell lines, HL-60 and U-937. These cell lines have different phenotypes, namely myeloid and monocytic, respectively. They were chosen in order to analyze the sensitivity of AML cells with different phenotypes to JS-K or P123/JS-K treatment. With HL-60 cells, IC50 values obtained from free JS-K treatment were similar to IC50 values obtained from the P123/JS-K formulation. P123 control showed negligible toxicity. In U-937 cells, IC50 values obtained from P123/JS-K was slightly lower than with free JS-K. The results (shown in Table I) indicate that Pluronics® do not diminish JS-K's cytotoxic properties.
Table I.
MTS cytotoxicity assay on HL-60 and U-937 cells comparing JS-K and P123/JS-K
| System | Formulation | IC50 ± SEM* (μM) |
|---|---|---|
| HL-60 Cells | P123/JS-K | 0.24 ± 0.059 |
| JS-K | 0.24 ± 0.055 | |
| U-937 Cells | P123/JS-K | 0.3 ± 0 |
| JS-K | 0.4 ± 0 |
Average and standard error mean of at least two separate experiments. Each experiment was performed in triplicates.
3.3 P123 micelle JS-K characterization
The P123/JS-K formulation was characterized by particle size measurement and morphology. These studies are important as particle size affects biodistribution and elimination. The mean diameter of blank P123 micelles was 29.2 ± 1.36 nm. We observed a slight reduction in size upon JS-K incorporation, suggesting stabilization of the micelles (Table II). TEM images demonstrated that P123/JS-K micelles are spherical in shape (Figure 2). TEM images of blank micelles could not be obtained. This could be due to the dynamic character of the Pluronics® changing from unimers to micelles. Therefore, JS-K incorporation seems to stabilize the micelles.
Table II.
Size measurement of blank micelles and P123 loaded with JS-K at different percentages
| System | Weight % drug loading | Size ± SEM (nm)* |
|---|---|---|
| Blank P123 | 0 | 29.24 ± 1.36 |
| P123/JS-K | 0.17 | 27.8 ± 4.9 |
| 1.7 | 17.6 ± 1.21 |
Averages and SEM of at least two separate measurements. Each measurement with 20 iterations.
Figure 2.

TEM image (667x) of 1 mM JS-K loaded in 2.25% P123 micelles.
3.4 Critical micelle concentration (CMC) determination
CMC is an important characterization parameter that influences micelle stability and solubilization characteristics. To measure CMC, we used a hydrophobic probe– Iodine (I2) as explained in the methods section. I2 gets solubilized in the hydrophobic micelle core. An excess KI solution aids conversion of triatomic I3− to I2, maintaining the saturated solution of I2 (21). The absorption intensity of I2 was plotted against different weight percent of Pluronic® and the CMC value was obtained from the point on the curve where an abrupt increase in absorbance was observed (Figure 3). We obtained an average CMC value of 5.6 ± 1.3 μM (average and SEM of two independent experiments with three replicates each). This value is consistent with the value reported by Kabanov et al. for P123 (23).
Figure 3.

Critical micelle concentration (CMC) value of Pluronic® P123 as determined by plotting Absorbance (AU) with log Pluronic® % weight/volume. The CMC was obtained from the break point calculated on R statistical software. CMC was calculated from the average of two independent experiments with each concentration prepared in triplicates.
3.5 Stability of P123/JS-K in different physiologic and biologic media
We compared the stability of unformulated JS-K and P123/JS-K in blood, glutathione, plasma and RPMI/10% FBS media. As shown in Table III, the P123 formulation extended the percentage recovery of JS-K as compared to the free drug. This likely reflects shielding of JS-K from proteins and nucleophiles by Pluronic® micelles. Also, the table highlights how different media affect drug recovery. Although all experiments were carried out up to 60 minutes, the drug was not recovered at that time point from some of the media like whole blood and a 4 mM GSH solution. The HPLC method we used for JS-K quantification detects intact JS-K only. JS-K degradation products were therefore not detected.
Table III.
Stability studies of JS-K. Percent recovery of JS-K or P123/JS-K calculated at the indicated time points
| Solution (time) | JS-K (% recovery ± SEM*) | P123/JS-K (% recovery ± SEM*) | p** |
|---|---|---|---|
| RPMI 1640/10% FBS (60 min.) | 23 ± 0.4 | 48 ± 4 | 0.0015 |
| Human plasma (60 min.) | 39 ± 1 | 59 ±1.5 | 0.012 |
| Whole blood (10 min.) | 0 ± 0 | 6 ± 0 | 0.0284 |
| 4 mM GSH (5 min.) | 8.5 ± 4.6 | 51 ± 10.8 | 0.003 |
Average and SEM of at least two independent experiments.
The Students t-test was used to obtain P values comparing both formulations. Results were considered statistically significant for P < 0.05.
3.6 HSA and AGP binding with JS-K and P123/JS-K
a) Equilibrium dialysis
Equilibrium dialysis studies were carried out to investigate the interaction of JS-K or P123/JS-K with HSA as explained in the material and methods section. JS-K showed affinity for HSA and the equilibrium time was obtained at two hours. Although at that time point some drug degradation/precipitation was seen with free JS-K, it was important to use the two hours to achieve steady state conditions. We measured drug levels from both “in the dialysis bag” and “out of the bag” in order to determine the binding constant. The values were derived using the ligand binding equation. The results are reported in Table IV.
Table IV.
Binding Constants (Ka) obtained from equilibrium dialysis and fluorescence quenching and Stern Volmer constant (Ksv) obtained from fluorescence quenching at room temperature. Concentration of drugs used in both studies ranged from 0.7 mM - 0.025 mM
| Formulation | Ka (equilibrium dialysis with HSA) (M−1 ± SEM)* | Ksv (fluorescence quenching) with HSA (M−1)*** | Ka (fluoroscence quenching with AGP) (M−1)*** | Ksv (fluorescence quenching) with AGP (M−1)*** |
|---|---|---|---|---|
| JS-K | 3.4 × 103 ± 2.2 Ns** = 2.2; R2 = 0.84 |
1.2 × 103 R2 = 0.59 |
15 × 103 R2 = 0.77 |
3 × 103 R2 = 0.91 |
| P123/JS-K | 3.0 × 103 ± 3.6 Ns** = 1.9; R2 = 0.89 |
2.2 × 103 R2 = 0.805 |
3.5 × 103 R2 = 0.99 |
2 × 103 R2 = 0.67 |
Average and standard error of mean (SEM) of at least three independent experiments.
Ns=No. of binding sites.
Average of at least three independent experiments.
R2 = goodness of fit
b) Fluorescence quenching study at room temperature
In preliminary work, both HSA and AGP quenching by JS-K and P123/JS-K were analyzed at room temperature. A Stern Volmer plot was generated for both formulations and proteins. Equation 1 was used to obtain the Stern Volmer quenching constant:
| (1) |
(where Fo: Fluorescence in the absence of external quencher; F: Quenched Fluorescence; Q: Conc. of quencher/drug; Ksv: Stern Volmer Constant).
Figures 4 and 5 show the Stern Volmer plots of HSA and AGP, respectively.
Figure 4.

Stern Volmer Plot for JS-K and HSA or P123/JS-K and HSA at room temperature. y-axis : Fluorescence of HSA/Fluorescence of drug; x-axis: JS-K or P123/JS-K concentration. Plots obtained from averages of 3 independent experiments.
Figure 5.

Stern Volmer plot for JS-K and AGP or P123/JS-K and AGP at room temperature. y-axis : Fluorescence of HSA/Fluorescence of drug; x-axis: JS-K or P123/JS-K concentration. Plots obtained from averages of 3 independent experiments.
The quenching data was used to determine the binding constant with HSA and compare the value obtained with equilibrium dialysis. Table IV shows the binding parameters obtained by the two techniques and the Stern Volmer constants obtained for HSA and AGP.
As shown in Table IV, the binding constants obtained from the two techniques are acceptably close. This confirmed the sensitivity and accuracy of both techniques. For subsequent experiments we chose fluorescence quenching over equilibrium dialysis because the former is a rapid and sensitive technique. It also requires less material (μL as compared to mL for dialysis) and is relatively quicker than dialysis. The latter factor is particularly important for our studies in order to avoid JS-K degradation.
c) Determination of quenching mechanism
Fluorescence quenching was performed at 25°C, 30°C and 37°C as explained in the material and methods section in order to obtain the Stern Volmer constant at different temperatures. As shown in Table V, a temperature effect was observed. With a rise in temperature, the Stern Volmer constant is reduced. Such an inverse relationship with temperature could explain the quenching behavior (Figures 6 and 7). Fluorescence quenching could be caused by a number of factors leading to molecular interactions such as excited state reactions, ground state complex formation and collisional quenching. The decrease in Ksv value with higher temperatures suggests a static quenching mechanism (24) i.e. formation of a complex between JS-K and HSA or P123/JS-K and HSA. The quenching rate constant (Kq) was calculated to further confirm formation of complex and static quenching mechanisms. The quenching rate constant can be calculated using Equation 2:
| (2) |
Table V.
Stern Volmer quenching constant (Ksv) and HSA association binding constant (Ka) of JS-K and P123/JS-K at three temperatures: 298, 303 and 310 K.
| Formulation | Temperature (K) | Ksv* (×103 M−1) | Kq* (×1011 M−1s−1) | Ka* (M−1) |
|---|---|---|---|---|
| JS-K | 298 | 6.6 R2 = 0.983 |
6.6 | 57.3 R2 = 0.96 |
| 303 | 6.9 R2 = 0.94 |
6.9 | 226.14 R2 = 0.98 |
|
| 310 | 3.1 R2 = 0.98 |
3.1 | 1052.3 R2 = 0.99 |
|
| P123/JS-K | 298 | 3.3 R2 = 0.99 |
3.3 | 608.2 R2 = 0.988 |
| 303 | 3.4 R2 = 0.79 |
3.4 | 615.2 R2 = 0.986 |
|
| 310 | 2.2 R2 = 0.93 |
2.2 | 681.2 R2 = 0.840 |
Calculated from the slope of the curve plotted. Two independent experiments were performed with nine replicates at each concentration.
R2 = goodness of fit
Figure 6.

Stern Volmer plot for JS-K and HSA at 298, 303 and 310 K. Each point and standard error of the mean (SEM) is a representative of two independent experiments with nine replicates.
Figure 7.

Stern Volmer plot for P123/JS-K at temperatures 298, 303 and 310 K. Each point and standard error of the mean (SEM) is a representative of two independent experiments with nine replicates.
[where τo is the average lifetime of HSA molecule in absence of JS-K (quencher) or any other drug]. It represents the average lifetime that a molecule spends in the excited state. The value of τo of HSA is 10−8 s (25). The Kq values calculated are shown in Table V. The Kq values obtained are higher than the maximum scatter collision quenching for HSA i.e. 2×1010 L mol−1s−1. We also performed a fluorescence study with HSA and P123 only. Interestingly, instead of quenching, fluorescence enhancement was observed (results not shown).
d) Analysis of Binding constants
The association binding constant (Ka) was calculated using a modified Stern Volmer equation (Equation 3):
| (3) |
Where Fo = original unquenched fluorescence of HSA; dF = difference in the fluorescence in the absence and presence of JS-K or P123/JS-K at the concentration Q; f = fraction of initial fluorescence accessible to quencher.
Binding constants obtained at different temperatures are shown in Table 5.
e) Thermodynamic constants analysis
Thermodynamic parameters are important for the determination of binding forces involved between a protein and a drug. The forces involved could be non-covalent bonds including hydrophobic interactions; electrostatic interactions; Van der Waals forces and/or hydrogen bonds. The thermodynamic parameters were evaluated using the Van't Hoff equation (Equations 4 and 5):
| (4) |
| (5) |
The enthalpy change (dH) and entropy change (dS) were calculated from the slope and intercept of the binding constant vs. temperature curve, respectively. Free energy (dG) was calculated from the derived enthalpy and entropy at different temperatures. The results are shown in Table VI.
Table VI.
Thermodynamic parameters for HSA and JS-K or HSA and P123/JS-K at three temperatures: 298, 303 and 310 K.
| Formulation | Temperature (K) | Ka (M−1) | dG° (kJ mol−1) | dH° (kJ mol−1) | dS° (J mol−1 K−1) |
|---|---|---|---|---|---|
| JS-K | 298 | 57.3 R2 = 0.96 |
−10.14 | 185.46 R2 = 0.99 |
656.39 R2 = 0.99 |
| 303 | 226.14 R2 = 0.98 |
−13.42 | |||
| 310 | 1052.3 R2 = 0.99 |
−18.02 | |||
| P123/JS-K | 298 | 608.2 R2 = 0.988 |
−15.93 | 7.4 R2 = 0.88 |
78.3 R2 = 0.88 |
| 303 | 615.2 R2 = 0.986 |
−16.32 | |||
| 310 | 681.2 R2 = 0.840 |
−16.87 |
Enthalpy and entropy calculated from the slope and intercept of the curve plotted between binding constant and temperature respectively as explained in the results section. Two independent experiments were performed with nine replicates at each concentration.
R2 = goodness of fit.
3.7 In vivo tumor regression analysis
We tested the in vivo efficacy of free JS-K and P123/JS-K using HL-60 cells implanted in NOD/SCID IL2Rγnull as detailed in the methods section. Five groups were compared as follows: no treatment control, P123 control, free JS-K (dissolved in PBS/20% DMSO) at 4 μmol/kg, P123/JS-K at 4 μmol/kg and P123/JS-K at 5 μmol/kg. There were 12 to 13 animals per variable. Mice were treated intravenously every other day for a total of 8 doses. Tumor volume curves for each variable are shown in Figure 8. When the curves were compared using the two way analysis of variance (ANOVA) method, they were not statistically different. However, when comparing tumor volumes using the Student's t-test at day 13, free JS-K at 4 μmol/kg and P123/JS-K at 5 μmol/kg induced a statistically significant reduction in tumor volume as compared to controls. Interestingly, at day 16, the difference between free JS-K-treated mice and controls was no longer significant. However, at day 16, P123/JS-K at 4 or 5 μmol/kg induced a statistically significant reduction in tumor volume. At day 16 tumor volume measurements were as follows (average and SEM): No treatment: 1.57 ± 0.23 cm3, P123 control: 1.62 ± 0.21 cm3, free JS-K (4 μmol/kg): 1.30 ± 0.13 cm3, P123/JS-K (4 μmol/kg): 0.89 ± 0.08 cm3, P123/JS-K (5 μmol/kg): 0.81 ± 0.06 cm3. Differences between P123/JS-K at 4 or 5 μmol/kg were not statistically significant. In the other set of analyses performed we calculated the cumulative area under the curve for tumor volumes plotted against the day of treatment. At day 16, we obtained statistically significant tumor reduction when all the treated groups were compared to control or no treatment. However, differences between P123/JS-K at 4 or 5 μmol/kg were not statistically significant. We could not carry the experiment beyond day 16 based on tumor volume guidelines of the University of Utah IACUC.
Figure 8.
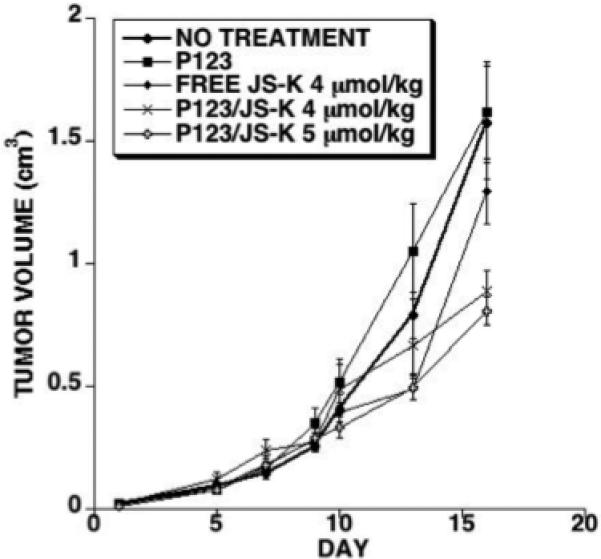
In vivo tumor regression analysis: NOD/SCID IL2Rγnull mice were injected with HL-60 cells subcutaneously. Treatments started after tumors became palpable. A total of 8 injections were given. Treatment groups consisted of no treatment control, Pluronic® P123 treatment control, free JS-K injected at 4 μmol/kg, P123/JS-K injected at 4 μmol/kg, or P123/JS-K injected at 5 μmol/kg.
4. Discussion
In this paper, we describe the effect of a Pluronic® formulation on the prospective anticancer agent JS-K. Pluronic® block copolymers have been widely used as they are comparatively non-toxic and offer ease of preparation (26). Polymeric micelles do not offer high loading capacity, but seemed to work well for our purposes. In preliminary studies, we compared Pluronics® F127, P105 and P123. All three Pluronics® are different with respect to their physical characteristics, HLB value and solubility (23). The results indicated that for JS-K, P123 was the best choice in terms of stability and enhancing solubility of this extremely hydrophobic drug. JS-K in P123 micelles was more stable than in the other 2 formulations. Although most of our studies were performed on HL-60 cells, we also tested P123/JS-K in U-937 cells at the same drug loading percentage and obtained similar in vitro cytotoxicity results.
All the studies were performed immediately in order to prevent any possible degradation. JS-K stocks were prepared and stored in amber vials. For experiments requiring extended hours, photo-degradation was prevented by covering the vials with aluminum foil.
We conducted studies to characterize P123/JS-K since it is a clinical candidate. As evident from the size measurements, there was a slight reduction in the micelle size observed when blank micelles were compared to P123/JS-K micelles. It is interesting to note that with increase in JS-K loading, size reduction became appreciable (Table II). Also, we could not observe the shape of the blank micelles with TEM but after JS-K incorporation, spherical shaped micelles were observed. These two observations point towards stabilization of the micellar structure due to JS-K incorporation. Although speculative at this point, it is likely that micelle stabilization by JS-K is due to hydrophobic interaction of the drug with the polymer. It has been suggested that for Pluronic® micelles, at concentrations significantly above the CMC micelle stability diminishes (27). Our studies support this observation as we were unable to get TEM images of blank micelles but with JS-K incorporation, spherical micelles were observed. Being extremely hydrophobic, JS-K likely stabilized the dynamic nature of the micelles with physical interactions. We also measured the CMC of the Pluronic® P123 using the KI/I2 method. The CMC value we obtained is consistent with what has been reported by other investigators (21, 23).
Upon testing free JS-K and P123/JS-K in various media, P123/JS-K showed enhanced recovery and extended stability as compared to JS-K alone. Human plasma, RPMI/FBS media and PBS were chosen as matrices of different complexity. Stability of JS-K was also tested in the presence of GSH because JS-K is cleaved in the presence of GSH. Higher stability of JSK in plasma as compared to whole blood could be attributed to higher GSH in blood as compared to plasma (28, 29). The relative stabilization of JS-K with P123 micelles could be explained by shielding of JS-K in the micelle, thus preventing interaction with GSH.
A drug's interaction with serum proteins is an important PK/PD parameter that needs to be studied as this provides the basis for dosage regimens and other related PK analyses. The two major serum proteins are HSA and AGP. While it is generally observed that acidic drugs tend to bind with HSA and basic drugs tend to bind with AGP, this observation is not absolute (30). We studied interaction of free JS-K with HSA and AGP and P123/JS-K with HSA and AGP. We compared results obtained from equilibrium dialysis with the results obtained from spectrometric analysis. Equilibrium dialysis is an old and reliable technique but suffers from drawbacks such as requirement of high volumes, extensive time for equilibrium, volume shift and the need for extensive preliminary studies. Nevertheless, it is the most widely used method and is still used to analyze plasma protein binding of various drugs (19). We initially performed the equilibrium dialysis study for eight hours. For such a long time, P123/JS-K was relatively stable but free JS-K degradation was observed. In the P123/JS-K system, equilibrium was observed within two hours. Complete equilibrium was not obtained for free JS-K probably due to drug instability. Nevertheless, the two-hour point was selected for the equilibrium dialysis for both formulations. Using SigmaPlot, we analyzed the binding constant and possible number of binding sites using a built-in ligand binding equation. As free JS-K is prone to degradation we validated our results using spectrophotometric analysis. Furthermore, none of the binding analysis techniques provide complete information. Therefore, a combination of two different techniques can provide valuable datasets (19). With the use of fluorescence quenching, we were able to derive the Stern Volmer constant, binding constant and thermodynamic constants such as enthalpy, entropy and free energy. Fluorescence quenching of tryptophan upon binding of drug can yield useful information. This technique is fast and reliable, and requires less sample (25). In a complex system where the drug (JS-K) is incorporated inside a micelle core, protein binding can take place between JS-K and tryptophan residues of protein, P123/JS-K micelle and the tryptophan residues and finally between the unimers/micelles and the tryptophan residues. As observed from the results, the association constant (Ka) obtained from the two techniques is slightly different. Comparing equilibrium constants at room temperature, for P123/JS-K, Ka obtained from equilibrium dialysis and fluorescence quenching was close whereas a nearly ten times higher Ka was calculated for free JS-K when fluorescence quenching analysis was performed. This difference could be explained by considering the stability of free JS-K. While P123/JS-K could attain equilibrium, free JS-K might have degraded during the experiment. Although it could be argued that the Stern Volmer plot obtained has various data points that could be considered outliers, this could have been due to the lack of technique optimization. The results were promising and therefore a more optimized temperature controlled study was performed. When a temperature controlled study was performed with the fluorescence quenching technique, both formulations showed an increase in binding constant or binding strength with temperature although the increase observed for free JS-K was more marked than P123/JS-K (Figures 6 and 7). This could be explained by reduced binding property of Pluronic® polymers.
In order to be effective, the drug should be released from the polymer to produce its therapeutic effect. How early JS-K is released from the micelle core needs to be established. The thermodynamic parameters obtained from the fluorescence quenching study hold significance as well. The negative free energy possessed by both free JS-K and P123/JS-K binding with HSA at different temperatures indicates a spontaneous reaction that is thermodynamically favorable. Again, both free JS-K and P123/JS-K possess positive enthalpy and entropy but the values are about ten times higher with free JS-K interaction with serum protein. It is hard to speculate about the relevance of that observation as the specific heat capacity is not considered. However, according to studies carried out by Ross and Subramanian (31) the positive value of both enthalpy and entropy indicate hydrophobic interactions.
Protein binding analysis can also be extended to hypothesize JS-K stability and interaction with HSA. As protein binding is occurring, it could be hypothesized that JS-K stability can be enhanced with HSA interaction. A depot can be formed between JS-K and HSA. Some studies have shown interaction of NO residues (such as nitrosothiols) with albumin (32, 33). Also, the major metabolite of JS-K, S-glutathione-2,4-dinitrophenyl can also interact with albumin via the GSH moiety. Therefore, either JS-K as a whole or its metabolites can interact with albumin to form a depot of either whole JS-K or the metabolites of JS-K that could be instrumental in producing the cytotoxic effects of the drug.
The in vivo tumor regression study carried out confirms the antineoplastic activity of JS-K. It is interesting that both JS-K and P123/JS-K showed nearly equal tumor regression up to seven injections but P123/JS-K-treated mice showed maximum regression after eight treatments. One could speculate that the P123 formulation allowed JS-K accumulation in the tumor to a greater extent and that a critical threshold of antineoplastic activity was reached as a result. Another point is that the micelles prevented the drug from interacting with proteins and as a result allowed more drug to reach leukemic cells. Finally, the micelles probably enhanced penetration of the drug in the leukemic cells as we have previously demonstrated (34). It would have been informative to be able to carry the experiment beyond that time point. However, ethical guidelines precluded us from doing so.
5. Conclusions
Overall, our study demonstrated that JS-K formulated in Pluronic® P123 has a more favorable in vitro and in vivo profile. The polymeric micelle formulation stabilizes the drug and diminishes its interaction with plasma proteins. The binding is thermodynamically favorable and points towards hydrophobic interactions. We also conclude that P123/JS-K is likely more therapeutically effective than free JS-K.
Acknowledgements
This work was supported by grant RO1 CA129611 from the National Cancer Institute.
Abbreviations
- AML
Acute Myeloid Leukemia
- NO
Nitric Oxide
- GSH
Glutathione
- HSA
Human serum albumin
- AGP
alpha 1 acid glycoprotein
- PEO
poly(ethylene oxide)
- PPO
poly(propylene oxide)
- HLB
Hydrophilic-lipophilic balance
- CMC
Critical micelle concentration
- TEM
Transmission electron microscopy
- AUC
Area under the curve
- KI
Potassium iodide
- mM
milimolar
- μM
micromolar
- mL
milliliter
- Fo
Fluorescence in the absence of external quencher
- F
Quenched Fluorescence
- Q
Conc. of quencher/drug
- Ksv
Stern Volmer Constant
- τo
average lifetime of HSA molecule in absence of JS-K (quencher) or any other drug
- Kq
quenching rate constant
- dF
difference in the fluorescence in the absence and presence of JS-K or P123/JS-K at the concentration Q
- f
fraction of initial fluorescence accessible to quencher
- Ka
association binding constant
- dH
enthalpy change
- dS
entropy change
- T
temperature in Kelvin
- dG
free energy
- HPLC
High performance liquid chromatography
- UPLC
Ultra performance liquid chromatography
Footnotes
8. Disclosures
Paul Shami is Scientific Founder, Chief Medical Officer and Chairman of the Board of Directors of JSK Therapeutics Inc.
Bibliography
- 1.Facts 2013. The Leukemia and Lymphoma Society. 2013 [Google Scholar]
- 2.Emadi A, Baer MR. Acute Myeloid Leukemia in Adults. In: Greer JP, editor. Wintrobe's Clinical Hematology. Thirtheenth Edition. Lippincott Williams and Wilkins; 2014. pp. 1577–1615. [Google Scholar]
- 3.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological reviews. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 4.Burgaud JL, Riffaud JP, Del Soldato P. Nitric-oxide releasing molecules: a new class of drugs with several major indications. Curr Pharm Des. 2002;8(3):201–213. doi: 10.2174/1381612023396357. [DOI] [PubMed] [Google Scholar]
- 5.Hibbs JB, Jr., Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun. 1988;157(1):87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- 6.Leon L, Jeannin JF, Bettaieb A. Post-translational modifications induced by nitric oxide (NO): implication in cancer cells apoptosis. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2008;19(2):77–83. doi: 10.1016/j.niox.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Magrinat G, Mason SN, Shami PJ, Weinberg JB. Nitric oxide modulation of human leukemia cell differentiation and gene expression. Blood. 1992;80(8):1880–1884. [PubMed] [Google Scholar]
- 8.Shami PJ, Sauls DL, Weinberg JB. Schedule and concentration-dependent induction of apoptosis in leukemia cells by nitric oxide. Leukemia. 1998;12(9):1461–1466. doi: 10.1038/sj.leu.2401131. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Malavya S, Wang X, Saavedra JE, Keefer LK, Tokar E, Qu W, Waalkes MP, Shami PJ. Gene expression profiling for nitric oxide prodrug JS-K to kill HL-60 myeloid leukemia cells. Genomics. 2009;94(1):32–38. doi: 10.1016/j.ygeno.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. JSK, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther. 2003;2(4):409–417. [PubMed] [Google Scholar]
- 11.Shami PJ, Saavedra JE, Bonifant CL, Chu J, Udupi V, Malaviya S, Carr BI, Kar S, Wang M, Jia L, Ji X, Keefer LK. Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. J Med Chem. 2006;49(14):4356–4366. doi: 10.1021/jm060022h. [DOI] [PubMed] [Google Scholar]
- 12.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110(2):709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maciag AE, Chakrapani H, Saavedra JE, Morris NL, Holland RJ, Kosak KM, Shami PJ, Anderson LM, Keefer LK. The nitric oxide prodrug JS-K is effective against non-small-cell lung cancer cells in vitro and in vivo: involvement of reactive oxygen species. J Pharmacol Exp Ther. 2011;336(2):313–320. doi: 10.1124/jpet.110.174904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiziltepe T, Anderson KC, Kutok JL, Jia L, Boucher KM, Saavedra JE, Keefer LK, Shami PJ. JS-K has potent anti-angiogenic activity in vitro and inhibits tumour angiogenesis in a multiple myeloma model in vivo. J Pharm Pharmacol. 2010;62(1):145–151. doi: 10.1211/jpp.62.01.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simeone AM, McMurtry V, Nieves-Alicea R, Saavedra JE, Keefer LK, Johnson MM, Tari AM. TIMP-2 mediates the anti-invasive effects of the nitric oxidereleasing prodrug JS-K in breast cancer cells. Breast Cancer Res. 2008;10(3):R44. doi: 10.1186/bcr2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. Journal of controlled release : official journal of the Controlled Release Society. 2008;130(2):98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi S, Sun Yantao, Qiao Chunyu, Zhang Hanqi, Liu Chunming. Binding of several anti-tumor drugs to bovine serum albumin: Fluorescence study. Journal of Luminescence. 2009;129(5):541–547. [Google Scholar]
- 18.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Advanced drug delivery reviews. 2009;61(6):428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vuignier K, Schappler J, Veuthey JL, Carrupt PA, Martel S. Drug-protein binding: a critical review of analytical tools. Analytical and bioanalytical chemistry. 2010;398(1):53–66. doi: 10.1007/s00216-010-3737-1. [DOI] [PubMed] [Google Scholar]
- 20.Saavedra JE, Srinivasan A, Bonifant CL, Chu J, Shanklin AP, Flippen-Anderson JL, Rice WG, Turpin JA, Davies KM, Keefer LK. The secondary amine/nitric oxide complex ion R(2)N[N(O)NO](−) as nucleophile and leaving group in S9N)Ar reactions. J Org Chem. 2001;66(9):3090–3098. doi: 10.1021/jo0016529. [DOI] [PubMed] [Google Scholar]
- 21.Wei Z, Hao J, Yuan S, Li Y, Juan W, Sha X, Fang X. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. International journal of pharmaceutics. 2009;376(1-2):176–185. doi: 10.1016/j.ijpharm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 22.Noni Husain RAA, Warner Isiah M. Spectroscopic analysis of the binding of doxorubicin to human .alpha.-1 acid glycoprotein. The Journal of Physical Chemistry. 1993;97(41):10857–10861. [Google Scholar]
- 23.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. Journal of controlled release : official journal of the Controlled Release Society. 2002;82(2-3):189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Liu H, Xu M, Li Z, Xie G, Huang L, Zeng Z. Spectroscopic studies on the interaction between an anticancer drug ampelopsin and bovine serum albumin. Spectrochimica acta Part A, Molecular and biomolecular spectroscopy. 2012;87:251–257. doi: 10.1016/j.saa.2011.11.048. [DOI] [PubMed] [Google Scholar]
- 25.Kandagal PB, Ashoka S, Seetharamappa J, Shaikh SM, Jadegoud Y, Ijare OB. Study of the interaction of an anticancer drug with human and bovine serum albumin: spectroscopic approach. Journal of pharmaceutical and biomedical analysis. 2006;41(2):393–399. doi: 10.1016/j.jpba.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 26.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. Journal of controlled release : official journal of the Controlled Release Society. 2005;109(1-3):169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 27.Cho HJ, Yoon HY, Koo H, Ko SH, Shim JS, Lee JH, Kim K, Kwon IC, Kim DD. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic(R) for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. doi: 10.1016/j.biomaterials.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 28.Eve Unt CK, Vaher Ivi, Zilmer Mihkel. Red blood cell and whole blood glutathione redox status in endurance-trained men following a ski marathon. Journal of Sports Science and Medicine. 2008;7:344–349. [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. The Journal of nutrition. 2004;134(3):489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 30.Tesseromatis C, Alevizou A. The role of the protein-binding on the mode of drug action as well the interactions with other drugs. European journal of drug metabolism and pharmacokinetics. 2008;33(4):225–230. doi: 10.1007/BF03190876. [DOI] [PubMed] [Google Scholar]
- 31.Ross PD, Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry. 1981;20(11):3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- 32.Quinlan GJ, Martin GS, Evans TW. Albumin: biochemical properties and therapeutic potential. Hepatology. 2005;41(6):1211–1219. doi: 10.1002/hep.20720. [DOI] [PubMed] [Google Scholar]
- 33.Kashiba-Iwatsuki M, Miyamoto M, Inoue M. Effect of nitric oxide on the ligandbinding activity of albumin. Archives of biochemistry and biophysics. 1997;345(2):237–242. doi: 10.1006/abbi.1997.0258. [DOI] [PubMed] [Google Scholar]
- 34.Kaur I, Terrazas M, Kosak KM, Kern SE, Boucher KM, Shami PJ. Cellular distribution studies of the nitric oxide-generating antineoplastic prodrug O(2) -(2,4-dinitrophenyl)1-((4-ethoxycarbonyl)piperazin-1-yl)diazen-1-ium-1,2-diolate formulated in Pluronic P123 micelles. J Pharm Pharmacol. 2013;65(9):1329–1336. doi: 10.1111/jphp.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]